

Abstract
Liver kinase B 1 (LKB1 or STK11) and phosphatase and tensin homologue deleted on chromosome 10 (PTEN) are two tumor suppressors that regulate the mammalian target of rapamycin signaling pathway. Deletion studies show that loss of either Lkb1 (Lkb+/–) or Pten (PtenloxP/loxP; Alb‐Cre+) leads to liver injury and development of hepatocarcinoma. In this study, we investigated the crosstalk of LKB1 and PTEN loss during tumorigenesis and liver development. We show that haplo‐insufficiency of Lkb1 in the liver leads to advanced tumor development in Pten‐null mice (PtenloxP/loxP; LkbloxP/+; Alb‐Cre+). Our analysis shows that LKB1 and PTEN interact with each other in their regulation of fatty acid synthase as well as p21 expression. The combined loss of LKB1 and PTEN (PtenloxP/loxP; LkbloxP/loxP; Alb‐Cre+) also leads to the inability to form zonal structures in the liver. The lack of metabolic zonal structures is consistent with the inability of the livers to store glycogen as well as elevated plasma bilirubin and alanine aminotransferase, indicative of liver dysfunction. These structural and functional defects are associated with cytoplasm distribution of a canalicular membrane protein multidrug resistant protein 2, which is responsible for clearing bilirubin. This observed regulation of multidrug resistant protein 2 by LKB1 likely contributes to the lack of cellular polarity and the early lethality phenotype associated with the homozygous loss of Lkb1 alone or in combination with Pten. Finally, Pten deletion does not rescue the precocious ductal plate formation reported for Lkb1‐deleted livers. Conclusion: Our study dissected the functional and molecular crosstalk of PTEN and LKB1 and elucidated key molecular targets for such interactions. (Hepatology Communications 2017;1:153‐167)
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AKT
protein kinase B
- ALT
alanine aminotransferase
- AMPK
adenosine monophosphate‐activated protein kinase
- ATP
adenosine triphosphate
- BSEP
bile salt export pump
- FASn
fatty acid synthase
- GS
glutamine synthase
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- LKB1
liver kinase B 1
- MEF
mouse embryonic fibroblast
- MRP2
multidrug resistant protein 2
- mTOR
mammalian target of rapamycin
- P21
cyclin‐dependent kinase inhibitor 1A
- p‐ACC
p‐acetyl‐CoA carboxylase
- PAS
periodic acid‐Schiff
- PI3K
phosphatidylinositol‐3 kinase
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- qPCR
quantitative polymerase chain reaction
- shRNA
short hairpin RNA
- TG
triglyceride
- Thr172
phospho‐AMPK alpha‐1
- TICs
tumor initiating cells
- TSC2
tuberin
Introduction
Liver kinase B1 (LKB1) is a serine/threonine protein kinase. Mutations in LKB1 are linked to the development of Peutz‐Jeghers syndrome (PJS),1 an autosomal dominant disorder in which patients develop benign hamartomatous polyps in the gastrointestinal tract. In addition, patients with PJS also possess a significant risk of developing cancers in multiple tissues.2 LKB1 has been identified as a tumor suppressor gene for its inhibitory effect on cell proliferation and has been shown to phosphorylate at least 13 members of the adenosine monophosphate‐activated protein kinase (AMPK) subfamily, many of which play a fundamental role in metabolic regulation.3 Among the downstream targets, AMPK is a well‐characterized kinase that is activated by phosphorylation by LKB1 of phospho‐AMPK alpha‐1 (Thr172) within a structure known as the T‐loop on the α subunit of the heterotrimeric protein.4 This phosphorylation results in mammalian target of rapamycin (mTOR) suppression via tuberous sclerosis protein 2 or tuberin (TSC2) to inhibit cell growth and metabolism.4
The regulation of mTOR by the LKB1/AMPK signal functionally and mechanistically parallels that of the phosphoinositide 3‐kinase (PI3K)/protein kinase B (AKT) signal where AKT activates mTOR via TSC2.4 Similar to LKB1, phosphatase and tensin homologue deleted on chromosome 10 (PTEN), the negative regulator of PI3K/AKT, functions as a tumor suppressor in a plethora of cancer types. In patients, familial loss of PTEN and LKB1 both result in hamartoma syndromes.5 In mouse models, germline deletion of either Pten or Lkb1 results in embryonic lethality at similar gestational stages, suggesting functional overlaps of the two tumor suppressor genes.6, 7 This evidence suggests that the two tumor suppressors may be functionally redundant through their regulation of the TSC–mTOR signaling pathway. Recent studies have begun to explore the potential interaction of the PTEN‐regulated and LKB1‐regulated signals during carcinogenesis, and synergy of the two tumor suppressors has been reported for ovarian, endometrium, lung, and bladder cancer.8, 9, 10, 11 Furthermore, PTEN also directly interacts with LKB1 where LKB1 phosphorylates PTEN, and this interaction plays a role in the cellular localization of LKB1.12 In the liver, either haplo‐insufficiency of LKB1 (Lkb1+/–) or PTEN loss leads to hepatocellular carcinoma (HCC).13, 14 Reduced function of PTEN and LKB1 are also both independently associated with liver malignancies,15, 16 suggesting that these two tumor suppressors may play important roles in liver carcinogenesis. However, the relationship between PTEN and LKB1 in liver biology and pathology has not been studied due to the perinatal lethality of the LkbloxP/loxP;Alb‐Cre+(LKO) mice.17 This, with the recent contradictory oncogenic activity reported for LKB1 in HCC18 makes it particularly important to address the interactions of PTEN and LKB1 in the liver. Our study demonstrated crosstalk of LKB1 and PTEN signals, suggesting that the two pathways interact with each other in promoting HCC development and regulation of liver morphogenesis. In addition, we identified a novel regulation of the efflux transporter multidrug resistant protein 2 (MRP2) by LKB1.
Materials and Methods
ANIMALS
The PtenloxP/loxP; Alb‐Cre+ (PKO) mice13, 19, 20, 21 were crossed with Lkb1flox/+ mice22 to generate the Lkbloxp/+;PtenloxP/loxP;Alb‐Cre+ (LhetPKO) and Lkbloxp/loxp;PtenloxP/loxP;Alb‐Cre+ (LPKO) mice. Controls were Alb‐Cre– from the same cohort of mice. In addition, hepatocytes were established from the Akt1lox/lox; Alb‐Cre+ (AKO) and PtenloxP/loxP; Akt1lox/lox; Alb‐Cre+ (APKO) mice and used in this study. All mice were kept on a 24‐hour light/dark cycle‐controlled environment and allowed free access to food and water with standard housing conditions. Animals were used at various time points for tissue collections, including plasma and liver. Livers were perfused with cold phosphate‐buffered saline with diethyl pyrocarbonate water and collected in formalin for histology. Parts of the livers were flash frozen in liquid nitrogen and stored for protein, and RNA analysis. All mouse experiments were carried out following protocols approved by the University of Southern California Institutional Animal Care and Use Committee. Data from male mice were used in this study.
CELL LINES
Mouse hepatocyte cell lines were established from Pten‐control and Pten‐null mice.23, 24 The use of mouse embryonic fibroblast (MEF) cells has been reported before.25 AMPK plasmids and short hairpin RNA for Lkb1 (shLkb1) transfections were conducted with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions as described.24 We plated 1 × 105 cells in each well of six‐well plates 24 hours before transfection. Then, 4 μg plasmid DNA and 10 μL lipofectamine were delivered into cells. The combination of 100 pmol short hairpin (sh)RNA and 5 μL lipofectamine was used for shRNA transfection. Wortmannin, LY294002, and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) (all from Cell Signaling Technology) were used to treat the cells in vitro.
PROTEIN GEL ELECTROPHORESIS
Liver tissues were homogenized in ice‐cold T‐PER Tissue Protein Extraction Reagent (Thermo Scientific) with the addition of a protease inhibitor cocktail (Roche) and phosphatase inhibitor (Roche). Standard western blotting was conducted as described25 using antibodies against p‐AKT(S473), p‐S6, PTEN, cyclin D, p‐AMPKα(T172), p‐AMPK (S485), p‐mTOR, p‐acetyl‐CoA carboxylase (p‐ACC), and cyclin‐dependent kinase inhibitor 1A (p21) from Cell Signaling Technology; LKB1 and fatty acid synthase from Upstate; MRP2 from Abcam; and actin from Sigma.
QUANTITATIVE POLYMERASE CHAIN REACTION ANALYSIS
Total RNA was extracted from mouse tissues and cell lines with Trizol reagent (Life Technologies) according to the manufacturer's protocol. Reverse transcription was performed from 2 μg of total RNA using M‐MLV Reverse Transcriptase (Promega) and random hexamer and deoxythymidine oligomer18 as primers. Quantitative polymerase chain reactions (qPCRs) were run using KAPA SYBR FAST qPCR kits, and the StepOnePlus Real‐Time PCR System was used to perform the amplification process. Primer sequences used here have been reported.13, 26, 27 Relative expressions of genes were normalized with 18S ribosomal RNA.
IMMUNOHISTOCHEMISTRY AND IMMUNOFLUORESCENCE
Mouse livers were fixed in 10% formalin for 12 hours and embedded in paraffin and processed to obtain 5‐μm‐thick sections. Immunostaining and immunofluorescence procedures were performed as reported.13, 20 For visualizing glycogen storage, liver sections were stained for periodic acid‐Shiff (PAS) stain as reported.27 All images were generated and analyzed using a ZEISS microscope with AxioVision software. Antibodies used for staining tissue sections were: Pan‐Keratin and Hep Par from Dako, glutamine synthase (GS) from Novus Biologicals (NB110‐41404), and MRP2 from Atlas.
BIOINFORMATAICS ANALYSIS
Total RNA from control and PKO livers (n = 5) were sent to the Penn State Hershey University Microarray Functional Genomics Core facility for mouse messenger RNA microarray analysis using the Illumina single‐color array platform. Data generated have been submitted to the Gene Expression Omnibus database (GSE70501). The data set was analyzed for a Gene Set Enrichment Score using online tools available from the Gene Set Enrichment Analysis (GSEA) website with the Notch signal gene set defined in the GSEA database.
PLASMA GLUCOSE, BILIRUBIN, AND ALANINE AMINOTRANSFERASE
Bilirubin levels were measured from plasma using the Bilirubin Assay kits (MAK126; Sigma‐Aldrich). Total and conjugated bilirubin were measured according to the manufacturer's instructions. Alanine aminotransferase (ALT) levels were measured from plasma using the ALT (GPT) kit from CIA Medical and according to the manufacturer's instructions.
STATISTICS
One‐way analysis of variance was performed by PRISM software, and comparative analyses of each group were done with Fisher's exact test and the Student t test. A P value of <0.05 was considered statistically significant.
Results
PTEN/PI3K/AKT SIGNAL REGULATES AMPK ACTIVITY TO CONTROL LIPOGENIC GENE EXPRESSION
To explore the interactions between the PTEN and AMPK signaling pathways, we isolated hepatocytes from the liver Pten‐deletion mice (Ptenl /l;Alb‐Cre+, PKO) and the respective controls (Ptenl/l;Alb‐Cre–, Con).21, 24 In these cells, phosphorylation of AMPK at T172, which indicates its activity, is reduced by more than 10‐fold in the PKO versus controls (Fig. 1A‐C). This AMPK(T172) phosphorylation is dependent on PI3K/AKT activity because inhibiting PI3K with either LY294002 or Wortmannin dose‐dependently induced AMPK phosphorylation (Fig. 1A,B). On the other hand, inducing AMPK phosphorylation by AICAR treatment had no effects on the phosphorylation of AKT or its downstream targets (Fig. 1C). To confirm this observation, we evaluated the phosphorylation of AMPK with hepatocytes isolated from Akt1‐deleted mice. Similar to previous publications,28 Akt1 deletion alone (AKO) or together with Pten (APKO) resulted in the inhibition of AMPK phosphorylation at S485 (Fig. 1D). The deletion of Akt1, however, did not result in the rescue of AMPK activity, as indicated by the low level of AMPK(T172) phosphorylation in the APKO samples. Thus, additional signaling, independent of AKT1, likely contributed to PTEN‐regulated AMPK phosphorylation.
Figure 1.
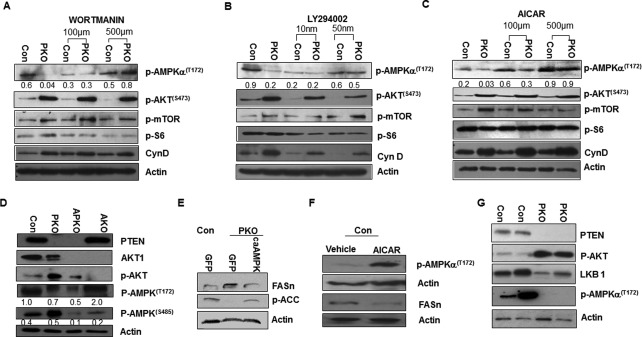
Crosstalk of LKB1‐regulated and PTEN‐regulated signals controls hepatic lipogenesis. Inhibition of PI3K signal by (A) Wortmannin and (B) LY294002 in Pten‐null hepatocytes restores down‐regulation of AMPK phosphorylation on T172 and its activation induced by PTEN loss. (C) Activation of AMPK by AICAR treatment does not alter AKT signaling. (D) The down‐regulation of AMPK due to PTEN loss is not mediated by AKT‐regulated phosphorylation of AMPK on S485. We used Pten‐null hepatocytes that also carry deletion of AKT1 (APKO) to address the role of AKT in the phosphorylation (T172) and activation of AMPK. Hepatocytes carrying the Akt1 deletion (AKO) are also included as another control. Deletion of AKT1 led to down‐regulation of AMPK phosphorylation at S485 but did not alter its phosphorylation on T172, which indicates AMPK activity. (E) Introduction of caAMPK leads to down‐regulation of FASn and ACC indicated by its phosphorylation. (F) Activation of AMPK by AICAR treatment down‐regulates FASn. (G) LKB1 down‐regulation by PTEN loss and AKT activation likely resulted in the inhibition of AMPK phosphorylation. Numbers below the gel image are densitometry quantification of the image shown. Abbreviations: ACC, acetyl‐CoA carboxylase; caAMPK, constitutively active AMPK; GFP, green fluorescent protein.
We have shown that Pten deletion in hepatocytes leads to AKT‐dependent, increased, de novo lipogenesis through the induction of fatty acid synthase (FASn).13, 26, 27 The inhibition of AMPK phosphorylation observed here appears to be necessary to allow lipogenesis to occur in the Pten‐null hepatocytes because induction of AMPK phosphorylation with either AICAR or the introduction of constitutively active AMPK reduced the levels of FASn (Fig. 1E,F). In addition, both LKB1 levels and AMPK phosphorylation were down‐regulated when Pten is deleted and AKT is induced in the Pten‐null livers (Fig. 1G), suggesting that inhibition of AMPK phosphorylation may have resulted from down‐regulation of LKB1. This initial analysis suggests that LKB1‐regulated and PTEN‐regulated signals have the potential to interact with each other and that this interaction may regulate liver pathogenesis.
HAPLO‐INSUFFICIENCY OF Lkb1 SYNERGIZES WITH PTEN LOSS TO PROMOTE LIVER CANCER DEVELOPMENT
To confirm the functional interaction of LKB1‐regulated and PTEN‐regulated signals in the liver, we crossed the PKO mice with LkbloxP/loxP to generate the LkbloxP/loxP;PtenloxP/loxP;Alb‐Cre+(LPKO) mice. As the LKO (LkbloxP/loxP;Alb‐Cre+) and LPKO mice are early postnatal lethal at around 3 weeks,17 we examined Lkblox/+;PtenloxP/loxP;Alb‐Cre+(LhetPKO) mice for lipid accumulation and tumor development, two prominent phenotypes observed when Pten is deleted.13, 20, 23, 26, 27 The LhetPKO, PKO, and control mice displayed similar body weights from 3‐12 months of age, whereas liver weight is higher when PTEN is lost (Supporting Fig. S1). The PKO mice develop nonalcoholic fatty liver disease up to 7‐8 months of age followed by spontaneous tumor development that reaches 100% by 12 months of age.13, 20, 23, 26, 27 To address the effects of haplo‐insufficiency of LKB1 on Pten deletion‐induced tumor development, we analyzed the LhetPKO mice and found that 20% of the mice had already developed tumors between 3‐6 months compared to the 7‐8 months onset that was reported previously for the PKO mice (Table 1). Significantly more hyperplasia at the periductal region was observed in the LhetPKO versus PKO livers at 3‐6 months of age (Fig. 2A). This phenotype is collaborated with the induced expression of alpha‐fetoprotein 2, epithelial cell adhesion molecule, and cytokeratin 19, particularly at 6 months of age, suggesting that the hyperplasia may come from liver tumor initiating cells (TICs) (Fig. 2B), similar to what we reported for tumors developed in the PKO mice.13, 29 By 9 months of age, all LhetPKO mice bore tumors in the liver (Fig. 2C). Both cholangiocarcinoma and HCC are observed in the LhetPKO (Fig. 2D), similar to what we reported for the PKO tumors at 12 months of age.13 The tumors developed in the LhetPKO livers are also dual‐lineage positive for both keratin and Hep Par‐1, suggesting the involvement of a TIC population with progenitor cell characteristics.13, 29
Table 1.
TUMOR SPECTRUM IN LhetPKO AND PKO MICE
| Genotype | 3‐6M | 6‐9M |
|---|---|---|
| LkbL/+; PtenL/L; Alb‐Cre‐ (Con) | 0/14 | 0/13 |
| Lkb+/+; PtenL/L; Alb‐Cre+ (PKO) | 0/13 | 4/10 |
| LkbL/+; PtenL/L; Alb‐Cre‐ (LhetCon) | 3/16 | 11/13 |
Figure 2.
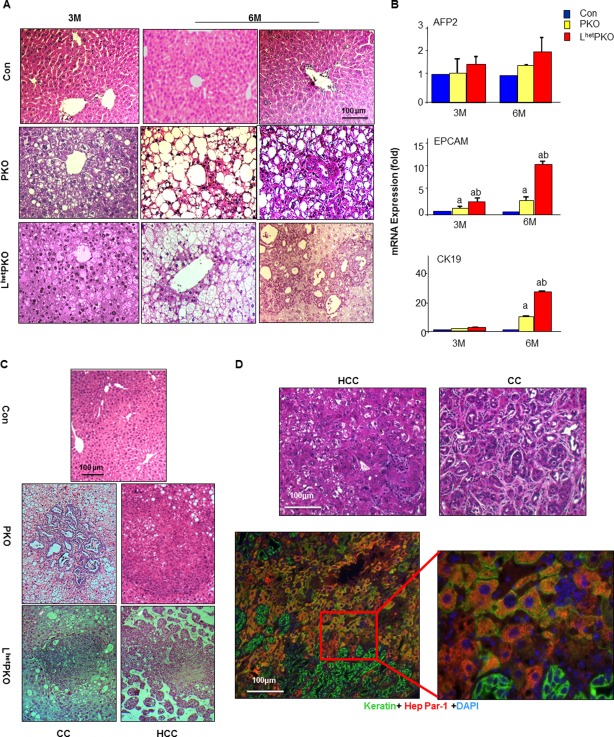
Heterozygous deletion of Lkb1 in the liver advances tumor development observed with Pten deletion. (A) H&E‐stained liver sections from 3‐month and 6‐month‐old mice of the respective genotypes. CON, Alb‐Cre‐ mice; PKO, PtenloxP/loxP; Alb‐Cre+; LhetPKO, Lkb1loxP/+; PtenloxP/loxP; Alb‐Cre+. (B) Expression of progenitor cell markers in liver lysates isolated from the respective mice. (C) H&E‐stained liver sections from 9‐month‐old mice of the respective genotypes showing development of CC and HCC. (D) Tumors developed in the LhetPKO livers. Top, H&E‐stained liver sections showing HCC and CC phenotype; bottom, costaining of keratin (green) and Hep Par‐1 (red) indicates the presence of bipotent progenitor cells in the tumors developed in the LhetPKO livers. Scale bar, 100 μm. Abbreviations: AFP2, alpha‐fetoprotein 2; CC, cholangiocarcinoma; CK19, cytokeratin 19; DAPI, 4',6‐diamidino‐2‐phenylindole; EPCAM, epithelial cell adhesion molecule; H&E, hematoxylin and eosin; mRNA, messenger RNA.
In the PKO livers, steatosis and lipotoxicity‐induced injuries led to death of hepatocytes that then established a niche for TIC growth and promoted tumorigenesis.13, 26, 27 We hypothesized that inhibition of AMPK phosphorylation resulting from LKB1 loss may have increased lipid accumulation in the liver, resulting in an earlier and more severe lipotoxic injury and therefore an earlier onset of tumors. Surprisingly, we failed to observe increased liver triglyceride (TG). Liver TG is reduced in all ages in the LhetPKO versus PKO livers, concurrent with a similar reduction of plasma ALT up to 9 months of age (Supporting Fig. S2A,C). In addition to hepatic TG content, plasma TG and hepatic and plasma cholesterol levels are all improved in the mice compared with the PKO mice (Supporting Fig. S2A,B). This reduction may have accounted for the smaller liver weight in LhetPKO versus PKO mice up to 9 months of age (Supporting Fig. S1) and suggests that signals other than increasing metabolic lipotoxicity likely led to the earlier tumor onset in the LhetPKO versus PKO mice. Supporting this argument, plasma insulin levels did not differ in either PKO or LhetPKO groups (Supporting Fig. S2D). Fasting glucose levels were reduced in PKO mice due to the improved insulin sensitivity as reported before.27 Loss of one allele of Lkb1 did not alter these reduced glucose levels (Supporting Fig. S2D).
LKB1 has been identified as a tumor suppressor.1 In the liver, we found that expression of Lkb1 is lower in HCC samples versus normal tissues based on an analysis of available Oncomine data (Supporting Fig. S3). To explore whether loss of LKB1 may represent a second genetic event that allowed for early onset for TIC transformation and tumor development, we explored the regulation of p21 by PTEN and LKB1. LKB1 is recruited to the promoter of p21 and participates in the p53‐regulated transcriptional activation of p21.30 PTEN regulates p21 through direct phosphorylation by AKT as well as transcriptional regulation through glycogen synthase kinase 3β and mTOR.31 Consistent with synergistic effects of PTEN and LKB1 on p21 transcription, deletion of both Pten and Lkb1 led to a significant inhibition of p21 (Fig. 3A), whereas no significant change was observed in MEFs lacking either PTEN or LKB1 alone. In vivo experiments showed that homozygous deletion of Lkb1 leads to smaller body size in both LPKO and LKO mice (Fig. 3B), and these mice die at around 3 weeks of age. In these mice, synergistic inhibition of AMPK phosphorylation is observed when both LKB1 and PTEN are simultaneously lost in the liver (Fig. 3C), similar to the in vitro observation (Fig. 1). Here, incomplete loss of PTEN32 in 3‐week‐old mice led to moderate down‐regulation of AMPK(T172) phosphorylation in the PKO group. Loss of both alleles of Lkb1 (LKO) resulted in a more significant reduction of AMPK(T172) phosphorylation, which is further down‐regulated when Pten is deleted simultaneously (LPKO) (Fig. 3B). In addition, deletion of both Pten and Lkb1 also induced more robust AKT phosphorylation than PTEN loss alone, suggesting that LKB1 activity also regulates PI3K/AKT signaling in vivo. Consistent with what we observed in MEFs (Fig. 3A), p21 levels were moderately inhibited in either LKO or PKO livers but further reduced in LPKO livers (Fig. 3C). Together, our data supported a synergistic effect of LKB1 and PTEN in vitro and in vivo in promoting liver tumorigenesis.
Figure 3.
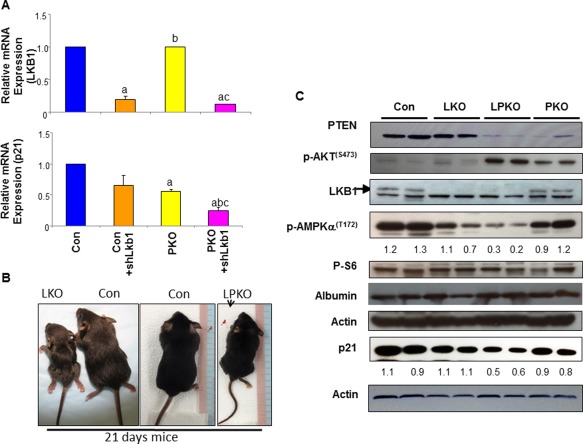
LKB1 and PTEN synergistically regulate p21 expression. (A) Introduction of shLkb1 to knockdown LKB1 leads to synergistic down‐regulation of p21 expression with PTEN loss. CON, wild‐type MEFs; PKO, Pten‐null MEFs; n = 3; P < 0.05. a, different from CON; b, different from CON+shLkb1; c, different from PKO. Data expressed as mean ± SEM. (B) Gross phenotype of 3‐week‐old mice lacking LKB1 in control and Pten‐null livers. CON, Alb‐Cre–; LKO, Lkb1loxP/loxP; Alb‐Cre+; LPKO, Lkb1loxP/loxP; PtenloxP/loxP; Alb‐Cre+. PKO, PtenloxP/loxP; Alb‐Cre+. (C) Synergistic regulation of p21 by PTEN and LKB1 loss in liver lysates isolated from the respective mouse models. Numbers below the gel image are densitometry quantification of the image shown. Abbreviation: mRNA, messenger RNA.
INTERACTION OF LKB1 AND PTEN DURING LIVER MORPHOGENESIS
Recently, it was reported that LKB1 loss leads to dysregulation of Notch and results in disorganized ductal structures.33 Ducts in the LKO mice were reported to be primitive ducts that lack symmetry with no frank disruption of parenchymal hepatocyte organization.17, 33 We analyzed the Notch signaling profiles in the Pten‐null livers (GSE70501) using GSEA. We found that the Gene Set Enrichment Score for Notch signaling is significantly higher in the Pten‐null livers and confirmed this with qPCR analysis of selective genes in the Notch signaling pathway (Fig. 4A,B). We therefore analyzed the ductal plate of the 3‐week‐old LPKO mice. While the ducts in the 3‐week‐old Con and PKO livers displayed completed ductal morphology, ducts in the LPKO livers were morphologically similar to that of the LKO mice (Fig. 4C). Whether this function of LKB on maintaining ductal integrity plays a role in the advanced tumorigenesis observed with LhetPKO mice is not clear.
Figure 4.
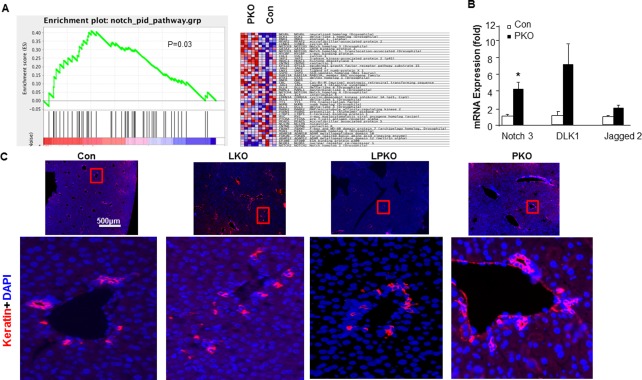
Effect of LKB1 and PTEN loss on ductal plate formation in the liver. (A) Gene enrichment analysis shows that expression of genes involved in the Notch signaling pathway is enriched in the Pten‐null livers versus controls. (B) qPCR analysis of selective genes in the Notch signaling pathway; n = 3; P < 0.05. (C) Keratin staining (red) showing immature ductal morphology of livers from the 3‐week‐old LKO and LPKO mice compared to the CON and PKO livers; blue, DAPI. Scale bar, 500 μm. Abbreviations: DAPI, 4',6‐diamidino‐2‐phenylindole; mRNA, messenger RNA.
In addition to the structural changes observed for the ductal hyperplasia, we noticed that the morphological development for the liver lobular structure is more distorted when LKB1 and PTEN are simultaneously lost. The PKO livers displayed normal lobular structures arranged with plates of hepatocytes radiating outward from a central vein. LKO livers also displayed similar but denser cellular structures. Cells in the LPKO livers were not arranged in cord structures like those in the other three genotype groups (Fig. 5A). Staining of GS confirmed this cooperative effect of PTEN and LKB1 on the loss of organization of liver structures (Fig. 5B). While PTEN loss alone has no significant effect on central venous cells expressing GS, LKB1 loss led to diffusion of these cells without proper formation of the central vein structure but no significant GS down‐regulation (Fig. 5B). Cells expressing GS were completely lost in livers lacking both LKB1 and PTEN. Staining for expression of cytochrome P450 2E1 also confirmed that LKB1 is necessary for maintaining the zonal structure of the liver (data not shown). These results suggest that PTEN and LKB1 functionally crosstalk in the regulation of GS expression and formation of the central venous zone in the liver.
Figure 5.
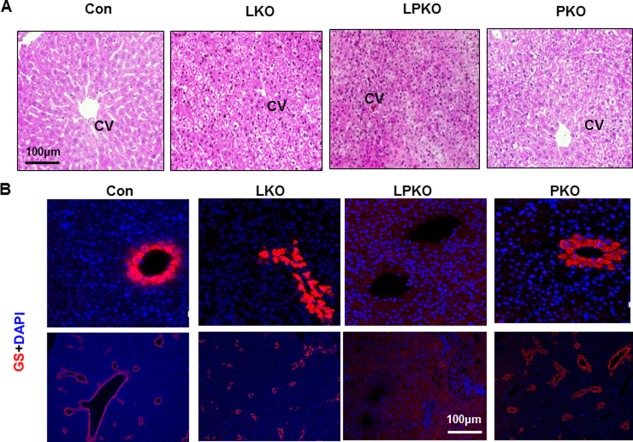
Effect of LKB1 and PTEN loss on liver zonation. (A) H&E staining of liver sections from 3‐week‐old mice shows lack of structure in LPKO livers. (B) Staining with GS (red) shows that central venous cells with GS staining are not formed in the LPKO livers. The GS‐positive cells are also scattered in the LKO livers instead of forming the central vein. Scale bar, 100 μm. Abbreviations: CV, central vein; DAPI, 4',6‐diamidino‐2‐phenylindole; H&E, hematoxylin and eosin.
LOSS OF PTEN CANNOT RESCUE THE EARLY LETHALITY OF LKO MICE
Despite the apparently normal parenchymal structure, LKO mice die soon after birth at 3 weeks of age with a postulated inability to clear bile salts and bilirubin.17 The mislocalization of the bile salt export pump (BSEP or ABCB11), a bile canalicular efflux transporter, is thought to result in the inability to clear bile salts. However, this lack of functional BSEP is unlikely to result in the lethal phenotype because the loss of BSEP is compensated by other bile salt species that are found in mouse.34 The hyperbilirubinemia in the LKO mice has not been characterized and may contribute to the early lethality phenotype. To explore the cause for this hyperbilirubinemia and the potential interaction of the PTEN and LKB1 pathways in the development of this phenotype, we determined the expression of the canalicular efflux transporter for conjugated bilirubin, MRP2. Our data indicated that levels of MRP2 were reduced in both LKO and LPKO livers (Fig. 6A,B). In addition to the lower protein levels, the typical canalicular localization of MRP2 was also lost in the LKO and LPKO livers, and MRP2 appeared to be diffusely distributed throughout the cell (Fig. 6B). The lowered expression level and mislocalization of MRP2 in the hepatocytes of LKO and LPKO mice correlated with the increases in plasma bilirubin levels (Fig. 6C). An elevated plasma bilirubin level was observed in both LKO and LPKO mice at 3 weeks of age, whereas its level was only moderately affected in the PKO mice (Fig. 6A,C). The combined loss of PTEN with LKB1 also led to elevated plasma ALT levels (Fig. 6C) and the damage likely due to hyperbilirubinemia.
Figure 6.
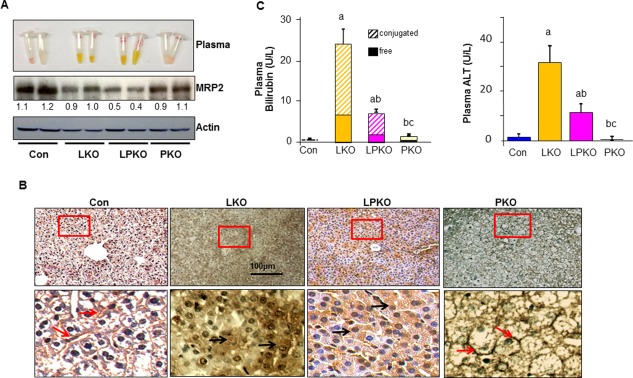
Effect of LKB1 and PTEN loss on liver function. (A) Expression of MRP2, a multidrug transporter responsible for clearance of bilirubin, is down‐regulated in LKO and LPKO livers. Numbers below the gel image are densitometry quantification of the image shown. (B) MRP2 is redistributed from the apical membrane (arrows) in the CON and PKO livers to a diffused cytoplasm distribution in the LKO and LPKO livers. Scale bar, 100 μm. Red square, areas magnified in the bottom panel. (C) Liver function index. Left, plasma bilirubin levels are also increased in LKO and LPKO livers at 3 weeks of age. Right, liver toxicity index plasma ALT is increased in LKO and LPKO livers, indicating liver injury; n = 3‐5; P < 0.05. a, different from CON; b, different from LKO; c, different from PKO. Data expressed as mean ± SEM.
While the levels of bilirubin and ALT were reduced in the LPKO mice compared with LKO mice (Fig. 6C), these reductions had no significant effects on the survival of the animals. LPKO mice died at 3 weeks after birth with a low liver and body weight that were about a third of the controls and PKO mice (Fig. 7A‐C). In addition to high bilirubin and bile salt buildup in the LKO and LPKO livers, the inability to store glycogen (Fig. 7D), a primary function of the liver, also likely contributed to the smaller body weight and early lethality phenotype.
Figure 7.
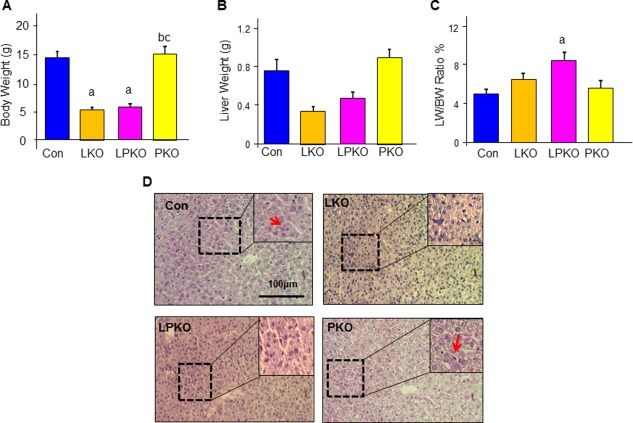
The effects of LKB1 and PTEN loss on liver metabolism. (A‐C) Body and liver weight of the LKO and LPKO mice are significantly reduced compared to the CON and PKO mice; n = 3‐5; P < 0.05. a, different from CON; b, different from LKO; c. different from PKO. Data expressed as mean ± SEM. (D) PAS staining (pink) indicate the LKO and LPKO livers are unable to store glycogen. Scale bar, 100 μm. Dashed box, areas magnified in the inset.
Discussion
The PI3K/AKT/mTORC1 pathway plays a key role in promoting cell survival and proliferation.35 Downstream of PI3K activation, phosphorylation and activation of AKT leads to activation of mTOR to regulate a number of cellular processes. This signaling pathway, however, is highly regulated by negative feedback, making targeting this pathway difficult for anticancer therapy, and highlights the need for further understanding of the fine‐tuning of this signaling pathway. PTEN and LKB1 are two negative regulators that lead to mTOR inhibition through different signaling pathways. While PTEN inhibits the activation of AKT and thus mTOR,35 LKB1 phosphorylates AMPK, which activates TSC to inhibit mTOR signaling.36 We demonstrated here that in addition to activating mTOR, PI3K activation due to PTEN loss also inhibits phosphorylation of AMPK, thus blocking the inhibitory signal from AMPK to mTOR and allowing full activation of mTOR. Inhibition of AKT signaling was shown previously to induce AMPK activity.28, 37 This function has been attributed to the role of AKT in regulating cellular metabolism and the AMP/adenosine triphosphate (ATP) ratio,28, 37 and phosphorylation of S485 on AMPK has been identified to be associated with this effect of AKT.28 Our data show that the PTEN‐regulated AMPK phosphorylation is independent of AKT‐regulated phosphorylation on S485. While S485 phosphorylation is fully inhibited by the loss of AKT1, it was unable to rescue the reduced phosphorylation (at T172) and activity of AMPK. In a yeast two‐hybrid screening, PTEN was identified as an LKB1‐interacting protein, and PTEN is phosphorylated by LKB1.38 Although the significance of this discovery and its impact on the PI3K/AKT signaling pathway remains to be elucidated, it may play a role in the lack of induction of AMPK activation when AKT and PTEN are both lost.
In addition, AMPK activation in cardiomyocytes leads to insulin‐independent phosphorylation and activation of the insulin receptor substrate and the subsequent activation of AKT.39 Such a relationship, however, was not observed in the hepatocytes under our cultured conditions. In hepatocytes lacking AKT1, we showed that AKT activity (and possibly phosphorylation of S485) can prevent the phosphorylation of T172 on AMPK, as reported.28 However, this effect can be overcome by other signals regulated by PTEN.
Like Pten, conventional deletion of Lkb1 is embryonic lethal, preventing tumor studies in these mice.14 With few exceptions, tissue‐specific deletion of Pten leads to tumor development in the respective tissues.25, 40, 41 In the liver, Pten loss leads to steatosis and cancer development.13, 19, 21, 26 Interestingly, heterozygous Lkb1 mice develop HCC in addition to the expected gastrointestinal polyps characteristic of PJS patients,14 and adult onset deletion of Lkb1 in the liver results in steatosis.22 The phenotypic similarities in mouse studies and the overlap of symptoms associated with germline Pten and Lkb1 loss in human patients5 led to the current study to investigate the crosstalk between Pten and Lkb1 during tumorigenesis. Our data demonstrated an advanced onset of HCC with haplo‐insufficiency of Lkb1 when Pten is deleted, while heterozygous loss of LKB1 alone has no obvious phenotype. Since adult onset deletion of Lkb1 also results in steatosis that cannot be ameliorated with treatment of metformin,22 we had hypothesized that more severe steatosis due to LKB1 loss likely drives the synergy between the two tumor suppressors. However, our data indicated that LKB1 loss did not further exacerbate the lipid burden or injury of the liver. This observation suggests that while the PI3K signal regulates AMPK activation and this crosstalk controls the lipid metabolic pathway, other signals are involved in vivo during tumorigenesis. Our analysis demonstrated a synergistic regulation of p21 by LKB1‐regulated and PTEN‐regulated signals. p21 CIP/WAF1 is a cell cycle inhibitor protein that regulates entry to the cell cycle. A number of mitogenic proteins regulate p21 at both transcriptional and translational levels. LKB1 was reported to be recruited to the promoter of p21 by p53 to control its expression42 and also regulates p21 by phosphorylating p21 protein‐activated kinase.43 p21 is recognized as a direct substrate for AKT and is also regulated by other signals downstream of AKT.35, 44 Our analysis suggests that these signals converge onto p21 to control liver carcinogenesis. Similar synergy between PTEN and LKB1 has been reported for other tumors. In Pten+/– mice where 50% of the mice develop tumors at 10 months of age, hypomorphic mutation of Lkb1 to reduces Lkb1 expression leading to a significant increase (80%) in mice developing tumors.38 Combined loss of LKB1 and PTEN leads to high‐grade papillary serous carcinoma in the ovary and early onset of bladder tumors, whereas neither Pten nor Lkb1 deletion alone results in tumorigenesis.9, 11 In the lung, this synergy is associated with the infiltration of polymorphonuclear cells.8 Whether the synergy of PTEN and LKB1 on p21 regulation also contributed to the development of these other tumors remains to be studied.
In addition to their synergy during tumorigenesis, our study also demonstrated an in vivo interaction of LKB1 and PTEN loss on morphological development of the liver. Zonation is a unique structure of the liver where molecules with gradient carry out multifaceted metabolic functions to achieve maximal efficiency. GS, which marks the central venous zone 2 and 3 of the liver, is completely lost when Lkb1 and Pten are simultaneously deleted. These results strongly suggest that PTEN and LKB1 are both pivotal and necessary when acting together in regulation of GS expression. Previous work found that the gradient of GS correlates with the trichloroacetic acid cycle.45 Within the GS positive zone, GS expression is positively correlated with the expression of genes involved in the trichloroacetic acid cycle and particularly that of complex V genes. Being the kinase that regulates AMPK, LKB1 is intimately involved with energy metabolism.36 In response to increased energy demand or a high AMP/ATP ratio, phosphorylation of AMPK by LKB1 switches on the catabolic pathways to generate ATP. This mechanism is targeted by the most prescribed diabetes medicine metformin to improve overall insulin sensitivity in patients.22 PTEN was shown to regulate both mitochondrial biogenesis and function in the liver. Through PI3K/AKT, PTEN regulates the orphan nuclear receptor estrogen receptor‐related receptor α and ATP synthase, a complex V enzyme.21, 46 Whether and how LKB1 and PTEN regulate GS expression through energy metabolic changes remain to be elucidated.
Loss of LKB1 was reported to result in precocious development of the ductal plate during embryo development.33 This abnormality is thought to be an effect of crosstalk between LKB1 and the Notch signaling pathway where down‐regulation of LKB results in inhibition of Notch‐regulated gene transcription. Our GSEA demonstrated that the Notch signaling pathway genes are differentially enriched when PTEN is lost in the liver, suggesting that PTEN and LKB1 may converge onto Notch signaling to regulate ductal plate formation. In mammary tissues and lung, the Notch signal is critically involved in branching morphogenesis during development.47, 48 Tumor development in these tissues is associated with the activation of this developmental pathway, suggesting an active role of Notch signaling in tumorigenesis, particularly in tissues primarily composed of topical epitheliums.
How developmental defects may contribute to liver carcinogenesis is not known. However, a role of Notch in asymmetric cell division may play a role in its function in both morphogenesis and tumor development. In Drosophila, Numb, an inhibitor of Notch, plays a key role in determining cellular polarity.49 This effect of Numb affects cell fate determination as well as maintenance of the stem cell compartment. The Drosophila and Caenorhabditis elegans homologue for LKB1 is PAR‐4, a protein necessary for establishing embryonal polarity shown by genetic studies.50, 51 The observation that LKB1 loss leads to the redistribution of MRP2 from canalicular plasma membrane to cytoplasm suggests that the regulation of cellular polarity by LKB1, possibly through Notch, likely plays a major role in hepatocyte maturation and development of the liver. MRP2 is an ATP‐binding cassette transporter located on the apical membrane of the hepatocytes and other tubular epitheliums. In the liver, MRP2 is responsible for transporting bilirubin as well as drugs the liver encounters. In breast and ovarian cancer cells, MRP2 localization to the nuclear envelope has been associated with a poor clinical outcome.52, 53 Whether such a correlation occurs in HCC remains to be studied.
In summary, we show here that PTEN and LKB1 are both pivotal regulators of the homeostasis of liver structure and function. Moreover, we also found that PTEN and LKB1 synergistically interact to regulate liver morphogenesis and promote tumorigenesis. Additionally, our study provides potential targets where these two signals may converge, providing a molecular understanding for how these two important tumor suppressors may collaborate in their function.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1027/suppinfo
Supporting Information Slide 2
Supporting Information Slide 6
Supporting Information Slide 7
Potential conflict of interest: Nothing to report.
Supported by the National Institutes of Health grants R01CA154986‐01 (B.L.S.), University of Southern California (USC) School of Pharmacy Bensussen Innovation Challenge Pilot Award (C.T.O.), National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK084241‐01 (B.L.S), USC Center for Liver Disease grant P30DK48522 (B.L.S.), International Postdoctoral Exchange Fellowship Program issued by The Office of Chinese Postdoctoral Council, sponsor number 20130012 (C.J).
REFERENCES
- 1. Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz‐Jeghers syndrome. Nature 1998;391:184‐187. [DOI] [PubMed] [Google Scholar]
- 2. Lim W, Olschwang S, Keller JJ, Westerman AM, Menko FH, Boardman LA, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004;126:1788‐1794. [DOI] [PubMed] [Google Scholar]
- 3. Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR‐1. EMBO J 2004;23:833‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz‐Jeghers syndrome. Genes Dev 2004;18:1533‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stratakis CA, Kirschner LS, Taymans SE, Tomlinson IP, Marsh DJ, Torpy DJ, et al. Carney complex, Peutz‐Jeghers syndrome, Cowden disease, and Bannayan‐Zonana syndrome share cutaneous and endocrine manifestations, but not genetic loci. J Clin Endocrinol Metab 1998;83:2972‐2976. [DOI] [PubMed] [Google Scholar]
- 6. Di Cristofano A, Pesce B, Cordon‐Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet 1998;19:348‐355. [DOI] [PubMed] [Google Scholar]
- 7. Ylikorkala A, Rossi DJ, Korsisaari N, Luukko K, Alitalo K, Henkemeyer M, et al. Vascular abnormalities and deregulation of VEGF in Lkb1‐deficient mice. Science 2001;293:1323‐1326. [DOI] [PubMed] [Google Scholar]
- 8. Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD‐L1 expression. Cancer Cell 2014;25:590‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanwar PS, Mohapatra G, Chiang S, Engler DA, Zhang L, Kaneko‐Tarui T, et al. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 2014;35:546‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanwar PS, Kaneko‐Tarui T, Zhang L, Tanaka Y, Crum CP, Teixeira JM. Stromal liver kinase B1 [STK11] signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian Target of Rapamycin Complex 1. PLoS Genet 2012;8:e1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shorning BY, Griffiths D, Clarke AR. Lkb1 and Pten synergise to suppress mTOR‐mediated tumorigenesis and epithelial‐mesenchymal transition in the mouse bladder. PLoS One 2011;6:e16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehenni H, Lin‐Marq N, Buchet‐Poyau K, Reymond A, Collart MA, Picard D, et al. LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet 2005;14:2209‐2219. [DOI] [PubMed] [Google Scholar]
- 13. Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, et al. Expansion of hepatic tumor progenitor cells in Pten‐null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 2010;139:2170‐2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res 2002;62:4549‐4553. [PubMed] [Google Scholar]
- 15. Bae JJ, Rho JW, Lee TJ, Yun SS, Kim HJ, Choi JH, et al. Loss of heterozygosity on chromosome 10q23 and mutation of the phosphatase and tensin homolog deleted from chromosome 10 tumor suppressor gene in Korean hepatocellular carcinoma patients. Oncol Rep 2007;18:1007‐1013. [PubMed] [Google Scholar]
- 16. Kim CJ, Cho YG, Park JY, Kim TY, Lee JH, Kim HS, et al. Genetic analysis of the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer 2004;40:136‐141. [DOI] [PubMed] [Google Scholar]
- 17. Woods A, Heslegrave AJ, Muckett PJ, Levene AP, Clements M, Mobberley M, et al. LKB1 is required for hepatic bile acid transport and canalicular membrane integrity in mice. Biochem J 2011;434:49‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia‐Rodriguez JL, Zubiete‐Franco I, Fernandez‐Ramos D, Fernandez‐Tussy P, Barbier‐Torres L, Lopitz‐Otsoa F, et al. Liver kinase B1 as an oncogenic driver in liver cancer. Journal of Hepatology supplemental issue: The International Liver Congress Tm 2015‐50th Annual Meeting of the European Association for the Study of the Liver 2015;62:P260. [Google Scholar]
- 19. Chen WT, Tseng CC, Pfaffenbach K, Kanel G, Luo B, Stiles BL, et al. Liver‐specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology 2014;59:947‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He L, Gubbins J, Peng Z, Medina V, Fei F, Asahina K, et al. Activation of hepatic stellate cell in Pten null liver injury model. Fibrogenesis Tissue Repair 2016;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C, et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen‐related receptor alpha (ERRalpha). J Biol Chem 2013;288:25007‐25024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005;310:1642‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, et al. Induction of intrahepatic cholangiocellular carcinoma by liver‐specific disruption of Smad4 and Pten in mice. J Clin Invest 2006;116:1843‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zeng N, Li Y, He L, Xu X, Galicia V, Deng C, et al. Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress‐induced cell death in Pten‐null hepatocytes. Mol Cancer Res 2011;9:1708‐1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW, et al. PTEN controls beta‐cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell 2013;12:1000‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He L, Hou X, Kanel G, Zeng N, Galicia V, Wang Y, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol 2010;176:2302‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver‐specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A 2004;101:2082‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF‐I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 2011;152:3143‐3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rountree CB, Ding W, He L, Stiles B. Expansion of CD133‐expressing liver cancer stem cells in liver‐specific phosphatase and tensin homolog deleted on chromosome 10‐deleted mice. Stem Cells 2009;27:290‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1). Hum Mol Genet 2002;11:1497‐1504. [DOI] [PubMed] [Google Scholar]
- 31. Rossig L, Jadidi AS, Urbich C, Badorff C, Zeiher AM, Dimmeler S. Akt‐dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol Cell Biol 2001;21:5644‐5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Postic C, Magnuson MA. DNA excision in liver by an albumin‐Cre transgene occurs progressively with age. Genesis 2000;26:149‐150. [DOI] [PubMed] [Google Scholar]
- 33. Just PA, Poncy A, Charawi S, Dahmani R, Traore M, Dumontet T, et al. LKB1 and Notch pathways interact and control biliary morphogenesis. PLoS One 2015;10:e0145400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, et al. Targeted inactivation of sister of P‐glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A 2001;98:2011‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol 2009;41:757‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hardie DG, Ross FA, Hawley SA. AMP‐activated protein kinase: a target for drugs both ancient and modern. Chem Biol 2012;19:1222‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hahn‐Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem 2005;280:32081‐32089. [DOI] [PubMed] [Google Scholar]
- 38. Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1‐AMPK pathway in suppressing tumorigenesis in PTEN‐deficient mice. Biochem J 2008;412:211‐221. [DOI] [PubMed] [Google Scholar]
- 39. Chopra I, Li HF, Wang H, Webster KA. Phosphorylation of the insulin receptor by AMP‐activated protein kinase (AMPK) promotes ligand‐independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012;55:783‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol 2004;273:175‐184. [DOI] [PubMed] [Google Scholar]
- 41. Zeng N, Bayan JA, He L, Stiles B. The role of PTEN in beta‐cell growth. Open Endocrinol J 2010;4:23‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng PY, Berger SL. LKB1 is recruited to the p21/WAF1 promoter by p53 to mediate transcriptional activation. Cancer Res 2006;66:10701‐10708. [DOI] [PubMed] [Google Scholar]
- 43. Deguchi A, Miyoshi H, Kojima Y, Okawa K, Aoki M, Taketo MM. LKB1 suppresses p21‐activated kinase‐1 (PAK1) by phosphorylation of Thr109 in the p21‐binding domain. J Biol Chem 2010;285:18283‐18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003;2:339‐345. [PubMed] [Google Scholar]
- 45. Gebhardt R, Matz‐Soja M. Liver zonation: novel aspects of its regulation and its impact on homeostasis. World J Gastroenterol 2014;20:8491‐8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li C, Li Y, He L, Agarwal AR, Zeng N, Cadenas E, et al. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med 2013;60:29‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. J Mammary Gland Biol Neoplasia 2004;9:145‐163. [DOI] [PubMed] [Google Scholar]
- 48. Collins BJ, Kleeberger W, Ball DW. Notch in lung development and lung cancer. Semin Cancer Biol 2004;14:357‐364. [DOI] [PubMed] [Google Scholar]
- 49. Pece S, Confalonieri S, R Romano P, Di Fiore PP. NUMB‐ing down cancer by more than just a NOTCH. Biochim Biophys Acta 2011;1815:26‐43. [DOI] [PubMed] [Google Scholar]
- 50. Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior‐posterior axis formation and epithelial polarity. Nature 2003;421:379‐384. [DOI] [PubMed] [Google Scholar]
- 51. Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par‐4 gene encodes a putative serine‐threonine kinase required for establishing embryonic asymmetry. Development 2000;127:1467‐1475. [DOI] [PubMed] [Google Scholar]
- 52. Maciejczyk A, Jagoda E, Wysocka T, Matkowski R, Gyorffy B, Lage H, et al. ABCC2 (MRP2, cMOAT) localized in the nuclear envelope of breast carcinoma cells correlates with poor clinical outcome. Pathol Oncol Res 2012;18:331‐342. [DOI] [PubMed] [Google Scholar]
- 53. Surowiak P, Materna V, Kaplenko I, Spaczynski M, Dolinska‐Krajewska B, Gebarowska E, et al. ABCC2 (MRP2, cMOAT) can be localized in the nuclear membrane of ovarian carcinomas and correlates with resistance to cisplatin and clinical outcome. Clin Cancer Res 2006;12:7149‐7158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1027/suppinfo
Supporting Information Slide 2
Supporting Information Slide 6
Supporting Information Slide 7