

Abstract
Neuroligins are postsynaptic cell-adhesion molecules that bind to presynaptic neurexins. Mutations in neuroligin-3 predispose to autism, but how such mutations affect synaptic function remains incompletely understood. Here we systematically examined the effect of three autism-associated mutations, the neuroligin-3 knockout, the R451C knockin, and the R704C knockin, on synaptic transmission in the calyx of Held, a central synapse ideally suited for high-resolution analyses of synaptic transmission. Surprisingly, germline knockout of neuroligin-3 did not alter synaptic transmission, whereas the neuroligin-3 R451C and R704C knockins decreased and increased, respectively, synaptic transmission. These puzzling results prompted us to ask whether neuroligin-3 mutant phenotypes may be reshaped by developmental plasticity. Indeed, conditional knockout of neuroligin-3 during late development produced a marked synaptic phenotype, whereas conditional knockout of neuroligin-3 during early development caused no detectable effect, mimicking the germline knockout. In canvassing potentially redundant candidate genes, we identified developmentally early expression of another synaptic neurexin ligand, cerebellin-1. Strikingly, developmentally early conditional knockout of cerebellin-1 only modestly impaired synaptic transmission, whereas in contrast to the individual single knockouts, developmentally early conditional double knockout of both cerebellin-1 and neuroligin-3 severely decreased synaptic transmission. Our data suggest an unanticipated mechanism of developmental compensation whereby cerebellin-1 and neuroligin-3 functionally occlude each other during development of calyx synapses. Thus, although acute manipulations more likely reveal basic gene functions, developmental plasticity can be a major factor in shaping the overall phenotypes of genetic neuropsychiatric disorders.
Introduction
Synapse formation and maintenance require precise apposition of pre- and postsynaptic specializations to produce the characteristic properties of a synapse. This coordinated apposition is thought to be mediated by trans-synaptic cell-adhesion molecules, such as neurexins, neuroligins and cerebellins.1–3
Neuroligins are evolutionarily conserved postsynaptic proteins that bind to presynaptic neurexins. Vertebrates express four neuroligin genes (referred to as NL1, NL2, NL3 and NL4), of which NL1 functions at excitatory synapses, NL2 at inhibitory synapses, NL3 at both excitatory and inhibitory synapses, and NL4 at glycinergic synapses.4–8 Genetic experiments in different circuits in vivo with conditional knockout mice showed essential roles of neuroligins in specifying the properties of synapses, but not in establishing synapses.9–11 Mutations in NL3 and other neuroligins in patients have been repeatedly linked to autism.12–14 However, although analyses of NL3 KO, R451C-knockin, and R704C-knockin mice genocopying some of these mutations confirmed their physiological significance with distinct effects on synaptic transmission in different brain regions,9,15–17 the precise effects of these mutations on synaptic transmission remain incompletely understood.
Cerebellins are a family of related secreted proteins (Cbln1–4) that are broadly expressed throughout brain, and that also bind to neurexins.3,18 In humans, mutations of cerebellins are associated with neurological disease.19–22 In mice, deletion of Cbln1, the best-studied cerebellin, causes a marked loss of parallel-fiber synapses in cerebellar Purkinje cells, suggesting that Cbln1 functions in parallel-fiber synapse formation and/or maintenance.18,23,24 Puzzlingly, however, deletion of Cbln1 increase synaptic spine density in striatral medium spiny neurons.25 Thus, the general functions of Cbln1 remain unclear.26,27
The calyx of Held is a giant glutamatergic synapse in the medial nucleus of the trapezoid body (MNTB) of the auditory brainstem. Presynaptic calyx terminals originate from neurons in the anterior ventral cochlear nucleus, and elaborate a single calyx synapse that contains hundreds of active zones covering the postsynaptic MNTB neuron. Because of its size and spherical architecture, the calyx of Held synapse allows studies of synaptic function with unprecedented precision. Moreover, calyx synapses exhibit a dramatic morphological and functional transformation during development,28–33 suggesting that they also serve as an accessible model system for synapse development and for studying the molecular basis of neuropsychiatry disorders.34–38
Here we have examined the effect of autism-associated NL3 mutations on synapse function at the calyx of Held. Unexpectedly, we uncovered a marked dependence of the phenotype on the developmental time point at which NL3 mutations were introduced, suggesting developmental plasticity. In surveying possible candidates for mediating such plasticity, we observed developmentally early expression of Cbln1 at calyx synapses, and confirmed that simultaneous conditional knockout of both NL3 and Cbln1 during early development elicits a massive phenotype that was absent from each of the single knockouts. Our data suggest that NL3 and Cbln1 perform developmentally segregated but functionally overlapping functions in calyx of Held synapses, which may occlude an otherwise more severe synaptic phenotype produced by NL3 mutations in autism patients and could account for the selective changes observed with such mutations in human subjects.
Materials and Methods
Mouse handling
Generation and genotyping of NL3 KO, PV-Cre, Ai14-Cre reporter, Krox-20-Cre, NL3 floxed mouse lines have been described previously. For details, see Supplementary Information. Cbln1 floxed mice were generated using 129svev strain embryonic stem cells in St. Jude Children's Research Hospital (Supplementary Figure S1 and Supplementary Methods). All procedures were approved by the Stanford IACUC.
Quantitative RT-PCR
Slices containing ventral cochlear nucleus or MNTB from postnatal days 1–18 (P1-P18) mice were carefully dissected under microscopy. Gene expression studies using reverse transcription PCR (RT-PCR) were performed as described previously.39 For details, see Supplementary Information.
Electrophysiology
Electrophysiological recordings at the calyx of Held synapses were performed essentially as described in acute brainstem slices from 9- to 13-day-old mutant and littermate control mice.40–42 Postsynaptic recordings were done in whole-cell voltage clamp mode. Miniature excitatory postsynaptic currents (mEPSCs) and evoked EPSC amplitude, rise-time, decay and so on. were analyzed using Clampfit 10.2 (Molecular Devices, Sunnyvale, CA, USA) or Mini Analysis software (Synaptosoft, Fort Lee, NJ, USA). For details, see Supplementary Information.
Immunohistochemistry
Immunohistochemistry was carried out as described before on 30 μm brainstem sections containing the MNTB.11 The primary antibody used was rabbit anti-Syt2 (dilution 1:500, A320), with a goat anti-rabbit Alexa488 (Invitrogen, Waltham, MA, USA) secondary antibody. Confocal images were acquired with an inverted Nikon IR+ microscope (Nikon, Tokyo, Japan) equipped with × 63 objective. Images were taken at 1024×1024 pixels and analyzed with Nikon analysis software.
Results
Autism-associated NL3 mutations differentially alter synaptic transmission at the calyx synapse
Previous studies indicated that the NL3 KO, R451C-knockin and R704C-knockin mutations produce distinct effects on synaptic transmission in different brain regions (cortex, hippocampus and striatum),9,15–17 but the precise mechanisms of these effects could not be defined due to the limitations of the preparations used. Therefore, we decided to systematically investigate how these mutations affect synaptic transmission at the calyx of Held synapse, which allows an unparalleled resolution of synaptic analysis (Figure 1a).
Figure 1.
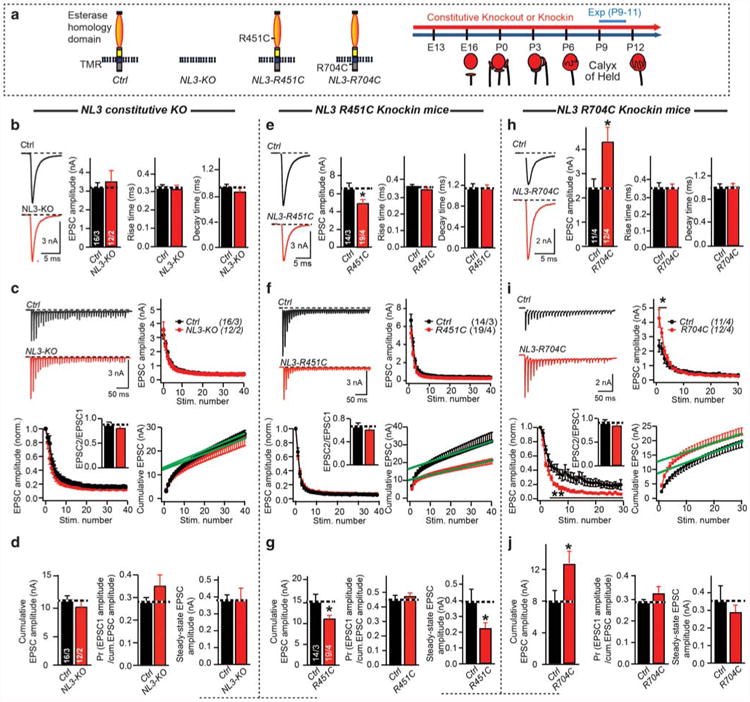
Constitutive NL3 knockout (KO) does not alter synaptic transmission at calyx of Held synapses, whereas the NL3 R451C knockin decreases, and the NL3 R704C knockin increases, their strength. (a) Schematic diagram of NL3 mutations (left), and developmental profile of the calyx of Held synapse (right). (b) AMPA receptors (AMPARs)-mediated EPSCs evoked by fiber stimulation in P9–P11 control and NL3 KO mice (left, sample traces; right, summary graphs of the amplitude, rise time and decay time). Stimulation artifacts were removed from all sample trace. (c) AMPAR-mediated EPSCs triggered by an action-potential train induced by fiber stimulation (40 AP at 100 Hz) in P9–P11 control and NL3 KO mice (top left, representative traces; top right, cumulative EPSC amplitude plots; bottom left and bottom right, absolute and normalized EPSC amplitudes as a function of stimulus number (green line = fitted linear regression line to calculate the estimated recovery-corrected readily releasable pool size by back-extrapolation to time zero of accumulative EPSCs)). Paired-pulse ratio was calculated as EPSC2/EPSC1 and inserted in bottom left. (d) Summary graphs of the cumulative EPSCs (left), release probability (middle), and the steady-state EPSC amplitude (mean amplitude of the last 10 EPSCs, right). (e–g), Same as (b–d), but for control and NL3-R451C knockin mice. (h–j), Same as (b–d), but for control and NL3-R704C knockin mice. Train stimulation of 30 AP at 100 Hz was used. Data are means ± s.e.m. Numbers in bars represent the numbers of cells/animals. Statistical significance was determined by two-tailed Student's t-test or by single-factor analysis of variance (ANOVA) (*P < 0.05; non-significant comparisons are not labeled). EPSC, excitatory postsynaptic currents; P, postnatal day.
We first studied the NL3 KO, which based on quantitative RT-PCR of total mRNA levels in the mouse MNTB exhibits a modest increase during postnatal development (Supplementary Figures S2A and S2B). We found that at P9-P11, the frequency and amplitude of mEPSCs were indistinguishable between NL3 KO and littermate control mice (Supplementary Figure S3A; note that NL3 is an X-chromosomal gene). We then analyzed EPSCs induced by afferent-fiber stimulation, and observed normal EPSC amplitudes, rise times, and decay times (Figure 1b). To probe for potential changes in the number and release probability of vesicles in the readily releasable pool, we used high-frequency afferent-fiber stimulation (100 Hz for 0.4 s).43 Short-term depression, assessed by normalizing the EPSCs of a train to the first EPSC, was similar between control and NL3 KO neurons (Figures 1c and d). Summation of evoked EPSC amplitudes during the stimulus train also failed to reveal changes in the cumulative EPSC amplitude in NL3 KO mice (Figure 1d). Similar to short-term synaptic plasticity, dividing the initial EPSC amplitude in the train by the cumulative EPSC, which provides an indirect measure of the presynaptic release probability, was also normal in NL3 KO mice. Finally, the steady-state EPSC amplitude (mean amplitude of the last 10 EPSCs) also was unchanged in NL3 KO mice (Figures 1c and d). Altogether, these data suggest that the constitutive NL3 KO has no effect on excitatory synaptic transmission at the calyx synapse.
We next performed analogous experiments with NL3-R451C-and NL3-R704C-knockin mice. Strikingly, we observed in R451C knockin mice during stimulus trains a reduced mEPSC amplitude and a smaller cumulative evoked EPSC amplitude without an apparent change in release probability, suggesting a reduced postsynaptic AMPAR content (Figures 1e–g and Supplementary Figure S3B). The reduction of the cumulative EPSC amplitude (an indirect measure of the pool size) appears to be bigger than the decrease of mEPSC amplitude, but the two parameters do not necessarily scale linearly and cannot be directly compared, although we cannot exclude a more subtle presynaptic phenotype.
In contrast to R451C knockin mice, we observed in R704C knockin mice a normal mEPSC amplitude, but an increase in the initial EPSC and in the cumulative EPSC amplitude during the train, accompanied by a corresponding change in the normalized EPSCs (Figures 1h–j, Supplementary Figure S3C). Again, the release probability as estimated indirectly via the paired-pulse ratio did not change in R704C-knockin mice (Figure 1i). Altogether, these data suggest an increase in the number of functional presynaptic release sites in R704C knockin mice.
Overall, our results show that the NL3 KO and the two NL3 knockin mutations differentially alter AMPAR-mediated synaptic transmission at calyx synapses. As the NL3 KO in itself did not produce a phenotype, the effects of the knockins likely arise by gain-of-function mechanisms. These effects, just like the absence of an effect in the NL3 KO, are surprisingly different from those observed previously,9,15–17,44,45 suggesting that developmental plasticity may have altered the phenotype.
Developmentally late but not early conditional NL3 KO impairs AMPAR-mediated synaptic transmission
To explore the potential for developmental plasticity as a basis for a lack of a phenotype in NL3 KO calyx synapses, we analyzed the effect of conditionally deleting NL3 during development. For this purpose, we needed Cre-driver mice with developmentally early and late Cre-recombinase expression in calyx synapses. Traditionally, conditional KO studies at the calyx synapse utilize Krox-20-Cre-driver mice that express Cre-recombinase relatively early (at ∼E10),46–48 which may allow for developmental plasticity. To identify a Cre-driver line with Cre-expression later in development, we examined mice in which Cre-recombinase expression is driven by the parvalbumin (PV) promoter (referred to as PV-Cre mice). When we analyzed PV-Cre mice crossed with Cre-dependent tdTomato reporter mice, we observed Cre-recombinase expression in both pre- and postsynaptic neurons of the calyx synapse (Figures 2a and b). Importantly, Cre-expression in principal neurons started postnatally, and was detectable in essentially all principal neurons at P12 (Figure 2c). We then crossed conditional NL3 KO mice with both Krox-20-Cre and PV-Cre driver mice to analyze the effects of developmentally early and late conditional NL3 KOs, respectively, on synaptic transmission at the calyx of Held.
Figure 2.
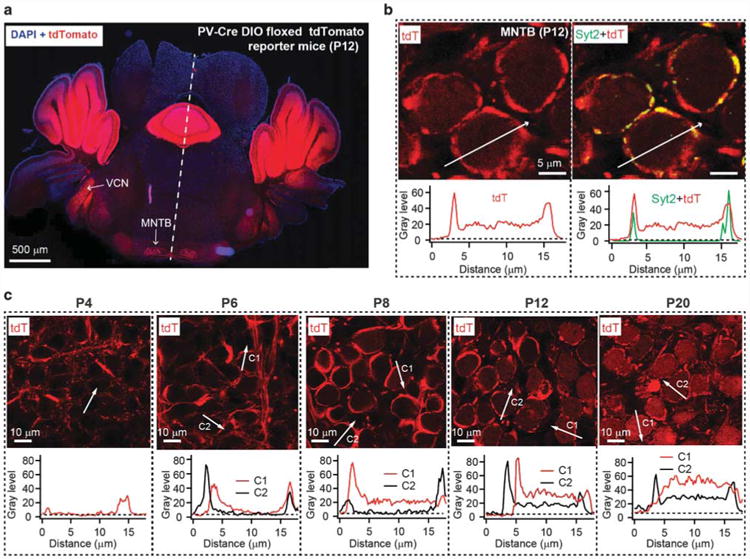
Cre-recombinase expressed under control of the parvalbumin promoter in PV-Cre mice manifests in a developmentally late pattern in both pre- and postsynaptic neurons of the calyx of Held synapse. (a) Low-magnification image of a single focal plane of a section of the brainstem and cerebellum from PV-Cre+ mice crossed with tdTomato reporter mice at P12 (red = tdTomato fluorescence; blue = DAPI staining). Note that tdTomato is robustly expressed in the ventral cochlear nucleus (VCN) and medial nucleus of the trapezoid body (MNTB). (b) High-magnification images of an MNTB section from a PV-Cre/tdTomato reporter mouse at P12, viewed in a single focal plane (red = tdTomato fluorescence; green = immunolabeling for synaptotagmin-2 (Syt2)). The white arrow depicts the position of the fluorescence intensity plot depicted below the images. (c) Analyses of Cre-expression in calyx synapses of PV-Cre mice as a function of developmental time analyzed in PV-Cre/tdTomato reporter mice. The age of the mice examined is listed above the panels. P, postnatal day.
We found that NL3 conditional KO mice with developmentally late NL3 deletion under control of the PV promoter, analyzed at P11-13, exhibited a dramatic impairment in synaptic transmission. We detected a ∼40% decrease in both spontaneous mEPSC and evoked EPSC amplitudes (Figure 3, Supplementary Figure S4A). Moreover, evoked EPSCs displayed altered kinetics with prolonged rise and decay times, and train stimulations revealed reduced cumulative EPSC amplitudes with a normal apparent release probability (Figures 3b–d). As we detected no change in the release probability, these data suggest that the developmentally late NL3 deletion causes a large decrease in postsynaptic AMPARs at the calyx synapse without a change in AMPAR composition,49 as suggested by the unaltered mEPSC kinetics (Supplementary Figure S4A).
Figure 3.
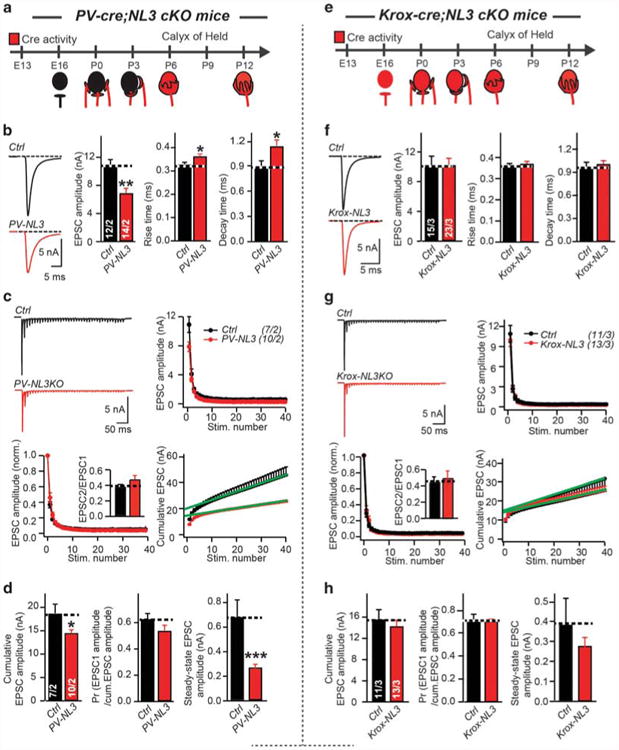
Developmentally late, but not developmentally early conditional NL3 KO reduces AMPAR-mediated synaptic transmission at the calyx of Held. (a) Cartoon of PV-Cre activity during development of MNTB. PV-20-Cre activity starts at early postnatal stages. Crossing PV-Cre mice with NL3-cKO mice will results in early postnatal NL3 deletion. (b–d), Sample traces and summary data of EPSCs (b), and EPSCs train (c, d) recorded from P11-13 MNTB neurons at control (black) or PV-NL3 (red) littermate. (e) Cartoon of Krox-20-Cre activity in development of MNTB. Krox-20-Cre activity starts at early embryonic stages, even before axons of calyx of Held cross the midline. Crossing Krox-20-Cre mice with NL3-cKO mice will results in early prenatal NL3 deletion. (f–h), Same as (b–d), but for P11-13 control and Krox-20-NL3 littermate. Data are mean ± s.e.m. Numbers in bars represent the numbers of cells/animals. Statistical significance was determined by two-tailed Student's t-test or by single-factor analysis of variance (ANOVA) (*P < 0.05; **P < 0.01; ***P < 0.001; non-significant comparisons are not labeled). AMPAR, AMPA receptor; EPSC, excitatory postsynaptic currents; P, postnatal day; MNTB, medial nucleus of the trapezoid body.
We next examined the Krox-20-Cre-induced conditional NL3 KO (Supplementary Figure S5A), which induces a developmentally earlier NL3 deletion (Supplementary Figure S2E, F). Strikingly, we found that similar to the NL3 KO, early Krox-20-Cre-induced deletion of NL3 failed to produce a phenotype (Figures 3e–h, Supplementary Figure S4B). Thus, developmentally early KO of NL3 likely enables compensation of the deleterious effect of the deletion, which does not occur with developmentally late KO of NL3. Possibly the developmentally late KO of NL3 does not cause compensation because there is insufficient time, or because the compensatory mechanism cannot be activated at a developmentally later time.
Developmentally early but not late Cbln1 deletion modestly impairs excitatory synaptic transmission
Could compensation of the Krox-20-Cre-induced conditional NL3 KO be mediated by NL1? To answer this question, we tested the effect of deleting NL1 in addition to NL3. However, we again observed no phenotype in NL1/NL3 double KO mice induced early in development (Supplementary Figure S4C), suggesting that NL1 does not compensate for the developmentally early NL3 KO. We then search for other candidate compensatory mechanisms, and observed by RT-PCR high-level expression of Cbln1 in the ventral cochlear nucleus, the origin of presynaptic terminals at the calyx of Held, and in the MNTB (Figure 4a, Supplementary Figure S1C). Since Cbln1 is a neurexin ligand similar to NL3 that may be functionally redundant with NL3, we hypothesized that Cbln1 may compensate for the effect of the developmentally early NL3 deletion. To test this hypothesis, we crossed conditional Cbln1 KO mice with PV-Cre and Krox-20-Cre mice (Figure 4b, Supplementary Figure S5B). Strikingly, we found that the PV-Cre-induced developmentally late conditional Cbln1 KO had no effect on synaptic transmission (Figures 4c–e, Supplementary Figure S6A), in contrast to the corresponding PV-Cre-induced conditional NL3 KO (Figure 3). The Krox-20-Cre-induced developmentally early conditional Cbln1 KO, however, produced a small but significant impairment in synaptic transmission (Figures 4f–h, Supplementary Figure S6B). In developmentally early Cbln1 conditional KO mice, evoked EPSC amplitudes were normal, but EPSC rise and decay times were increased; moreover, although cumulative EPSC amplitudes were normal and exhibited unchanged short-term plasticity, the slope of the cumulative EPSC amplitudes was reduced (Figures 4f–h). Thus, Cbln1 plays a minor but significant role in enabling early synaptic development of the calyx synapse.
Figure 4.
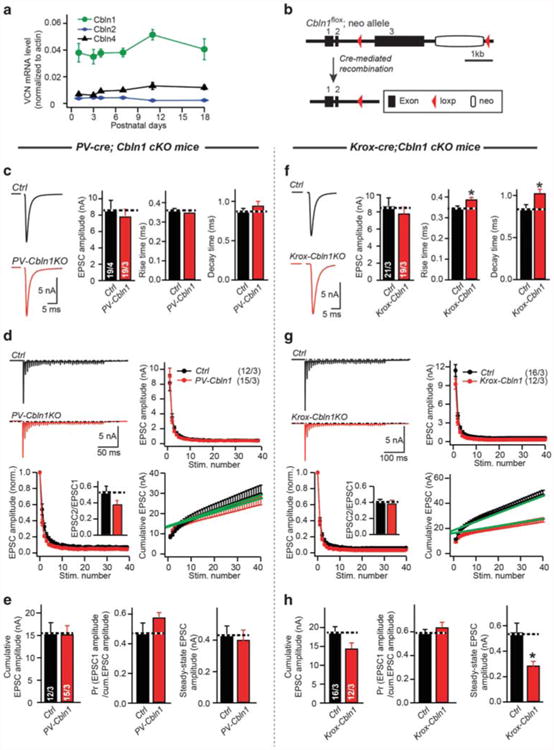
Developmentally early but not developmentally late conditional Cbln1 knockout (KO) causes a modest impairment of synaptic transmission at the calyx of Held. (a) Measurement of mRNA of Cbln1, -2 and -4 at ventral cochlear nucleus (VCN) during development. The amounts of mRNA were normalized to actin. (b) Schema of generating the conditional Cbln1 KO mice. After introduction of Cre recombinase, the Cbln1 gene will be conditionally deleted. (c–e), Sample traces and summary data of EPSCs (c) and EPSCs train by 40 stimuli at 100 Hz (d, e) recorded from P11-13 MNTB neurons at control (black) or PV-Cbln1 (red) littermate. Consistent with the minimal phenotype produced by postnatal deletion of Cbln1 (PV-Cbln1), incubation of Cbln1 peptide at P11-13 calyx of Held brain slice also did not alter the EPSC amplitude (Supplementary Figure S4D). (f–h), Same as (c–e), but for P11-13 control and Krox-20-Cbln1 littermate. Data are mean ± s.e.m. Numbers in bars represent the numbers of cells/animals Statistical significance was determined by two-tailed Student's t-test or by single-factor analysis of variance (ANOVA) (*P < 0.05; non-significant comparisons are not labeled). EPSC, excitatory postsynaptic currents; P, postnatal day.
Developmentally early double-conditional KO of both NL3 and Cbln1 induces severe impairments of excitatory synaptic transmission
In order to test whether NL3 and Cbln1 perform complementary, developmentally segregated functions at the calyx synapse, we generated double NL3/Cbln1 conditional KO mice, and crossed them with Krox-20-Cre or PV-Cre mice. When we analyzed synaptic transmission at the calyx in NL3/Cbln1 KO vs littermate control mice (comparisons were with homozygous mutant conditional NL3 and Cbln1 KO mice without or with the Krox-20-Cre or PV-Cre allele; Supplementary Figure S5C), we observed a marked phenotype in Krox-20-Cre-driven double-conditional NL3/Cbln1 KO mice. This phenotype consisted of a decrease in the evoked EPSC amplitude, a retardation of EPSC kinetics, and a large impairment in the cumulative EPSC amplitude during stimulus trains (Figures 5a–c, Supplementary Figure S7A). This phenotype mimicked the phenotype observed in PV-Cre-driven conditional NL3 KO mice (Figures 3a–d), and was similarly present in the PV-Cre-driven double-conditional NL3/Cbln1 KO mice as far analyzed (Figure 5d, Supplementary Figure S7B), suggesting that Cbln1 expression occludes the NL3 KO phenotype when NL3 is deleted developmentally early.
Figure 5.
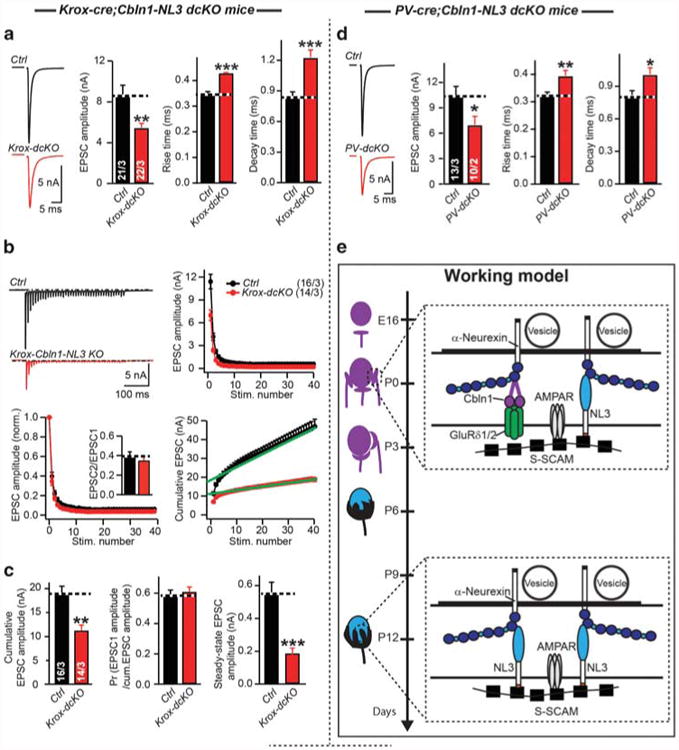
Developmentally early conditional double KO of Cbln1 and NL3 severely impairs synaptic transmission at the calyx of Held. (a–c), Sample traces and summary data of EPSCs (a), and EPSCs train by 40 stimuli at 100 Hz (b, c) recorded from P11-13 MNTB neurons in control (black) or Krox-Cbln1-NL3 (red) littermate. (d) Sample traces and summary data of EPSCs recorded from P11-13 MNTB neurons in control (black) or PV-Cbln1-NL3 KO (red) littermate. (e) Model of how AMPAR-mediated transmission was regulated by NL3 and Cbln1 during development at calyx of Held. Cbln1 and NL3 work sequentially at calyx of Held, possibly because of down-regulation of postsynaptic GluRδ1/2 receptor during development (Supplementary Figure S1D). NL3 functions primarily postnatally, whereas Cbln1 functions primarily prenatally. Interaction of NL3 with AMPAR was mediated by synaptic scaffolding molecule (S-SCAM). Data are mean ± s.e.m. Numbers in bars represent the numbers of cells/animals. Statistical significance was determined by two-tailed Student's t-test or by single-factor analysis of variance (ANOVA) (*P < 0.05; **P < 0.01; ***P < 0.001; non-significant comparisons are not labeled). AMPAR, AMPA receptor; EPSC, excitatory postsynaptic currents; P, postnatal day; MNTB, medial nucleus of the trapezoid body.
Discussion
Neuroligins have been the objects of intense interest, in large part because of their likely important functions at synapses and their genetic association with autism-spectrum disorders.1,4–8,12–14,50 Examining the function of neuroligins proved challenging, however, because mammals express multiple isoforms (NL1-4),51–53 which may perform only partly overlapping functions and whose mutations may cause developmental compensation. A better understanding of how mutations in neuroligin genes predispose to neuropsychiatric disorders is made additionally difficult by the presence of these mutations throughout an individual's development,12–14 and the phenotypic consequences of such mutations are likely the combined result of both mutation-induced functional changes and of developmental plasticity and compensation.50
To address these challenges at least in part, we here have analyzed the effects of autism-associated NL3 mutations on synaptic transmission as a function of development. We performed these analyses in what is arguably the best preparation to study synaptic transmission at high resolution, the calyx of Held synapse. We found that the constitutive NL3 KO surprisingly exhibited no phenotype, whereas two NL3 point mutations, the R451C- and R704C-substitutions analyzed as knockins, produced nearly opposite phenotypes (Figure 1). Hypothesizing that this puzzling result could be due to developmental plasticity that might occlude the NL3 KO phenotype, but not prevent dominant-positive knockin phenotypes, we tested whether developmentally later conditional KO of NL3 could uncover a phenotype. We identified PV-Cre mice as a tool that enables developmentally late, postnatal Cre-recombinase expression in MNTB neurons, in contrast to Krox-20-Cre mice which mediate developmentally early Cre-recombinase expression in these neurons (Figure 2, also see ref. 54). We found that the developmentally late conditional NL3 KO produced a marked impairment of synaptic transmission, whereas the developmentally early conditional NL3 KO, just like the constitutive NL3 KO, caused no phenotype (Figure 3).
These results strongly support the hypothesis of developmental plasticity, leading us to search for candidate genes for such compensation. After ruling out NL1, we focused on Cbln1 as a lead candidate because it functions as a neurexin ligand similar to NL3,3 and because it is expressed developmentally early in pre- and postsynaptic calyx neurons (Figure 4a, Supplementary Fig. S2C, see also ref. 55). When we analyzed conditional Cbln1 KO mice, we found that the developmentally early and late conditional Cbln1 KO caused only modest or no changes in synaptic transmission, respectively, suggesting that by itself, Cbln1 performs a minor essential role (Figure 4). However, when we examined the developmentally early conditional double KO of both NL3 and Cbln1, we observed a large impairment in synaptic transmission, consistent with the hypothesis that developmentally early Cbln1 expression functionally compensates for a loss of NL3 as a mechanism of developmental plasticity (Figure 5e).
To the best of our knowledge, our results represent the first instance at which functional redundancy between neuroligins and cerebellins is demonstrated, and the first description of a molecular mechanism of developmental plasticity that compensates for a synaptic phenotype. Apart from highlighting the importance of conditional manipulations at different stages of development in order to deconstruct the functions of a gene, our results illustrate the need for not only recognizing, but also understanding the mechanisms of developmental plasticity for further insight into how a mutation predisposes to a neuropsychiatric disorder. Such developmental plasticity is likely region-and synapse-specific, since the same mutations that we studied here often exhibit different phenotypes in other types of synapses.9,15–17 This plasticity has major implications for disease pathogenesis. For example, it is well documented that the same or similar mutations in a particular gene can produce different clinical manifestations (for example, see refs 12,50,56–72). Among others, this observation was revealed in the initial description of NL3 mutations in autism, which showed that brothers with the same NL3 R451C mutation had different clinical presentations.12 A plausible basis for such differences in presentations is that different processes of developmental plasticity may be differentially effective in the affected patients, such as small changes (polymorphisms) in the responsible genes that may cause variations in their expression. It seems to us that such differences in compensating gene expression could account for much of the clinical variation that is observed, making it imperative to better understand the underlying processes.
Supplementary Material
Acknowledgments
We thank Drs Sungjin Lee and Salome Botelho for the help of Cbln1 purification and RT-PCR, and members of the Südhof labs for valuable discussions. This study was supported by a grant from the Simons Foundation Autism Research Initiative (307762, to TCS) and from NIMH (R37MH052804, to TCS).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuro-ligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuzaki M. Cbln1 and its family proteins in synapse formation and maintenance. Curr Opin Neurobiol. 2011;21:215–220. doi: 10.1016/j.conb.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Song JY, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- 6.Budreck EC, Scheiffele P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur J Neurosci. 2007;26:1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x. [DOI] [PubMed] [Google Scholar]
- 7.Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt KF, et al. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci USA. 2011;108:3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takács VT, Freund TF, Nyiri G. Neuroligin 2 is expressed in synapses established by cholinergic cells in the mouse brain. PLoS ONE. 2013;8:e72450. doi: 10.1371/journal.pone.0072450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang J, Xu W, Hsu YT, Yee A, Chen L, Südhof T. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol Psychiatry. 2015;20:850–859. doi: 10.1038/mp.2015.31. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, et al. Neuroligins sculpt cerebellar purkinje-cell circuits by differential control of distinct classes of synapses. Neuron. 2015;87:781–796. doi: 10.1016/j.neuron.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy D, Ronemus M, Yamrom B, Lee Yha, Leotta A, Kendall J, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Etherton M, Földy C, Sharma M, Tabuchi K, Liu X, Shamloo M, et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci USA. 2011;108:13764–13769. doi: 10.1073/pnas.1111093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, et al. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci. 2005;8:1534–1541. doi: 10.1038/nn1576. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno Y, Takahashi K, Totsune K, Ohneda M, Konno H, Murakami O, et al. Decrease in cerebellin and corticotropin-releasing hormone in the cerebellum of olivopontocerebellar atrophy and Shy-Drager syndrome. Brain Res. 1995;686:115–118. doi: 10.1016/0006-8993(95)00467-5. [DOI] [PubMed] [Google Scholar]
- 20.Boghosian-Sell L, Comings DE, Overhauser J. Tourette syndrome in a pedigree with a 7;18 translocation: identification of a YAC spanning the translocation breakpoint at 18q22.3. Am J Hum Genet. 1996;59:999–1005. [PMC free article] [PubMed] [Google Scholar]
- 21.Abalkhail H, Mitchell J, Habgood J, Orrell R, de Belleroche J. A new familial amyotrophic lateral sclerosis locus on chromosome 16q12.1-16q12.2. Am J Hum Genet. 2003;73:383–389. doi: 10.1086/377156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke Ra, Lee S, Eapen V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including Autism. Transl Psychiatry. 2012;2:e163. doi: 10.1038/tp.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, et al. Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- 24.Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science (80-) 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- 25.Kusnoor SV, Parris J, Muly EC, Morgan JI, Deutch aY. Extracerebellar role for Cerebellin1: modulation of dendritic spine density and synapses in striatal medium spiny neurons. J Comp Neurol. 2010;518:2525–2537. doi: 10.1002/cne.22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miura E, Iijima T, Yuzaki M, Watanabe M. Distinct expression of Cbln family mRNAs in developing and adult mouse brains. Eur J Neurosci. 2006;24:750–760. doi: 10.1111/j.1460-9568.2006.04950.x. [DOI] [PubMed] [Google Scholar]
- 27.Wei P, Smeyne RJ, Bao D, Parris J, Morgan JI. Mapping of Cbln1-like immunoreactivity in adult and developing mouse brain and its localization to the endolysosomal compartment of neurons. Eur J Neurosci. 2007;26:2962–2978. doi: 10.1111/j.1460-9568.2007.05913.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu WM, Goodrich LV. Morphological and physiological development of auditory synapses. Hear Res. 2014;311:3–16. doi: 10.1016/j.heares.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borst JGG, Soria van Hoeve J. The calyx of held synapse: from model synapse to auditory relay. Annu Rev Physiol. 2012;74:199–224. doi: 10.1146/annurev-physiol-020911-153236. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura PA, Cramer KS. Formation and maturation of the calyx of Held. Hear Res. 2011;276:70–78. doi: 10.1016/j.heares.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LY, Fedchyshyn MJ, Yang YM. Action potential evoked transmitter release in central synapses: insights from the developing calyx of Held. Mol Brain. 2009;2:36. doi: 10.1186/1756-6606-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi T. Strength and precision of neurotransmission at mammalian presynaptic terminals. Proc Jpn Acad Ser B Phys Biol Sci. 2015;91:305–320. doi: 10.2183/pjab.91.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Gersdorff H, Borst JGG. Short-term plasticity at the calyx of held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- 34.Holcomb PS, Hoffpauir BK, Hoyson MC, Jackson DR, Deerinck TJ, Marrs GS, et al. Synaptic inputs compete during rapid formation of the calyx of held: a new model system for neural development. J Neurosci. 2013;33:12954–12969. doi: 10.1523/JNEUROSCI.1087-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao L, Michalski N, Kronander E, Gjoni E, Genoud C, Knott G, et al. BMP signaling specifies the development of a large and fast CNS synapse. Nat Neurosci. 2013;16:856–864. doi: 10.1038/nn.3414. [DOI] [PubMed] [Google Scholar]
- 36.Abdul-Latif ML, Salazar JAA, Marshak S, Dinh ML, Cramer KS. Ephrin-A2 and ephrin-A5 guide contralateral targeting but not topographic mapping of ventral cochlear nucleus axons. Neural Dev. 2015;10:27. doi: 10.1186/s13064-015-0054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang YM, Fedchyshyn MJ, Grande G, Aitoubah J, Tsang CW, Xie H, et al. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca(2+) influx to neurotransmitter release at a central synapse. Neuron. 2010;67:100–115. doi: 10.1016/j.neuron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michalski N, Babai N, Renier N, Perkel DJ, Chédotal A, Schneggenburger R. Robo3-driven axon midline crossing conditions functional maturation of a large commissural synapse. Neuron. 2013;78:855–868. doi: 10.1016/j.neuron.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell. 2013;154:75–88. doi: 10.1016/j.cell.2013.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang B, Sun L, Yang YM, Huang HP, Zhu FP, Wang L, et al. Action potential bursts enhance transmitter release at a giant central synapse. J Physiol. 2011;589:2213–2227. doi: 10.1113/jphysiol.2010.200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acuna C, Liu X, Gonzalez A, Südhof TC. RIM-BPs mediate tight coupling of action potentials to Ca2+-triggered neurotransmitter release. Neuron. 2015;87:1234–1247. doi: 10.1016/j.neuron.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 42.Forsythe ID, Barnes-Davies M. The binaural auditory pathway: membrane currents limiting multiple action potential generation in the rat medial nucleus of the trapezoid body. Proc Biol Sci. 1993;251:143–150. doi: 10.1098/rspb.1993.0021. [DOI] [PubMed] [Google Scholar]
- 43.Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- 44.Etherton MR, Tabuchi K, Sharma M, Ko J, Südhof TC. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 2011;30:2908–2919. doi: 10.1038/emboj.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chanda S, Aoto J, Lee SJ, Wernig M, Südhof TC. Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol Psychiatry. 2015;21:169–177. doi: 10.1038/mp.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voiculescu O, Charnay P, Schneider-Maunoury S. Expression pattern of a Krox-20/Cre knock-in allele in the developing hindbrain, bones, and peripheral nervous system. Genesis. 2000;26:123–126. doi: 10.1002/(sici)1526-968x(200002)26:2<123::aid-gene7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 47.De S, Shuler CF, Turman JE. The ontogeny of Krox-20 expression in brainstem and cerebellar neurons. J Chem Neuroanat. 2003;25:213–226. doi: 10.1016/s0891-0618(03)00011-5. [DOI] [PubMed] [Google Scholar]
- 48.Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang YM, Aitoubah J, Lauer AM, Nuriya M, Takamiya K, Jia Z, et al. GluA4 is indispensable for driving fast neurotransmission across a high-fidelity central synapse. J Physiol. 2011;589:4209–4227. doi: 10.1113/jphysiol.2011.208066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 51.Ichtchenko K, Nguyen T, Südhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 52.Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Comoletti D, Flynn RE, Boucard AA, Demeler B, Schirf V, Shi J, et al. Gene selection, alternative splicing, and post-translational processing regulate neuroligin selectivity for β-neurexins. Biochemistry. 2006;45:12816–12827. doi: 10.1021/bi0614131. [DOI] [PubMed] [Google Scholar]
- 54.Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci USA. 2014;111:1138–1143. doi: 10.1073/pnas.1321990111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolson DR, Wan J, Wu J, Dehoff M, Brandebura AN, Qian J, et al. Temporal patterns of gene expression during calyx of held development. Dev Neurobiol. 2016;76:166–189. doi: 10.1002/dneu.22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 57.Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;10:8192–8192. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol. 2014;74:113–122. doi: 10.1002/dneu.22128. [DOI] [PubMed] [Google Scholar]
- 60.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Group C, Consortium PG. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2014;19:872–879. doi: 10.1038/mp.2013.127. [DOI] [PubMed] [Google Scholar]
- 63.Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet Part C Semin Med Genet. 2003;123C:48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- 64.Maier W, Lichtermann D, Minges J, Hallmayer J, Heun R, Benkert O, et al. Continuity and discontinuity of affective disorders and schizophrenia: Results of a controlled family study. Arch Gen Psychiatry. 1993;50:871–883. doi: 10.1001/archpsyc.1993.01820230041004. [DOI] [PubMed] [Google Scholar]
- 65.Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N. Autism spectrum disorders and childhood-onset schizophrenia: clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry. 2009;48:10–18. doi: 10.1097/CHI.0b013e31818b1c63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.King BH, Lord C. Is schizophrenia on the autism spectrum? Brain Res. 2011;1380:34–41. doi: 10.1016/j.brainres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- 68.Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, et al. Copy number variants in schizophrenia: Confirmation of five previous finding sand new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ronald A, Simonoff E, Kuntsi J, Asherson P, Plomin R. Evidence for overlapping genetic influences on autistic and ADHD behaviours in a community twin sample. J Child Psychol Psychiatry Allied Discip. 2008;49:535–542. doi: 10.1111/j.1469-7610.2007.01857.x. [DOI] [PubMed] [Google Scholar]
- 70.Rommelse NNJ, Franke B, Geurts HM, Hartman CA, Buitelaar JK. Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur Child Adolesc Psychiatry. 2010;19:281–295. doi: 10.1007/s00787-010-0092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lichtenstein P, Carlström E, Råstam M, Gillberg C, Anckarsäter H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010;167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- 72.Williams NM, Franke B, Mick E, Anney RJL, Freitag CM, Gill M, et al. Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: The role of rare variants and duplications at 15q13.3. Am J Psychiatry. 2012;169:195–204. doi: 10.1176/appi.ajp.2011.11060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.