

Summary
Early onset epileptic encephalopathies (EOEE) are a debilitating spectrum of disorders associated with cognitive impairments. We present a clinical report of a KCNT2 mutation in an EOEE patient. The de novo heterozygous variant Phe240Leu SLICK was identified by exome sequencing and confirmed by Sanger sequencing. Phe240Leu rSlick and hSLICK channels were electrophysiologically heterologously characterized to reveal three significant alterations to channel function. First, [Cl−]i-sensitivity was reversed in Phe240Leu channels. Second, predominantly K+-selective WT channels were made to favor Na+ over K+ by Phe240Leu. Third and consequent to altered ion selectivity, Phe240Leu channels had larger inward conductance. Further, rSlick channels induced membrane hyperexcitability when expressed in primary neurons, resembling the cellular seizure phenotype. Taken together, our results confirm that Phe240Leu is a ‘change-of-function’ KCNT2 mutation, demonstrating unusual altered selectivity in KNa channels. These findings establish pathogenicity of the Phe240Leu KCNT2 mutation in the reported EOEE patient.
Keywords: Seizures, Epileptic Encephalopathy, Ion Channels, Potassium channels, Slick, KCNT2, Selectivity, Xenopus oocytes, Slack, KCNT1
eTOC Blurb
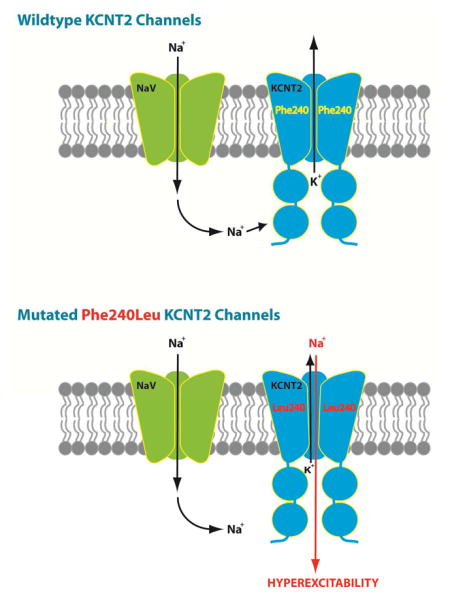
Gururaj et al. report a KCNT2 mutation in a patient with epileptic encephalopathy and employ electrophysiological analyses to establish channel properties that could underlie epileptogenesis: namely inhibition by high [Cl−]i and loss of exclusive selectivity to K+. Furthermore, primary neurons expressing Ph240Leu display a hyperexcitable phenotype.
Introduction
Early infantile epileptic encephalopathy (EOEE) is characterised by intractable seizures and severe cognitive impairment and/or developmental delay (Berg et al., 2010). The incidence of EE is estimated to be 4.3 per 10,000 live births per year (Hino-Fukuyo et al., 2009) and the diagnosis is a devastating one, with a high burden of care for families and the health service, a significant risk of comorbidities and shortened life-span (Khan and Al Baradie, 2012). Over 200 divergent genetic causes have been identified including pathogenic variants in ion channels, synaptic, regulatory and developmental proteins (McTague et al., 2016, Tamsett et al., 2009). Despite next generation sequencing approaches, which have the power to interrogate multiple genes, over 60% of children with EOEE remain without a genetic diagnosis (Helbig et al., 2016). Identification of additional genetic causes is therefore critically important, as a molecular diagnosis allows personalized management such as guidance of choice of antiepileptic or metabolic treatment, appropriate health surveillance for recognized co-morbidities, accurate estimation of recurrence risk in the family, provision of ‘closure’ and access to specific support groups for families (Berkovic, 2015).
KCNT1, encoding the KNa channel SLACK (“sequence like calcium-activated K+”, Slo2.2), has recently emerged as a key genetic cause of EOEE, with gain-of-function variants increasing peak current amplitudes of SLACK channels 3–12 fold (Barcia et al., 2012, Heron et al., 2012, Martin et al., 2014). Variants of KCNT2 (SLICK, “sequence like an intermediate conductance K+“, Slo2.1) have not previously been linked to a human phenotype despite ~74% gene sequence homology and postulated hetero-tetramerization between the two subunits in at least some brain regions (Lim et al., 2014, Chen et al., 2009, Bhattacharjee et al., 2003, Lim et al., 2016). Slick is set apart by its rapid activation kinetics, sensitivity to intracellular chloride and cell volume variations, slight sodium permeability as well as selective localization in brain regions such as the sub-plate of the cerebral cortex in utero and the hippocampus and cortex in the adult, suggesting that the channel plays an ongoing role in cerebral function (Oeschger et al., 2012, Bhattacharjee et al., 2003, Tejada et al., 2014, Rizzi et al., 2016, Bhattacharjee et al., 2002).
Results and Discussion
We report a KCNT2 variant in a male child with an EOEE phenotype consisting of neonatal hypotonia, profound developmental delay and an intractable infantile-onset seizure disorder. The male proband is the only child of non-consanguineous parents with no pertinent family history. He was delivered at term following an uneventful pregnancy, although in retrospect his mother reported fetal hiccoughs. Growth parameters have been normal, and he has no dysmorphic features or congenital anomalies.
From 3 months of age, he had multiple daily episodes of staring with eye deviation to the left and single isolated jerks. At 4 months he developed daily clusters of epileptic spasms lasting up to 8 minutes, and there was further regression in his development. His epileptic encephalopathy has been a lifelong feature (clinical and electrical). At the current age of 4 years, he has multiple daily seizures of mixed semiology that have remained resistant to polypharmacy as detailed in the Supplementary Clinic Information. The predominant symptom is a prolonged tonic seizure; he also has myoclonic jerks and atypical absences.
The electroencephalogram (EEG) has been persistently abnormal with a disorganised background, decrements, multifocal epileptogenic activity or hypsarrhythmia (Fig 1A and B). Brain MRI demonstrated a generalised reduction in white matter and thinning of the corpus callosum (Fig. 1C and D).
Fig. 1. Clinical presentation.

Representative electroencephalograms: longitudinal bipolar EEG montages (Scale 150μV/cm) demonstrating: (A) high-voltage, posterior-dominant epileptiform activity, disorganised background and decrements (5 months) (B) high-voltage disorganised background, multifocal epileptiform activity consistent with hypsarrhythmia (1 year). MRI Brain Images (5 months): (C) Axial T2-weighted MRI demonstrating enlarged ventricles and extra-axial CSF spaces with thin corpus callosum (D) Sagittal T1 MRI showing thin corpus callosum. (E) Pedigree (F) Sanger sequencing demonstrating de novo KCNT2 variant (NM_001287819.1:c.[720T>A];p.[(Phe240Leu)];[=]) in proband (IV:1) Abbreviations: EEG, electroencephalogram; MRI, magnetic resonance imaging. CSF, cerebrospinal fluid.
Prior extensive neurometabolic and genetic investigations were non-diagnostic. Exome sequencing (ES) identified a non-synonymous variant in KCNT2 (NM_001287819.1:c.[720T>A];p.[(Phe240Leu)];[=]) (Fig. 1E), confirmed as de novo by bidirectional Sanger sequencing (Fig. 1F). ES detected no other plausible variants in known EOEE genes. The affected residue is highly conserved, not listed in ExAC (last accessed July 2016) and in silico tools predicted pathogenicity (Supplementary Clinical Information). At the structural level, the residue Phe-240 is situated in the channel pore helix between transmembrane domains S5 and S6; it has been demonstrated by Sanguinetti and co-workers as critical to normal Slick channel gating (Garg et al., 2013, Suzuki et al., 2016). Based on a SLO2.1 pore helix homology model, salient interactions between Phe-240 and the S5 and S6 domains of the Slick channel were predicted and subsequently, electrophysiologically confirmed (Garg et al., 2013). Several Phe-240 variants showed constitutive activity as a function of substitute residue hydrophilicity and further, hydrophobic interactions between Phe-240 and S5 residues were essential to the stable closed resting state of the channel (Suzuki et al., 2016). We therefore hypothesized that the Phe240Leu mutation alters channel function.
To test the functional effect of the p.(Phe240Leu) variant, we recorded Wild Type (WT) and Phe240Leu rSlick currents in Chinese Hamster Ovary (CHO) cells. Rat Slick channels in the pTRACER vector were used for this study following the failure of human SLICK pTRACER expression to produce detectable currents. The high sensitivity of rSlick channels to intracellular Cl− was recapitulated by Wild Type (WT) channels with a significant Cl− dependent four-fold increase in current density from ~15 mM to ~139 mM [Cl−]i in the presence of 13.2 mM [Na+]i (Fig. 2A) (Bhattacharjee et al., 2003). Surprisingly, Phe240Leu channels showed a three-fold decrease in current density from ~15 to ~139 mM [Cl−]i (Fig. 2B). The model postulated by Garg et al. attributes constitutive activity of Phe-240 mutant channels to uncoupling of the C-terminus from the channel pore helix (Garg et al., 2013). Although the region that confers Cl− sensitivity is unknown, it could be speculated that the uncoupling of an intracellular site for Cl− sensitivity from the activation gate would render the channel insensitive to Cl− or uncover an inhibitory mechanism for Cl−. An alternative possibility is simply that WT and mutant channels undergo opposite regulation by Cl−.
Fig. 2. Phe240Leu alters [Cl−]i sensitivity and K+ selectivity of rSlick channels.

Representative whole-cell current traces evoked by depolarizing steps in pipette chloride concentration of ~15 mM (left) or ~139 mM (right) of (A) WT rSlick and (C) F240L rSlick. Effect of increasing [Cl−]i concentration on current-voltage relationship of (B) WT and (D) F240L ISlick (n=7 per group). Unpaired t-test, *p<.005. Shown are mean ± SEM. (E) Scatter plot of reversal potentials (ERev) recorded in WT-, F240L- and 1:1 WT+F240L-expressing CHO cells. Bars represent mean value per group (n=12 per group). Data points reflect ERev values across both ~15 and ~139 mM [Cl−]i.
Most strikingly, we observed a ~60 mV shift in reversal potential (ERev) (0 mV) of Phe240Leu channels compared to WT (−60 mV) (Fig. 2C), independent of [Cl−]i. This indicates that the predominantly K+-selective WT channel is converted into a channel of multi-ion nature by Phe240Leu, non-selective to K+ at the very least and presumably permissive to Na+ (Joiner et al., 1998, Bhattacharjee et al., 2003). Of note, the −60 mV ERev of WT Slick channels denotes the slight selectivity (1/20th of that of potassium) to sodium ions that Slick channels were originally characterized to possess, as an exclusively potassium-selective channel would have an ERev much closer to predicted EK of ~−73 mV (Bhattacharjee et al., 2003). When a 1:1 ratio of WT and Phe240Leu rSlick was expressed, a variable distribution of ERev values was observed (Fig. 2E). We speculate that this could imply assembly of variable stoichiometries of WT and Phe240Leu subunits into tetramers; the range of ERev values corresponds to WT or Phe240Leu Slick homomers as well as WT: Phe240Leu heteromers, with the possible configurations being 1:3, 3:1 and 1:1. Nonetheless, since homomeric WT channels unvaryingly reverse at more negative potentials, heterozygous expression of Phe240Leu Slick, and presumably the Phe240Leu KCNT2 allele, appears to be sufficient for altered channel properties and pathogenicity.
In Xenopus laevis oocytes, altered selectivity of Phe240Leu hSLICK channels was demonstrated by a ~20 mV ERev shift towards ENa (WT: −40 mV, Phe240Leu: −22 mV) and increased inward current (Fig. 3B). Rat Phe240Leu Slick channels also displayed these altered properties in Xenopus oocytes (Fig. S1). Membrane biotinylation assays showed high surface expression for WT hSLICK channels whereas Phe240Leu channels were only detected upon over-exposure of signal (Fig. 3C). In addition, Phe240Leu had low total protein levels, consistent with reduced membrane trafficking, translation of cRNA and/or rapid protein degradation. Phe240leu ISlick being comparable to WT ISlick despite poor expression suggests fewer but larger conducting mutant channels at the oocyte membrane. Whether this is a result of mutation-induced alterations in pore helix structure facilitating increased conductance or open probability of single channels is unresolved from the present experiment design. Poorly expressed Phe-240 substitutions (Phe240Ser, Phe240Asn and Phe240Cys) have been previously reported in Xenopus oocytes (Garg et al., 2013). Phe-240 is therefore critical not only to gating but also to normal membrane expression of hSLICK channels.
Fig. 3. Phe240Leu alters K+ selectivity of hSLICK channels.

(A) Representative whole-cell oocyte current traces evoked by depolarizing steps. (B) Effect of the F240L mutation on ISlick current amplitudes recorded in injected oocytes (n=5 per group per family). Unpaired t-test, *p<.05. Shown are mean ± SEM. (C) Top- Representative western blot for membrane hSLICK in Xenopus oocytes. Actin was used as a loading control; Bottom- Densitometric analysis of represented western blots for Slack quantified as normalized to Actin. Data is expressed as mean ± SEM (n=3 for all groups). Statistics performed using One Way ANOVA followed by multiple comparisons using Tukey’s method (*p < 0.05). (D) Relative reversal potentials (ERev) recorded in injected oocytes in ND-96 bath solution containing 2 mM, 50 mM or 94 mM KCl during depolarizing voltage steps (n=5 per group for water and WT hSLICK groups, n=6 per group for Phe240leu hSLICK group). (E) Representative ISlick current amplitudes. Unpaired t-test, **p<.005 between the 2 mM KCl and 50 mM KCl WT and F240L hSLICK groups, 2 mM KCl and 94 mM KCl WT F240L hSLICK groups, 50 mM KCl and 94 mM KCl WT and F240L hSLICK groups, *p<.05 between the 2mM KCl and 94 mM KCl F240L hSLICK groups. Shown are mean ± SEM. All recordings were performed in 500 μM NFA.
The exclusive selectivity of KNa channels to K+ is vital to their physiological functions. Altered Phe240Leu ion selectivity was tested in Xenopus oocytes by partially replacing external Na+ with K+ over a range of concentrations (Fig. 3D,E). WT channels demonstrated a significant depolarizing shift in ERevs from −40 mV (in 2 mM KCl) to −22 mV (in 50 mM KCl) to 0 mV (in 94 mM KCl) (mean of n=5 for all groups), the latter most ERev in high K+ being evidence of a channel predominantly selective to potassium. In contrast, Phe240Leu channels reversed at positive voltages of 10 mV in 94 mM KCl bath solution (mean of n=6 for all groups). Upon comparing empirically obtained ERev values with theoretical predictions of the Nernst Potential, we found that in high external K+, the WT ERev of ~0 mV is similar to the predicted value of ~−3 mV whereas the F240L ERev of ~+10 mV is significantly depolarized in comparison, indicative of an altered selectivity of mutant channels favouring sodium ions over potassium. Together, these findings indicate loss of predominant selectivity to K+ in Phe240Leu channels and likely, permeability to Na+.
To demonstrate physiological causality of the Phe240Leu mutation towards the epilepsy phenotype, membrane properties of primary neurons overexpressing WT or F240L rSlick were examined. Primary embryonic rat dorsal root ganglion (DRG) neurons were chosen for this experiment as they possess homogenous firing properties resembling adult primary neurons and express the complete subset of membrane proteins necessary for intrinsic membrane plasticity and action potential propagation (Tamsett et al., 2009, Grigaliunas et al., 2002). Our first observation was that F240L rSlick induced neuronal toxicity and resulted in exclusion of the majority of GFP-positive neurons from analysis, wherein exclusion criteria were cell death or inability to successfully seal or break in during patch clamp experiments. This is reflected in that fact that a total of 88 individual attempts (~6.8 %) to record GFP-positive cells were made to obtain the reported n of 6 (Fig. 4D). In comparison, empty pTRACER- and WT rSlick-transfected neurons showed lesser toxicity (n=6 of 25 individual attempts (24%) to record cells), demonstrating pathogenicity of mutant channel over-expression (Fig. 4B and 4C). Secondly, we found the mutation to induce distinct alterations in firing properties. WT rSlick-transfected neurons were unable to fire complete action potentials as would be predicted for the overexpression of a large conductance, rapidly-activating K+ channel (Bhattacharjee et al., 2003); indeed, the rapid activation kinetics of Slick were predicted to affect action potential formation using neuronal simulations (Bhattacharjee et al., 2005). F240L rSlick induced repetitive firing during current injection which was in clear contrast to un-transfected and empty vector-transfected neurons that unequivocally fired a single action potential, as has previously been established as characteristic of the primary DRG neuronal model (Grigaliunas et al., 2002, Toman et al., 2004, Nuwer et al., 2010). These results indicate that the Phe240Leu mutation causes membrane hyperexcitability- a cellular hallmark of the epilepsy phenotype- in DRG neurons. Our in vitro observations in combination with the in vivo patient information demonstrate severe pathogenicity of heterozygous expression of the mutation, limiting further physiological testing of the mutation in an in vivo animal model. Nonetheless, it is conceivable that the functional neuronal findings of increased membrane excitability contribute to the cellular manifestation of the epileptic phenotype.
Fig. 4. Over-expression of the KCNT2 Phe240Leu mutation causes membrane hyperexcitability in primary neurons.

Representative action potential recordings of (A) un-transfected, (B) empty pTRACER-, (C) WT rSlick- and (D) F240L rSlick-transfected neurons. Equal amounts (0.5 μg) of wild-type rSlick or Phe240Leu rSlick or empty pTRACER vector were transfected into embryonic rat DRG neurons plated on 18 mm glass coverslips. Whole cell current clamp was performed 24–48h post-transfection. Current-clamp recordings were performed using an injection of a supra threshold stimulus of 400 pA for 1000ms. Shown are individual traces for a total n=6 for the un-transfected, pTRACER-, WT rSlick- and F240L rSlick-transfected groups.
This work characterizes Phe240Leu as a ‘change-in-function’ de novo mutation. The variant alters a K+-channel up-regulated by Cl− into a Na+-channel down-regulated by Cl−. We present genetic and functional evidence that supports the pathogenicity of the p.(Phe240Leu) variant in the proband. Non-selectivity to K+ has previously been reported with a mutation in the inward rectifying GIRK2 channel in the weaver mouse (Slesinger et al., 1996), however, the present study reports a KCNT2 mutation in humans.
Despite high sequence homology and structural similarities, KCNT1 and KCNT2 channels appear to have very different roles in physiological as well as pathophysiological conditions, likely resulting from distinct, often non-overlapping, patterns of localization within the central and peripheral nervous system (Bhattacharjee et al., 2002, Bhattacharjee et al., 2003, Rizzi et al., 2016, Tomasello et al., 2015). Thus, it follows that while gain-of-function KCNT1 epilepsy mutations reported thus far are proposed to selectively enhance K+ currents resulting in inhibitory neuronal suppression (Barcia et al., 2012, Kim and Kaczmarek, 2014), the Phe240Leu KCNT2 mutation suggests a uniquely contrasting mechanism wherein increased inward INa may affect the precisely synchronized Na+ and K+ exchange that is crucial to normal intrinsic neuronal excitability. Interestingly a gain-of-function mutation in the voltage-gated sodium channel NaV1.2 encoding SCN2A has been shown to cause hyper-excitability in utero (Scalmani et al., 2006) and SCN2A variants-are an important genetic cause of EOEE.
In conclusion, we present empirical evidence for functional pathogenicity of a KNa channel mutation associated with epilepsy in a physiologically relevant cellular model, namely primary cultured neurons. It is therefore readily conceivable that increased SLICK (Phe240Leu)-dependent Na+ currents in selective neuronal populations, such as in excitatory neurons of the hippocampus, contributes to epileptogenesis.
Experimental Procedures
Subjects
The proband was enrolled, with his parents, in a trio exome sequencing project performed at the Garvan Institute as part of a cohort of 31 patients with epileptic encephalopathy, and coordinated by SEALS molecular genetics laboratory and the Departments of Clinical Genetics and Paediatric Neurology at Sydney Children’s Hospital as previously described (Palmer et al., 2015). Informed consent for exome sequencing was obtained from all participants or their guardians as approved by the ethics committee from The Sydney Children’s Hospital Network and the Prince of Wales Hospital Campus, Sydney, Australia (HREC ref no 13/094 and LNR/13/SCHN/112).
Exome sequencing and Sanger confirmation
DNA was extracted from peripheral blood and Next Generation Sequencing was performed using a Nextera rapid capture expanded exome kit, with libraries analysed on an Illumina HiSeq2500. Reads were aligned to Human Genome Reference Sequence Hg19/GRCh37 using BWA MEM, and single nucleotide and short insertion/deletion variants were identified using HaplotypeCaller from GATK. Data filtering and variant prioritization were performed using the GEMINI platform as described previously (Palmer et al., 2015). Bidirectional Sanger sequencing was performed on DNA from the proband and both parents using standard methodology to verify and segregate candidate variants. The ClinVar accession number associated with the Phe240Leu variant is SCV000281970.
Cell Culture
Chinese Hamster Ovary (CHO) cells were cultured at 37 °C in 5% CO2 in an IMDM medium supplemented with 10% FBS, 1% HT supplement (Life Technologies) and 1% penicillin-streptomycin. One day after plating at ~70% confluency, CHO cells were transiently transfected with 1 μg of either WT or mutated rSlick DNA in the expression plasmid pTRACER via lipofectamine (Invitrogen) and cultured for 48 hours. For the 1:1 WT: F240L Slick co-expression experiments, CHO cells were co-transfected with equal amounts (0.5 μg) of WT and mutated rSlick DNA. Cells were plated in 35 mm dishes for all electrophysiology and in 6-well plates for all biochemistry experiments.
Mutagenesis and RNA Preparation
Site-directed mutagenesis at Phe240 was performed using the QuikChange site-directed mutagenesis kit (Stratagene). The mutation Phe240Leu was engineered into rat Slick cDNA in pTRACER, rat Slick cDNA in pOX, human Slick cDNA in psGEM using specific primer sets (Table S1). Pfu Turbo mediated PCR incorporation of the mutation(s) was followed by DpnI digestion to eliminate non-amplified cDNA. Remaining PCR products were transformed into XL-10 Gold Ultracompetent Cells (Agilent Technologies) and mini-prepped (Qiagen Spin mini-prep kit). The final products were sequence verified to confirm the sole mutation in the sequence to be Phe240Leu. Rat and human Slick cRNA was then prepared using the T3 and T7 mMessage kit, respectively (Ambion). Rat Kcnt2 cDNA in the pOX vector was kindly provided by J. Kronengold (Yale University) and human KCNT2 cDNA in the psGEM vector was kindly provided by M. Sanguinetti (University of Utah).
Preparation of Oocytes
Xenopus laevis oocytes from mature females were obtained and digested in OR3 solution containing collagenase. De-folliculated oocytes (stages V–VI) were injected with 75 ng of rSlick (WT or Phe240Leu) cRNA or 150 ng hSLICK (WT or Phe240Leu) cRNA or water alone with the Nanoject Microinjection system (Drummond Scientific Co.). Oocytes were incubated at 18°C in OR3 medium for 2–3 days prior to functional analyses.
DRG neuronal culture
Female and adult timed pregnant Sprague Dawley rats (Harlan Laboratories) were used for all experiments. Animals were housed singly in a temperature and humidity controlled animal facility on a 12 h: 12 h light: dark schedule with food and water freely available. All procedures were approved by the University at Buffalo Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the use of laboratory animals in research. On the day of the dissection, rats were euthanized by CO2 asphyxiation and E15 embryos were extracted. DRG were dissected from the embryos and enzymatically digested with Trypsin (2.5 mg/ml) at 37 C for 50 min, followed by mechanical dissociation and plating. DRG neurons were plated onto poly-D-lysine (Sigma; 100 mg/ml) and laminin (Invitrogen; 3 mg/ml) coated coverslips. Cells were maintained at 37 °C in a 7% CO2 humidified incubator in serum-free medium, comprised of the trophic factors N2 (Gemini Bio products; 1%), L-Glutamine (Invitrogen; 200 mg/ml), and Nerve Growth Factor (NGF) (Harlan Laboratories; 100 ng/ml) in 50% DMEM and 50% F-12 (Nuwer et al., 2010). For two days after the day of the dissection, cells were cultured in C2 media containing the anti-mitotic cytosine β-D-arabinofuranoside hydrochloride (Sigma; 3 mM), followed by two days of recovery before use in experiments. Transient transfection of neurons with empty pTRACER vector, WT rSlick pTRACER or F240L rSlick pTRACER was done on day 5 of culture using lipofectamine 2000 (Invitrogen). Recordings were taken 24–48 hours after transfection.
Electrophysiology
Two-Electrode Voltage-Clamp
Oocytes were voltage-clamped in a whole-cell mode configuration using a two-microelectrode oocyte clamp amplifier (OC-725A, Warner Instruments Corp.), and currents were digitized and recorded at room temperature. Microelectrodes with resistances of 0.5–2 MΩ (when filled with 1 M KCl) were fabricated from 1.5 mm o.d. borosilicate glass tubing (TW150-4, World Precision Instruments) using a two-stage puller (Kopf Instruments) and filled with 1 M KCl. The bath solution ND-96 contained (mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, pH 7.4. All recordings were performed in ND-96 containing 500 μM Niflumic acid (NFA) to exclude endogenous [Cl−]i conductance expressed in Xenopus oocytes; NFA is known to maximally activate Slick KNa channels (EC50 ~2.1 mM), but at much lower concentrations, it blocks Ca2+-activated [Cl−]i channels endogenously expressed by Xenopus oocytes (IC50 ~17 μM) (White M M., 1990, Dai et al., 2010). Oocytes were voltage clamped at a holding potential of −80 mV, and voltage steps from −100 mV to +40 mV were applied in 10 mV steps. Data were digitized and analysed using pCLAMP 10.2 and analysed using Clampex (Molecular Devices). Current-voltage relationships were determined by measuring currents at the end of the test pulses. Current traces were not leak or capacitance subtracted. Oocytes with resting membrane potentials more positive than −30 mV or with leaky currents were excluded.
Whole Cell Patch Clamp
Glass electrodes were pulled using a horizontal pipette puller (Sutter Instrument Company) and fire polished to be of 5–12 MΩ resistance. Whole-cell voltage clamp recordings were performed on transfected CHO cells; whole-cell current clamp recordings were performed on transfected DRG neurons. Currents were recorded in voltage clamp mode at a holding potential of −70 mV, and voltage steps from −120 mV to +120 mV were applied in 20 mV steps. A current clamp protocol consisting of depolarizing steps in increments of 10 pA from −10 to 200 pA (20 ms duration) was used to record action potentials. Firing frequency was examined by measurement of repetitive discharge of each cell upon injecting a supra threshold stimulus of 400 pA for 1000ms. Pipette offset was zeroed to account for tip potentials and capacitance compensation was used to limit series resistance errors.
The following solutions were used in these experiments.
For CHO cells:
Low-chloride pipette solution (in mM): 124 K-Gluconate, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP at pH 7.2.
High-chloride pipette solution (in mM): 124 KCl, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP at pH 7.2.
Bath Solution (in mM): 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 Glucose.
For DRG neurons:
Pipette solution (in mM): 124 K-gluconate, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP, pH 7.2.
Bath solution (in mM): 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 15.6 HEPES, and 10 glucose, pH 7.4.
All data was acquired using the Axopatch 200B amplifier (Molecular Devices) and Multiclamp-700B (Molecular Devices), digitized and filtered at 2 kHz. Data acquisition was monitored and controlled using pClamp 10.2 and analysed using Clampex (Molecular Devices). Current-voltage relationships were determined by measuring currents at the end of the test pulses. Leaky currents were excluded from analysis.
Membrane Biotinylation Assay
Cell surface biotinylation on Xenopus oocytes was performed with the Pierce Cell Surface Protein Isolation Kit (Thermo Scientific) according to the manufacturer’s protocol. Briefly, 20 oocytes per group were washed with phosphate-buffered solution (PBS) and incubated with EZ-LINK Sulfo-NHS-SS-biotin for 30 min at 4 °C followed by quenching solution. Oocytes were homogenized in lysis buffer (500 μl) containing protease inhibitor cocktail. An aliquot of the lysate was saved for Western blotting for total protein. Biotinylated SLICK was isolated with NeutrAvidin agarose gel, eluted by sample buffer containing dithiothreitol. The resulting elute was loaded onto a Ready Gel and western analysis was performed to detect membrane SLICK with monoclonal anti-Slick (UC Davis/NIH NeuroMab Clone NC 11/33). Signal was detected using horseradish peroxidase-conjugated mouse secondary antibody and chemiluminescent substrate kit (KPL). β-actin was used as a loading control.
Statistics
All electrophysiological analysis was performed using Clampfit 10 (Axon Instruments) and Origin 8.0 (Microcal Software Inc.). All statistical tests were performed using Prism (GraphPad). Data are shown as mean ± SEM. Confidence levels were calculated using Student’s t test and ANOVA. Sample size for all experiments was least recordings required to detect significant differences and ensure reproducibility.
Supplementary Material
Highlights.
We report a variant in the KNa channel KCNT2 that causes a human phenotype.
Phe240Leu channels are no longer predominantly permissive to only K+.
Expressing Phe240Leu in primary neurons induces membrane hyperexcitability.
‘Change-of-function’ establishes pathogenicity of mutation in epileptic encephalopathy.
Acknowledgments
We thank the patient and his family for participating in the study; the clinical neurology, general paediatric and clinical genetics teams involved with his clinical care and support; Michael Buckley from SEALS Genetics Laboratory and the technical assistance of Glenda Mullan, William Lo, George Elakis and Corrina Walsh from SEALS Genetics Laboratory; E. Myers, A. Marshall and M.D. Parker (University at Buffalo) for providing us with Xenopus oocytes; J. Kronengold (Yale University) for the rat SLICK construct in pOX; M. Sanguinetti (University of Utah) for the human SLICK construct in psGEM. Grant acknowledgements The research was supported by the National Institute of Health, Grant NS078184 (to A.Bh.), the Kinghorn Foundation, and the National Health and Medical Research Council Grant GNT11149630 (to E.P.) and GNT0512123 (to T.R.).
Footnotes
Author Contributions
S.G. performed all experiments in CHO cells, Xenopus oocytes and primary neuronal cultures. E.P. performed the exome analysis, identified the KCNT2 variant and initiated collaboration with A.Bh. and team. G.D.S. helped perform electrophysiological recordings in CHO cells. T.R., A.By, R.S., E.K., E.P and T.K. designed the exome study, and recruited patients. T.R., M.D., M.C. and Y.Z. designed the exome sequencing protocols and defined bioinformatic analysis pathways. K.Y., P.M., J.T. and KR.D performed exome sequencing. T.R., R.S. and E.K. assisted in performing genomic analyses. E.P. and T.K. collected phenotypic information. R.M. recruited patients. E.K. performed Sanger sequencing and supervised assessment of exome sequencing analysis. T.R. developed genomic consent forms and ensured IRB accreditation for the study. M.E.D helped perform all Xenopus oocyte experiments. A.Bh. designed experiments in CHO cells and Xenopus oocytes. The initial draft of the manuscript was written by A.Bh. S.G. and E.P. and edited by A.By, E.K., R.S.,T.R and M.E.D. All authors critically revised and gave final approval to this manuscript.
Accession Numbers
The ClinVar accession number associated with the Phe240Leu variant is SCV000281970.
Competing Financial Interest
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barcia G, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–9. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Berkovic SF. Genetics of Epilepsy in Clinical Practice. Epilepsy Curr. 2015;15:192–6. doi: 10.5698/1535-7511-15.4.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol. 2002;454:241–54. doi: 10.1002/cne.10439. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. 2003;23:11681–91. doi: 10.1523/JNEUROSCI.23-37-11681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, von Hehn CA, Mei X, Kaczmarek LK. Localization of the Na+-activated K+ channel Slick in the rat central nervous system. J Comp Neurol. 2005;484:80–92. doi: 10.1002/cne.20462. [DOI] [PubMed] [Google Scholar]
- Chen H, et al. The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J Neurosci. 2009;29:5654–65. doi: 10.1523/JNEUROSCI.5978-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Garg V, Sanguinetti MC. Activation of Slo2.1 channels by niflumic acid. J Gen Physiol. 2010;135:275–95. doi: 10.1085/jgp.200910316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Gardner A, Garg V, Sanguinetti MC. Structural basis of ion permeation gating in Slo2.1 K+ channels. J Gen Physiol. 2013;142:523–42. doi: 10.1085/jgp.201311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigaliunas A, Bradley RM, MacCallum DK, Mistretta CM. Distinctive neurophysiological properties of embryonic trigeminal and geniculate neurons in culture. J Neurophysiol. 2002;88:2058–74. doi: 10.1152/jn.2002.88.4.2058. [DOI] [PubMed] [Google Scholar]
- Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, Tang S, Helbig I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016 doi: 10.1038/gim.2015.186. [DOI] [PubMed]
- Heron SE, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188–90. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- Hino-Fukuyo N, Haginoya K, Iinuma K, Uematsu M, Tsuchiya S. Neuroepidemiology of West syndrome and early infantile epileptic encephalopathy in Miyagi Prefecture, Japan. Epilepsy Res. 2009;87:299–301. doi: 10.1016/j.eplepsyres.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci. 1998;1:462–9. doi: 10.1038/2176. [DOI] [PubMed] [Google Scholar]
- Khan S, Al Baradie R. Epileptic encephalopathies: an overview. Epilepsy Res Treat. 2012;2012:403592. doi: 10.1155/2012/403592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GE, Kaczmarek LK. Emerging role of the KCNT1 Slack channel in intellectual disability. Front Cell Neurosci. 2014;8:209. doi: 10.3389/fncel.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. 2016;53:217–25. doi: 10.1136/jmedgenet-2015-103508. [DOI] [PubMed] [Google Scholar]
- Lim ET, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HC, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. 2014;23:3200–11. doi: 10.1093/hmg/ddu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. doi: 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- Nuwer MO, Picchione KE, Bhattacharjee A. PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J Neurosci. 2010;30:14165–72. doi: 10.1523/JNEUROSCI.3150-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeschger FM, Wang WZ, Lee S, Garcia-Moreno F, Goffinet AM, Arbones ML, Rakic S, Molnar Z. Gene expression analysis of the embryonic subplate. Cereb Cortex. 2012;22:1343–59. doi: 10.1093/cercor/bhr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer EE, et al. Asparagine Synthetase Deficiency causes reduced proliferation of cells under conditions of limited asparagine. Mol Genet Metab. 2015;116:178–86. doi: 10.1016/j.ymgme.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi S, Knaus HG, Schwarzer C. Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J Comp Neurol. 2016;524:2093–116. doi: 10.1002/cne.23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalmani P, Rusconi R, Armatura E, Zara F, Avanzini G, Franceschetti S, Mantegazza M. Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci. 2006;26:10100–9. doi: 10.1523/JNEUROSCI.2476-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA, Patil N, Liao YJ, Jan YN, Jan LY, Cox DR. Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron. 1996;16:321–31. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Hansen A, Sanguinetti MC. Hydrophobic interactions between the S5 segment and the pore helix stabilizes the closed state of Slo2.1 potassium channels. Biochim Biophys Acta. 2016;1858:783–92. doi: 10.1016/j.bbamem.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–34. doi: 10.1523/JNEUROSCI.0859-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada MA, Stople K, Hammami Bomholtz S, Meinild AK, Poulsen AN, Klaerke DA. Cell volume changes regulate slick (Slo2.1), but not slack (Slo2.2) K+ channels. PLoS One. 2014;9:e110833. doi: 10.1371/journal.pone.0110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toman RE, Payne SG, Watterson KR, Maceyka M, Lee NH, Milstien S, Bigbee JW, Spiegel S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J Cell Biol. 2004;166:381–92. doi: 10.1083/jcb.200402016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello DL, Gancarz-Kausch AM, Dietz DM, Bhattacharjee A. Transcriptional Regulation of the Sodium-activated Potassium Channel SLICK (KCNT2) Promoter by Nuclear Factor-kappaB. J Biol Chem. 2015;290:18575–83. doi: 10.1074/jbc.M115.643536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MMAM. Niflumic and Flufenamic Acids are Potent Reversible Blockers of Ca2ı-Activated C1 Channels in Xenopus Oocytes. Molecular Pharmacology. 1990;37:720–724. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.