

Summary
Influenza A virus (IAV) is an RNA virus that is cytotoxic to most cell types in which it replicates. IAV activates the host kinase RIPK3, which induces cell death via parallel pathways of necroptosis, driven by the pseudokinase MLKL, and apoptosis, dependent on the adaptor proteins RIPK1 and FADD. How IAV activates RIPK3 remains unknown. We report that DAI (ZBP-1/DLM-1), previously implicated as a cytoplasmic DNA sensor, is essential for RIPK3 activation by IAV. Upon infection, DAI recognizes IAV genomic RNA, associates with RIPK3, and is required for recruitment of MLKL and RIPK1 to RIPK3. Cells lacking DAI or containing DAI mutants deficient in nucleic acid binding are resistant to IAV-triggered necroptosis and apoptosis. DAI-deficient mice fail to control IAV replication and succumb to lethal respiratory infection. These results identify DAI as a link between IAV replication and RIPK3 activation, and implicate DAI as a sensor of RNA viruses.
Graphical abstract
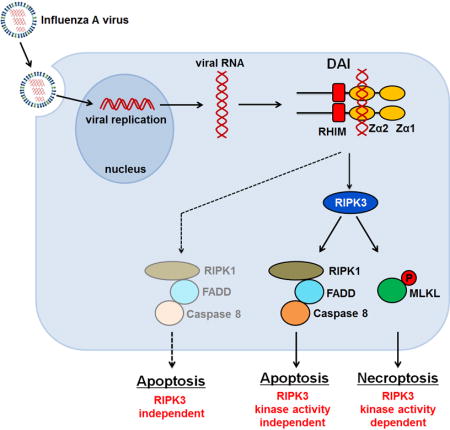
Introduction
Influenza A virus (IAV) replication in cultured cells and in vivo is typically accompanied by the death of the infected cell. We have recently shown that, in IAV-infected murine fibroblasts and airway epithelial cells, cell death is driven by RIPK3 (Nogusa et al., 2016). Upon IAV infection, RIPK3 nucleates a ‘necrosome’ complex, minimally containing MLKL, RIPK1, and FADD, which then activates both necroptosis, mediated by MLKL, and apoptosis, via a RIPK1-FADD-caspase-8 axis. While the formation of the RIPK3 necrosome and consequent activation of cell death require active IAV replication, the mechanism by which virus biology stimulates assembly of the necrosome and triggers cell death remains unknown. Our previous work (Nogusa et al., 2016) has ruled out roles for known RNA virus sensing pathways, indicating that some as-yet unknown host- or virus-encoded factor bridges replicating IAV to RIPK3 activation.
While conducting experiments to identify this factor, we discovered that the host protein DAI (also known as ZBP1/DLM-1) was required for RIPK3 activation and cell death in IAV-infected murine cells. DAI contains two tandem Z-form nucleic-acid binding domains (Zα domains; Zα1 and Zα2) towards its N-terminus, and is one of four known vertebrate proteins with a RIP homology interaction motif (RHIM; the other three are RIPK1, RIPK3 and the TLR3/4 adaptor protein TRIF) (Kaiser et al., 2008; Rebsamen et al., 2009). DAI was first implicated in host antiviral innate-immune responses as a cytosolic DNA sensor in the pathway leading to production of type I IFNs (Takaoka et al., 2007). However, DAI is not essential for the type I IFN response to most DNA viruses (Ishii et al., 2008); instead, we have previously found that DAI is a potent effector of necroptosis following infection of cells with murine cytomegalovirus (MCMV, a β-herpesvirus with a DNA genome), when its virus-encoded inhibitor of necroptosis, vIRA, is disabled (Upton et al., 2010, 2012).
Here, we show that DAI recognizes IAV RNA by a mechanism requiring the second of its Zα domains, and nucleates a RHIM-dependent RIPK3-containing necrosome. DAI also mediates IAV-induced RIPK3-independent apoptosis. Consequently, cells lacking DAI are remarkably resistant to IAV-triggered lysis, and DAI-deficient mice are hyper-susceptible to lethal infection by this virus. These findings identify DAI as a central mediator of IAV-driven cell death, and implicate this protein as a sensor of RNA viruses.
Results
DAI is required for IAV-induced cell death
In a focused screen for mediators of IAV-activated cell death, we discovered that murine embryo fibroblasts (MEFs) from DAI-deficient (zbp1−/−) animals were extraordinarily resistant to death triggered by this virus. When evaluated over a period of 24 hr, near-confluent monolayers of primary, early-passage MEFs from two separately-housed zbp1−/− mouse colonies uniformly displayed >85% viability when infected with the IAV strain A/Puerto Rico/8/1934 (PR8, H1N1), while similarly-infected zbp1+/+ MEFs manifested extensive cell death by this time (Fig. 1A, B). Notably, zbp1−/− MEFs were also resistant to cell death activated by seasonal H1N1 and H3N2 strains of IAV, as well as by influenza B virus (IBV), but not by another virus with a negative-sense RNA genome (vesicular stomatitis virus; Rhabdoviridae) (Fig. S1A). Zbp1−/− MEFs displayed levels of death effector proteins equivalent to controls (Fig. S1B), and remained susceptible to necroptosis induced by the combination of TNF-α, cycloheximide, and zVAD (TCZ) (Fig. 1A,B). IAV entry, as measured by GFP-positivity 18 hr post-infection (p.i.) with recombinant PR8 expressing GFP [PR8-GFP; (Manicassamy et al., 2010)], was equivalent between wild-type and zbp1−/− MEFs (Fig. 1C). Virus proteins NP and NS1 were also produced at similar levels and with equivalent kinetics in PR8-infected wild-type and zbp1−/− MEFs (Fig. 1D).
Figure 1. DAI is essential for IAV-induced cell death in MEFs and alveolar epithelial cells.
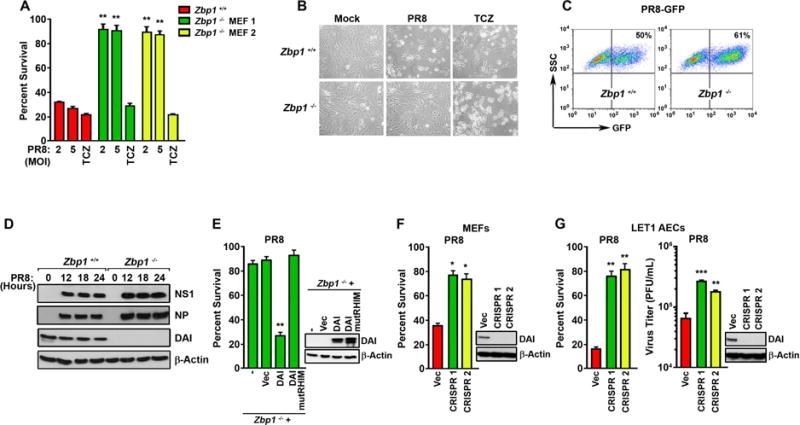
(A) Zbp1+/+ or zbp1−/− MEFs from two separately-maintained colonies (MEF 1 and MEF 2) were infected with PR8 (MOI=2 and 5), or treated with the combination of TNF-α [50ng/ml] + cycloheximide [250ng/ml] + zVAD [50uM] (TCZ) and cell viability was determined 24 hr p.i. (B) Photomicrographs of zbp1+/+ and zbp1−/− MEFs infected with PR8 (MOI=2) or treated with TCZ for 24 hr. (C) FACS analysis of zbp1+/+ and zbp1−/− MEFs infected with PR8-GFP (MOI=2) for 18 hr. The y-axis shows side scatter. Mock-infected cells showed negligible (<0.1%) GFP-positivity. (D) Lysates from zbp1+/+ and zbp1−/− MEFs infected with PR8 (MOI=2) were examined for expression of NS1, NP, and DAI. (E) Immortalized zbp1−/− MEFs reconstituted with empty vector (Vec), full-length murine DAI (DAI), or murine DAI with mutant RHIM domain (amino acids 192–195 IQIG to AAAA) (DAI mutRHIM) were infected with PR8 (MOI=2) and cell viability was determined 24 hr p.i. Expression of WT or mutant DAI in these cells is shown to the right. (F) WT MEFs in which DAI expression was ablated by CRISPR/Cas9 targeting were infected with PR8 (MOI=2) and cell viability was determined 24 hr p.i. Ablation of DAI expression was confirmed by immunoblotting (right). (G) LET1 cells in which DAI expression was ablated by CRISPR/Cas9 targeting were infected with PR8 (MOI=2) and cell viability was determined 12 hr p.i. (left). In parallel, progeny virion output from these cells was determined 30 hr p.i. (right). Viability data shown in this figure are representative of at least three independent experiments. Error bars represent mean +/− SD. *p<0.05, **p<0.005, ***p<0.0005. See also Fig. S1.
While immortalized zbp1−/− MEFs were resistant to IAV-induced cell death, reintroduction of wild-type DAI, but not a mutant of DAI (DAI mutRHIM), carrying a tetra-alanine substitution of the core RHIM sequence IQIG (aa 192–195) (Kaiser et al., 2008) into these cells fully restored susceptibility to IAV-mediated death (Fig. 1E). In a corollary experiment, CRISPR/Cas9-based ablation of zbp1 expression in wild-type MEFs rendered these cells resistant to IAV-induced cell death (Fig. 1F). In neither case was susceptibility to TNF-α-induced necroptosis affected (Fig. S1C,D).
To extend these findings to a cell type relevant to IAV replication in vivo, we ablated zbp1 expression in murine LET1 cells and infected them with IAV. The LET1 cell line (Rosenberger et al., 2014) is derived from type I alveolar epithelium, a primary early target of IAV in the lung. These cells, unlike MEFs, support the complete IAV lifecycle, and produce progeny virions upon infection (Rosenberger et al., 2014). Two distinct sgRNAs to murine zbp1 both reduced IAV-triggered cell death by ~60% in LET 1 cells (Fig.1G, left), comparable to the protection afforded by ablation of ripk3 itself (Nogusa et al., 2016). LET1 cells lacking zbp1 also produced significantly more progeny IAV than controls over a 30 hr timeframe (Fig. 1G, right). Notably, unlike cells lacking ripk3 (Nogusa et al., 2016), zbp1-ablated LET1 cells were still susceptible to TNF-α-induced necroptosis (Fig. S1E).
DAI associates with RIPK3 and mediates both apoptosis and necroptosis in IAV-infected cells
IAV, uniquely among viruses studied thus far, activates both apoptosis and necroptosis downstream of RIPK3 (Nogusa et al., 2016). It does so by nucleating a RIPK3-containing ‘necrosome’ complex that also comprises MLKL, which mediates necroptosis, as well as RIPK1 and FADD, which activate apoptosis (Nogusa et al., 2016). We found that IAV infection induced the robust association of DAI with RIPK3, and that DAI was essential for recruitment of both MLKL and RIPK1 to RIPK3 (Fig. 2A). Strikingly, zbp1−/− MEFs were completely defective in both MLKL and caspase-8 activation upon IAV infection (Fig. 2B), although necrosome assembly (not shown) and activation of MLKL (Fig. 2B) in response to TNF-α occurred normally in the absence of DAI. LET1 cells in which expression of zbp1 was ablated by CRISPR/Cas9 gene-targeting also failed to support either MLKL or caspase-8 activation following infection by IAV, although activation of MLKL by TNF-α was unaffected (Fig. S2A). Loss of RIPK3, expectedly, resulted in abolishment of MLKL activation in both MEFs and LET1 cells following either infection by IAV or exposure to TNF-α (Figs. 2B, S2A). Reintroduction of wild-type DAI, but not DAI mutRHIM, into immortalized zbp1−/− MEFs restored both phosphorylation of MLKL and cleavage of caspase-8 in response to IAV (Fig. 2C). In accordance with these findings, wild-type DAI, but not DAI mutRHIM, robustly complexed with RIPK3 following IAV infection (Fig. 2D). Together, these results demonstrate that DAI is essential for activation of RIPK3 in the pathways leading to stimulation of MLKL and caspase-8, and requires its RHIM to associate with RIPK3 and activate these pathways.
Figure 2. DAI is required for IAV induced formation of RIPK3-containing necrosome and activation of MLKL and caspase-8.
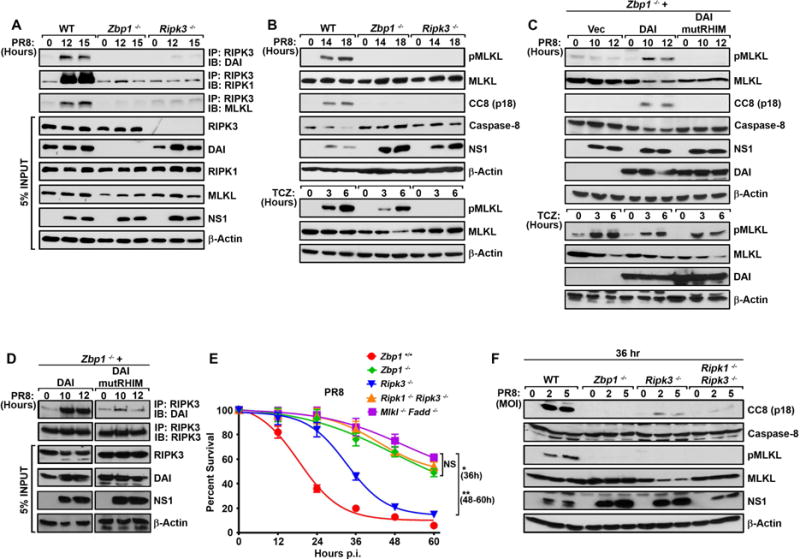
(A) Wild-type (WT), zbp1−/−, and ripk3−/− MEFs were infected with PR8 (MOI=2) and anti-RIPK3 immunoprecipitates were examined for DAI, RIPK1 and MLKL. Whole-cell extract (5% input) was examined in parallel for RIPK3, DAI, RIPK1, MLKL and IAV NS1 proteins. (B) WT, zbp1−/−, and ripk3−/− MEFs were infected with PR8 (MOI=2) and examined for phosphorylated (p) MLKL and cleaved caspase-8 (CC8) p18 subunit at the indicated times p.i. In parallel, WT, zbp1−/−, and ripk3−/− MEFs were treated with TCZ and examined for phosphorylated MLKL. (C) Zbp1−/− MEFs reconstituted with empty vector (Vec), full-length murine DAI (DAI), or DAI with a mutated RHIM domain (DAI mutRHIM) were infected with PR8 (MOI=2) and examined for phosphorylated MLKL and cleaved caspase-8 at the indicated times p.i. In parallel, reconstituted cells were treated with TCZ and examined for expression of phosphorylated MLKL. (D) Anti-RIPK3 immunoprecipitates from PR8 (MOI=2)-infected zbp1−/− MEFs reconstituted with DAI or DAI mutRHIM were examined for presence of DAI. Whole-cell extract (5% input) was examined in parallel for RIPK3, DAI and IAV NS1 proteins. (E) Kinetics of cell death after PR8 infection (MOI=2) of WT, zbp1−/−, ripk3−/−, ripk1−/−ripk3−/− double knockout and mlkl−/− fadd−/− double knockout MEFs. Data are representative of three independent experiments. (F) Whole cell extracts from WT, zbp1−/−, ripk3−/− and ripk1−/−ripk3−/− double knockout MEFs infected with PR8 (MOI=2 or 5) were examined for cleaved caspase-8 (CC8). Error bars represent mean +/− SD. *p<0.05, **p<0.005. See also Figure S2.
We have previously shown that IAV triggers a delayed RIPK3-independent pathway of apoptosis that relies on FADD and caspase-8 and is activated between 24–36 hr p.i (Nogusa et al., 2016). Cells deficient in both RIPK3 and FADD, RIPK3 and caspase-8, or MLKL and FADD, continue to survive past 36 hr, whereas >50% of ripk3−/− MEFs die by this time point. In fact, these double knockout MEFs (e.g., fadd−/−mlkl−/− MEFs) survive for up to 60 hr p.i with no obvious abatement of virus replication (Nogusa et al., 2016) (Fig. 2E). We noticed that zbp1−/− MEFs were significantly more resistant to IAV than ripk3−/− MEFs, phenocopying the resistance of MEFs doubly-deficient in apoptosis and necroptosis pathways (Fig. 2E) and suggesting that DAI may lie upstream of RIPK3-independent apoptosis as well. In such a scenario, we reasoned that, when RIPK3 is absent, DAI may instead employ RIPK1 as a RHIM-containing adaptor to link replicating IAV to FADD and caspase-8; thus, co-ablation of RIPK1 with RIPK3 would be needed to abrogate all major pathways of IAV-triggered programmed cell death. To test this idea, we evaluated the kinetics of IAV-induced cell death in ripk1−/−ripk3−/− double knockout MEFs, and found that these cells were as resistant to IAV-induced death as zbp1−/− MEFs or fadd−/−mlkl−/− double knockout MEFs, each of which survived IAV beyond 36 hr p.i. (Fig. 2E). Moreover, delayed caspase-8 activity seen in ripk3−/− MEFs and completely absent in zbp1−/− MEFs was largely abolished in ripk1−/−ripk3−/− double knockout MEFs (Fig. 2F). In agreement with these findings, RIPK1 associated with DAI in IAV-infected ripk3−/− MEFs between 24 and 36 hr p.i. (Fig. S2B).
DAI senses IAV genomic RNA
As DAI can bind nucleic acids (Takaoka et al., 2007), we theorized it may directly sense viral RNAs and link proliferating IAV to RIPK3. DAI possesses two tandem nucleic acid-binding Zα domains towards its N-terminus which, for convenience, we call Zα1 (a.a. 8–72) and Zα2 (aa 84–147) (Fig. 3A). Zα2 is sometimes referred to as ‘Zβ’ in previous studies, but Zα2 is a more authentic descriptor of this domain that distinguishes it from the functionally-distinct Zβ domain of ADAR1 (Athanasiadis, 2012). These domains are followed by the RHIM (aa 184–200) and a C-terminal half containing a region (aa 314–411) shown to interact with TBK-1/IRF-3 in the pathway leading to production of type I IFNs (Takaoka et al., 2007) (Fig. 3A). While this manuscript was in revision, Kuriakose and colleagues (Kuriakose et al., 2016) reported an association between DAI and the IAV proteins NP and PB1 that also mapped to a section of DAI (a.a. 311–377) almost completely contained within its TBK-1/IRF-3 binding region. Deletion of the C-terminal TBK-1/IRF-3/NP/PB-1 binding module of DAI did not significantly diminish the magnitude of cell death (Fig. 3B) or capacity of RIPK3 to phosphorylate MLKL (Fig. 3C) in IAV-infected cells at 24 hr. Similarly, singly deleting Zα1 had little effect on the extent of IAV-induced cell death (Fig. 3B) or RIPK3 activation (Fig 3C). Each deletion, however, modestly impeded the kinetics with which IAV killed the infected cell over this time course (Fig. S3A). In contrast, deleting both the Zα1 and Zα2 domains abolished cell death and RIPK3 activity (Fig. 3B,C). Indeed, point mutations in two amino acids (N122 and Y126) in Zα2 of DAI, shown to be essential for binding to Z-DNA (Ha et al., 2008), and analogous to residues in ADAR1 Zα known to contact Z-RNA (Placido et al., 2007), completely nullified the ability of DAI to induce cell death (Fig. 3B) or stimulate RIPK3 (Fig. 3C) upon IAV infection. Each of these mutants was expressed at levels comparable to wild-type DAI (Fig. S3B), and none of the mutations affected TNF-α-induced necroptosis signaling in these cells (Fig. 3C, S3C). Caspase-8 activity downstream of RIPK3 also required a functional Zα2 domain (not shown). Together, these results implicate the second Zα domain of DAI as critical for IAV-driven cell death responses, and strongly suggest that this domain interacts with IAV RNA to initiate activation of DAI and subsequent signaling to RIPK3.
Figure 3. DAI senses IAV genomic RNA.
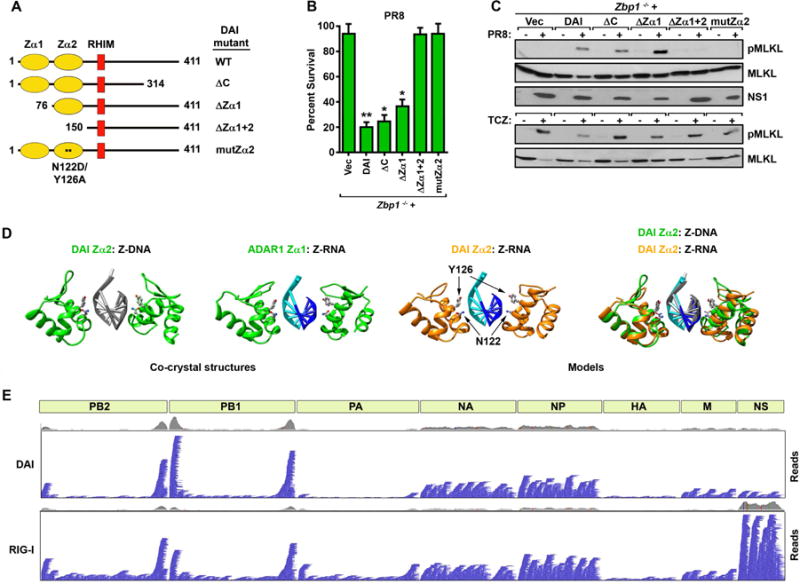
(A) Schematic of DAI and its mutants used in this figure. (B) Immortalized zbp1−/− MEFs reconstituted with wild-type (WT) DAI or with the indicated mutants were infected with PR8 (MOI=2) and cell viability was measured 24 hr p.i. Data are representative of three independent experiments. (C) Immortalized zbp1−/− MEFs reconstituted with WT DAI or with the indicated mutants were infected with PR8 for 12 hr (MOI=2, top) or treated with TCZ for 6 hr (bottom) and examined for phosphorylated (p) MLKL. (D) Similarity of Zα1 and Zα2 domains bound to either Z-DNA or Z-RNA substrates. The known structures of human DAI Zα2 dimers (green ribbons) bound to Z-DNA (PDB code 3EYI, first panel) and ADAR1 Zα1 dimers (green ribbons) bound to Z-RNA (PDB code 2GXB, second panel). A homology model of murine DAI Zα2 dimers (orange ribbons) bound to Z-RNA, based on the above co-crystal structures, is shown in the third panel, with the location of N122 and Y126 depicted by arrows. A superposition of mDAI Zα2 (orange ribbons) bound to Z-RNA (cyan and blue ribbons) compared to the known structure of hDAI Zα2 (green ribbons) bound to Z-DNA (gray ribbons) is shown in the fourth panel. The conserved N and Y residues of the mDAI Zα2:Z-RNA complex are shown in ball-and-stick representation, while the corresponding residues in the hDAI Zα2:Z-DNA complex are displayed as sticks. (E) Integrated Genome Viewer representation of captured IAV genomic (negative polarity) reads from FLAG-immunoprecipitations of PR8-infected 293T cells expressing either FLAG-DAI (top) or FLAG-RIG-I (bottom). Each horizontal blue bar represents a single 150nt read and the position where it aligns relative to an IAV gene segment, schematically shown as light green rectangles. The grey histogram is a synopsis of total coverage in any given position. Error bars represent mean +/−SD. *p<0.05, **p<0.005. See also Fig. S3.
To our knowledge, DAI has not previously been shown to recognize RNA. But earlier studies have revealed that the Zα domain, although first identified as a DNA-binding module (Herbert et al., 1997), can also bind dsRNA in its left-handed (Z-) conformation (Brown et al., 2000; Placido et al., 2007). As a prelude to determining if DAI bound RNA, we first modeled a putative interaction between DAI Zα2 and RNA using as templates the published co-crystal structures of Zα domains bound to Z-form DNA or RNA. The Zα:Z-DNA and Zα:Z-RNA structures bear remarkable similarity to each other (Fig 3D, left two panels). The conserved residues in these structures that correspond to N122 and Y126 in the nucleic acid recognition helix of mDAI Zα2 are in identical positions, and very similar rotamers make contact with both Z-DNA and Z-RNA (Fig. 3D, two left panels). Correspondingly, our model of mDAI Zα2 complexed with Z-RNA strongly indicates that N122 and Y126 of mDAI Zα2 are capable of making identical contacts to either Z-form nucleic acid (Fig.3D, third panel). Indeed, when we superimposed our mDAI Zα2:RNA model (orange) over the known structure of hDAI Zα2:DNA (green), both N122 and Y126 and were in comparable rotamer conformations to the analogous residues in the hDAI Zα2:DNA structure (Fig. 3D, fourth panel). Our models are in general agreement with previous comparisons of Zα domains bound to Z-DNA or Z-RNA (Ha et al., 2008; Placido et al., 2007), indicating that the DAI Zα2 domain is similarly suited to bind either left-handed Z-form nucleic acid.
To test if DAI bound IAV RNA, and to identify these RNAs, we eluted RNA co-precipitating with FLAG-tagged DAI from IAV-infected cells, and examined eluates for the presence of IAV RNA. In parallel, we evaluated RNA co-precipitating with FLAG-RIG-I, a known sensor of IAV RNA, as a positive control. We readily detected IAV-specific vRNA mapping to all eight IAV gene segments in DAI immunoprecipitates (Fig. 3E, top), in a pattern that bore striking resemblance to the spectrum of vRNAs co-precipitating with RIG-I (Fig. 3E, bottom) and previously shown to be enriched in IAV genomic material packaged into defective interfering (DI) particles (Baum et al., 2010).
It is noteworthy that, while we were able to obtain significant RNA yields from IAV-infected cells expressing wild-type (or mutRHIM) DAI, the amount of eluted RNA from similarly-infected cells expressing Zα2 mutants of DAI was far lower, and comparable to background yield from vector controls (Fig. S3D), despite equivalent expression of each FLAG-tagged DAI construct (Fig. S3E). Importantly, only wild-type DAI and DAI mutRHIM, but not DAI Zα2 mutants, associated with IAV sub-genomic vRNA seen in DI particles (Fig. S3F). By RT-qPCR, we also found that wild-type DAI and DAI mutRHIM bound IAV genomic RNAs (e.g., NA, NP vRNAs) equivalently to RIG-I, while binding to these RNAs by Zα2 mutants of DAI was markedly (~90%) lower (Fig. S3G). Taken together, these results demonstrate that DAI binds IAV genomic RNA in a manner requiring its Zα2 domain, and likely senses both IAV DI particle sub-genomes, as well as full-length segment vRNAs, that are also recognized by RIG-I.
DAI is required for protection against IAV in vivo
To examine the role of DAI in host defense to IAV, we infected zbp1−/− mice with PR8 at a dose (1000 EID50) that was not lethal to wild-type littermate controls, and monitored the survival of these mice. All wild-type controls lost weight over the first week, but eventually recovered from infection by 15–18 days p.i. (d.p.i.) (Fig. 4A,B). In contrast, ~80% of zbp1−/− mice succumbed to IAV infection between 9 and 12 d.p.i. (Fig. 4A,B). Next, we measured virus replication in lungs of mice infected with an even lower dose of PR8 (750 EID50) to delay some the lethality associated with zbp1 loss. Progeny virion production was not notably different between lungs of wild-type or zbp1−/− mice 6 d.p.i., whereas virus was essentially cleared from wild-type lungs by 9 d.p.i,. titers remained markedly elevated in lungs from zbp1−/− animals (Fig. 4C).
Figure 4. DAI is required for protection against IAV in vivo.
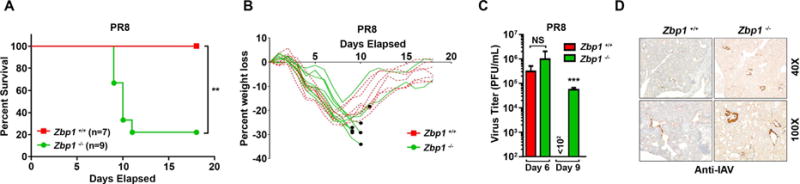
(A) Survival and (B) weight loss analysis of 8–12 week-old sex-matched zbp1−/− and littermate-control zbp1+/+ mice infected with PR8 (1000 EID50/mouse i.n.). Dead mice are represented by black circles in B. Six of nine zbp1−/− mice succumbed overnight to PR8 without need for humane intervention, while a seventh was euthanized upon reaching the designated 35% weight-loss cut-off. (C) Virus titers from zbp1+/+ and zbp1−/− mice infected with PR8 (750 EID50/mouse i.n.) were determined by plaque assay (n=3–5 mice/condition). (D) Representative images of lungs from zbp1+/+ and zbp1−/− mice stained with anti-IAV antibodies 9 d.p.i. Error bars represent mean +/− SD. **p<0.005, ***p<0.0005. See also Fig. S3.
The susceptibility of zbp1−/− mice to PR8-induced virus replication and lethality was notably greater than that of ripk3−/− mice, and paralleled the vulnerability of mlkl−/−/fadd−/− double knockout mice to IAV replication and consequent mortality, in agreement with a role for DAI upstream of both RIPK3-dependent and –independent cell death pathways. DAI was also required for activation of NLRP3 inflammasome-driven IL-1β production in PR8-infected bone marrow-derived macrophages (Fig. S3H). Surprisingly, Kuriakose and colleagues (Kuriakose et al., 2016), observed decreased lethality in IAV-infected zbp1−/− mice, despite these mice manifesting higher lung virus titers and slower recovery times than controls. The reason(s) underlying this difference in survival outcomes between their study and ours is currently unclear. We observed numerous virus-positive bronchiolar epithelial cells in zbp1−/− lungs 9 d.p.i., by which time wild-type lungs had largely cleared virus, and only cellular debris in the lumens of certain airways remained virus antigen-positive (Fig 4D). We interpret these results to signify that infected zbp1−/− bronchiolar epithelial cells fail to undergo apoptosis or necroptosis, and continue to produce virus, events that greatly compromise lung function and result in mortality.
Discussion
In this study, we provide evidence implicating DAI as the host sensor protein linking IAV replication to activation of RIPK3-dependent necroptosis and apoptosis. We posit a simple ‘induced-proximity’ model for how DAI senses IAV and stimulates RIPK3. In this model, DAI recognizes nascent IAV genomic RNA following nuclear export, an event that we propose induces the multimerization of DAI. We base this proposition on our in silico model of a DAI:RNA complex (Fig. 3D, third panel), which suggests that the Zα2 domain of DAI associates with dsRNA as a dimer, bringing into proximity two or more molecules of DAI. This model is based on the published co-crystal structures of Zα domains in complex with either Z-DNA and Z-RNA, both of which reveal dimers of Zα associating with a single Z-form nucleic acid double helix (Ha et al., 2008; Placido et al., 2007) (Fig. 3D). Once juxtaposed, RHIM-based associations between DAI monomers would promote further oligomerization of DAI, and subsequent recruitment of RIPK3. Previous work has shown that oligomerization of RIPK3 is sufficient for induction of cell death (Cook et al., 2014; Orozco et al., 2014), so multimerization of DAI by IAV RNA may, at a minimum, suffice to recruit RIPK3, cluster this kinase, and initiate downstream death signaling. DAI also mediates RIPK3-independent cell death during IAV infection, shown previously by us to proceed via a FADD/caspase 8 axis (Nogusa et al., 2016). This pathway is at least partially dependent on RIPK1, and our evidence suggests that DAI utilizes RIPK1 as an adaptor to activate this alternative, delayed axis of RIPK3-independent cell death.
Our sequence analyses of IAV RNAs bound to DAI revealed several interesting features. DAI appears to preferentially associate with shorter vRNAs, including internally-deleted variants of the polymerase gene segments previously identified as DI particle RNAs (Baum et al., 2010). Such subgenomic DI particle RNAs are produced when the IAV RNA-dependent RNA polymerase (RdRp) falls off its genomic template and re-engages further downstream (Nayak et al., 1985). These RNA segments can form DI particles, as they retain the packaging signals found within the 3′ and 5′ terminal ends of each vRNA and include the polymerase binding site found in the non-coding regions. The partial complementarity of these conserved nucleotides allows the formation of secondary ‘corkscrew’ structures that are recognized by the IAV RdRp (Tomescu et al., 2014), and offer an alternative explanation for the interaction between DAI and IAV RdRp/RNP components observed by Kuriakose and colleagues (Kuriakose et al., 2016): DAI-bound vRNAs that also associate with PB1 and NP indirectly ‘bridge’ these IAV proteins to DAI. Interestingly, these corkscrew structures have also been shown to serve as ligands for the RNA sensor RIG-I (Baum et al., 2010; Liu et al., 2015). In fact, the profile of DAI-associated vRNA species bears remarkable similarity to vRNAs that associate with RIG-I during the course of IAV infection (Baum et al., 2010). These findings support the idea that both DAI and RIG-I may recognize similar vRNA species produced by IAV, with a propensity for subgenomic DI particle RNAs that may be improperly packaged and therefore more prone to detection by the host innate-immune machinery.
DAI also associates with some of the shorter IAV gene segments (most notably NA and NP) across their entire length, indicating that dsRNA complexes of these genomic RNAs may serve as additional ligands for this sensor. These IAV vRNAs may adopt the Z-conformation, perhaps as a result of torsional stress induced by negative supercoiling during viral replication, as has been shown to occur with dsDNA during cellular transcription (Wittig et al., 1991). Alternatively, it is possible that double-stranded vRNA or DI segments may undergo an A-Z transition upon interaction with DAI. Notably, Z-RNA has previously been detected in the cytoplasm and nucleolus of eukaryotic cells (Zarling et al., 1987), and circular dichroism studies have shown that the Zα domain facilitates the transition from A-RNA to Z-RNA under near-physiological conditions (Brown et al., 2000), indicating that dsRNAs in the left-handed Z conformation can serve as physiological substrates for DAI, and for other proteins with Zα domains.
Besides DAI, the only known vertebrate proteins with Zα domains are ADAR1 and PKZ (Athanasiadis, 2012). ADAR1 mediates immunity to multiple RNA viruses (Samuel, 2011) and the fish protein PKZ, an eIF2α kinase reminiscent of PKR but with two tandem N-terminal Zα-domains instead of canonical dsRNA binding domains, functions much like PKR in the cytoplasm to inhibit mRNA translation (Bergan et al., 2008; Rothenburg et al., 2005). Our finding that DAI responds to an RNA virus infection to activate cell death thus indicates that proteins with Zα domains may play broader roles in innate immune responses to RNA viruses than was previously appreciated.
Experimental Procedures
Mice, cells, viruses, and reagents
Mice were housed in SPF facilities at the Fox Chase Cancer Center and the University of Texas, Austin, and all in vivo experiments were conducted under protocols approved by the Committee on Use and Care of Animals at these institutions. Primary MEFs were generated from E14.5 embryos and used within five passages in experiments. LET1 cells have been described before (Rosenberger et al., 2014). All IAV and IBV strains were propagated by allantoic inoculation of embryonated hen's eggs with diluted (1:106) seed virus. Stock virus titers were determined as 50% egg infectious dose (EID50). Cell culture, in vivo infections, and lung virus titers were performed as described previously (Nogusa et al, 2016). Anti-murine DAI antibody (clone Zippy-1) was obtained from Millipore and Adipogen. All other biological and chemical reagents were obtained from sources described previously (Nogusa et al, 2016).
Purification and analysis of DAI-associated IAV RNA
HEK 293T cells were transfected with FLAG-tagged RIG-I or DAI constructs and infected with PR8 (MOI=2) for 12 hr. Cells were disrupted in lysis buffer (50 mM HEPES, 150 mM KCl, 2 mM EDTA, 1mM NaF, 0.5% NP40, 0.5 mM DTT, protease inhibitor cocktail, 25 units RNasin), and lysates were then incubated with 30 μL/sample anti-FLAG agarose bead slurry (Clone M2, Sigma) overnight with rotation at 4°C. Beads were collected by centrifugation, washed ten times with NT2 buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP40), resuspended in 250 μL of DNase digestion buffer (40 mM Tris pH 8.0, 10 mM MgSO4, 1 mM CaCl2) and treated with 25U RNasin (Promega) and 2U DNAse I (NEB) at 37°C for 20 min. Beads were then washed and resuspended in 100 μL NT2 buffer, and treated with 4 units proteinase K at 55°C for 30 minutes. 1 mL Tri-reagent (Sigma) was added to each sample, and RNA was harvested according to the manufacturer’s instructions. RNA was eluted from beads and examined by RT-qPCR or RNA-Seq as described in extended experimental procedures.
Statistics
Statistical significance was determined by use of either Student’s t-test or ANOVA. Significance of in vivo survival data was determined by the log-rank (Mantel-Cox) test. P-values of 0.05 or lower were considered significant. Graphs were generated using GraphPad Prism 6.0 software.
Supplementary Material
Acknowledgments
We gratefully acknowledge Adolfo-Garcia Sastre for PR8-GFP, Chris Dillon and Douglas Green for ripk1−/−ripk3−/− double knockout MEFs, and Tony Lerro and Simon Tarpanian for animal husbandry. We thank Edward Mocarski for comments on the manuscript. This work was supported by NIAID contract HHSN272201400006C (St. Jude Center of Excellence for Influenza Research and Surveillance) to P.G.T., Cancer Prevention & Research Institute of Texas (CPRIT) Scholar Award R1202 to J.W.U., and NIH grants AI093571 to B.R.t, AI110130 to AAB, and CA168621, CA190542, and AI113469 to S.B. Additional funds were provided by NIH Cancer Center Support Grant P30CA006927 and an appropriation from the Commonwealth of Pennsylvania to S.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
RJT, JPI, KBR, SN, DFB, AAB, HS, RK, MS, VJL, MA and PV performed experiments and analyzed data. LJS supplied critical reagents. B.R.t, PGT, JWU and SB designed and oversaw experiments. SB conceived the study and wrote the manuscript. All authors participated in editing the manuscript.
References
- Athanasiadis A. Zalpha-domains: at the intersection between RNA editing and innate immunity. Seminars in cell & developmental biology. 2012;23:275–280. doi: 10.1016/j.semcdb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A. 2010;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergan V, Jagus R, Lauksund S, Kileng O, Robertsen B. The Atlantic salmon Z-DNA binding protein kinase phosphorylates translation initiation factor 2 alpha and constitutes a unique orthologue to the mammalian dsRNA-activated protein kinase R. The FEBS journal. 2008;275:184–197. doi: 10.1111/j.1742-4658.2007.06188.x. [DOI] [PubMed] [Google Scholar]
- Brown BA, 2nd, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A. The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc Natl Acad Sci U S A. 2000;97:13532–13536. doi: 10.1073/pnas.240464097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM, Vaux DL. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21:1600–1612. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SC, Kim D, Hwang HY, Rich A, Kim YG, Kim KK. The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc Natl Acad Sci U S A. 2008;105:20671–20676. doi: 10.1073/pnas.0810463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A, Alfken J, Kim YG, Mian IS, Nishikura K, Rich A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci U S A. 1997;94:8421–8426. doi: 10.1073/pnas.94.16.8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–6434. doi: 10.4049/jimmunol.181.9.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardana S, Place DE, Neale G, Vogel P, Kanneganti TD. ZBP1/DAI is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Science Immunology. 2016;1:aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J Virol. 2015;89:6067–6079. doi: 10.1128/JVI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DP, Chambers TM, Akkina RK. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr Top Microbiol Immunol. 1985;114:103–151. doi: 10.1007/978-3-642-70227-3_3. [DOI] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–1521. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placido D, Brown BA, 2nd, Lowenhaupt K, Rich A, Athanasiadis A. A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure. 2007;15:395–404. doi: 10.1016/j.str.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10:916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger CM, Podyminogin RL, Askovich PS, Navarro G, Kaiser SM, Sanders CJ, McClaren JL, Tam VC, Dash P, Noonan JG, et al. Characterization of innate responses to influenza virus infection in a novel lung type I epithelial cell model. J Gen Virol. 2014;95:350–362. doi: 10.1099/vir.0.058438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenburg S, Deigendesch N, Dittmar K, Koch-Nolte F, Haag F, Lowenhaupt K, Rich A. A PKR-like eukaryotic initiation factor 2alpha kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc Natl Acad Sci U S A. 2005;102:1602–1607. doi: 10.1073/pnas.0408714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Tomescu AI, Robb NC, Hengrung N, Fodor E, Kapanidis AN. Single-molecule FRET reveals a corkscrew RNA structure for the polymerase-bound influenza virus promoter. Proc Natl Acad Sci U S A. 2014;111:E3335–3342. doi: 10.1073/pnas.1406056111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig B, Dorbic T, Rich A. Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc Natl Acad Sci U S A. 1991;88:2259–2263. doi: 10.1073/pnas.88.6.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling DA, Calhoun CJ, Hardin CC, Zarling AH. Cytoplasmic Z-RNA. Proc Natl Acad Sci U S A. 1987;84:6117–6121. doi: 10.1073/pnas.84.17.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.