

Summary
Progressive multifocal leukoencephalopathy (PML) is a lethal brain disease caused by uncontrolled replication of JC polyomavirus (JCV). JCV strains recovered from the brains of PML patients carry mutations that prevent the engagement of sialylated glycans, which are thought to serve as receptors for the infectious entry of wild-type JCV. In this report, we show that non-sialylated glycosaminoglycans (GAGs) can serve as alternative attachment receptors for the infectious entry of both wild-type and PML-mutant JCV strains. After GAG-mediated attachment, PML-mutant strains engage non-sialylated non-GAG co-receptor glycans, such as asialo-GM1. JCV-neutralizing monoclonal antibodies isolated from patients who recovered from PML appear to block infection by preventing the docking of post-attachment co-receptor glycans in an apical pocket of the JCV major capsid protein. Identification of the GAG-dependent/sialylated glycan-independent alternative entry pathway should facilitate the development of infection inhibitors, including recombinant neutralizing antibodies.
eTOC blurb
Geoghegan et al show that JC polyomavirus strains that cause brain disease infect cells via a pathway involving a heparin-like attachment receptor and a non-sialylated co-receptor. Candidate therapeutic human monoclonal antibodies neutralize by blocking co-receptor engagement.
Introduction
Serological studies indicate that a majority of healthy adults are chronically co-infected with both JC polyomavirus (JCV) and BK polyomavirus (BKV) (Gossai et al., 2016; Kean et al., 2009; Knowles et al., 2003). In most individuals, lifelong infection with the two closely related viruses is thought to be primarily restricted to the urinary epithelium. Although infection is not typically associated with known symptoms in healthy individuals, a JCV-induced brain disease called progressive multifocal leukoencephalopathy (PML) affects about 5% of patients with HIV/AIDS (Collazos, 2003; Steiner and Berger, 2012). BKV is only rarely associated with brain disease (Lopes da Silva, 2011), but it is a common cause of nephropathy following kidney transplantation (Lopes da Silva, 2011; Reploeg et al., 2001). Although the incidence of PML in HIV-infected individuals has decreased with the advent of combination antiretroviral therapy, PML remains a persistent threat (Casado et al., 2014; Collazos, 2003). More recently, it has become apparent that treatment with various therapeutic immunomodulatory monoclonal antibodies (mAbs), including natalizumab and rituximab, is associated with PML (Berger and Fox, 2016; Carson et al., 2009; Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005). There is currently no effective treatment for PML except to attempt to restore immune function, which can, in turn, lead to immune reconstitution inflammatory syndrome (another potentially fatal outcome) (Bauer et al., 2015; Steiner and Berger, 2012). Immunomodulatory therapies hold a great deal of promise for treating a wide variety of diseases such as multiple sclerosis, rheumatoid arthritis, Crohn’s disease, lupus, lymphoid cancers, and many other diseases, but the risk of PML continues to hamper widespread use of some of these therapies (Berger and Fox, 2016; Carson et al., 2009; Diotti et al., 2013; Steiner and Berger, 2012; Tur and Montalban, 2014).
It is well established that many polyomavirus species, including JCV, BKV, the Rhesus monkey BKV/JCV-related virus SV40, and Merkel cell polyomavirus (MCV), require sialylated glycans for infectious entry into cells (DeCaprio and Garcea, 2013; Stroh and Stehle, 2014). MCV is unique among polyomaviruses in that it is known to require sequential engagement of non-sialylated glycosaminoglycan (GAG) receptors for attachment to the cell surface and sialylated co-receptor glycans for post-attachment steps in the infectious entry process (Schowalter et al., 2011). GAGs are long, unbranched glycans made up of repeating disaccharide units that are typically O-linked to proteins. Heparan sulfate (HS), dermatan sulfate, chondroitin sulfate, and keratan sulfate are common classes of GAGs (Kamhi et al., 2013). Cells deficient in either GAGs or sialylated glycans do not support MCV infection (Schowalter et al., 2011). An initial goal of the current study was to examine the hypothesis that JCV, BKV and SV40 can, like MCV, employ GAGs as attachment receptors.
In addition to further exploring the potential role of GAGs in polyomavirus entry, we sought to address a lingering puzzle applicable to JCV. While it is well established that JCV strains found in the urine of healthy subjects bind the sialic acid-bearing pentasaccharide LSTc (Neu et al., 2010; Ströh et al., 2015), JCV strains found in the serum, cerebrospinal fluid, and brains of PML patients often contain mutations in residues lining the LSTc-binding pocket on the apical surface of the major capsid protein VP1 (Gorelik et al., 2011; Reid et al., 2011; Sunyaev et al., 2009). Most PML-associated mutations render the virus incapable of binding LSTc or other sialylated glycans (Gorelik et al., 2011; Maginnis et al., 2013). Interestingly, some PML-associated mutants have been shown to remain capable of binding to the non-sialylated glycan asialo-GM1 (Gorelik et al., 2011).
Several of the most common PML-mutant strains of JCV have been predicted to be under positive selection during the development of PML (Ray et al., 2015; Sunyaev et al., 2009). This prediction is consistent with results using a human glial cell-engrafted chimeric mouse model of PML recently described by Kondo and colleagues (Kondo et al., 2014). In the chimeric mouse model, infection with a native wild-type JCV strain leads to PML-like pathology and, significantly, disease progression is marked by the appearance of viral strains with VP1 mutations adjacent to the apical LSTc-binding pocket, including some of the hallmark mutations observed in the brains of PML patients (Kondo et al., 2014). Mice challenged with JCV carrying either of the two most common PML mutations (VP1 L55F or S269F) showed robust infection of engrafted human oligodendrocytes, astrocytes, and glial progenitor cells in vivo and infection occurred at an equivalent or greater frequency compared to the wild-type virus. Thus, despite their inability to bind the known sialylated receptor glycan LSTc, PML-mutant JCV strains remain fully infectious in various brain cell types in vivo. A possible explanation for these findings is that PML-mutant JCVs engage a previously unrecognized alternative infectious entry pathway that does not depend on known sialylated receptor glycans, such as LSTc. Specifically, we hypothesized that the sialylated glycan-independent alternative infectious entry pathway engaged by PML-mutant JCV strains involves GAG-mediated attachment.
Jelcic and colleagues recently reported a panel of JCV-neutralizing human mAbs cloned from individuals who survived PML (Jelcic et al., 2015). Although a subset of the mAbs were shown to bind virus-like particles (VLPs) carrying PML-associated mutations, their ability to effectively neutralize PML-mutant JCV strains was not evaluated. Using a panel of common wild-type and PML-mutant JCV strains, we examined the neutralizing activity of these mAbs. The mAbs also proved to be useful tools for dissecting the entry pathways JCV uses to infect cells.
Results
Attachment of PML-mutant JCV to cultured cell lines is primarily mediated by GAGs
JCV genotypes 2 and 3 were selected as common wild-type backgrounds and two previously reported PML-associated JCV isolates, JCV2-269F and JCV3-55F, were used as model PML-associated mutant strains previously shown to be defective for engagement of various sialylated glycans (Gorelik et al., 2011; Ray et al., 2015; Reid et al., 2011). A commonly studied BKV genotype Ia isolate was chosen as a representative reference virus. A previously reported BKV VP1 F76W mutation designed to prevent engagement of sialic acid (SA) (Neu et al., 2013) was constructed in this background. We collectively refer to the PML-mutant JCV strains and the BKV-76W mutant as “SA-mutant” viruses.
To begin to examine the question of whether JCV and BKV might, like MCV, use heparin-like GAGs for infectious entry into cells, cultured cell lines were treated with a blend of the heparan sulfate-degrading enzymes (heparinase I and heparinase III) and probed for changes in the ability to bind JCV or BKV VLPs. For SFT cells (a gliosarcoma line that has previously been shown to support the infectious entry of PML-mutant JCV (Ray et al., 2015)), the heparinase treatment resulted in an 80% decrease in the binding of MCV VLPs, confirming that the GAG-degrading enzyme treatment was effective (Figure 1).
Fig. 1. SA-mutant VLPs require GAGS for binding to SFT gliosarcoma cells.
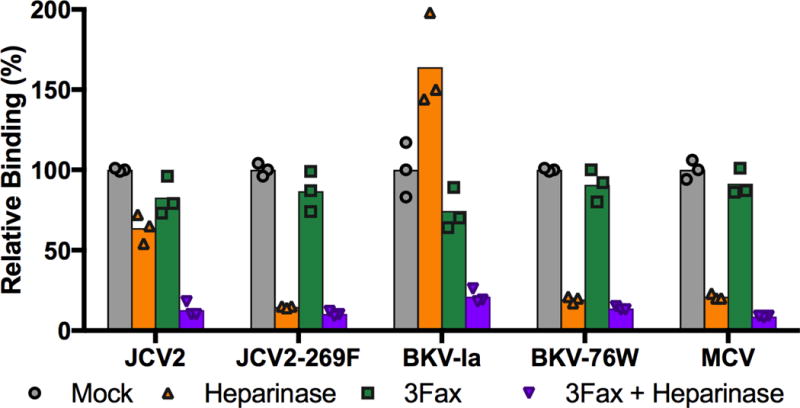
Flow cytometry was used to measure the binding of wild-type and SA-mutant JCV and BKV VLPs to cells treated with heparinase I/III (orange), the sialyltransferase inhibitor 3Fax (green), or a combination of the two treatments (purple). MCV VLPs, which are known to require GAGs for cell attachment, were used as a control. Results are standardized to mock-treated SFT cells. One of two independent experiments is shown.
Although heparinase treatment showed only modest effects on the binding of wild-type JCV or BKV, the SA-mutant viruses showed a dramatically reduced ability to attach to heparinase-treated cells (Figure 1). Similar results were observed using heparinase-treated ART cells (data not shown), which are likewise able to support the infectious entry of PML-mutant JCV strains (Ray et al., 2015). Additionally, SA-mutant JCV and BKV VLPs (but not wild-type VLPs) showed a ~99% reduction in binding to GAG-deficient CHO pgsA-745 cells (Esko et al., 1985) relative to wild-type CHO cells (Supplemental Figure 1). The results show that, while wild-type JCV and BKV VLPs can attach to cultured cells via heparinase-resistant moieties, attachment of SA-mutant viruses is mediated primarily by GAGs.
Wild-type JCV binds both GAGs and sialylated glycans
To confirm the finding that PML-mutant JCVs do not engage sialylated glycans, we used 3Fax-Neu5Ac (3Fax), a drug that is converted intracellularly into a CMP-Neu5Ac mimetic that competitively blocks sialyltransferase activity (Rillahan et al., 2012). Cells treated with 3Fax produce non-sialylated or hyposialylated glycans. The drug was applied to a panel of cell lines we have previously shown to be permissive for the infectious entry of PML-mutant JCV reporter pseudovirions (Ray et al., 2015). After three days of drug treatment, cells were stained using a blend of lectins specific for α-2,3- and α-2,6-linked sialic acids and analyzed using flow cytometry. The 3Fax treatment showed variable effectiveness, depending on the cell line (Supplemental Table 1). The drug was particularly effective for reducing cell-surface sialylated glycan expression on SFT cells, with an average of 81% decrease in lectin-based staining of cell surface α-2,3 and α-2,6 sialic acids (Supplemental Figure 2A). The results confirm that 3Fax treatment effectively blocks surface expression of sialylated glycans on some cell types.
Treatment of SFT cells with 3Fax resulted in only a minimal decrease in the cellular attachment of all tested VLPs representing wild-type or SA-mutant strains (Figure 1). Consistent with the prior findings of Gorelik and colleagues (Gorelik et al., 2011) our results suggest that JCV and BKV, like MCV, do not require sialylated glycans for initial attachment to cells. The binding data leave open the possibility that, like MCV, wild-type JCV and BKV require sialylated glycans for a post-attachment infectious entry step.
To investigate the role that GAGs play in the cell attachment of wild-type and SA-mutant VLPs under conditions where sialylated glycans are unavailable, cells were treated with both heparinase and 3Fax. Simultaneous removal of both sialylated glycans and GAGs dramatically reduced the binding of all wild-type and SA-mutant VLPs. Together, the data are consistent with a model in which the wild-type virus can make facultative use of either GAGs or sialylated glycans for initial attachment to cells, while SA-mutant strains are restricted to the exclusive use of GAGs for attachment.
Inhibition of infectivity by the model GAG heparin
A previously described reporter pseudovirion system (Ray et al., 2015) was used to monitor the effect of various inhibitors on the infectious entry process of JCV and BKV. Although the effects of heparinase on virion attachment to cells can readily be monitored in short-term experiments, longer-term infectivity experiments are more challenging because the enzymatic activity of heparinases eventually decays and GAGs begin to reappear on the cell surface. We therefore tested the infection-inhibitory effects of heparin, a freely soluble GAG used clinically as an anticoagulant. The infectivity of wild-type JCV2, JCV3, and BKV on ART and SFT cells was partially inhibited by heparin, but the inhibition curves plateaued at roughly 50% inhibition (Figure 2 and Supplemental Figure 3). A possible explanation for this observation might be that, for the wild-type pseudoviruses, the known sialylated glycan-dependent infectious entry pathway remains operational even in the presence of high doses of heparin. In contrast to the wild-type pseudoviruses, the infectivity of the SA-mutant pseudoviruses (which cannot engage the sialylated glycan-dependent entry pathway) was completely blocked by heparin. This suggests that infectious entry of the SA-mutant viruses relies entirely on a GAG-dependent entry pathway that can be competitively blocked with exogenous heparin.
Fig. 2. SA-mutant pseudoviruses are more sensitive to heparin inhibition than wild-type pseudoviruses.
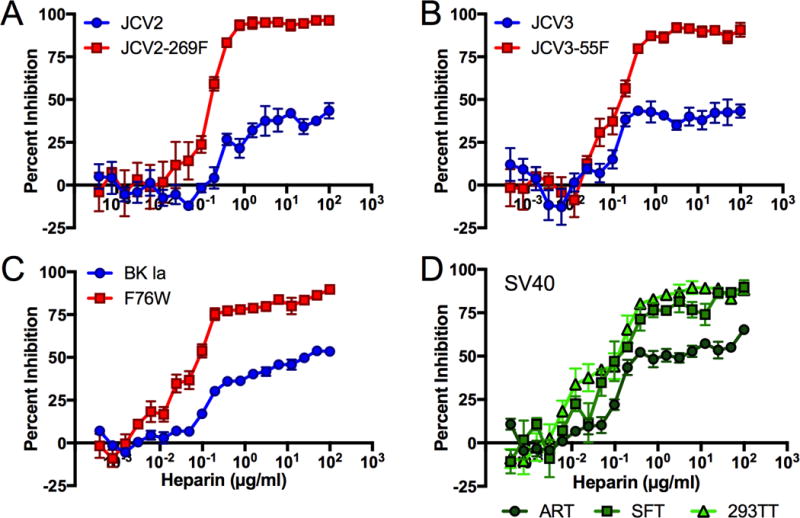
Infectivity of the indicated pseudovirus was measured in the presence of a two-fold dilution series of heparin. Quadruplicate values were averaged and expressed as percent inhibition compared to untreated virus control. Panels (A–C) show results for ART cells, while panel (D) was performed using wild-type SV40 pseudovirus on the indicated cell types. Similar results were obtained for panels (A–C) using SFT cells (Supplemental Figure 3) and 293TTs (data not shown). Results are representative of four to six independent experiments. Error bars represent SEM.
SV40, which is closely related to BKV and JCV, is known to require the sialylated ganglioside GM1 for infectious entry into many (but not all) cell types (Magaldi et al., 2012). Prior work has not addressed the possibility that GAGs might be involved in SV40 infectious entry. An SV40 pseudovirus was inhibited by heparin in the same manner as wild-type JCV and BKV on ART cells, suggesting SV40 is also at least partially dependent on GAGs for entry into this cell type (Figure 2D). On 293TT and SFT cells, SV40 was almost completely inhibited by heparin, suggesting that GAG engagement is a required step in SV40 infectious entry into these cell types.
The concept that SA-mutants require GAGs for infectious entry was confirmed using a GAG-blocking mAb, HS20 (Gao et al., 2014)(Supplemental Figure 4). A separate set of experiments using cells treated with chlorate suggest that, in contrast to MCV, neither JCV nor BKV require GAG sulfate modifications for infectious entry (Schowalter et al., 2011)(Supplemental Figure 5).
3Fax treatment unexpectedly enhances PML-mutant infectivity
SFT cells were treated with 3Fax to further investigate the role of sialylated glycans in infectious entry. The infectivity of MCV, which is known to require a sialylated glycan co-receptor for infectious entry, was completely blocked by 3Fax treatment (Figure 3). In contrast, the infectivity of HPV16, which is not known to require sialylated glycans for infectious entry, was slightly enhanced by 3Fax treatment. The HPV16 pseudovirus results confirm that 3Fax does not cause general cytotoxicity for SFT cells. One possible explanation for the modest enhancement of HPV16 infectivity might be that 3Fax treatment results in increased expression of longer GAG-like glycans due to a lack of terminating sialic acid modifications (Rillahan et al., 2012). To investigate this idea, we performed staining using HPV16 VLPs or the HS20 mAb. As seen in Supplemental Figure 2 panels B and C, the staining results do not support the concept of increased GAG expression on 3Fax-treated cells.
Fig. 3. JCV and BKV pseudovirions make facultative use of either sialylated glycans or by heparin-like GAGs for infectious entry.
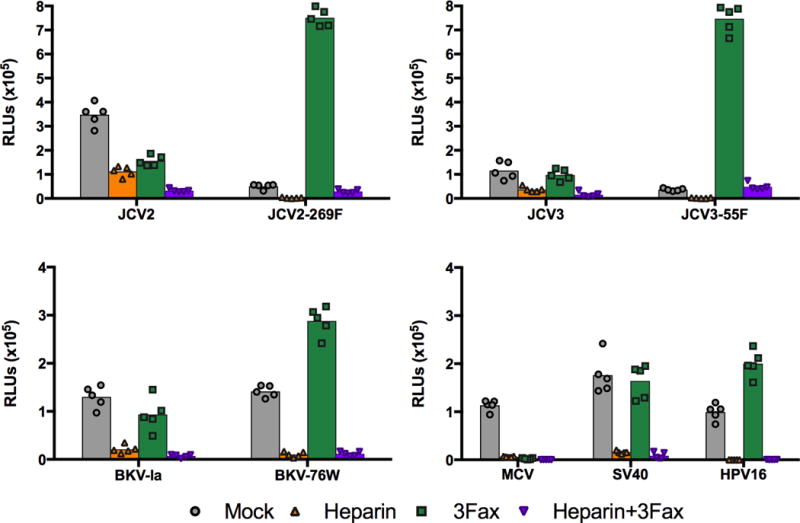
SFTs were cultured with or without 3Fax and a single dilution of heparin was added to cells prior to addition of indicated reporter pseudoviruses. Similar results were obtained in three independent experiments.
For wild-type JCV2, JCV3, BKV, and SV40 pseudoviruses, 3Fax treatment either had no effect or slightly inhibited infection compared to untreated cells (Figure 3). These infectivity results are comparable to the VLP-based cell binding results shown in Figure 1, suggesting that the wild-type viruses are able to infect cells via a GAG-dependent and sialylated glycan-independent entry pathway. Surprisingly, 3Fax treatment resulted in a dramatic increase in the infectivity of SA-mutant pseudoviruses. The JCV2-269F mutant showed a 15-fold increase in reporter signal and the JCV3-55F mutant showed a 21-fold increase in reporter signal on 3Fax-treated SFT cells. The enhancement of the infectivity of the BKV SA-mutant F76W was more modest (only about 2–3 fold). All tested virus types were susceptible to competitive inhibition with 100 μg/ml heparin on 3Fax-treated cells. The results are consistent with the idea that the infectious entry of both wild-type and SA-mutant pseudoviruses is entirely dependent on GAG-like glycans when sialylated glycans are unavailable.
Six additional cell lines were treated with 3Fax to determine whether the enhancement of PML mutants on SFT cells is generalizable. The cell lines exhibited a range of sensitivities to 3Fax treatment, as measured by decreases in lectin staining (Supplemental Table 1). There was a generally positive correlation between the degree of 3Fax efficacy for reducing sialylated glycan expression and the level of enhancement of infectivity of the JCV2-269F and JCV3-55F PML-mutant pseudoviruses. This suggests that the availability of hypo-sialylated glycans plays an important role in the infectious entry of PML-mutants.
The concept that PML-mutant JCV infectivity is improved by the appearance of hyposialylated GAGs on the cell surface was confirmed using neuraminidase-treated cells. Neuraminidase treatment caused an 89% decrease in lectin staining as measured by flow cytometry (Supplemental Figure 4E), and resulted in a roughly 3-fold increase in PML-mutant infectivity (Supplemental Figure 6). Similarly, siRNA-mediated knockdown of two genes involved in the production of sialylated glycans, SLC35A1 and CMAS, resulted in a 40% decrease in lectin staining and a 4.5- and 8-fold increase, respectively, in PML-mutant infectivity (Supplemental Figure 7).
Supplying cells with a non-sialylated ganglioside enhances PML-mutant infectivity
We hypothesized that removal of sialic acid residues from cellular glycans might be “revealing” a spectrum of non-sialylated glycans that are not normally present at high levels on cultured cells, and that such non-sialylated glycans might serve as post-GAG-attachment co-receptors required for efficient infectious entry of PML-mutant viruses. Since 3Fax treatment did not significantly affect the overall binding of PML-mutant JCV strains to cells (Figure 1), non-sialylated co-receptor glycans would presumably play a role as post-attachment co-receptors. A candidate glycan in this category might be the naturally occurring non-sialylated form of the ganglioside GM1 (asialo-GM1), a glycolipid that Gorelik and colleagues previously showed is capable of binding various PML-mutant JCV strains (Gorelik et al., 2011).
Exogenous gangliosides can conveniently be loaded onto the surface of cultured cells (Pastrana et al., 2013). Consistent with the fact that JCV is not known to use the primary sialylated form of GM1 as an entry receptor (Ströh et al., 2015), GM1 pre-treatment of cells had no effect on the infectivity of wild-type or PML-mutant pseudoviruses (Figure 4). In contrast to JCV, SV40 has previously been shown to use GM1 for infectious entry into most cell types and, accordingly, GM1 pre-treatment increased the infectivity of an SV40 pseudovirus by about five-fold (data not shown)(Ewers et al., 2010; Neu et al., 2008; Tsai et al., 2003). The SV40 results confirm that the ganglioside loading procedure was effective. Although treating cells with exogenous asialo-GM1 did not affect the infectivity of wild-type JCV pseudoviruses, it substantially increased the infectivity of PML-mutant pseudoviruses (Figure 4). Specifically, JCV2-269F was enhanced by 4-fold and JCV3-55F was enhanced by 5-fold using ART cells. The results confirm the hypothesis that the availability of hypo-sialylated co-receptor glycans can be a limiting factor in the infectious entry of PML-mutant pseudoviruses.
Fig. 4. Addition of asialo-GM1 enhances the infectivity of PML-mutant JCV strains.
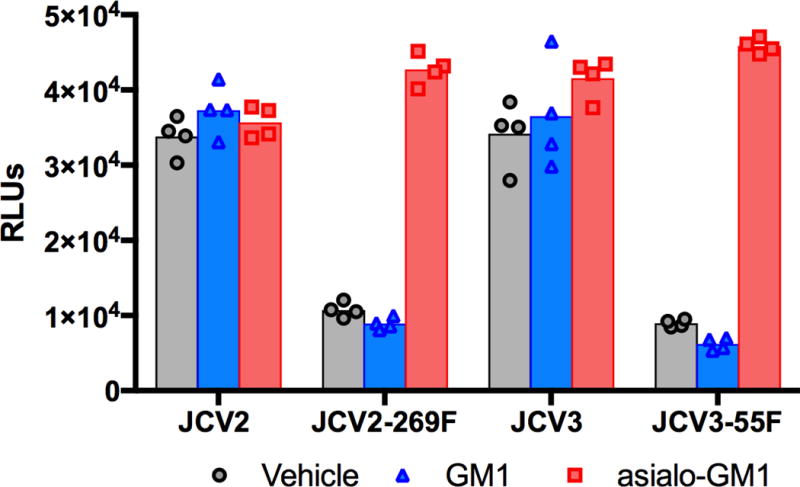
ART cells were treated with exogenous GM1 or asialo-GM1 for two hours, trypsinized, replated in 96- well plates, then transduced with indicated pseudoviruses. Similar results were obtained in three independent experiments using SFT cells (data not shown).
Characterization of JCV-neutralizing human mAbs
Jelcic and colleagues have recently reported a panel of JCV VP1-specific mAbs cloned from PML patients (Jelcic et al., 2015). Although Jelcic and colleagues demonstrated that many mAbs in the panel potently neutralize the infectivity of wild-type JCV-1A, at the time of the prior study it was not technically feasible to directly test the mAbs for neutralization of PML-mutant strains. Our observation that wild-type and PML-mutant JCV strains can use different infectious entry pathways raises the possibility that some mAbs might neutralize wild-type JCV but fail to neutralize PML-mutant JCV. To experimentally address this caveat, we performed high-throughput pseudovirus-based neutralization assays with a panel of the mAbs reported by Jelcic and colleagues. Assays were performed using ART and SFT cells, as well as SFT cells pre-treated with 3Fax.
Antibodies 50H4, 98H1, 27C11, 26A3, 72F10, and 27C2 potently neutralized all tested JCV strains on all tested cell types with 50% neutralization (EC50) values in the low picomolar or high femtomolar range (Figure 5). The results indicate that the alternative infectious entry pathway used by PML-mutant pseudoviruses remains sensitive to mAbs that neutralize wild-type JCV.
Fig 5. Neutralization properties of anti-JCV mAbs on 3Fax-treated SFT cells.
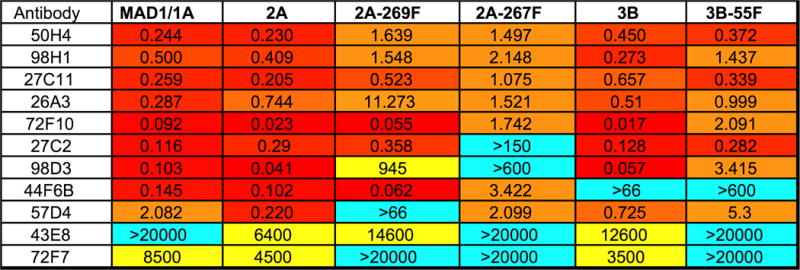
The neutralizing EC50 (picomolar) for each antibody against indicated pseudoviruses was measured on 3Fax-treated SFT cells. Similar results were observed using untreated SFT cells and ART cells.
JCV-neutralizing human mAbs do not inhibit attachment
In principle, neutralizing antibodies can function either by blocking the initial attachment of virions to cells or by interfering with a post-attachment entry step, such as engagement of a co-receptor. Cell attachment assays were used to distinguish between these possible mechanisms. The experiments used JCV reporter VLPs carrying a recombinant fusion of human histone subunit H2B to NanoLuc (Promega) reporter enzyme. The fusion protein, which is encapsidated alongside the native host histone proteins typically found within polyomavirus virions, allows the monitoring of VLP attachment to cells using NanoLuc luminometry, thus obviating the need for use of secondary antibody reagents.
Heparin and HS20 both blocked the attachment of PML-mutant VLPs and only partially prevented binding of wild-type JCV2 (Figure 6). The result confirms that competitive antagonists of cell surface GAG engagement interfere with infectivity at least in part by blocking the initial cell-attachment step. In contrast to heparin and HS20, none of the anti-JCV mAbs prevented cell-attachment of wild-type or PML-mutant VLPs. Puzzlingly, mAbs 98D3, 50H4, and 27C11 caused increased binding of JCV2-269F VLPs to both SFT and ART cells. The effect does not appear to be due to the presence of Fc receptors or due to the antibody recognizing a cellular epitope and then bridging the cell and the VLP, as the three mAbs showed little or no binding to cells in flow cytometric analyses (data not shown). A possible explanation for the increase in binding observed with these antibodies is that they induce a conformational shift that makes the GAG-binding site more accessible, thereby promoting cell-binding. The full neutralization curves for mAbs 98D3, 50H4, and 27C11 did not show any evidence of antibody-dependent enhancement of infectivity (Dowd and Pierson, 2011).
Fig. 6. Neutralizing mAbs do not prevent JCV attachment to cells.
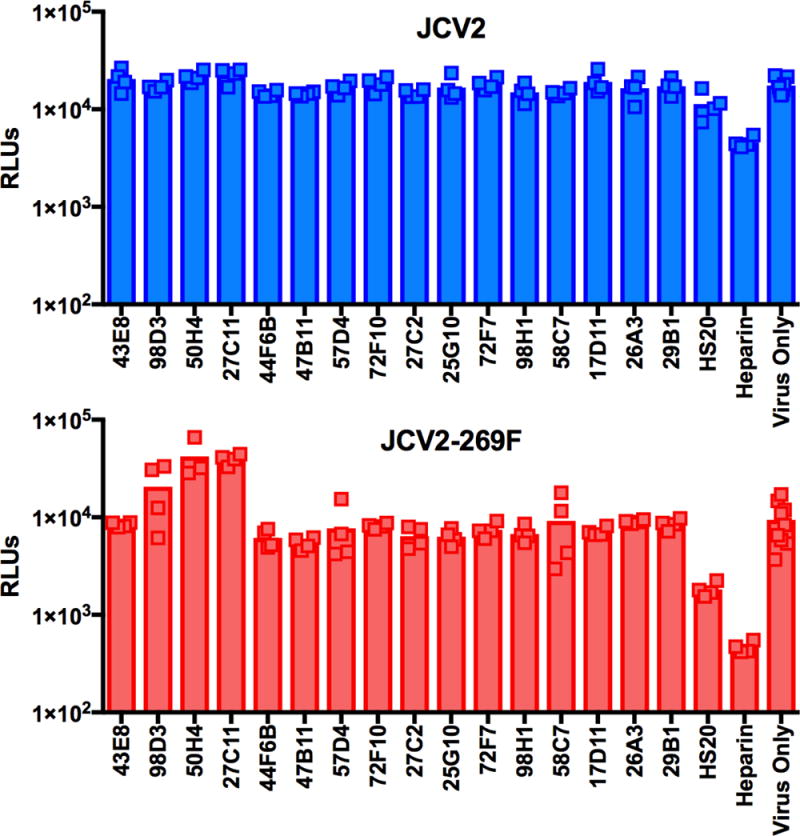
JCV pseudovirions carrying a histone-NanoLuc reporter protein were incubated with indicated anti-VP1 mAb, HS20, or heparin and then added to a suspension of SFT cells. After a 30 minute incubation, cells were washed twice, and then transferred to white luminometry plate to measure NanoLuc signal. Results are representative of three independent experiments. Similar results were observed using ART cells (data not shown).
JCV-neutralizing human mAbs block hemagglutination
Although wild-type JCV virions and VLPs readily hemagglutinate human red blood cells (RBCs), PML-mutant VLPs are unable to mediate hemagglutination (data not shown)(Gorelik et al., 2011; Maginnis et al., 2013; Sunyaev et al., 2009). Taken together with prior results showing that neuraminidase pre-treatment of RBCs prevents JCV-mediated hemagglutination (Liu et al., 1998), it seems clear that hemagglutination is primarily mediated by interactions between the virion and sialylated glycans on the RBC surface.
Hemagglutination testing revealed that all antibodies that neutralized JCV-1A (against which the mAbs were initially screened) also blocked hemagglutination. Only two of the tested mAbs, 43E8 and 72F7, failed to block hemagglutination, consistent with the fact that they do not neutralize JCV-1A. The results indicate that all of the neutralizing mAbs occlude the known LSTc-binding pocket found on the apical surface of the wild-type VP1 capsomer. This model is consistent with the mapped epitopic footprint of a previously reported JCV-neutralizing mAb known as GRE1 (Diotti et al., 2014) and is also consistent with the prior observation that mAbs 98D3, 44F6B, and 57D4 fail to bind VLPs with PML-associated VP1 mutations that map to the apical pocket (Jelcic et al., 2015).
Although PML-mutants could not be tested in the HAI assay, we note that the 269F and 55F mutants were efficiently neutralized by many of these antibodies. A simple way to explain the findings is that the putative non-sialylated glycan co-receptor PML-mutant strains use for infectious entry docks in the same apical pocket where sialylated glycans are known to engage on wild-type JCV strains. The neutralizing mAbs would thus block access to sialylated co-receptor glycans required by wild-type JCV, as well as the non-sialylated co-receptor glycans apparently required by PML-mutant JCV strains.
Discussion
Past observations showing that PML-associated JCV strains remain infectious, despite being unable to engage known sialylated receptor glycans, suggested that these strains might use a sialylated glycan-independent infectious entry pathway. Our results show that PML-mutant JCV strains require non-sialylated GAGs for initial attachment to cells. Cell attachment is followed by engagement of non-sialylated non-GAG co-receptor glycans (Figure 7). Wild-type JCV strains are also able to make facultative use of the GAG-dependent entry pathway, allowing them to remain partially infectious when either GAG or sialic acid engagement is blocked. The existence of this previously unknown sialylated glycan-independent infectious entry pathway could help explain why JCV can robustly infect human astrocytes and oligodendrocytes in vivo (Kondo et al., 2014), despite the fact that these cell types appear to lack previously identified sialylated glycan receptors for the virus (Haley et al., 2015).
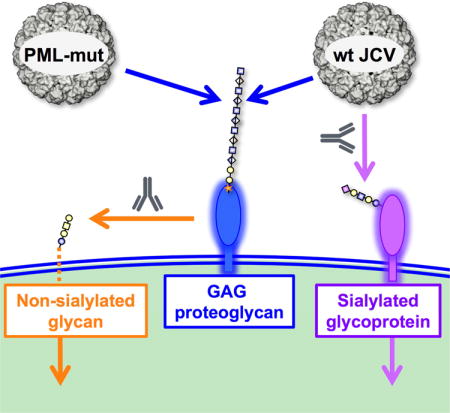
Fig. 7. Model for infectious entry of JCV.
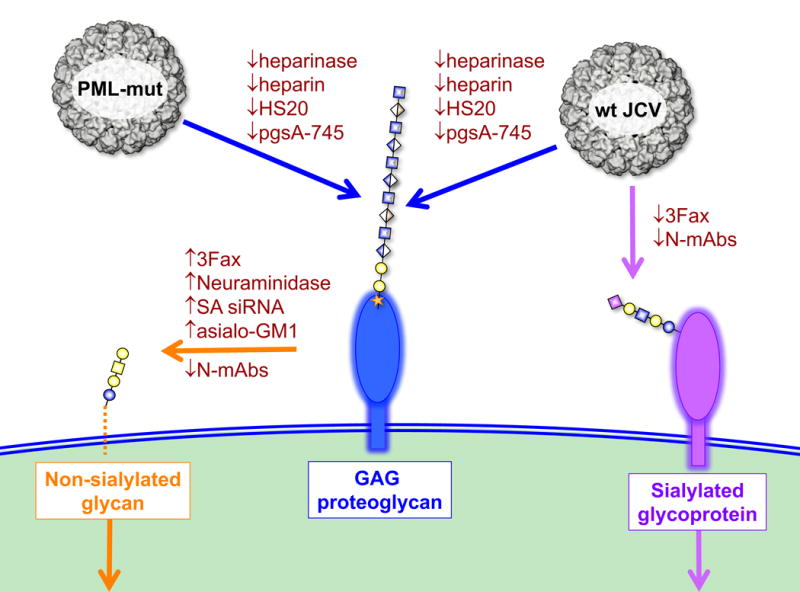
Wild-type JCV is capable of facultative engagement of either GAGs or sialylated glycans to initiate entry, PML mutants require GAGs for attachment to cells. Following attachment to cells, PML-mutants are transferred to a non-sialylated co-receptor glycan. Standard glycan symbols (Varki et al., 2015a; Varki et al., 2015b) are used to depict the glycan headgroup of asialo-GM1 (orange), an example of a GAG proteoglycan (blue) and the known sialylated wild-type JCV co-receptor LSTc (purple). SA = sialic acid; siRNA = siRNA targeting sialic acid pathway genes; N-mAbs = neutralizing monoclonal antibodies.
Although previous analysis of PML-mutant evolution has suggested that these strains are under positive selection during disease progression (Sunyaev et al., 2009), it has been uncertain what selective advantages the mutations might confer. It appears that the virus may make a tradeoff between the ability to use the preferred sialylated receptor glycans and escaping the effects of neutralizing antibodies (Ray et al., 2015). Modulation of glycan-binding specificity may be a general mechanism that viruses can use to escape from neutralizing antibodies, with potentially pathogenic consequences. For example, influenza viruses have been shown to change their glycan usage in response to selective pressure from neutralizing antibodies, resulting in changes in tissue tropism and increased pathogenicity (Hensley et al., 2009; O’Donnell et al., 2012). The hypervirulent LID strain of mouse polyomavirus, which harbors a VP1 V296A mutation (homologous to the PML-associated S269F mutation in JCV) engages an altered spectrum of sialylated glycans, shows different tissue tropism, and is more rapidly lethal than wild-type mouse polyomavirus strains (Bauer et al., 1999; Carroll et al., 2007). Likewise, there is a link between sialylated glycan affinity and pathogenicity for parvoviruses, including canine parvovirus and minute virus of mouse (Huang et al., 2014). It would be interesting to learn whether virulence-associated mutations in mouse polyomavirus and animal parvoviruses also allow escape from antibody-mediated neutralization.
Gangliosides are known receptors for many other members of the polyomavirus family (DeCaprio and Garcea, 2013) but have not been clearly established as functional receptors for JCV. Although several studies have demonstrated that JCV is capable of binding a variety of gangliosides (Gorelik et al., 2011; Komagome et al., 2002; Ströh et al., 2015), and that pre-treating wild-type JCV virions with gangliosides can block infectivity (Komagome et al., 2002; Ströh et al., 2015), pre-loading cultured cells with exogenous sialylated gangliosides neither enhanced the infectivity of wild-type JCV in permissive cells nor potentiated infection in a non-permissive cell line (Ströh et al., 2015). Additionally, there is substantial evidence that glycoproteins bearing the pentasaccharide LSTc, rather than glycolipids, can serve as the main receptors for JCV infectious entry into some cultured cell lines (Dugan et al., 2008; Liu et al., 1998; Neu et al., 2010; Ströh et al., 2015). We demonstrate that addition of the ganglioside asialo-GM1 to permissive cells increases the infectivity of PML-mutant strains. It remains uncertain whether the asialo-GM1 enhancement we observed is simply attributable to the increased abundance of the non-sialylated co-receptor glycan headgroup on the cell surface or whether the presentation of the glycan in the specific context of a glycolipid is important. 3Fax treatment of cells, neuraminidase treatment, and siRNA-induced decreases in sialylated glycan biosynthesis should affect glycolipids and glycoproteins similarly, so the relative importance of the two classes of glycan cannot be inferred from our data. Asialo-GM1 has been reported to be a specific marker of oligodendrocytes and myelin-producing cells, raising the possibility that this glycan motif may play a central role in PML-mutant infectivity and pathogenesis in the brain (Dasgupta et al., 2007; Dasgupta and Hogan, 1993; Dasgupta et al., 1992; Dasgupta et al., 2000; Kusunoki et al., 1985). Additionally, there are numerous examples of heparan sulfate proteoglycans, including glypicans and syndecans, that could potentially serve as GAG attachment receptors on brain cell types known to be infected by JCV (Farhy Tselnicker et al., 2014; Maeda, 2015; Yim et al., 1993).
While a VLP-based vaccine against JCV might provide a broadly cross-neutralizing antibody response that would durably protect at-risk patients against PML (Ray et al., 2015), broadly neutralizing mAbs are also promising as potential therapeutic agents, particularly for patients treated with immunomodulatory drugs or immunocompromised patients who may not respond robustly to vaccination. In this work, we evaluated a previously published panel of wild-type JCV-neutralizing antibodies cloned from patients who recovered from PML (Jelcic et al., 2015) for neutralization of PML-mutant strains. Our functional analysis of these antibodies has identified several candidates for further development, in particular antibodies 50H4, 98H1, 27C11, 26A3, 27C2, and 72F10, which effectively cross-neutralize all tested wild-type genotypes, including variants representing the most commonly occurring PML mutations. Structural studies will be necessary to directly identify where these antibodies bind the virus, but our results strongly suggest that all known JCV-neutralizing antibodies bind to the apical pocket on the top surface of the VP1 capsomer knob, where sialylated glycans, such as LSTc, are known to be engaged by wild-type JCV (Neu et al., 2010). Although the PML-mutants do not engage sialylated glycans, they are nevertheless potently neutralized by some of the tested mAbs. A potential explanation for these findings is that PML-mutant strains continue to use the apical pocket to bind non-sialylated co-receptor glycans, and antibody-mediated interference with the function of this pocket can thus effectively neutralize the PML-mutant strains. It is encouraging that this apical glycan binding pocket appears likely to be an Achilles heel for both wild-type and PML-mutant strains, strengthening the clinical potential for treating or preventing PML with neutralizing mAbs.
Antibodies that prevent cell binding through interference with GAG engagement might also be an effective way to decrease the infectivity of JCV and BKV, although our results suggest that wild-type viruses bound by such antibodies might remain capable of utilizing the sialylated glycan-dependent pathway (in much the same way that wild-type viruses remain partially infectious in the presence of heparin or HS20). Although no mAbs that prevent cell binding have yet been identified for JCV or BKV, we have previously characterized mAbs capable of blocking the GAG-mediated attachment of MCV to cultured cells (Pastrana et al., 2010). Although the attachment-blocking mAbs fully neutralize MCV, it does not appear that MCV is able to make facultative use of GAG-independent entry pathways, as wild-type JCV and BKV do.
Our results using Jelcic and colleagues’ mAb panel demonstrate that the GAG-binding site is distinct from the apical pocket that engages sialylated glycans in wild-type strains. Binding studies performed with recombinant JCV VP1 capsomers that lack the C-terminus and are unable to assemble into VLPs indicate that binding of free capsomers to cells is completely dependent on the presence of sialylated glycans (Maginnis et al., 2013; Neu et al., 2013). In contrast to the truncated capsomers, assembled capsids are able to bind cells in a sialic acid-independent manner (Gorelik et al., 2011)(Figure 1 and Supplemental Figure 1). This suggests that GAG-attachment motifs are formed by inter-capsomer interactions in the canyons between the capsomer knobs.
In addition to establishing that GAGs play a role in infectious entry of JCV, we demonstrate that GAGs are involved in BKV and SV40 entry. Wild-type BKV and the BKV-76W mutant behaved in the same manner as wild-type JCV and PML-mutant strains, respectively, in terms of binding to cells and infectivity when various treatments were employed to prevent engagement of GAGs or sialylated glycans. Although the closely related SV40 is generally thought to use the sialylated ganglioside GM1 as a receptor for infectious entry (Ewers et al., 2010; Neu et al., 2008; Tsai et al., 2003), several SV40 mutants have been identified that are unable to bind GM1 and yet remain infectious, suggesting that there is a GM1-independent entry pathway (Luo et al., 2016; Magaldi et al., 2012). Although we did not construct an SA-mutant for SV40, we were still able to evaluate the ability of SV40 to use the GAG-dependent pathway in isolation using 293TT cells, which can be infected by SV40 despite very low levels of GM1 expression (Magaldi et al., 2012). Additionally, SFT cells treated with GM1 supported higher levels of SV40 infection (data not shown), suggesting that untreated SFTs likewise have limiting amounts of GM1. Infectious entry of SV40 into untreated 293TT and SFT cells thus seems likely to be occurring through a GM1-independent pathway. Our data demonstrate that SV40 transduction of 293TTs and SFTs is almost entirely inhibited by heparin, similar to JCV and BKV SA-mutants. This indicates that SV40 is highly dependent on GAGs for entry into some cell types when the preferred GM1 receptor is unavailable. In contrast to BKV and JCV, SV40 infectivity showed sensitivity to chlorate treatment, suggesting that it may require a different spectrum of GAG modifications than BKV and JCV.
There is indirect evidence from clinical studies that the GAG pathway may be medically relevant for polyomavirus infections. A clinical study involving four patients suggested, speculatively, that systemic heparin treatment might be useful for halting the progression of PML (Major et al., 1992). GAGs have also been investigated as possible treatments for BKV-induced hemorrhagic cystitis following bone marrow transplant (Arthur et al., 1986; Reploeg et al., 2001) and for a potentially BKV/JCV-associated chronic bladder syndrome known as interstitial cystitis (Van der Aa et al., 2014; Winter et al., 2015). GAGs and GAG analogs, such as pentosan polysulfate, heparin, and chondroitin sulfate, are currently recommended treatments for interstitial cystitis (Al-Zahrani and Gajewski, 2011; Cervigni, 2015; Madersbacher et al., 2013; Parsons and Mulholland, 1987; Tutolo et al., 2016), and there is also evidence that treatment with hyaluronic acid (a non-sulfated GAG) can relieve symptoms and achieve remission of hemorrhagic cystitis (Iavazzo et al., 2007; Isik et al., 2014; Miodosky et al., 2006; Shao et al., 2012). Although previous justifications for these treatments have focused on GAGs restoring the barrier function of the bladder epithelium, it is possible to imagine that treatment with GAGs or GAG analogs may actually be inhibiting polyomavirus infectious entry. The observation that hyaluronic acid may sometimes be an effective treatment for hemorrhagic cystitis is particularly intriguing, given our observation that blocking sulfate modifications through chlorate treatment of cells did not affect JCV or BKV transduction.
The recently reported chimeric mouse model to evaluate JCV infectivity and PML mutant pathogenesis could serve as a tool for pre-clinical evaluation of intervention strategies (Kondo et al., 2014). Having an improved understanding of the PML-mutant entry process should allow for new treatment and prevention options for patients at risk of developing PML and for greater usage of immunomodulatory therapeutic agents for management of autoimmune diseases and lymphoid cancers.
Experimental Procedures
Pseudovirus and VLP Production
Methods used to produce pseudovirions and VLPs have been described previously (Buck et al., 2004; Pastrana et al., 2009; Ray et al., 2015). Plasmid maps and detailed methods are available at our lab website, http://home.ccr.cancer.gov/LCO/. See Supplemental Experimental Procedures for additional details.
Synthesis and use of 3Fax-Peracetyl Neu5Ac
3Fax-Peracetyl Neu5Ac (3Fax) was synthesized as previously described (Rillahan et al., 2012). Cells were pre-cultured with 200 μM 3Fax for 2–4 days prior to re-plating for infectivity inhibition assays. See Supplemental Experimental Procedures for further details.
VLP Binding Assay
50,000 SFT cells treated with 3Fax (or DMSO control) were suspended in 0.1 ml of wash medium (DPBS with 1% FBS, 10 mM HEPES, and PSF) and treated with 0.02 units of heparinase I/III (Sigma H3917) or mock-treated at 37 °C for 1 h. Cells were washed once then incubated with 50 ng of VLPs for 1 h at 4 °C. Cells were then washed and incubated with primary antibody (1:1000) for 30 min at 4 °C followed by washing and addition of Alexa Fluor 488-conjugated secondary antibody (Invitrogen) Primary antibodies used include: mouse-anti-JCV polyclonal sera (Ray et al., 2015), mouse-anti-BKV polyclonal sera (Pastrana et al., 2013), rabbit-anti-MCV polyclonal sera (Pastrana et al., 2009), mouse-anti-HPV polyclonal sera (a generous gift from Hanna Seitz and John Schiller, NCI). Additionally, anti-heparan sulfate mAb HS20 (0.5 μg/ml) or a combination of biotinylated SNA (2 μg/ml) and MAL-II (5 μg/ml) lectins were used to measure the effect of the 3Fax treatment on surface expression of heparan sulfate and sialic acid, respectively. Fluorescent signal was measured by flow cytometry using a BD FACSCanto II. Cytometry was analyzed using FlowJo software.
Infectivity Inhibition Assays
Inhibition of the infectivity of pseudovirions by heparin, HS20, and anti-JCV mAbs was measured as previously described (Ray et al., 2015; Schowalter et al., 2011). See Supplemental Experimental Procedures for additional details.
Neuraminidase Treatment
ART or SFT cells were trypsinized and counted, then resuspended at 100,000 cells/ml in wash medium. Neuraminidase (Sigma N2876) was added at 0.4 U/ml for enzyme treated cells, and both enzyme and mock treated cells were incubated at 37°C for 45–60 minutes. Cells were washed once in wash medium and then plated in 96-well plates at 5,000 cells/well for infectivity inhibition assay.
Ganglioside Treatment
ART or SFT cells were pre-plated in 6-well plates the day before use at 500,000 cells/well. GM1 and asialo-GM1 were purchased from Sigma and dissolved in PBS (GM1) or 1:1 methanol:chloroform (asialo-GM1). Gangliosides were diluted to 50 μM in RPMI, sonicated 4× 30 seconds, then transferred to new separate 6-well plates and left in a laminar flow hood with the lid off for 20 minutes to allow evaporation of methanol and chloroform from the asialo-GM1 containing media. Plate-bound cells were washed twice with serum free RPMI and then 2 ml of ganglioside-containing medium was added to wells. Plates were returned to 37°C for 2 hours, and then cells were trypsinized, counted, and re-plated for inhibition assay.
siRNA Knockdown of Sialylated Glycans
SMARTpool ON-TARGET plus siRNA pools targeting SLC35A1 (L-007537-00-0005) and CMAS (L-009780-01-0005), or a non-targeting pool (D-001810-10-05) were purchased from Dharmacon (Lafayette, CO). Cells were plated in 6-well plates at 165,000 cells/well, and the next day transfected with 400 pg of siRNA pools using 2.5 μl of Lipofectamine 2000. After 24 hours, cells were trypsinized, counted, and replated at 165,000 cells/well in a fresh 6-well plate overnight. The culture was then re-transfected with the same siRNA pool. After 24 hours, cells were trypsinized, counted, and re-plated for inhibition assay.
Cell Binding Assay with H2B-NanoLuc (H2BN) VLPs
JCV VLPs carrying an encapsidated histone H2B-NanoLuc fusion protein (H2BN) were generated by inclusion of plasmid pH2BN in the VLP transfection mix. The pH2BN plasmid was generated using PCR to recombine segments of pNL1.1CMV (Promega), plasmid pwM (Tolstov et al., 2009), and pBOS-H2BGFP (BD Pharmingen)(Kanda et al., 1998). Sequencing revealed that the H2B gene in pBOS-H2BGFP contained two non-silent mutations (D26G and V119I). The errors were reverted to the wild-type H2B sequence by overlap PCR.
For the cell binding assay, 1 ng of H2BN-containing VLPs were diluted in wash medium then incubated with 10 μg/ml mAbs or 100 μg/ml heparin in a round bottom untreated 96-well plate for 30 minutes at room temperature. ART or SFT cells were dislodged using PBS with10 mM EDTA, counted, and 20,000 cells were added per well in a separate round bottom plate. Cells were spun for 5 minutes at 500 × g, washed once, resuspended in the antibody/virus mixture, and incubated at room temperature with rocking for 30 minutes. Cells were washed twice then transferred to a white luminometry plate for measurement of NanoLuc signal at a gain of 3200.
Hemagglutination Assay
Hemagglutination assays using JCV-1A VLPs were performed previously described (Pastrana et al., 2013). See Supplemental Experimental Procedures for additional details.
Statistical Analysis
Graphpad Prism software was used to estimate EC50 values by fitting a sigmoidal dose-response curve with variable slope. The top and bottom of the curve were fixed at 100% neutralization or 0% neutralization, respectively.
Supplementary Material
Highlights.
Wild-type JC polyomavirus makes facultative use of two parallel infectious entry pathways
Pathogenic mutants are restricted to a glycosaminoglycan-dependent pathway
Neutralizing antibodies act by blocking engagement of co-receptor glycans
Acknowledgments
General: The authors would like to thank members of the Laboratory of Cellular Oncology (NCI) for helpful suggestions and insights, and we are grateful to Drs. Dan DiMaio and Thomas Magaldi for valuable initial discussions regarding SV40 infectious entry.
Funding: The work was funded by the Intramural Research Program of the NIH, with support from the Center for Cancer Research, NCI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, EMG and CBB; Investigation, EMG, DVP, CK; Resources, EMG, DVP, RMS, UR, WG, MH, GTP, DMS, CK, ECM, BC, JG; Writing – Original Draft, EMG, CBB; Writing – Review & Editing, all authors; Funding Acquisition, CBB.
Competing interests: C.K. and E.C-M are employees of Biogen, Inc and B.C. and J.G. are employees of Neurimmune. M.H., C.B.B, W.G. and E.M.G. have filed an NCI Employee Invention Report.
Data and materials availability: All data and materials are available upon request. Anti-JCV mAb reagents will be covered by a Material Transfer Agreement.
References
- Al-Zahrani Aa, Gajewski JB. Long-term efficacy and tolerability of pentosan polysulphate sodium in the treatment of bladder pain syndrome. Canadian Urological Association journal = Journal de l’Association des urologues du Canada. 2011;5:113–118. doi: 10.5489/cuaj.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur RR, Shah KV, Baust SJ, Santos GW, Saral R. Association of BK viruria with hemorrhagic cystitis in recipients of bone marrow transplants. The New England journal of medicine. 1986;315:230–234. doi: 10.1056/NEJM198607243150405. [DOI] [PubMed] [Google Scholar]
- Bauer J, Gold R, Adams O, Lassmann H. Progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome (IRIS) Acta neuropathologica. 2015;130:751–764. doi: 10.1007/s00401-015-1471-7. [DOI] [PubMed] [Google Scholar]
- Bauer PH, Cui C, Liu WR, Stehle T, Harrison SC, DeCaprio JA, Benjamin TL. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J Virol. 1999;73:5826–5832. doi: 10.1128/jvi.73.7.5826-5832.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JR, Fox RJ. Reassessing the risk of natalizumab-associated PML. Journal of neurovirology. 2016 doi: 10.1007/s13365-016-0427-6. [DOI] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors. Journal of Virology. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Dey D, Kreisman L, Velupillai P, Dahl J, Telford S, Bronson R, Benjamin T. Receptor-Binding and Oncogenic Properties of Polyoma Viruses Isolated from Feral Mice. PLoS pathogens. 2007;3:e179. doi: 10.1371/journal.ppat.0030179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. The Lancet Oncology. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- Casado JL, Corral I, Garcia J, Martinez-San Millan J, Navas E, Moreno A, Moreno S. Continued declining incidence and improved survival of progressive multifocal leukoencephalopathy in HIV/AIDS patients in the current era. Eur J Clin Microbiol Infect Dis. 2014;33:179–187. doi: 10.1007/s10096-013-1941-6. [DOI] [PubMed] [Google Scholar]
- Cervigni M. Interstitial cystitis/bladder pain syndrome and glycosaminoglycans replacement therapy. Translational andrology and urology. 2015;4:638–642. doi: 10.3978/j.issn.2223-4683.2015.11.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collazos J. Opportunistic infections of the CNS in patients with AIDS: diagnosis and management. CNS drugs. 2003;17:869–887. doi: 10.2165/00023210-200317120-00002. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Bhat NR, Spicer SS, Hogan EL, Furuya S, Hirabayashi Y. Cell-specific expression of neutral glycosphingolipids in vertebrate brain: immunochemical localization of 3-O-acetyl-sphingosine-series glycolipid(s) in myelin and oligodendrocytes. J Neurosci Res. 2007;85:2856–2862. doi: 10.1002/jnr.21419. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Hogan EL. Molecular characterization of gangliotetraosylceramide (GA1) in normal human brain and its developmental change. Indian J Biochem Biophys. 1993;30:341–345. [PubMed] [Google Scholar]
- Dasgupta S, van Halbeek H, Hogan EL. Ganglio-N-tetraosylceramide (GA1) of bovine and human brain. Molecular characterization and presence in myelin. FEBS Lett. 1992;301:141–144. doi: 10.1016/0014-5793(92)81234-d. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, van Halbeek H, Spicer S, Hogan EL. Molecular characterization and immunohistochemical localization of IV(4)GalNAcGgOse(4)Cer: a naturally occurring novel neutral glycosphingolipid in bovine brain. Glycobiology. 2000;10:1–9. doi: 10.1093/glycob/10.1.1. [DOI] [PubMed] [Google Scholar]
- DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nature Reviews Microbiology. 2013;11:264–276. doi: 10.1038/nrmicro2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diotti RA, Mancini N, Clementi N, Sautto G, Moreno GJ, Criscuolo E, Cappelletti F, Man P, Forest E, Remy L, et al. Cloning of the first human anti-JCPyV/VP1 neutralizing monoclonal antibody: epitope definition and implications in risk stratification of patients under natalizumab therapy. Antiviral Res. 2014;108:94–103. doi: 10.1016/j.antiviral.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Diotti RA, Nakanishi A, Clementi N, Mancini N, Criscuolo E, Solforosi L, Clementi M. JC Polyomavirus (JCV) and Monoclonal Antibodies: Friends or Potential Foes? Clinical and Developmental Immunology. 2013;2013:1–11. doi: 10.1155/2013/967581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd KA, Pierson TC. Antibody-mediated neutralization of flaviviruses: a reductionist view. Virology. 2011;411:306–315. doi: 10.1016/j.virol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan AS, Gasparovic ML, Atwood WJ. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus) Journal of virology. 2008;82:2560–2564. doi: 10.1128/JVI.02123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Stewart TE, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers H, Romer W, Smith AE, Bacia K, Dmitrieff S, Chai W, Mancini R, Kartenbeck J, Chambon V, Berland L, et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat Cell Biol. 2010;12:11–18. doi: 10.1038/ncb1999. sup pp 11-12. [DOI] [PubMed] [Google Scholar]
- Farhy Tselnicker I, Boisvert MM, Allen NJ. The role of neuronal versus astrocyte-derived heparan sulfate proteoglycans in brain development and injury. Biochemical Society transactions. 2014;42:1263–1269. doi: 10.1042/BST20140166. [DOI] [PubMed] [Google Scholar]
- Gao W, Kim H, Feng M, Phung Y, Xavier CP, Rubin JS, Ho M. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology. 2014;60:576–587. doi: 10.1002/hep.26996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, et al. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. The Journal of infectious diseases. 2011;204:103–114. doi: 10.1093/infdis/jir198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossai A, Waterboer T, Nelson HH, Michel A, Willhauck-Fleckenstein M, Farzan SF, Hoen AG, Christensen BC, Kelsey KT, Marsit CJ, et al. Seroepidemiology of Human Polyomaviruses in a US Population. American journal of epidemiology. 2016;183:61–69. doi: 10.1093/aje/kwv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley Sa, O’Hara Ba, Nelson CDS, Brittingham FLP, Henriksen KJ, Stopa EG, Atwood WJ. The American Journal of Pathology. 2015. Human Polyomavirus Receptor Distribution in Brain Parenchyma Contrasts with Receptor Distribution in Kidney and Choroid Plexus; pp. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley SE, Das SR, Bailey AL, Schmidt LM, Hickman HD, Jayaraman A, Viswanathan K, Raman R, Sasisekharan R, Bennink JR, et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science. 2009;326:734–736. doi: 10.1126/science.1178258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LY, Halder S, Agbandje-McKenna M. Parvovirus glycan interactions. Curr Opin Virol. 2014;7:108–118. doi: 10.1016/j.coviro.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavazzo C, Athanasiou S, Pitsouni E, Falagas ME. Hyaluronic acid: an effective alternative treatment of interstitial cystitis, recurrent urinary tract infections, and hemorrhagic cystitis? European urology. 2007;51:1534–1540. doi: 10.1016/j.eururo.2007.03.020. discussion 1540-1531. [DOI] [PubMed] [Google Scholar]
- Isik P, Ozbek N, Azik F, Arman Bilir O, Baylan B, Tunc B. Successful treatment of hemorrhagic cystitis after hematopoietic stem cell transplantation with intravesical hyaluronic acid. Pediatric transplantation. 2014;18:780–781. doi: 10.1111/petr.12354. [DOI] [PubMed] [Google Scholar]
- Jelcic I, Combaluzier B, Jelcic I, Faigle W, Senn L, Reinhart BJ, Ströh L, Nitsch RM, Stehle T, Sospedra M, et al. Broadly neutralizing human monoclonal JC polyomavirus VP1-specific antibodies as candidate therapeutics for progressive multifocal leukoencephalopathy. Science translational medicine. 2015;7:306ra150–306ra150. doi: 10.1126/scitranslmed.aac8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamhi E, Joo EJ, Dordick JS, Linhardt RJ. Glycosaminoglycans in infectious disease. Biological Reviews. 2013 doi: 10.1111/brv.12034. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS pathogens. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. The New England journal of medicine. 2005;353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, Miller E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. Journal of medical virology. 2003;71:115–123. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- Komagome R, Sawa H, Suzuki T, Suzuki Y, Tanaka S, Atwood WJ, Nagashima K. Oligosaccharides as receptors for JC virus. Journal of virology. 2002;76:12992–13000. doi: 10.1128/JVI.76.24.12992-13000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, et al. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. The Journal of clinical investigation. 2014:1–14. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusunoki S, Tsuji S, Nagai Y. Ganglio-N-tetraosylceramide (asialo GM1), an antigen common to the brain and immune system: its localization in myelin. Brain Res. 1985;334:117–124. doi: 10.1016/0006-8993(85)90573-6. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. The New England journal of medicine. 2005;353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Liu CK, Wei G, Atwood WJ. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal alpha(2–6)-linked sialic acids. Journal of virology. 1998;72:4643–4649. doi: 10.1128/jvi.72.6.4643-4649.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Silva R. Polyoma BK virus: an emerging opportunistic infectious agent of the human central nervous system. Braz J Infect Dis. 2011;15:276–284. doi: 10.1016/s1413-8670(11)70189-1. [DOI] [PubMed] [Google Scholar]
- Luo Y, Motamedi N, Magaldi TG, Gee GV, Atwood WJ, DiMaio D. Interaction between Simian Virus 40 Major Capsid Protein VP1 and Cell Surface Ganglioside GM1 Triggers Vacuole Formation. MBio. 2016;7 doi: 10.1128/mBio.00297-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madersbacher H, van Ophoven A, van Kerrebroeck PEVA. GAG layer replenishment therapy for chronic forms of cystitis with intravesical glycosaminoglycans-A review. Neurourology and Urodynamics. 2013;32:9–18. doi: 10.1002/nau.22256. [DOI] [PubMed] [Google Scholar]
- Maeda N. Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front Neurosci. 2015;9:98. doi: 10.3389/fnins.2015.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaldi TG, Buch MHC, Murata H, Erickson KD, Neu U, Garcea RL, Peden K, Stehle T, DiMaio D. Mutations in the GM1 binding site of simian virus 40 VP1 alter receptor usage and cell tropism. Journal of Virology. 2012;86:7028–7042. doi: 10.1128/JVI.00371-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Ströh LJ, Gee GV, O’Hara BA, Derdowski A, Stehle T, Atwood WJ. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio. 2013;4:e00247–00213. doi: 10.1128/mBio.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clinical microbiology reviews. 1992;5:49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miodosky M, Abdul-Hai A, Tsirigotis P, Or R, Bitan M, Resnick IB, Gesundheit B, Zilberman I, Ioffe L, Leubovic A, et al. Treatment of post-hematopoietic stem cell transplantation hemorrhagic cystitis with intravesicular sodium hyaluronate. Bone marrow transplantation. 2006;38:507–511. doi: 10.1038/sj.bmt.1705474. [DOI] [PubMed] [Google Scholar]
- Neu U, Allen S-aA, Blaum BS, Liu Y, Frank M, Palma AS, Ströh LJ, Feizi T, Peters T, Atwood WJ, et al. A structure-guided mutation in the major capsid protein retargets BK polyomavirus. PLoS pathogens. 2013;9:e1003688–e1003688. doi: 10.1371/journal.ppat.1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Maginnis MS, Palma AS, Ströh LJ, Nelson CDS, Feizi T, Atwood WJ, Stehle T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell host & microbe. 2010;8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Woellner K, Gauglitz G, Stehle T. Structural basis of GM1 ganglioside recognition by simian virus 40. Proc Natl Acad Sci U S A. 2008;105:5219–5224. doi: 10.1073/pnas.0710301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell CD, Vogel L, Wright A, Das SR, Wrammert J, Li GM, McCausland M, Zheng NY, Yewdell JW, Ahmed R, et al. Antibody pressure by a human monoclonal antibody targeting the 2009 pandemic H1N1 virus hemagglutinin drives the emergence of a virus with increased virulence in mice. MBio. 2012;3 doi: 10.1128/mBio.00120-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CL, Mulholland SG. Successful therapy of interstitial cystitis with pentosanpolysulfate. The Journal of urology. 1987;138:513–516. doi: 10.1016/s0022-5347(17)43243-5. [DOI] [PubMed] [Google Scholar]
- Pastrana DV, Pumphrey KA, Cuburu N, Schowalter RM, Buck CB. Characterization of monoclonal antibodies specific for the Merkel cell polyomavirus capsid. Virology. 2010;405:20–25. doi: 10.1016/j.virol.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana DV, Ray U, Magaldi TG, Schowalter RM, Çuburu N, Buck CB. BK polyomavirus genotypes represent distinct serotypes with distinct entry tropism. Journal of virology. 2013;87:10105–10113. doi: 10.1128/JVI.01189-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana DV, Tolstov YL, Becker JC, Moore PS, Chang Y, Buck CB. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS pathogens. 2009;5:e1000578–e1000578. doi: 10.1371/journal.ppat.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray U, Cinque P, Gerevini S, Longo V, Lazzarin A, Schippling S, Martin R, Buck CB, Pastrana DV. JC polyomavirus mutants escape antibody-mediated neutralization. Science translational medicine. 2015;7:306ra151–306ra151. doi: 10.1126/scitranslmed.aab1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, McAuliffe M, Tonkin C, Simon K, Goelz S, et al. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. The Journal of infectious diseases. 2011;204:237–244. doi: 10.1093/infdis/jir256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reploeg MD, Storch GA, Clifford DB. Bk virus: a clinical review. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2001;33:191–202. doi: 10.1086/321813. [DOI] [PubMed] [Google Scholar]
- Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, Paulson JC. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nature Chemical Biology. 2012;8:661–668. doi: 10.1038/nchembio.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowalter RM, Pastrana DV, Buck CB. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS pathogens. 2011;7:e1002161–e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Lu G-l, Shen Z-j. Comparison of intravesical hyaluronic acid instillation and hyperbaric oxygen in the treatment of radiation-induced hemorrhagic cystitis. BJU international. 2012;109:691–694. doi: 10.1111/j.1464-410X.2011.10550.x. [DOI] [PubMed] [Google Scholar]
- Steiner I, Berger JR. Update on progressive multifocal leukoencephalopathy. Current neurology and neuroscience reports. 2012;12:680–686. doi: 10.1007/s11910-012-0313-4. [DOI] [PubMed] [Google Scholar]
- Ströh LJ, Maginnis MS, Blaum BS, Nelson CDS, Neu U, Gee GV, O’Hara BA, Motamedi N, DiMaio D, Atwood WJ, et al. Increased Affinity of JC Polyomavirus Capsid for LSTc Over Other Sialylated Glycans Is a Major Determinant of Infectivity. Journal of Virology, JVI. 2015:00489–00415. doi: 10.1128/JVI.00489-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroh LJ, Stehle T. Glycan Engagement by Viruses: Receptor Switches and Specificity. Annu Rev Virol. 2014;1:285–306. doi: 10.1146/annurev-virology-031413-085417. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML) PLoS genetics. 2009;5:e1000368–e1000368. doi: 10.1371/journal.pgen.1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, Chang Y, Buck CB, Moore PS. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays - Tolstov - 2009 - International Journal of Cancer - Wiley Online Library. International Journal of Cancer. 2009;125:1250–1256. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. Gangliosides are receptors for murine polyoma virus and SV40. The EMBO journal. 2003;22:4346–4355. doi: 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tur C, Montalban X. Natalizumab: risk stratification of individual patients with multiple sclerosis. CNS drugs. 2014;28:641–648. doi: 10.1007/s40263-014-0168-0. [DOI] [PubMed] [Google Scholar]
- Tutolo M, Ammirati E, Castagna G, Klockaerts K, Plancke H, Ost D, Van der Aa F, De Ridder D. A prospective randomized controlled multicentre trial comparing intravesical DMSO and chondroitin sulphate 2% for painful bladder syndrome/interstitial cystitis. Int Braz J Urol. 2016 doi: 10.1590/S1677-5538.IBJU.2016.0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. The New England journal of medicine. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Van der Aa F, Beckley I, de Ridder D. Polyomavirus BK–a potential new therapeutic target for painful bladder syndrome/interstitial cystitis? Medical hypotheses. 2014;83:317–320. doi: 10.1016/j.mehy.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T, et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology. 2015a;25:1323–1324. doi: 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Stanley P, Hart G, Aebi M, Darvill A, Kinoshita T, Packer NH, Prestegard JJ, et al. In: Essentials of Glycobiology In Essentials of Glycobiology. Varki A, Cummings RD, Esko JD, Stanley P, Hart G, Aebi M, Darvill A, Kinoshita T, Packer NH, Prestegard JJ, et al., editors. Cold Spring Harbor; NY: 2015b. [PubMed] [Google Scholar]
- Winter BJ, O’Connell HE, Bowden S, Carey M, Eisen DP. A Case Control Study Reveals that Polyomaviruria Is Significantly Associated with Interstitial Cystitis and Vesical Ulceration. PloS one. 2015;10:e0137310–e0137310. doi: 10.1371/journal.pone.0137310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim SH, Sherin JE, Szuchet S. Oligodendrocyte proteoglycans: modulation by cell-substratum adhesion. J Neurosci Res. 1993;34:401–413. doi: 10.1002/jnr.490340405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.