

SUMMARY
The blood-tumor barrier (BTB) is a major obstacle for drug delivery to malignant brain tumors such as glioblastoma. Disrupting the BTB is therefore highly desirable, but complicated by the need to maintain the normal blood-brain barrier (BBB). Here, we show that targeting glioma stem cell (GSC)-derived pericytes specifically disrupts the BTB and enhances drug effusion into brain tumors. We found that pericyte coverage of tumor vasculature is inversely correlated with GBM patient survival after chemotherapy. Eliminating GSC-derived pericytes in xenograft models disrupted BTB tight junctions and increased vascular permeability. We identified BMX as an essential factor for maintaining GSC-derived pericytes. Inhibiting BMX with ibrutinib selectively targeted neoplastic pericytes and disrupts the BTB, but not the BBB, thereby increasing drug effusion into established tumors and enhancing chemotherapeutic efficacy of drugs with poor BTB penetration. These findings highlight the clinical potential of targeting neoplastic pericytes to significantly enhance treatment of brain tumors.
Keywords: pericytes, glioma stem cells, blood-tumor barrier (BTB), blood-brain barrier (BBB), BMX, ibrutinib
eTOC Blurb
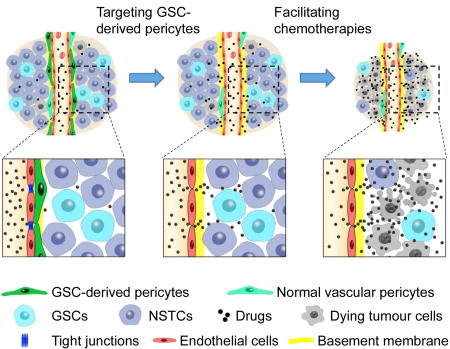
The blood-brain barrier (BBB) blocks entry of harmful materials into normal brains, but the blood-tumor barrier (BTB) prevents anti-cancer drugs from penetrating GBM tumors. Targeting GSC-derived neoplastic pericytes selectively disrupted the BTB but not BBB and potently enhanced drug delivery to effectively improve GBM treatment.
INTRODUCTION
Glioblastoma (GBM) is a highly lethal brain tumor resistant to current therapies. Despite a significant advance in cancer treatment, there has been little improvement in the prognosis of GBM patients over decades (Chinot et al., 2014; McNamara et al., 2014). This striking failure could be ascribed to multiple factors, but the blood-tumor barrier (BTB) represents a formidable obstacle. As a filtering barrier of capillaries, the BTB and the blood-brain barrier (BBB) prevent most anti-cancer agents from penetrating GBM tumors and limit therapeutic efficacy (Fellner et al., 2002; Neuwelt et al., 1986). The BTB and BBB exclude most compounds except highly lipidized small molecules of less than 400 daltons, rendering potentially powerful anti-cancer drugs impotent for GBM treatment (Pardridge, 2002). Thus, breaking the BTB or BBB will significantly impact GBM treatment. However, as the BBB serves as an indispensable vascular barrier to protect normal brain by blocking entry of harmful materials (Bergers and Song, 2005), abnormal breakdown of the BBB could result in neuronal degenerative changes or diseases (Bell et al., 2010). Thus, it is crucial to selectively disrupt the BTB in GBM tumors while sparing the BBB in normal brain tissues during chemotherapy.
The BBB and BTB are composed of several components including tight junctions, vessel basement membrane, and astrocyte end-feet that cover endothelial cells to form a physical barrier (Watkins et al., 2014; Yao et al., 2014). Besides, molecular effusion through endothelial cells is actively regulated by transcytosis, the vesicular transport of macromolecules across cells (Shue et al., 2008; Zhao et al., 2015). Despite their similarities, the BBB and BTB differ from many aspects. The BTB is formed by highly proliferative, morphologically abnormal tumor capillaries (Pries et al., 2010; Verbeek et al., 1994). These tumor vessels may have aberrant expression of transcytotic proteins (Strickland et al., 2005). In addition, the perivascular components such as astrocytic end-feet on tumor vessels are altered (Watkins et al., 2014). These facts suggest a possibility to selectively disrupt the BTB in the tumor without affecting the BBB in normal brain tissue, which will enhance delivery of the poor BTB-penetrating but potent anti-cancer drugs into brain tumors to improve chemotherapy efficacy but limit the unwanted toxicity to the normal brain.
Vascular pericytes that attach on endothelial cells play a pivotal role in maintaining the BBB. Pericyte depletion disrupted the BBB and elevated vascular permeability in Pdgfb knock-out mice (Armulik et al., 2010; Daneman et al., 2010). Consistent with the essential functions of pericytes in maintaining the BBB, loss of pericytes has been observed in brain disorders including Alzheimer’s disease and ischemia, which is believed to exacerbate the diseases (Hall et al., 2014; Sagare et al., 2013). Interestingly, in GBM tumors, the majority of vascular pericytes are derived from glioma stem cells (GSCs) through trans-differentiation (Cheng et al., 2013). These GSC-derived pericytes bear tumor-specific genetic alterations that distinguish them from normal pericytes (Cheng et al., 2013), indicating a possibility to selectively target these neoplastic pericytes. However, the functional significance of the GSC-derived pericytes in maintaining the structure and function of the BTB in GBMs remains elusive. This study aims to determine the role of GSC-derived pericytes in the BTB maintenance and the effects of genetic and pharmacological targeting of tumor pericytes on vascular permeability, drug delivery and chemotherapy efficacy. Because the majority of neoplastic pericytes in GBM tumors originate from GSCs, these pericytes likely share some essential molecular signatures with GSCs. Among the multiple regulators critical for GSC maintenance, we identified the bone marrow and X-linked (BMX) non-receptor tyrosine kinase to be highly expressed in the GSC-derived pericytes but not in normal pericytes in the brain.
BMX is a member of the Tec tyrosine kinase family (Saharinen et al., 1997). BMX kinase can be activated by multiple signaling pathways and several chemokines, VEGF receptors, ErbB3 and integrins (Chen et al., 2001; Jiang et al., 2007; Pan et al., 2002). BMX is highly expressed in multiple cancers and contributes to cell transformation, proliferation and resistance to therapies (Dai et al., 2010; Guo et al., 2007). We found that BMX is preferentially expressed in GSCs and activates STAT3 to maintain the self-renewal and tumorigenic potential of GSCs (Guryanova et al., 2011). Interestingly, we and others detected BMX expression in the perivascular regions (Ekman et al., 1997; Guryanova et al., 2011). As GSCs are able to differentiate into pericytes but not endothelial cells (Cheng et al., 2013), GSC-derived pericytes may maintain BMX expression. In this study, we found that BMX is preferentially expressed in GSC-derived pericyte, which makes selective targeting and pharmacological inhibition of neoplastic pericytes possible. Importantly, the kinase activity of BMX could be inhibited by ibrutinib (PCI-32765), an inhibitor initially developed to inhibit the Bruton’s tyrosine kinase (BTK) but showed a similar affinity to BMX (Honigberg et al., 2010). Recently approved by the FDA, ibrutinib has been used clinically to effectively treat lymphomas and chronic lymphocytic leukemia through targeting BTK (Bernard et al., 2015; Herman et al., 2014; Lionakis et al., 2017; Wilson et al., 2015). Moreover, ibrutinib has been shown to penetrate the BBB (Bernard et al., 2015; Lionakis et al., 2017). Therefore, there is significant clinical potential in repurposing ibrutinib for BMX inhibition to target GSC-derived pericytes for selective disruption of the BTB.
In this study, we assessed the therapeutic impact of targeting neoplastic pericytes on the BTB disruption to enhance drug delivery into GBM tumors. We investigated the potential relationship between vascular pericyte coverage and the survival of GBM patients treated with chemotherapy. In an orthotopic GBM model, we demonstrated that selective targeting of GSC-derived pericytes potently disrupted the BTB tight junctions and increased vascular permeability to enhance drug effusion into GBM tumors. Importantly, we found that inhibition of BMX by ibrutinib in vivo potently disrupted tumor vascular pericytes, selectively impaired the BTB but not the BBB, and enhanced delivery of poor BTB-penetrating drugs into tumors to inhibit malignant growth. Our preclinical studies identified ibrutinib as a BTB-disrupting drug that can synergize with the poor BTB-penetrating anti-cancer drugs to improve chemotherapy efficacy for GBM patients.
RESULTS
Pericyte Coverage Is Inversely Correlated with Prognosis of GBM Patients Treated with Chemotherapy
Because high pericyte coverage is associated with low vascular permeability, we speculated that the degree of pericyte coverage in GBM tumors may impact drug delivery in chemotherapy. We initially examined the potential link between pericyte coverage in GBM specimens and the survival of patients treated with or without chemotherapy. Immunofluorescent staining of α-SMA, a tumor pericyte marker that is expressed in vascular pericytes in brain tumors and smooth muscle cells on extracranial vessels (Armulik et al., 2011), and the endothelial cell (EC) marker CD31 in human primary GBMs revealed that pericyte coverage (pericyte density/EC density) on tumor vessels varies across GBM specimens (Figures 1A and S1A). To explore the potential association between pericyte coverage and chemotherapy efficacy, we examined pericyte coverage in a cohort of human GBM specimens (Table S1), and investigated its correlation with the survival of GBM patients treated with or without chemotherapy. Pericyte coverage in each GBM was determined by immunohistochemical staining of the tumor pericyte marker α-SMA and the EC marker CD34 that labels ECs better than CD31 in the formalin-fixed and paraffin-embedded tumor sections (Figures 1B and S1B–C). GBM patients were stratified into high and low pericyte coverage groups by using the average value of pericyte coverage of all examined GBMs as a threshold. Statistical analysis confirmed a significant difference in tumor pericyte coverage between these two groups of GBMs (Figure S1D). We then determined whether pericyte coverage correlates with the patient’s response to chemotherapy. Kaplan-Meier survival analyses revealed an inverse correlation between pericyte coverage and the overall survival and progression-free survival of GBM patients treated with chemotherapy (n = 40) (Figures 1C and 1D). However, in the group of GBM patients without chemotherapy (n = 26), the pericyte coverage showed no significant correlation with the prognosis (Figures 1E and 1F). In addition, regression analyses further confirmed a significant inverse correlation between pericyte coverage and the prognosis of GBM patients treated with chemotherapy (Figure 1G), but such correlation was not significant in GBM patients without chemotherapy (Figure 1H). Collectively, these data demonstrate that the extent of pericyte coverage in GBM is inversely associated with patient response to chemotherapy, suggesting that neoplastic pericytes in GBM may regulate the BTB to impact the efficiency of drug delivery.
Figure 1. High Pericyte Coverage Predicts Poor Prognosis of GBM Patients in Response to Chemotherapy.

(A) Representative immunofluorescent images of human primary GBMs with high or low pericyte coverage stained for the tumor pericyte marker α-SMA (green), the endothelial marker CD31 (red), and nuclei with DAPI (blue) (scale bar, 80µm).
(B) Representative immunohistochemical images of human primary GBMs with high or low pericyte coverage. Consecutive paraffin sections were stained for α-SMA or the endothelial marker CD34 (scale bar, 80µm).
(C and D) Kaplan-Meier survival curves show an inverse correlation between pericyte coverage and overall survival (C) or progression-free survival (D) of GBM patients treated with chemotherapy (n = 40). Pericyte coverage was defined as the ratio of α-SMA intensity to the CD34 intensity. The average pericyte coverage of all GBMs was used as the cut-off to divide GBM patients into high and low pericyte coverage groups. (n = 27 for low pericyte coverage, n = 13 for high pericyte coverage; ***, p < 0.001, two-tailed log-rank test).
(E and F) Kaplan-Meier survival curves show no correlation between pericyte coverage and overall survival (E) or progression-free survival (F) of GBM patients without chemotherapy (n = 26). Similar analyses described in (C and D) were performed in a cohort of GBM patients who did not receive any chemotherapy. (n = 18 for low pericyte coverage, n = 8 for high pericyte coverage; two-tailed log-rank test).
(G and H) Linear regression analysis of the correlation between pericyte coverage and progression-free survival of GBM patients treated with chemotherapy (G) (n = 40) or without chemotherapy (H) (n = 26). Pericyte coverage is inversely correlated to progression-free survival of patients treated with chemotherapy (G) (n = 40, p = 0.032, r = −0.341). No correlation is observed between pericyte coverage and progression-free survival of GBM patients without chemotherapy (H) (n = 26, p = 0.28, r = −0.22).
See also Figure S1 and Table S1.
Targeting GSC-derived Pericytes Disrupts the BTB and Increases Vascular Permeability
Next we addressed whether targeting neoplastic pericytes disrupts the BTB to increase vascular permeability in GBM xenografts. Our previous study demonstrated that the majority of vascular pericytes in GBM tumors are derived from GSCs (Cheng et al., 2013). We took advantage of the herpes simplex virus thymidine kinase (HsvTK) targeting system (Culver et al., 1992) with the Desmin promoter-driven HsvTK (DesPro-HsvTK) (Figure 2A) to selectively eliminate the GSC-derived pericytes in GBM xenografts upon ganciclovir (GCV) treatment (Cheng et al., 2013). Briefly, sorted GSCs were transduced with DesPro-HsvTK and then transplanted into brains of immunocompromised mice. Mice bearing the tumors were then treated with GCV to selectively eliminate the GSC-derived pericytes expressing HsvTK. We used this selective targeting system to investigate whether elimination of GSC-derived pericytes alters vascular permeability in the intracranial GBM xenografts. Twenty days after implantation of the GSCs transduced with DesPro-HsvTK or the DesPro-Vec control, mice were confirmed to have similarly sized tumors and then treated with GCV for 3–5 days. Because long-term depletion of GSC-derived pericytes by GCV treatment severely disrupted vessels (Cheng et al., 2013), we tested whether a short-term GCV treatment could largely eliminate GSC-derived pericyte but preserve vessels. GCV treatment for 4 days reduced pericyte-coverage from 80% to 10% but retained vessels as measured by co-staining of the pericyte marker Desmin and the endothelial marker Glut1 in the xenografts derived from GSCs (T4121 and CCF2045) transduced with DesPro-HsvTK (Figures S2A and S2B). Meanwhile, the vessel density was not affected by the short-term GCV treatment (Figures S2A and S2C). Then we performed the same experimental procedure followed by tail vein injection of fluorescent tracers to examine vascular permeability (Figure 2B). Cadaverine is a small molecular (1 kDa) fluorescent tracer commonly used to measure the integrity of the BBB or BTB and thus serves as an indicator of vascular permeability in vivo (Armulik et al., 2010; Watkins et al., 2014). Two hours after cadaverine injection, mice were sacrificed and the intracranial tumors were analyzed for cadaverine effusion. Selective targeting of GSC-derived pericytes significantly elevated (>40 fold) cadaverine effusion into tumors derived from the GSCs transduced with DesPro-HsvTK but not into the matched normal brain tissue (Figures 2C and 2D; Figures S2D and S2E), whereas cadaverine effusion was negligible in tumors derived from the GSCs transduced with DesPro-Vec after GCV treatment. However, targeting GSC-derived pericytes did not increase the entry of a large fluorescent tracer dextran (10 kDa) into tumors (Figures 2E and 2F). Furthermore, we found that the effusion of a tracer of 3 kDa was slightly increased, whereas tracers larger than 3 kDa were impeded from entering the tumor mass (Figures 2E and 2F; Figures S2F–I). Importantly, the elevated tracer effusion was detected only in tumor tissues but not in the matched normal brain tissues after targeting GSC-derived pericytes (Figures 2C–F), suggesting a selective disruption of the BTB but not the BBB. These results demonstrate that selective disruption of GSC-derived pericytes significantly increases vascular permeability, allowing for efficient effusion of small molecules. Thus, selective targeting of GSC-derived pericytes may specifically disrupt the BTB and facilitate drug delivery into GBM tumors to improve chemotherapy efficacy.
Figure 2. Selective Elimination of GSC-derived Pericytes Increased Vascular Permeability and Allowed Small Molecule Effusion into Intracranial GBM Xenografts.

(A) A schematic presentation of the Desmin promoter-driven HsvTK construct (DesPro-HsvTK) for selective targeting of GSC-derived pericytes. The CMV promoter of the pCDH-CMV-MCS-EF1-Puro lentiviral vector was replaced by the Desmin promoter. The herpes simplex virus thymidine kinase (HsvTK) coding sequence was inserted behind the Desmin promoter.
(B) A workflow of the study to test the effect of selective targeting of GSC-derived pericytes on vascular permeability. GSCs transduced with DesPro-HsvTK or DesPro-Vec were implanted into athymic mice to generate intracranial GBM xenografts. 20 days after implantation, mice were treated with GCV (1mg/20g) through intraperitoneal injection for 4 days. Then the fluorescent tracer cadaverine (1 kDa) or dextran (10 kDa) was introduced into the mice through tail vein injection (250µg/20g). 2 hours after tracer injection, mice were sacrificed and the mouse brains were harvested for subsequent analyses.
(C) Detection of vascular permeability with the autonomous fluorescent tracer cadaverine (green) and immunofluorescent staining of CD31 (red) in xenografts derived from T4121 GSCs transduced with DesPro-HsvTK or DesPro-Vec after GCV treatment (scale bar, 80µm).
(D) Statistical analysis of cadaverine effusion in xenografts derived from the GSCs transduced with DesPro-HsvTK or DesPro-Vec after GCV treatment (n = 5 tumors / group).
(E) Detection of vascular permeability with the autonomous fluorescent tracer dextran (green) and immunofluorescent staining of CD31 (red) in xenografts derived from the GSCs transduced with DesPro-HsvTK or DesPro-Vec after GCV treatment (scale bar, 80µm).
(F) Statistical quantification of dextran effusion into xenografts derived from the GSCs transduced with DesPro-HsvTK or DesPro-Vec after GCV treatment. (n = 5 tumors / group).
Data are presented as mean ± s.e.m. ** p < 0.01 and ns p > 0.05 as assayed by Mann Whitney test.
See also Figure S2.
Targeting GSC-derived Pericytes Impairs BTB Tight Junctions
We next investigated the impact of disrupting GSC-derived pericytes on the BTB integrity in tumor vessels. The BBB or BTB contains several components including tight junctions, vessel basement membrane, and astrocyte end-feet. In addition, transcytosis across endothelial cells may regulate the BBB or BTB. Vascular tight junctions block the free exchange/diffusion of materials between vessels and the surrounding tissues (Tietz and Engelhardt, 2015) and thus provide the fundamental structural basis for the BBB or BTB. Through immunofluorescence staining of two tight junction markers ZO-1 and Occludin, we found that disruption of GSC-derived pericytes severely impaired the tight junctions (Figures 3A and 3B; Figures S3A and S3B). Less than 5% of tumor vessels marked by CD31 staining had tight junctions after elimination of GSC-derived pericytes (Figures 3C and 3D; Figures S3C and S3D). In contrast, the tight junctions were intact in the control xenografts without pericyte disruption (Figures 3A and 3B; Figures S3A and S3B). More than 60% of tumor vessels were positive for ZO-1 and Occludin in the control xenografts (Figures 3C and 3D; Figures S3C and S3D). Moreover, transmission electron microscopy demonstrated the loss of tight junction structures between endothelial cells on tumor vessels in xenografts after disruption of GSC-derived pericytes (Figure 3E), but disruption of the neoplastic pericytes did not alter the tight junctions on vessels in the matched normal brain tissue (Figure 3E). These data indicate that disruption of GSC-derived pericytes caused a significant loss of tight junctions to affect BTB integrity. Next, we examined whether targeting GSC-derived pericytes impacts other BTB components (Figures S3E–H; Figure S4). Surprisingly, disruption of GSC-derived pericytes had little effect on the vascular basement membrane according to immunofluorescent staining of the vascular basement membrane marker Laminin (Figures S3E and S3F; Figures S4A and S4B). Likewise, expression of Plvap that associated with endothelial vesicle transport and the BBB or BTB transcytosis (Shue et al., 2008) was not changed in the xenografts after disruption of GSC-derived pericytes (Figures S3G and S3H; Figures S4C and S4D), indicating the unchanged endothelial transcytosis after pericyte disruption in the tumors. Of note, Plvap was not detected on normal vessels in mouse brains but highly expressed on tumor vessels (Figures S4E and S4F). This phenomenon was reminiscent of the elevated Plvap expression in human gliomas (Strickland et al., 2005), suggesting that the regulation of Plvap expression in endothelial cells may be different between tumor and normal brain tissues. Analyses of endothelial vesicles by electron microscopy further confirmed the unaltered vascular endothelial transcytosis after disruption of GSC-derived pericytes (Figures S4G and S4H). In addition, few astrocytes showed contact with vessels in the intracranial GSC-derived xenografts, which was not altered by the disruption of GSC-derived pericytes (Figures S4I and S4J). Collectively, these data demonstrate that targeting GSC-derived pericytes mainly disrupted tight junctions to increase vascular permeability in GBM tumors.
Figure 3. Selective Targeting of GSC-derived Pericytes Disrupted the BTB Tight Junctions.

(A and B) Immunofluorescent analysis of the tight junction markers ZO-1 (A) and Occludin (B) (green) and the endothelial marker CD31 (red) in xenografts derived from T4121 GSCs transduced with DesPro-Vec or DesPro-HsvTK after GCV treatment for 4 days (scale bar, 80µm).
(C and D) Statistical quantification of (A) and (B) to determine the ZO-1 (C) and Occludin (D) intensity on vessels. ZO-1 or Occludin intensity was determined by the numbers of ZO-1-positive or Occludin-positive cells normalized to the numbers of CD31-positive cells (n = 5 tumors / group; **, p < 0.01; mean ± s.e.m.; Mann Whitney test).
(E) Transmission electron microscopy images of vessels in GBM tumors and matched brain tissues from mice bearing intracranial GBMs derived from T4121 GSCs transduced with DesPro-HsvTK or DesPro-Vec after GCV treatment for 5 days. Red arrows indicate tight junctions between endothelial cells on vessels. Blue arrows indicate the gaps lacking tight junctions between endothelial cells on vessels in tumors after disruption of GSC-derived pericytes. Endothelial cells (EC) were highlighted by red dot lines. Pericytes (PC) were highlighted by green dot lines. No pericyte was detected on vessels in the tumors derived from DesPro-HsvTK-transduced GSCs after GCV treatment (scale bar, 400nm).
See also Figures S3 and S4.
Disruption of the BTB by Targeting GSC-derived Pericytes Improves GBM Chemotherapy Efficacy
Because high pericyte coverage is significantly correlated with poor survival of GBM patients treated with chemotherapy (Figures 1C and 1D), the low vascular permeability mediated by high pericyte coverage on tumor vessels is likely to hamper the chemotherapeutic efficacy. We hypothesized that targeting GSC-derived pericytes should enhance drug delivery to benefit chemotherapy. To test this hypothesis, we established intracranial GBM xenografts using the GSCs transduced with luciferase and the DesPro-HsvTK or DesPro-Vec control. We then treated the mice with GCV and etoposide, a poor BBB-penetrating anti-cancer drug. Etoposide is a cytotoxic agent that inhibits topoisomerase and is an integral component in the treatment of multiple cancers (Alifrangis et al., 2013; de Wit et al., 2012; Hande, 1998), but it is relegated to third-line therapy for GBMs partially due to its poor BBB penetration (Wijnholds et al., 2000). We examined whether targeting GSC-derived pericytes could enable etoposide to be more effective in GBM treatment. Because complete depletion of pericytes would collapse vessels and reduce perfusion, we applied pulsed-dose GCV for two consecutive days with a two-day interval to disrupt the pericytes while maintaining the underlying vasculature. Etoposide was administered daily (Figure 4A). This procedure resulted in similar hypoxia formation in tumors with or without pericyte disruption (Figures S5A and S5B), suggesting that it maintained perfusion in the tumor tissue. Bioluminescent imaging of orthotopic xenografts demonstrated that while etoposide treatment alone had little effect on tumor growth, disruption of GSC-derived pericytes by GCV treatment enabled etoposide to effectively retard tumor growth in mice bearing the xenografts derived from the DesPro-HsvTK-transduced GSCs relative to other groups (Figure 4B). Of note, in the group of mice bearing the xenografts derived from the DesPro-Vec-transduced GSCs, GCV plus etoposide treatment showed little effect on tumor growth (Figure 4B). Importantly, Kaplan-Meier survival curves demonstrated that disrupting GSC-derived pericytes in combination with etoposide treatment significantly extended the survival of mice (Figure 4C). These data indicate that disruption of GSC-derived pericytes is critical for the effective delivery of etoposide into tumors to inhibit malignant growth. Chromatography confirmed a significant increase of etoposide concentration in tumor tissues versus matched normal brain tissues after disruption of GSC-derived pericytes (Figure S5C). The relative etoposide effusion increased by around 12 fold in tumor tissues after selective disruption of the neoplastic pericytes (Figure S5D). These data demonstrated that targeting GSC-derived pericytes truly elevated the etoposide delivery into tumor tissues specifically. Immunofluorescent staining of Desmin validated the disruption of vascular pericytes in tumors derived from DesPro-HsvTK-transduced GSCs but not in those derived from DesPro-Vec-transduced GSCs (Figures 4D; Figures S5E–G). Furthermore, an increased apoptosis was detected in the GCV plus etoposide-treated tumors derived from the DesPro-HsvTK-transduced GSCs relative to other groups of xenografts (Figures 4D and 4E), validating that disruption of GSC-derived pericytes effectively enhanced etoposide effusion into tumors and thus significantly elevated its anti-cancer activity. Taken together, these data establish a proof-of-principle, namely that disrupting GSC-derived pericytes facilitates the delivery of a poor BTB-penetrating drug into GBM tumors and significantly improves the chemotherapeutic efficacy.
Figure 4. Targeting GSC-derived Pericytes Improved Chemotherapy Efficacy for Intracranial GBMs.

(A) A schedule of chemotherapy in combination with disruption of GSC-derived pericytes in orthotopic GBM xenografts. GSCs transduced with luciferase and DesPro-HsvTK or DesPro-Vec were implanted into mouse brains. 5 days after GSC implantation, mice were treated with pulse administration of GCV (1mg/20g) for two consecutive days with a two-day interval. Etoposide (60µg/20g) was administered daily. Mice were monitored by bioluminescent imaging and maintained until manifestation of neurological signs.
(B) In vivo bioluminescent imaging of intracranial tumor growth in mice bearing GBM xenografts derived from T4121 GSCs transduced with DesPro-HsvTK or DesPro-Vec after treatment with GCV in combination with etoposide or DMSO control. Representative images on day 15 and day 25 post-transplantation of GSCs are shown.
(C) Kaplan-Meier survival curves of mice bearing GBMs derived from the GSCs transduced with DesPro-HsvTK or DesPro-Vec and treated with GCV in combination with etoposide or DMSO (n = 5 mice / group; p = 0.0278; two-tailed log-rank test).
(D) Immunofluorescent analysis of the apoptotic marker cleaved caspase-3 (red) and the pericyte marker Desmin (green) in xenografts derived from GSCs transduced with DesPro-Vec or DesPro-HsvTK after treatment with GCV plus etoposide or DMSO (scale bar, 80µm).
(E) Statistical quantification of (D) shows relative apoptosis in xenografts derived from the GSCs transduced with DesPro-Vec or DesPro-HsvTK after treatment with GCV plus etoposide or DMSO (n = 5 tumors / group; *, p < 0.05; **, p < 0.01; mean ± s.e.m.; Mann Whitney test).
See also Figures S5.
BMX is Highly Expressed in GSC-derived Pericytes and is Essential for the Maintenance of these Neoplastic Pericytes
We next sought to identify potential molecular targets of GSC-derived pericytes for pharmacological targeting. As the majority of neoplastic pericytes in GBMs are derived from GSCs (Cheng et al., 2013), these tumor pericytes and GSCs may share some key molecular signatures. Among the key molecular regulators (L1CAM, BMX, SOX2, CD15, CD133, etc.) of GSCs (Bao et al., 2006; Cheng et al., 2011; Guryanova et al., 2011; Singh et al., 2003; Son et al., 2009), we identified that the non-receptor tyrosine kinase BMX is differentially expressed in the neoplastic pericytes relative to normal pericytes in the brain (Figures 5A and 5B). Interestingly, BMX was previously reported to be expressed in the endothelium (Ekman et al., 1997) and in the perivascular regions (Guryanova et al., 2011). However, as pericytes are localized in close physical proximity to endothelial cells on vessels, it is critical to determine BMX expression in the neoplastic pericytes with a pericyte-specific marker. Immunofluorescent analyses showed that the majority of the α-SMA-marked pericytes expressed BMX in human primary GBMs (Figures 5A and 5B). In contrast, pericytes in the brain tissues from epilepsy patients did not show BMX expression (Figures 5A and 5B), indicating that BMX is preferentially expressed in neoplastic pericytes but not in normal pericytes. Importantly, relatively higher expression of BMX was detected in human GBM specimens with high pericyte coverage than GBMs with low pericyte coverage (Figures S6A–C). The majority of BMX signal was observed in α-SMA-marked pericytes (Figure S6A), indicating that expression of BMX is largely confined to pericytes rather than endothelial cells in GBMs. Besides, BMX was highly expressed in the Desmin-marked pericytes derived from GSCs through differentiation in vitro (Figures 5C and 5D; Figures S6D and S6E). We further examined BMX expression in GSC-derived pericytes in GBM xenografts derived from the DesPro-HsvTK-tranduced GSCs. When the majority of GSC-derived pericytes was disrupted by GCV treatment (Figures S2A–C), there was a significant reduction of BMX signal in the perivascular region of these tumors relative to the control tumors derived from the DesPro-Vec-transduced GSCs (Figure S6F and S6G). Importantly, the reduction of BMX signal occurred mostly along the vessels, where pericytes were localized (Figure S6F). However, the BMX signals in the perivascular area (but not on vessels) remained unchanged, and most likely corresponded to GSCs in the xenografts after disruption of GSC-derived pericytes (Figure S6F). These results strongly supported that BMX is expressed in both GSCs and GSC-derived pericytes. To determine whether BMX is required for the maintenance of GSC-derived pericytes, GSCs transduced with shBMX or control non-targeting shRNAs (shNT) were implanted into mouse brains to generate GBMs. Although disruption of BMX significantly delayed tumor growth, eventually the shBMX-transduced GSCs gave rise to tumors (Guryanova et al., 2011). Immunofluorescent analyses demonstrated a significant reduction of pericytes in the tumors derived from shBMX-transduced GSCs relative to the control tumors derived from shNT-transduced GSCs (Figures 5E and 5F), suggesting that disruption of GSC-derived pericytes can be achieved by targeting BMX. Moreover, immunofluorescent analyses showed a significant reduction of the tight junction markers ZO-1 and Occludin on tumor vessels in the xenografts derived from shBMX-transduced GSCs relative to the control tumors (Figures S6H–K), indicating that targeting BMX affected the BTB tight junctions. This finding was reminiscent of the loss of tight junctions in xenografts after disruption of GSC-derived pericytes (Figures 3A–D). Taken together, these data demonstrate that BMX is highly expressed in GSC-derived pericytes and is critical for maintaining these neoplastic pericytes in GBM tumors.
Figure 5. BMX Kinase Is Expressed in GSC-derived Neoplastic Pericytes and Is Essential for their Maintenance.

(A) Immunofluorescent staining of BMX (green) and α-SMA (red) in human primary GBM tumors and epilepsy brain tissues (scale bar, 40µm).
(B) Statistical quantification of (A) shows the percentage of BMX positive staining in α-SMA+ pericytes in GBMs and epilepsy brain tissues (n = 5 sections / group).
(C) Immunofluorescent staining of BMX (green) and Desmin (red) in the differentiated cells derived from GSCs. GSCs were cultured in DMEM supplemented with 10% FBS for 5 days to achieve differentiation. BMX signals were detected in the pericyte-like cells (Desmin+) but rarely in other differentiated cells (scale bar, 40µm).
(D) Statistical quantification of (C) shows the percentage of BMX-positive staining in Desmin+ pericyte-like cells in vitro in differentiated cells from GSCs (n = 100 cells / group; three independent repeats).
(E) Immunofluorescent staining of Desmin (green) and the vascular endothelial cell marker Glut1 (red) in xenografts derived from shNT- or shBMX-transduced GSCs (scale bar, 40µm).
(F) Statistical analysis of the pericyte coverage in xenografts derived from the shNT- or shBMX-transduced GSCs (n = 5 tumors / group).
Data are presented as mean ± s.e.m. *** p< 0.001, ** p < 0.01, and * p < 0.05 as assayed by Mann Whitney test.
See also Figure S6.
BMX Inhibition by Ibrutinib Disrupted GSC-derived Pericytes and the BTB to Increase Vascular Permeability in GBMs
As BMX is required for the maintenance of GSC-derived pericytes, we hypothesized that functional inhibition of BMX would target these neoplastic pericytes to disrupt the BTB. We utilized the BMX/BTK inhibitor ibrutinib to achieve the pharmacological targeting of GSC-derived pericytes. Mice bearing GSC-derived tumors were treated with ibrutinib for 3 days and the xenografts were analyzed by immunofluorescent staining of Desmin and the apoptotic marker cleaved caspase-3. Whereas apoptotic signals were hardly detected in pericytes in the control xenografts from mice treated with DMSO, the majority of vascular pericytes underwent apoptosis in the xenografts from mice treated with ibrutinib (Figures 6A and 6B), indicating the efficient targeting of tumor pericytes by ibrutinib treatment. To determine the effect of ibrutinib on vascular permeability, mice bearing GSC-derived tumors were treated with ibrutinib for 5 days followed by tail vein injection of cadaverine. Ibrutinib treatment significantly reduced pericyte coverage in the resultant tumors (Figures 6C and 6D). Consequently, effusion of cadaverine was elevated in tumors treated with ibrutinib relative to the control tumors treated with DMSO (Figures 6C and 6E). Of note, neither pericyte coverage nor cadaverine effusion in the matched normal brain tissues was affected by ibrutinib treatment (Figures 6C–E), which was consistent with the fact that BMX is not expressed in normal vascular pericytes in the brain (Figures 5A and 5B). Further analyses of BTB tight junctions showed that ibrutinib treatment significantly reduced ZO-1 and Occludin signals on tumor vessels (Figures 6F–I). These data demonstrate that BMX inhibition by ibrutinib treatment selectively targets GSC-derived pericytes in tumors but not normal pericytes in brain tissues. Consequently, ibrutinib treatment potently enhances effusion of small molecules into tumor tissues but not normal brain tissues, indicating that pharmacological inhibition of BMX by ibrutinib could potently break the BTB to increase vascular permeability.
Figure 6. Inhibition of BMX by Ibrutinib Disrupted Neoplastic Pericytes and the BTB Tight Junctions to Increase Vascular Permeability in GBMs.

(A) Immunofluorescent staining of cleaved caspase-3 (green) and Desmin (red) in xenografts derived from T4121 GSCs after treatment with ibrutinib or DMSO for 3 days (scale bar, 80µm).
(B) Statistical quantification of (A) shows relative fraction of apoptotic pericytes in GBM xenografts after treatment with ibrutinib or DMSO. Percentage of apoptotic pericytes was defined by the numbers of pericytes (Desmin+) with cleaved caspase-3 positive staining normalized to the total numbers of pericytes (n = 5 tumors / group).
(C) Detection of vascular permeability with the autonomous fluorescent tracer cadaverine (green, 1kDa), and immunofluorescent staining of Desmin (red) and the endothelial marker Glut1 (gray) in xenografts derived from T4121 GSCs after ibrutinib or DMSO treatment for 5 days (scale bar, 40µm).
(D) Statistical analysis of the pericyte coverage in xenografts and the adjacent brain tissues from mice treated with ibrutinib or DMSO (n = 5 tumors / group).
(E) Statistical analysis of cadaverine effusion in xenografts and the adjacent brain tissues from mice treated with ibrutinib or DMSO (n = 5 tumors / group).
(F and G) Immunofluorescent staining of the tight junction markers ZO-1 (F) and Occludin (G) (green) and the endothelial marker CD31 (red) in GSC-derived GBMs and the adjacent brain tissues from mice treated with ibrutinib or DMSO control for 5 days (scale bar, 80µm).
(H and I) Statistical quantification to determine the ZO-1 (H) and Occludin (I) signals on vessels in xenografts and the adjacent brain tissues from mice treated with DMSO or ibrutinib (n = 5 tumors / group).
Data are presented as mean ± s.e.m. ** p < 0.01, * p < 0.05 and ns p > 0.05 as assayed by Mann Whitney test.
See also Figure S6.
Targeting GSC-derived Pericytes by Ibrutinib Significantly Improves Chemotherapy Efficacy for GBMs and Extends Survival of Tumor-bearing Mice
To demonstrate the clinical value of ibrutinib for BTB disruption, we next investigated the potential of ibrutinib as an adjuvant drug to improve chemotherapy efficacy for GBMs. GSCs expressing luciferase were implanted into mouse cerebrums to generate GBMs. 7 days after implantation, mice bearing GSC-derived GBMs were treated with ibrutinib and/or the poor BTB-penetrating drug etoposide daily through intraperitoneal injection. Bioluminescent imaging demonstrated that ibrutinib treatment alone significantly retarded tumor growth (Figure 7A), which may be ascribed to the targeting of both GSCs and GSC-derived pericytes. However, while etoposide treatment alone had no effect, the combination of ibrutinib and etoposide treatment achieved the greatest inhibition of tumor growth among the four treatment groups (Figure 7A). Consistently, Kaplan-Meier survival curves demonstrated that whereas etoposide alone had no effect and ibrutinib alone had significant but moderate effect, the combination of ibrutinib and etoposide treatment largely extended the mouse survival (Figure 7B). These data highlight the clinical value of targeting BMX-expressing neoplastic pericytes by ibrutinib in disrupting the BTB to improve chemotherapy efficacy. Immunofluorescent staining of Desmin confirmed the disruption of neoplastic pericytes in the ibrutinib-treated xenografts (Figures 7C and 7D). Moreover, whereas ibrutinib treatment alone induced some apoptosis in tumors, the combined treatment of ibrutinib and etoposide resulted in devastating apoptosis in tumors (Figures 7E and 7F), indicating that targeting GSC-derived pericytes by ibrutinib effectively enhanced etoposide effusion into tumors and thus significantly elevated the anti-cancer activity of etoposide. Collectively, these data clearly demonstrate that targeting neoplastic pericytes by ibrutinib treatment selectively disrupts the BTB to specifically enhance drug delivery into GBM tumors, which can significantly improve the efficacy and specificity of chemotherapy for GBM patients.
Figure 7. Inhibition of BMX by Ibrutinib Improved Efficacy of Chemotherapy with Poor BTB-penetrating Drugs in GBM Xenografts.

(A) In vivo bioluminescent imaging of intracranial tumor in mice bearing T4121 GSC-derived GBMs after treatment with etoposide, ibrutinib, ibrutinib plus etoposide, or DMSO control on indicated days after GSC implantation.
(B) Kaplan-Meier survival curves of mice bearing GSC-derived xenografts with indicated treatments (n = 5 mice / group; **, p < 0.01; two tailed log-rank test).
(C) Immunofluorescent analyses of Desmin (green) and Glut1 (red) in GSC-derived xenografts from mice with indicated treatments (scale bar, 40µm).
(D) Statistical quantification of pericyte coverage in GSC-derived xenografts from mice with indicated treatments (n = 5 tumors / group, **, p < 0.01; mean ± s.e.m.; Mann Whitney test).
(E) Immunofluorescent staining of cleaved caspase-3 (red) in GSC-derived xenografts from mice with indicated treatments (scale bar, 40µm).
(F) Statistical quantification of (E) shows relative apoptosis in GSC-derived xenografts with indicated treatments (n = 5 tumors / group; **, p < 0.01; mean ± s.e.m.; Mann Whitney test).
See also Figure S7.
Because varied human GBMs display differential pericyte coverage on tumor vessels (Figure 1), we could mimic the varied tumor vascular pericyte coverage in mouse models using GSCs isolated from different GBM patients (Figures S5E–G). We next examined the effectiveness of pericyte disruption on the improvement of chemotherapy in the UCG163 GSC-derived GBM xenografts with relatively low pericyte coverage. Interestingly, etoposide treatment alone inhibited tumor growth and extended the survival of animals bearing the xenografts with low pericyte coverage (Figure S7), which was not observed in the GBMs with high pericyte coverage (Figures 4 and 7). This difference suggests that tumors with low pericyte coverage exhibit increased vascular permeability for drug delivery relative to tumors with high pericyte coverage. Pericytes themselves seemed to be more critical for tumors with low pericyte coverage, because disruption of pericytes more significantly inhibited tumor growth of the xenografts with low pericyte coverage than those with high pericyte coverage (Figure 4; Figure S7). However, targeting GSC-derived pericytes by ibrutinib or genetic ablation in combination with etoposide treatment further benefited the mice bearing orthotopic GBMs with either high or low pericyte coverage (Figures 4 and 7; Figure S7). These data highlight the potency and importance of therapeutic targeting of neoplastic pericytes by ibrutinib to effectively improve chemotherapy efficacy for a wide spectrum of GBM patients.
DISCUSSION
Our study demonstrates that targeting GSC-derived pericytes by genetic or pharmacological approaches disrupts BTB tight junctions and increases vascular permeability, leading to enhanced drug delivery and improved chemotherapeutic efficacy for GBMs. We uncovered that GSC-derived pericytes are essential for maintaining the BTB that blocks effusion of most anti-cancer drugs into GBM tumors. While complete depletion of GSC-derived pericytes resulted in the collapse of vessel structure and inhibited tumor growth (Cheng et al., 2013), short-term disruption of GSC-derived pericytes elevated vascular permeability without affecting the density of tumor vessels (Figures S2A–C), thus facilitating the delivery of chemotherapeutic drugs. Most importantly, we identified BMX kinase as a molecular target highly expressed in GSC-derived pericytes but not in normal pericytes in the brain. Inhibition of BMX by ibrutinib disrupted neoplastic pericytes and the BTB, enhanced effusion of a poor BTB-penetrating drug into tumor tissues, and significantly improved chemotherapeutic efficacy. Since many potent anti-cancer drugs are ineffective in GBM treatment due to their poor BTB or BBB penetration, ibrutinib as an adjuvant drug targeting neoplastic pericytes to selectively disrupt the BTB may greatly expand the spectrum of anti-GBM drugs. Moreover, because disruption of the BTB potently enhances drug delivery, lower doses of anti-cancer drugs may be used to achieve a greater therapeutic efficacy with fewer side effects. Thus, targeting GSC-derived pericytes and GSCs by ibrutinib will not only suppress autonomous tumor growth but also effectively improve therapeutic efficacy of many anti-cancer agents, particularly those with low BTB-penetrating properties.
Our data showed that GBM tumors from different patients could have varied vascular pericyte coverage that is inversely correlated with the survival of GBM patients treated with chemotherapy (Figure 1; Figure S1). Importantly, pericyte coverage in the GBM surgical specimens is closely associated with patient response to chemotherapy, indicating that pericyte coverage could be a valuable preclinical index to predict outcome of chemotherapy for GBM patients. These findings raise interesting questions about the cause for the variation in pericyte coverage. Since the majority of pericytes in GBMs originate from GSCs (Cheng et al., 2013), the varied GSC populations in different GBM patients may contribute to the corresponding wide range of pericyte production and pericyte coverage across patients. However, pericyte coverage could be affected by other factors in the tumor microenvironment. Multiple cytokines, including TGF-β, SDF-1, and Notch1, could regulate pericyte generation and migration (Aghi et al., 2006; Guichet et al., 2015; Verbeek et al., 1994). In addition, other types of cells including endothelial cells and astrocytes may regulate pericyte coverage (Aghi et al., 2006; Hong et al., 2015; Ramsauer et al., 2002). Furthermore, the oxygen concentration in the tumor microenvironment may affect pericyte production (Du et al., 2008). It would be interesting to determine the potential association between these factors and the BTB in brain tumors. As the tumor microenvironment evolves during tumor progression and treatment, pericyte coverage may also change accordingly. Thus, analysis of pericyte coverage in pre- and post-treatment specimens would be an important approach to address the mechanisms associated with the BTB and tumor resistance to chemotherapy. The production of neoplastic pericytes is likely a continuous process during tumor progression and treatment. However, our preliminary study emphasizes GSC trans-differentiation rather than proliferation of pre-existing pericytes as a main cause of pericyte production in GBM tumors. Thus, it is critical to explore the mechanisms regulating GSC differentiation into pericytes in order to develop more efficient approaches to disrupt neoplastic pericytes and improve GBM chemotherapy.
Our study in GBMs and other studies in normal brains indicated that pericytes play a critical role in maintaining the tight junctions on vessels (Armulik et al., 2010; Daneman et al., 2010). Vascular tight junctions restrict free passage of molecules through the space between plasma membranes of adjacent endothelial cells (Tietz and Engelhardt, 2015). As key components of the BBB and BTB, vascular pericytes directly attach to endothelial cells and promote the formation of tight junction structures between adjacent endothelial cells (Armulik et al., 2010; Daneman et al., 2010). Despite the abnormality of tight junction structures on vessels in the brain depleted of pericytes, reduction of tight junction proteins was not observed in brains of Pdgfb knock-out mice (Armulik et al., 2010; Daneman et al., 2010). Our study showed a decrease of tight junction proteins and the disruption of the tight junction structure on intracranial tumor vessels after elimination of GSC-derived pericytes, suggesting a different process of tight junction formation in tumor neovascularization. During tumor angiogenesis, tumor-derived factors act on pre-existing vessel endothelial cells to induce cell migration and proliferation (Eelen et al., 2015; Schneider et al., 2004). In addition, the interaction between astrocytes and endothelial cells at the BBB may regulate tight junctions (Abbott et al., 2006), but the astrocyte distribution in GBMs is often disordered (Figures S4I and S4J). Because tumor and normal vascular endothelial cells are localized in different microenvironments, the influence of pericytes on tight junction structures and proteins could be different in GBM tumors and in normal brain tissues, although pericyte coverage is critical for maintaining the function of both BTB and BBB.
In summary, our findings provide a critical insight into the failure or low efficacy of current chemotherapy in GBM treatment and offer a targeting approach to effectively improve the chemotherapy efficacy for GBM patients. Targeting GSC-derived pericytes through BMX inhibition by the FDA approved drug ibrutinib could selectively disrupt the tumor BTB but not normal BBB to specifically enhance drug delivery into GBM tumors. Repurposing ibrutinib as a BTB-breaking drug provides a critical therapeutic approach that may potently synergize with current therapies to effectively improve survival of GBM patients. In addition, this targeting strategy may be used to enhance therapeutic efficacy and specificity for patients with other malignant brain tumors, brain metastases or other lethal malignancies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shideng Bao (baos@ccf.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human GBM Specimens and Glioma Stem Cells (GSCs)
De-identified GBM surgical specimens were collected from The Brain Tumor and Neuro-Oncology Center at Cleveland Clinic in accordance with an Institutional Review Board-approved protocol. Additionally, a cohort of the formalin-fixed, paraffin-embedded (FFPE), de-identified GBM specimens and the related patient follow-up information were collected from the Southwest Hospital in Chongqing in accordance with an Institutional Review Board-approved protocol (Table S1). Informed consent was obtained from all subjects. IDH1 and MGMT status of the FFPE human GBM specimens were indicated in Table S1. GSCs were isolated from primary GBMs or xenografts and cultured as previously described (Bao et al., 2006; Cheng et al., 2013; Guryanova et al., 2011; Huang et al., 2011; Zhou et al., 2015). In brief, tumor cells were isolated from GBM tumors with Papain Dissociation System (Worthington Biochemical). After recovery in the stem cell medium (Neurobasal-A medium with B27 supplement, 10 ng/ml EGF, 10 ng/ml bFGF, 1% penicillin/streptomycin, 1 mM sodium pyruvate, and 2 mM L-glutamine) in a humidified incubator with 5% CO2 for at least 6 hours for the re-expression of surface markers, the isolated cells were then labeled with a phycoerythrin (PE)-conjugated anti-CD133 antibody (Miltenyi Biotec, 130-098-826) and a FITC-conjugated anti-CD15 antibody (Millipore, CBL144F) followed by fluorescence-activated cell sorting (FACS) to sort the GSC population (CD15+/CD133+). A series of functional assays were then applied to validate the cancer stem cell phenotypes of the isolated GSCs, including self-renewal (serial neurosphere formation), multipotent differentiation (serum induction of multi-lineage differentiation in vitro), and tumor-initiation (in vivo limiting dilution assay) as previously described (Bao et al., 2006; Cheng et al., 2013; Guryanova et al., 2011; Huang et al., 2011; Zhou et al., 2015). The validated GSCs were cultured in the stem cell medium in a humidified incubator with 5% CO2 for a short period (<5 passages) before further experiments. Specifically, T4121 GSCs were derived from a GBM from a 53-year old male patient. CCF2045 GSCs were isolated from a GBM from 35-year old male patient. UCG163 GSCs were derived from a GBM from a 58-year old male patient. CCF3264 GSCs were derived from a recurrent GBM from a 65-year old female patient. T3832 GSCs were derived from a GBM from a 75-year female patient.
Intracranial Tumor Formation and Treatment in vivo
Intracranial transplantation of GSCs to establish orthotopic GBM xenografts was performed as described (Bao et al., 2006; Cheng et al., 2013; Guryanova et al., 2011; Huang et al., 2011; Zhou et al., 2015). GSCs were transduced with constructs of Desmin promoter driven GFP and/or herpes simplex virus thymidine kinase (DesPro-GFP, DesPro-HsvTK, DesPro-HsvTK-GFP) or the vector control (DesPro-Vec) along with luciferase through lentiviral infection. Cells were selected with puromycin (1 µg/mL, Fisher Scientific) for 48 hours after infection. 500 to 2000 cells were then engrafted intracranially into immunocompromised mice (NSG or nude mice). Immune-deficient C57BL/6 athymic Crl:NU-Foxn1nu nude mice (obtained from Charles River Laboratories) and immune-deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ NSG mice (obtained from Jackson Laboratories) were used for the animal experiments. Animals were not involved in any previous procedures and were drug/test naïve when used for the animal experiments. Animal experiments were performed in accordance with Cleveland Clinic-approved Institutional Animal Care and Use Committee protocols and Southwest Hospital-approved Institutional Animal Care and Use Committee protocols. Five mice were housed per cage, with a 12-hour light/dark cycle, and were provided food and water ad libitum. 6–8 weeks old mice were randomly assigned to one of the experimental groups. Animals were monitored by bioluminescent imaging or maintained until manifestation of neurological signs. For ganciclovir treatment, mice were treated with ganciclovir (InvivoGen, sud-gcv) at 1mg/20g through intraperitoneal injection. For etoposide treatment, mice were intraperitoneally injected with etoposide (Santa Cruz, sc-3512A) at 60µg/20g daily. For ibrutinib treatment, mice were intraperitoneally injected with ibrutinib (LC Laboratories, I-3311; Medkoo, 202171) at 0.2mg/20g daily. For drug preparation, ganciclovir (GCV) was dissolved in 0.1M HCl at 10mg/mL. Etoposide was dissolved in DMSO at 3mg/mL. Ibrutinib was dissolved in DMSO at 10mg/mL.
METHOD DETAILS
Immunofluorescent and Immunohistochemical (IHC) Staining
Immunofluorescent and immunohistochemical staining were performed as described (Cheng et al., 2013; Zhou et al., 2015). Surgical human GBM biopsy specimens were fixed overnight in 4% PFA at 4°C, stored in 30% sucrose solution overnight at 4°C, embedded in OCT at −20°C overnight, and cryosectioned at a thickness of 7 microns. Sections were blocked with a PBS solution containing 1% BSA (Sigma) plus 0.03% TWEEN20 for 1 hour at room temperature. The following primary antibodies (1:200 dilution) were incubated overnight at 4°C. Specific antibodies against endothelial cell marker CD31 (Dako M0823 or Bethyl IHC-00055), CD34 (Biolegend, 343502) or Glut1 (Thermo Scientific RB-9052-P, Santa Cruz sc-1605), BMX (Abcam, ab59360 or BD Biosciences, 610792), pericyte marker Desmin (Dako M0760) or α-SMA (Dako M0851), GSC marker SOX2 (Millipore AB5603; Santa Cruz sc-17320) or Olig2 (R&D systems AF2418), the tight junction marker ZO-1 (Invitrogen 40–2300) or Occludin (Santa Cruz sc-5562), vessel basement membrane marker laminin (Novus Biologicals NB300-144), cell apoptotic marker cleaved caspase-3 (Cell Signaling Tech. 9661), hypoxia marker CA9 (Novus, NB100–417), astrocyte marker GFAP (Covance, PRB-571C-100) and the transcytosis marker Plvap (ABD Serotec MCA2539T) were used for the staining on GBM tumor sections as indicated. For immunofluorescent staining, the second day, sections were washed with PBS and then incubated with fluorescent second antibodies (1:200 dilution) at room temperature for 2 hours. The sections were then washed with PBS, mounted on glass slides, and subjected to microscopy. Otherwise, for IHC staining, sections were incubated with biotinylated secondary antibodies (1: 2000 dilution) for 30 minutes at room temperature and then washed with PBS. Target proteins were detected with the ABC kit and DAB kit (Vector Laboratories) according to the manufacturer’s instructions. The FFPE specimens were deparaffinized by incubating the slides at 55 for 1 hour. Rehydration was performed by incubating the slides with xylene, 100% ethanol, 95% ethanol, 80% ethanol, 75% ethanol, and PBS sequentially, each reagent for 10 minutes. Staining on tissue samples were performed on sections from at least 5 tumors and 5 random fields in each section were used for analyses. Staining on cultured cells were repeated at least three times and analyses were performed on at least 100 random cells every time.
Measurement of Pericyte Coverage on Tumor Vessels
Pericyte coverage on tumor vessels was determined by immunohistochemical staining of formalin-fixed, paraffin-embedded (FFPE) human primary GBM specimens with the endothelial marker CD34 and the tumor pericyte marker α-SMA. In brief, sequential sections of FFPE GBM samples were stained with CD34 or α-SMA. Average density of CD34-positive or α-SMA-positive staining was determined out of 5 random 750×750 µm fields for each sample with Image J. Pericyte coverage was calculated by the ratio of the average density of α-SMA to that of CD34 for each tumor case. For the stratification of high and low pericyte coverage groups, the average pericyte coverage of all the GBM patients was used as the cut-off. Tumor samples that have pericyte coverage higher than or equal to the average value were stratified into the high pericyte coverage group. Otherwise, the tumor specimens with pericyte coverage lower than the average value were assigned to the low pericyte coverage group. The treatment regimen and the survival of patients were then associated with the tumor pericyte coverages.
For determination of pericyte coverage in GBM xenografts by immunofluorescent analyses, tissue sections were co-stained with the pericyte markers and the endothelial cell markers. Pericyte coverage was defined as the number of endothelial cells covered by pericytes normalized to the total number of endothelial cells. Immunofluorescent staining was performed on sections from at least 5 tumors and 5 random fields in each section were used for analysis.
Determination of Tracer Effusion in Tumor and Brain Tissues
GBM xenografts were established by intracranial implantation of 500 to 2000 GSCs. Twenty days after implantation, mice were confirmed to have similarly sized tumors by the bioluminescent imaging and then subjected to ganciclovir or ibrutinib treatment for 3–5 days. The mice were then subjected to tail vein injection of fluorescent tracers (250µg/20g) including cadaverine (1kDa) (Invitrogen, A30676) or dextran (3kDa, 10kDa, and 40kDa) (Thermo Fisher, D3306 and D1820; Sigma, 42874). Two hours after tracer injection, mice were anaesthetized and perfused with PBS followed by 4% paraformaldehyde. The intracranial tumors were fixed overnight at 4°C before cryosection. Effusion of tracers in tumor and brain tissues was determined by fluorescence microscopy. Tracer effusion was determined by the sizes of tracer positive areas normalized to the vessel density in the same areas, both analyzed by the Image J software. The tracer effusion experiments were repeated three times. Sections from at least 5 tumors for each group were used for analysis.
DNA Constructs and Lentivirus Production
The DesPro-Vec was constructed by replacing the CMV promoter of the pCDH-CMV-MCS-EF1-Puro vector (System Biosciences CD510B-1) with Desmin promoter. DesPro-HsvTK or DesPro-GFP was constructed by insertion of herpes simplex virus thymidine kinase (HsvTK) coding sequence or GFP into the multiple cloning site of DesPro-Vec as described in our previous study (Cheng et al., 2013). For lentivirus production, 293FT cells were transfected with DesPro-Vec or DesPro-HsvTK plus two helper plasmids pCI-VSVG and ps-PAX2. 48–72 hours after transfection, lentiviral supernatant was collected and titered as described previously (Bao et al., 2006; Cheng et al., 2013; Guryanova et al., 2011; Huang et al., 2011; Zhou et al., 2015). GSCs were then infected with lentivirus at a MOI around 3.
Electron Microscopy
GBM xenografts were established by intracranial implantation of 2 000 GSCs. Twenty days after implantation, mice were confirmed to have similarly sized tumors by the bioluminescent imaging and then subjected to ganciclovir treatment for 5 days. Mice were then anaesthetized, perfused with PBS followed by Electron Microscopy (EM) Buffer (2.5% glutaraldehyde, 4% paraformaldehyde in 0.1 M cacodylate buffer). Then the xenografts and matched brain tissues were fixed in the EM buffer overnight at 4°C. Samples were embedded with Eponate 12 medium. Sections (85 nm) were cut using a diamond knife, stained with uranyl acetate and lead citrate, and then examined with a Tecnai G2 SpiritBT electron microscope operated at 60 k V.
To examine the vascular ultrastructure of tumor tissue and the corresponding normal brain tissue, 4 brains from each treatment group were subjected to the EM analyses. For each brain, at least 12 tumor vessels and 12 normal vessels were analyzed by the EM. For quantification of endothelial vesicles, the small vesicles (20 to 100 nm in diameter) inside vascular endothelial cells, which presented as circular organelles having lighter electron density within, were counted and quantified. For each group, 12 tumor vessels and 12 normal vessels were analyzed for endothelial vesicles.
Determination of Etoposide Effusion in vivo by Chromatography
Orthotopic GBMs were established by intracranial implantation of T4121 GSCs transduced with DesPro-Vec or DesPro-HsvTK. 5 mice of each group were used for the determination of etoposide effusion in vivo. 20 days after implantation, mice were treated with ganciclovir for 4 days followed by intraperitoneal injection of etoposide (60µg/20g). 2 hours after etoposide injection, mice were anaesthetized, perfused with 10 mL PBS twice, and sacrificed. Tumor and the corresponding normal brain tissues were collected, homogenized in PBS, and subjected to chromatography by using HPLC On-line Electrospray Ionization Tandem Mass Spectrometry (LC/ESI/MS/MS). Briefly, tissue homogenate was solved in 80% methanol and centrifuged. Supernatant (5 µl) was injected onto a C18 column (2.1 × 150mm, ODS (2), 5 µm, Phenomenex) for HPLC separation using mobile phases A (water containing 5 mM 2-aminoacridone) and B (acetonitrile containing 5 mM 2-aminoacridone). The HPLC eluent was directly injected into a triple quadrupole mass spectrometer (Shimadzu LCMS-8050) and the etoposide was ionized at ESI positive mode. Etoposide was quantified using Selected Reaction Monitoring (SRM), and three SRM transitions (m/z) at 606 > 229, 606 > 185 and 606 > 435 were used to monitor the etoposide. The software Labsolutions LCMS was used to get the peak area for etoposide and external calibration standard curve was used to calculate the quantity of etoposide. The relative fold changes of etoposide effusion were determined by dividing the etoposide concentration in the tumor tissue by the etoposide concentration in the matched normal brain tissue.
QUANTIFICATION AND STATISTICAL ANALYSIS
For bar graphs, the level of significance was determined by the non-parametric Mann Whitney test, which does not require the assumption of normal distributions but is nearly as efficient as the t-test on normal distributions. For Kaplan-Meier survival curves, the level of significance was determined by the two-tailed log-rank test with α = 0.05 (the probability of making a Type I error, i.e. the probability of rejecting the null hypothesis when in fact it is true), which is commonly used for survival analysis (Bao et al., 2006; Cheng et al., 2013; Guryanova et al., 2011; Huang et al., 2011; Zhou et al., 2015). All analyses were performed with the GraphPad Prism 5 software. P values < 0.05 were considered significant. All quantitative data presented are the mean ± s.e.m. from at least three samples or experiments per data point. No statistical method was used to predetermine the samples size. Due to the nature of the experiments, the investigators were not blinded. No data inclusion/exclusion criteria was applied in this study. Precise experimental details (number of animals or cells and experimental replication) are provided in the figure legends.
Supplementary Material
Highlights.
High pericyte coverage compromises chemotherapeutic efficacy in GBM patients
Targeting GSC-derived pericytes disrupts the blood-tumor barrier (BTB)
Inhibiting BMX kinase with ibrutinib disrupts GSC-derived pericytes and the BTB
Ibrutinib enhances effusion and activity of drugs with poor BTB penetration
Acknowledgments
We thank the Brain Tumor and Neuro-Oncology Centers at Cleveland Clinic for providing GBM specimens for this study. We also thank the Institute of Pathology and Southwest Cancer Center, Southwest Hospital, The Third Military Medical University (Chongqing, China), for the FFPE GBM specimens and the follow-up information of the GBM patients. We are grateful to members in Dr. Rich’s laboratory for their assistance and scientific discussion. We also thank the Flow Cytometry Core, Imaging Core, Central Cell Services, and Mass Spectrometry Core at Cleveland Clinic Lerner Research Institute for their assistance. This work was supported by the Cleveland Clinic Foundation and NIH R01 grants (CA184090, NS091080, NS099175 and CA169117) to S. Bao; grants from the National Natural Science Foundation of China (Nos. 81230062 and 61327902-4) and the National Key Research and Development Program of China (Stem Cell and Translational Research, 2016YFA0101200) to X. Bian; NIH R01 grants (NS092641 and NS094199) to J. S. Yu. This work utilized the FEI Tecnai G2 Spirit transmission electron microscope and the IVIS system (Spectrum CT) that were purchased with NIH SIG grants (1S10RR031536-01 and S10OD018205).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.B. and W.Z. developed the working hypothesis and scientific concept. W.Z., X.B., and S.B. designed the experiments, analyzed data and prepared the manuscript. W.Z., C.C., Y.S., Q.W., R.C.G., Z.H, X.F. K.Z., and S.Q.K. performed the experiments. C.C. and Y.S. performed database analyses. H.F. provided GBM surgical specimens. Y.S., Y.P., C.C., and X.B. performed pathological analyses. J.N.R. and J.S.Y. provided scientific inputs for the manuscript.
The authors declare no competing financial interests.
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006;66:9054–9064. doi: 10.1158/0008-5472.CAN-05-3759. [DOI] [PubMed] [Google Scholar]
- Alifrangis C, Agarwal R, Short D, Fisher RA, Sebire NJ, Harvey R, Savage PM, Seckl MJ. EMA/CO for high-risk gestational trophoblastic neoplasia: good outcomes with induction low-dose etoposide-cisplatin and genetic analysis. J Clin Oncol. 2013;31:280–286. doi: 10.1200/JCO.2012.43.1817. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard S, Goldwirt L, Amorim S, Brice P, Briere J, de Kerviler E, Mourah S, Sauvageon H, Thieblemont C. Activity of ibrutinib in mantle cell lymphoma patients with central nervous system relapse. Blood. 2015;126:1695–1698. doi: 10.1182/blood-2015-05-647834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Kim O, Li M, Xiong X, Guan JL, Kung HJ, Chen H, Shimizu Y, Qiu Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat Cell Biol. 2001;3:439–444. doi: 10.1038/35074500. [DOI] [PubMed] [Google Scholar]
- Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Wu Q, Huang Z, Guryanova OA, Huang Q, Shou W, Rich JN, Bao S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. The EMBO journal. 2011;30:800–813. doi: 10.1038/emboj.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, Li W, Guo Z, Xu K, Kim O, et al. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70:5587–5596. doi: 10.1158/0008-5472.CAN-09-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit R, Skoneczna I, Daugaard G, De Santis M, Garin A, Aass N, Witjes AJ, Albers P, White JD, Germa-Lluch JR, et al. Randomized phase III study comparing paclitaxel-bleomycin, etoposide, and cisplatin (BEP) to standard BEP in intermediate-prognosis germ-cell cancer: intergroup study EORTC 30983. J Clin Oncol. 2012;30:792–799. doi: 10.1200/JCO.2011.37.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman N, Lymboussaki A, Vastrik I, Sarvas K, Kaipainen A, Alitalo K. Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation. 1997;96:1729–1732. doi: 10.1161/01.cir.96.6.1729. [DOI] [PubMed] [Google Scholar]
- Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhanel M, Spruss T, Bernhardt G, Graeff C, Farber L, Gschaidmeier H, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichet PO, Guelfi S, Teigell M, Hoppe L, Bakalara N, Bauchet L, Duffau H, Lamszus K, Rothhut B, Hugnot JP. Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells. 2015;33:21–34. doi: 10.1002/stem.1767. [DOI] [PubMed] [Google Scholar]
- Guo L, Guo Y, Xiao S. Expression of tyrosine kinase Etk/Bmx and its relationship with AP-1- and NF-kappaB-associated proteins in hepatocellular carcinoma. Oncology. 2007;72:410–416. doi: 10.1159/000113491. [DOI] [PubMed] [Google Scholar]
- Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, MacSwords J, Eyler CE, McLendon RE, Heddleston JM, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19:498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, Farooqui M, Wiestner A. Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123:3286–3295. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, Garcia-Ibanez Y, Qin H, Paulsson J, Zeitelhofer M, Adzemovic MZ, et al. Role of Tumor Pericytes in the Recruitment of Myeloid-Derived Suppressor Cells. J Natl Cancer Inst. 2015:107. doi: 10.1093/jnci/djv209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu Q, Guryanova OA, Cheng L, Shou W, Rich JN, Bao S. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol. 2011;13:142–152. doi: 10.1038/ncb2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP. Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J Biol Chem. 2007;282:32689–32698. doi: 10.1074/jbc.M703412200. [DOI] [PubMed] [Google Scholar]
- Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, Yang Y, Cole DE, Melani C, Higham CS, et al. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell. 2017;31:833–843. e835. doi: 10.1016/j.ccell.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara MG, Lwin Z, Jiang H, Chung C, Millar BA, Sahgal A, Laperriere N, Mason WP. Conditional probability of survival and post-progression survival in patients with glioblastoma in the temozolomide treatment era. J Neurooncol. 2014;117:153–160. doi: 10.1007/s11060-014-1368-7. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Howieson J, Frenkel EP, Specht HD, Weigel R, Buchan CG, Hill SA. Therapeutic efficacy of multiagent chemotherapy with drug delivery enhancement by blood-brain barrier modification in glioblastoma. Neurosurgery. 1986;19:573–582. doi: 10.1227/00006123-198610000-00011. [DOI] [PubMed] [Google Scholar]
- Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002;22:7512–7523. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. Drug and gene targeting to the brain with molecular Trojan horses. Nat Rev Drug Discov. 2002;1:131–139. doi: 10.1038/nrd725. [DOI] [PubMed] [Google Scholar]
- Pries AR, Hopfner M, le Noble F, Dewhirst MW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer. 2010;10:587–593. doi: 10.1038/nrc2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsauer M, Krause D, Dermietzel R. Angiogenesis of the blood-brain barrier in vitro and the function of cerebral pericytes. FASEB J. 2002;16:1274–1276. doi: 10.1096/fj.01-0814fje. [DOI] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saharinen P, Ekman N, Sarvas K, Parker P, Alitalo K, Silvennoinen O. The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cdelta. Blood. 1997;90:4341–4353. [PubMed] [Google Scholar]
- Schneider SW, Ludwig T, Tatenhorst L, Braune S, Oberleithner H, Senner V, Paulus W. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol. 2004;107:272–276. doi: 10.1007/s00401-003-0810-2. [DOI] [PubMed] [Google Scholar]
- Shue EH, Carson-Walter EB, Liu Y, Winans BN, Ali ZS, Chen J, Walter KA. Plasmalemmal vesicle associated protein-1 (PV-1) is a marker of blood-brain barrier disruption in rodent models. BMC Neurosci. 2008;9:29. doi: 10.1186/1471-2202-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell stem cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland LA, Jubb AM, Hongo JA, Zhong F, Burwick J, Fu L, Frantz GD, Koeppen H. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF) J Pathol. 2005;206:466–475. doi: 10.1002/path.1805. [DOI] [PubMed] [Google Scholar]
- Tietz S, Engelhardt B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J Cell Biol. 2015;209:493–506. doi: 10.1083/jcb.201412147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek MM, Otte-Holler I, Wesseling P, Ruiter DJ, de Waal RM. Induction of alpha-smooth muscle actin expression in cultured human brain pericytes by transforming growth factor-beta 1. Am J Pathol. 1994;144:372–382. [PMC free article] [PubMed] [Google Scholar]
- Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat Commun. 2014;5:4196. doi: 10.1038/ncomms5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnholds J, deLange EC, Scheffer GL, van den Berg DJ, Mol CA, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. The Journal of clinical investigation. 2000;105:279–285. doi: 10.1172/JCI8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano J, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–926. doi: 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. doi: 10.1038/ncomms4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170–182. doi: 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.