

Summary
The tumor suppressing transcription factor p53 is highly conserved at the protein level and plays a key role in the DNA damage response. One important aspect of p53 regulation is its dynamics in response to DNA damage, which include oscillations. Here, we observe that while the qualitative oscillatory nature of p53 dynamics is conserved across cell lines derived from human, monkey, dog, mouse and rat, the oscillation period is variable. Specifically, rodent cells exhibit rapid p53 oscillations, whereas dog, monkey and human cells show slower oscillations. Computational modeling and experiments identify stronger negative feedback between p53 and MDM2 as the driver of faster oscillations in rodents, suggesting that an oscillation's specific period is a network-level property. In total, our study shows that despite highly conserved signaling, the quantitative features of p53 oscillations can diverge across evolution. We caution that strong amino acid conservation of proteins and transcriptional network similarity do not necessarily imply conservation of time dynamics.
Graphical abstract
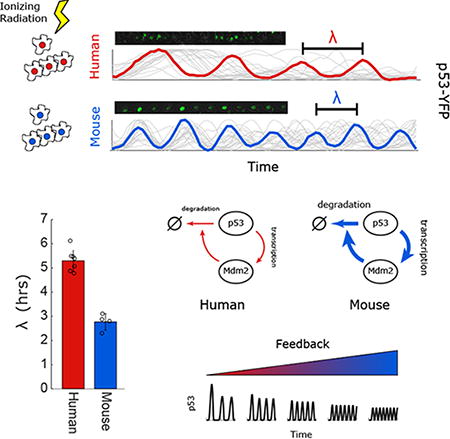
Introduction
The dynamics of signaling molecules are encoded in the structure and weights of regulatory networks. Genomic studies have shown that many signaling pathways are highly conserved, and that specific feedback links are often also conserved across species. The NFκB and p53 regulatory circuits, for example, are ancient and highly conserved (Brandt et al., 2009; Lane et al., 2011; Ghosh et al., 2002; Wang et al., 2006). In both systems feedback regulation by the transcription factor's negative regulator—IKKB and MDM2, respectively—is conserved through at least mammals. In principle, these basic regulatory circuits can provide a wide range of different dynamical behaviors depending on the parameters that govern their function (Ma et al., 2009; Hunziker et al., 2010; Caicedo-Casso et al., 2015). Parameters such as protein degradation rate, catalytic efficiency, or promoter strength and affinity are difficult to obtain from high throughput measurements. This means that while the array of sequenced genomes makes it feasible to precisely and quantitatively define the evolution of protein identity, it does not enable tracing the evolution of regulatory networks' properties, such as their dynamical behaviors.
Protein levels of p53 fluctuate dynamically in response to stress, with temporal patterns of activity ranging from oscillatory to a single pulse of varying width (Lahav et al., 2004; Batchelor et al., 2011). Pharmacological modulation of these dynamics results in altered fate outcomes in response to DNA damage (Purvis et al., 2012; Paek et al., 2016). Although the dynamics of p53 and other oscillatory systems, such as NFκB, have been qualitatively found to oscillate in both human and mouse cells, they have not been quantitatively compared across these and other species (Tay et al., 2010; Stewart-Ornstein et al., 2016; Hamstra et al., 2006; Bar-Or et al., 2000). Since the dynamics of p53 were shown to be crucial for its function (Purvis et al., 2012; Paek et al., 2016), one would expect that specific dynamic features will be quantitatively conserved.
Here we focused on the conservation of the dynamics of the tumor suppressor protein p53 across species. We compared its oscillations in single cells in response to DNA double strand breaks in humans, monkeys, dogs, rats and mice. While the period of p53 oscillations was similar in humans, monkeys and dogs, cells derived from rodents showed substantially faster oscillations. Using model guided experiments we found that mouse p53 oscillates more rapidly due to faster degradation of p53 and stronger transcription of p53 target gene and negative regulator MDM2. These results suggest that the dynamics of p53 are structurally conserved across species, but that fine characteristics such as period vary. Further, this study argues that even in cases of strong amino acid conservation of proteins and transcriptional network similarity, conservation of time dynamics should not be assumed.
Results
p53's oscillations are conserved across species while their periods vary
To compare p53 oscillations in mouse and human cells we irradiated MCF7 (p53 wild-type human breast) and NIH3T3 (p53 wild-type mouse fibroblasts) cells that were previously shown to induce p53 in response to ionizing radiation (IR; Bar-Or et al., 2000). We collected protein every hour for 10hrs after IR and probed for endogenous human or mouse p53 on a western blot (Fig. 1A). We note that while both cell lines show clear oscillatory abundance of p53, the mouse line oscillated substantially more rapidly. To gain a more quantitative understanding of p53 oscillations across species we turned to well established single cell reporters of p53 abundance. We constructed human, mouse, dog, monkey, and rat kidney derived cell lines expressing p53-YFP. We selected kidney cells to compare across species as cells from this lineage are available from a wide range of species. When treated with neocarzinostatin (NCS, a radiomimetic drug) p53-YFP abundance increased rapidly in all species, and a large fraction of cells (20%-95%) showed p53 oscillations (Fig. 1B). Although oscillations were observed in all species, the period of these oscillations varied, consistent with our population measurements in mice and human cells (Fig.1A). Mouse and rat cells showed noticeably fast oscillations with a period of ∼3hrs, whereas human, monkey, and dog cells show oscillations with a period just over 5hrs (Fig. 1C). Although the period of these oscillations varied, their shape was similar. Human p53 pulses had a width of 0.54+/-0.18 periods, compared to 0.58+/-0.23 periods for mouse cells, and in both cases the pulses were symmetrical (leading:trailing edge ratios 1.04 and 1.08 respectively). The distribution of p53's periods in single cells was moderate and exhibited a minimal overlap between the fast oscillating species (mouse and rat) and the slow oscillating species (human, monkey and dog). Within each group, monkey, human, and dog cells did not show distinguishable period differences, but rat cells oscillated slightly slower than mouse cells (Fig 1C; p-value < 0.05 t-test). These results show that while oscillations of p53 are conserved across species, the quantitative features of these oscillations are not. Specifically, the p53 period is conserved between human and dogs (∼90mya diverged; Hedges 2002) but varies between rodent and humans (∼75mya diverged; Waterston et al., 2002).
Figure 1. p53 oscillates more rapidly in mice and rats.
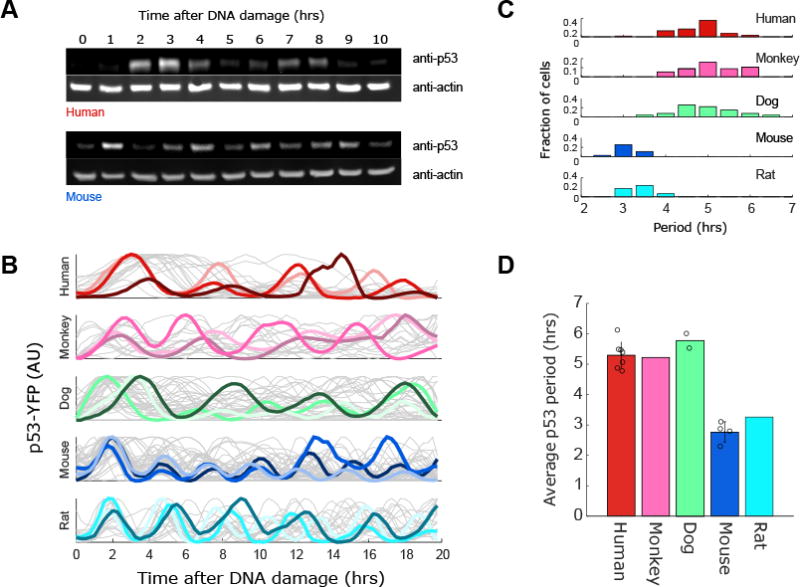
(A) Western blot comparing p53 levels over time after DNA damage (8Gy of γ-irradiation) in Mouse (NIH3T3) and Human (MCF7) cell lines. (B) Kidney cell lines from human (UO-31), monkey (Vero), dog (MDCK), mouse (MAK), and rat (NRK52E) were transduced with a p53-YFP reporter. These cells were exposed to DNA damage (NCS, 100ng/ml) and YFP levels were quantified in individual cells over time (N>30 for each species). Three cells from each cell line are highlighted. (C) Distributions of the period of p53 oscillations in single cells for each species. (D) Average period for each species. Each dot represents a single cell line. For mouse (N=4 [NIH3T3, HEPA1C1C7, RAW264.3, MAK), dog (N=2 [MDCK, D17]), and human (N=7 [MCF7, UO31, UACC257, UACC62, A549, H460, U2OS]). Error bars represents std. dev across lines within a species.
To determine the effect of cell type on p53 oscillatory period we measured p53 oscillations in four mouse cell lines (representing fibroblast, hematopoietic, and epithelial linages), reanalyzed data from a previous study that included seven human oscillating cell lines (Stewart-Ornstein and Lahav, 2017), and obtained and analyzed a second dog cell line (Fig. 1D). These analyses revealed that the p53 oscillatory period is tightly constrained within a species, varying by no more than half an hour in mouse and ∼1hr in humans across cell lines. The difference in oscillation periods between human and mouse cells therefore is preserved across different cell types within each species. Other features of p53 dynamics did vary across cell lines within a species. For example, the stability of signaling, measured by the mean magnitude of the autocorrelation peak, overlapped between and varied across human (0.15-0.31, mean 0.23) and mouse cell lines (0.22-0.47, mean 0.34) (Data not shown). These cell line-cell line variations in signal waveform and duration of p53 signaling reflect in part different aspects of the DNA damage signaling and repair process as has been previously explored (Loewer et al., 2013; Stewart-Ornstein and Lahav, 2017).
The different periods of p53 oscillations between mice and human are derived from the core p53/Mdm2 feedback loop
We next sought to identify the molecular mechanism that determine the period of p53 oscillations by focusing on mice and human cells. Mouse and human p53 and MDM2 are similarly and substantially conserved sharing 87% and 89% amino acid similarity, respectively. The different frequency in p53 dynamics can result from differences in the p53 protein sequence itself or other mechanisms independent of the p53 sequence. To distinguish between these two options we expressed a human p53-YFP in mouse cells and a mouse p53-YFP in human cells and tested the resultant oscillation frequencies in response to DNA damage. We found that the frequency is determined by the species background; human p53-YFP expressed in mice showed the rapid mice-like period, while mice p53-YFP expressed in humans showed the slow human-like period (Fig. 2A). This suggests that the sequence of the p53 protein itself is not responsible for the differences in oscillations frequencies observed between species.
Figure 2. Faster oscillations of p53 in mouse cells are not due to the p53 sequence, general metabolic state or DNA damage signaling.
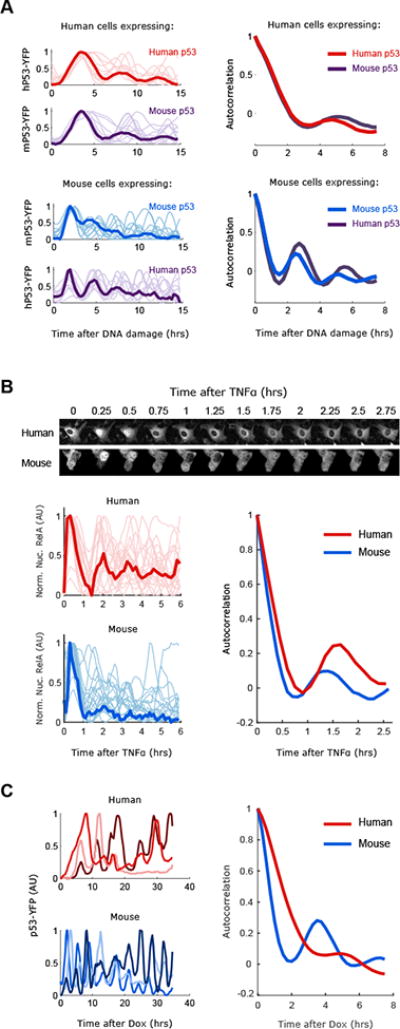
(A) Human (MCF7) or Mouse (NIH3T3) cells expressing either human or mouse p53-YFP were DNA damaged (NCS 100ng/ml) and imaged for 15hrs (faint lines indicate single cells, bold lines indicate the average). Oscillations of p53 were quantified and the period was calculated with autocorrelation function (N>20 cells). (B) Human (MCF7) or Mouse (NIH3T3) cells expressing RELA-CFP were treated with TNFα (10ng/ml) and imaged for 6hrs. RelA-CFP nuclear signal was quantified and autocorrelations was used to calculate the oscillatory period (N>20 cells). (C) Human (MCF7) or Mouse (HEPA1c1c7) cells expressing a dox inducible p53-YFP construct were imaged after addition of doxycycline (50ng/ml). Three examples traces are shown and the quantified periods (N>10 cells).
Higher frequency in mouse might result from gross changes in metabolic state. In this scenario other oscillating systems should also show rapid oscillations in mouse. We applied live cell reporters for a second oscillatory system, NFκB, and quantified its dynamics in response to Tumor Necrosis Factor. We found that the period of NFκB oscillations did not substantially differ between human and mouse cells (Fig. 2B), both oscillating with periods consistent with previous reports (Hoffmann et al., 2002; Tay et al., 2010). Other oscillatory systems in mice and human cells, such as the cell cycle duration or circadian rhythms are not significantly different between mice and humans (Amundson et al., 2008; Czeisler et al., 1999). These results suggest that the difference in the period of p53 oscillations between mice and human is specific to the p53 system.
The difference in the period of p53 oscillations between mice and human can result from the p53 regulatory core circuit that includes p53's negative regulator MDM2, or from other DNA damage dependent pathways upstream of p53 and Mdm2. To distinguish between these two scenarios we expressed a p53-YFP reporter under a doxycycline inducible promoter and measured the resultant dynamics of p53 following induction. We found that dox-induced p53 shows rapid oscillations in mouse cells compared to human cells even in the absence of DNA damage. Note that in both cases the periods were slightly longer than damage induced oscillations (Fig. 2C). This shows that while DNA damage regulates p53 oscillations, the difference in p53 period between mice and human is driven by specific features of the core p53/Mdm2 loop independent of DNA damage.
We next asked what features of the p53/Mdm2 regulatory circuit might be responsible for faster oscillations in mouse cells. We applied a simple mathematical model of the p53/Mdm2 regulatory circuit (Geva-Zatorsky et al., 2006, Fig S1A) and asked what parameters govern the oscillatory period. We identified two major parameters; Mdm2-mediated degradation rate of p53 (Fig. S1; k1) and the regulation of MDM2 production (Fig. S1; k2 & k3). Specifically, this model suggests that p53 oscillates faster in mice than humans due to stronger transcription of MDM2 and faster degradation of p53 (Fig. S1B, C). Other parameters had either minimal effect on the period of p53 oscillations (p53 transcription, k5, Mdm2-independent degradation of p53, k6 and the hill function between p53 and Mdm2, k7), or a modest non-monotonic effect (Mdm2 degradation, k4) (Fig.S1C).
Faster degradation rates of p53 in mice contribute to its rapid oscillations
We first focused on the degradation rate of p53 and examined if it is indeed faster in mouse cells as suggested by our model (Fig. S1). We treated mouse and human p53 reporter lines with a competitive inhibitor of MDM2 binding to p53 (Nutlin3) for 0.5 hours and then washed out the drug (Fig. 3A). This treatment caused p53 levels to rise in both species with similar kinetics (Fig. 3A). When the drug was washed out p53 levels dropped rapidly to baseline. Quantification of the time at which p53 reached half its peak value (T1/2) in single cells showed that on average p53 degradation was more rapid in mouse cells (∼0.4hrs) compared to human cells (∼0.7hrs) (Fig. 3B), albeit with significant overlap.
Figure 3. The degradation rate of p53 tunes the period of p53 oscillations between mouse and human cells.
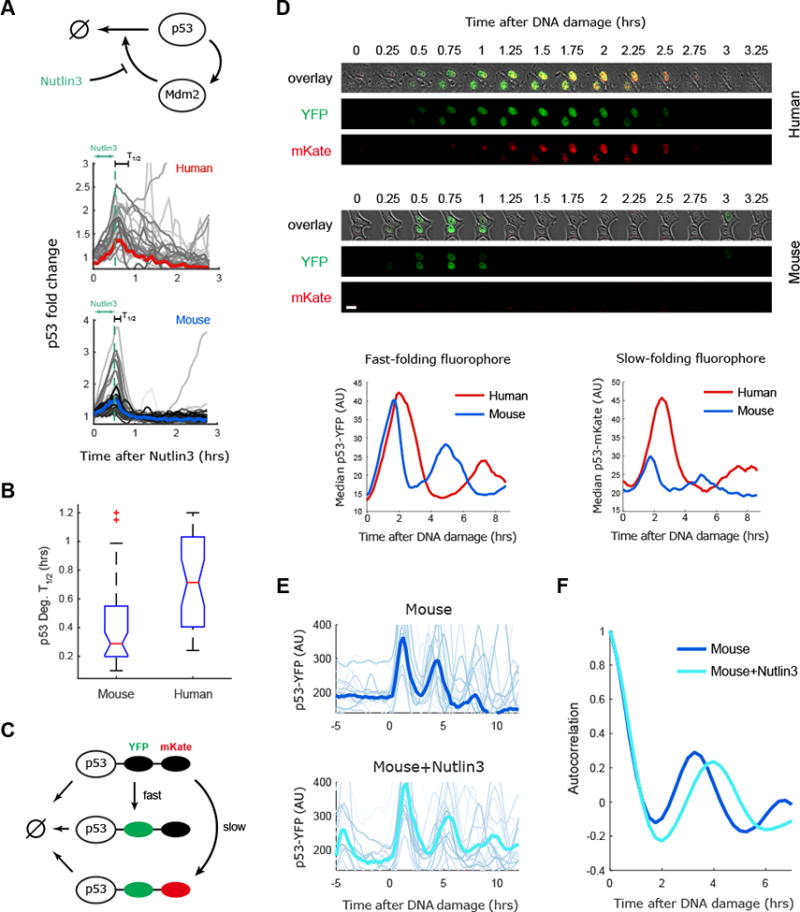
(A) Mouse and human cells expressing the p53 reporter were treated with an inhibitor of p53 degradation (Nutlin3) for 30 minutes and p53 levels were quantified in single cells, the bold line gives the median of the trajectories. (B) Quantification of p53 degradation rate from the experiment shown in (A). The boxplots indicate the distribution of single cell degradation times, showing faster degradation of p53 in mouse than human cells (N>30). (C) A two color p53 reporter is used to quantify the half-life of p53 during the response to DNA damage (NCS). (D) Top: Images of mice and human cells expressing p53-YFP and p53-mKate in response to DNA damage. Bottom: Averaged dynamics of p53-YFP and p53-mKate2 in human and mouse cells responding to DNA damage (N>30). The amplitude of the fast folding fluorophore p53-YFP is comparable between human and mouse cell lines, but the levels of the slow folding fluorophore p53-mKate2 are higher in human cells, indicating a longer half-life of p53 in human cells. (E) Mouse cells (RAW264.3) expressing p53-YFP were treated with NCS with or without pre-treatment with the MDM2 inhibitor Nutlin3. NCS was applied at time 0. Bold lines represent the averaged behavior (N>30). (F) Autocorrelation of the data presented in (E) showing that reduced p53 degradation by Nutlin3 in mouse cells slows the p53-YFP oscillatory period.
Direct measurement of degradation rate in dynamic circumstances is challenging by conventional assays. We therefore employed a two color tandem-fluorescent timer system by fusing a fast (Venus/YFP) and slower (mKate2) folding fluorophore to p53 (Fig. 3C), and expressing them in mouse and human cells (Khmelinskii et al., 2014). Treatment of these lines with DNA damage led to the expected rise in p53-YFP, followed with a slight delay by the p53-mKate reporter (Fig. 3D). Comparing the two species, human cells showed similar levels of mKate2 and YFP, whereas mouse cells showed a similar amount of YFP to human cells, but much lower levels of mKate2 (Fig. 3D). The reduction of mKate2 (the slower folding fluorescence protein) in mice is consistent with faster degradation of p53 in mouse than in human cells. Taken together these results confirmed that, as suggested by our model (Fig. S1) mouse cells have faster degradation rate of p53 than human cells, suggesting that the rapid p53 oscillations in mouse cells might be driven in part by faster degradation of p53. Additionally, we noted that mouse cells responded more transiently to Nutlin3 addition compared to human cells that showed sustained p53 signaling in response to Nutlin3 treatment (Fig. S2A). Such a transient behavior is expected from a faster degraded p53 in mice cells.
We next tested if slowing the p53 degradation rate in mouse cells can alter the period of its oscillations. We again used the Mdm2 inhibitor Nutlin3. This time we pre-treated mouse cells with the inhibitor and then induced DNA damage without washing it from the media, therefore increasing p53 half-life throughout the experiment. Pre-treatment with Nutlin3 led to an extension in the period of p53 oscillations in mouse from three to four hours (Fig. 3E, F). This supports our model's prediction that slower degradation rates of p53 contribute to the slower period observed in human versus mouse cells. Consistent with this, it was recently shown that increased degradation of a mutant p53 could result in faster oscillations in human cells (Borcherds et al., 2014).
Slower Mdm2 transcription contributes to slower oscillations of p53 in human cells
The second parameter that was suggested to control the period of p53 oscillations was Mdm2 transcription. We treated the mouse and human p53 reporter lines with an inhibitor of transcription (Flavopiridol) for 3 hours and tested the resultant p53 dynamics in single cells. As previously described, Flavopiridol caused p53 levels to rise (Demidenko and Blagosklonny, 2004). After washout of the drug, p53 levels continued to rise before subsequently falling to baseline (Fig. 4A). The delay between the washout time and the time at which p53 levels start to decrease depends on the rate of MDM2 production in each cell line. This delay was ∼1hr in mouse cells and ∼2.25hrs in human cells (Fig. 4B), consistent with the model suggestion that Mdm2 transcription is slower in human cells (Fig. S1). In agreement with these results, qPCR measurements of Mdm2 mRNA levels following high (10μM) doses of Nutlin3 revealed faster induction of Mdm2 in mouse cell lines compared to human cell lines (Fig. 4C). Taken together these results show that Mdm2 transcription is faster in mouse than human, a behavior predicted to shorten the period of p53 oscillations (Fig. S1).
Figure 4. The transcription rate of MDM2 is faster in mice contributing to shorter period of p53 oscillations.
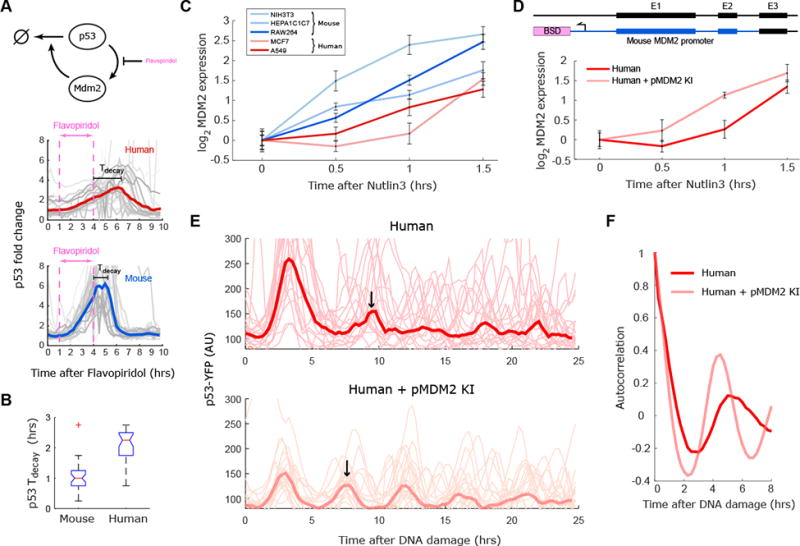
(A) Mouse and human cells with p53 reporters were treated with an inhibitor of transcription (Flavopiridol) for three hours and p53 levels were quantified. (B) Quantification of the delay between Flavopiridol washout and p53 degradation from the experiment shown in (A). The boxplots indicate the distribution of single cell delays (N>30). (C) Three mouse (red) and two human (blue) cell lines were treated with the MDM2 inhibitor Nutlin3, and MDM2 transcription was quantified using qPCR. The mouse lines show faster and stronger induction of Mdm2 transcript than the human lines. (D) Knock-in of MDM2 promoter (pMDM2KI) into the MDM2 locus in the human MCF7 cells results in rapid induction of MDM2 RNA quantified by qPCR after Nutlin3 treatment. (E) Single cell traces of p53-YFP in response to DNA damage (NCS) in parental human MCF7 cells and in cells knocked in with the mouse Mdm2 promoter (N>30). Bold lines represent the averaged behavior, with an arrow indicating the timing of the second pulse. (F) Autocorrelation of the data presented in (E) indicating that human cells knocked in with the mouse MDM2-promoter show more rapid oscillation than non-altered human cells. Error bars for C and D indicate SEM (N=3).
Can modification of Mdm2 activation by p53 in human cells accelerate the period of p53 oscillations? We constructed human cells expressing a p53 reporter and knocked in the mouse MDM2 promoter into the human MDM2 locus (Fig. 4D). We first confirmed that this genetic alteration leads to more rapid induction of Mdm2 transcript compared to non-altered human cells (Fig. 4D). We then measured p53 dynamics in these altered cells and compared them with their wild type counter parts. We found that, in response to DNA damage, the human cell line with the mouse MDM2 promoter exhibited faster p53 oscillations than the parental cells that have the original human Mdm2 promoter (Fig. 4E, F). We also note that, consistent with our model, this lower period is accompanied by lower amplitude oscillations. This shows that the rate of p53 activation of MDM2 transcription regulates the period of p53 oscillations with faster/stronger induction of Mdm2 transcription leading to faster oscillations and shorter periods in mice cells compared to human cells.
Discussion
Tracing the evolution of transcriptional circuits has generally focused on changes in transcription factor binding sites. Studies of the budding yeast mating pathway showed how gradual replacement of transcription factors, and changes in the recognition site, can alter the structure of mating pathway signaling without greatly altering its function (Baker et al., 2011; Sorrells et al., 2015). Measurements of evolution of signaling pathways across mammals and drosophila have similarly focused on loss and gain of transcription factor binding sites (Schmidt, et al., 2010; He et al., 2011; Stefflova et al., 2013).
Here we focused on the dynamics of signaling, a potentially sensitive measure of conservation of not only gene-transcription factor connections, but also of the relative weights of these connections. We found that while the overall oscillatory behavior of the p53 circuit is preserved across species, its period is not conserved. Using mathematical modeling and experiments in single cells we found that the change in period is driven by p53 degradation rates and Mdm2 transcription. It is interesting to consider these results given our recent study which compared multiple human cell lines and noted that the period of p53 oscillations is one of the few conserved features of p53 signaling (Stewart-Ornstein and Lahav, 2017). These results suggest that tissue-tissue variation in signaling and species-species variation may act differentially, with species level variation tuning features of the core oscillator, while tissues alter the magnitude of its input, for example via altering ATM activity.
While the function of the p53 protein as a tumor suppressor is conserved between mouse and humans other aspects of p53 function appear to be more divergent. Studies comparing p53 binding between mouse and human cell lines have identified substantial divergence in p53 targets (Akdemir et al., 2013; Kenzelmann et al., 2014). Our own meta-analysis of p53 binding in mouse and human cell lines using published ChIP-Seq data and motif analysis revealed that 30-40% of p53 bindings sites are conserved between mice and humans (Fig. S3A, B). It should, however, be noted that the majority of the ‘core’ p53 targets related to cell cycle arrest and apoptosis are conserved (Horvath et al., 2007; Jegga et al., 2008). Unfortunately, we do not have appropriate computational tools as of yet to more comprehensively compare functional elements of promoters between species, however, we note that the p53 binding site in the MDM2 promoter differs by only a single base-pair between human and mouse. This difference is predicted to allow p53 to bind more strongly in mice (Fig. S3C) in agreement with our model prediction (Fig. S1) and experimental results (Fig. 4). Notably, this single base pair change is present in mouse and rats, but absent in other mammals that showed the human-like p53 oscillatory period (Fig 1C-D, S3C).
The question of what is the functional role of the difference in p53 period between species remains open. It is possible that the elements under selection are not oscillations per se, but rather strong feedback controls. In this scenario, variation in p53 period is simply a neutral drift of this feedback system. Alternatively, the stronger feedback and faster oscillation in rodents might be a mechanism to adapt to a shorter or more metabolically active lifespan. For example, a higher metabolic rate might lead to higher rates of spontaneous DNA damage, requiring stronger feedback and reduced and more transient p53 activity to prevent excess cell death or senescence. A comprehensive comparison of p53 activity across species and particularly rodents—which have a wide range of lifespans and lifestyles—is needed to address this. Regardless of the potential evolutionary benefit of a particular p53 oscillatory period, the fact that mouse and human lines differ significantly in their p53 period raises concerns regarding tests of chemo-toxicity in mice models harboring human tumors, and specifically the response to DNA damage agents and Mdm2 inhibitors. More generally, our work demonstrates the sensitivity of protein dynamics to subtle and potentially non-coding sequence changes in signaling networks and highlights the need to consider conservation of dynamics when studying signaling pathways across species.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Galit Lahav (Galit@hms.harvard.edu).
Experimental Model and Subject Details
Cell lines are as follows: MCF7 (Human, female, Invasive ductal carcinoma), UO31 (Human, female, Renal cell carcinoma), A549 (Human, male, Lung adenocarcinoma), MAK (mouse, female, spontaneously immortalized primary kidney), NIH3T3 (mouse, female, spontaneously immortalized primary fibroblasts), HEPA1C7 (mouse, female, hepatocellular carcinoma), RAW264 (mouse, male, leukemia), MDCK (dog, female, spontaneously immortalized primary kidney), D-17 (dog, female, osteosarcoma), Vero (Chlorocebus sabaeus, female, spontaneously immortalized primary kidney), NRK52e (Rat, gender unknown-not reported in original publication, spontaneously immortalized primary kidney).
All cell lines were thawed and propagated in RPMI (GIBCO) with 5% FBS, except NIH3T3 cells which were grown in DMEM + 10% FCS.
A subset of these cell lines (A549, MCF7, NIH3T3, UO31) were authenticated by Short-Tandem-Repeats at the Dana Farber Molecular Diagnostics Laboratory.
Method Details
Cell culture and cell line construction
MCF7, UO-31, Vero, MDCK, and D-17 cell lines were obtained from ATTC. NIH3T3 cells were a gift from Prof. Peter Sorger (Harvard Medical School). HEPA1C1C7 cells were a gift from Prof. Charles Weitz (Harvard Medical School). Mouse kidney cells were obtained by dissecting and spontaneously immortalizing by serial propagation cells from an adult mouse kidney (C57BL/6). Lines were thawed and propagated in RPMI (GIBCO) with 5% FBS, except NIH3T3 cells which were grown in DMEM + 10% FCS. For microscopy RPMI lacking phenol red and riboflavin was used. For viral production 293T cells were grown in DMEM (GIBCO) + 10% FBS. All media was supplemented with 1% antibiotic and antimycotic (Corning). Cells from each line were infected at <0.1 MOI with p53-YFP or RelA-CFP lenti-virus in 35mm dishes, selected with G418 (Sigma), Puromycin or Hygromycin (Thermo), and split into 96 well plates to select for clones expressing the transgene.
For the knock-in of the MDM2 promoter we constructed a cassette with 1300bp of the mouse MDM2 promoter adjacent to a blasticidin resistance gene (BLAST-prmMDM2). We PCRed this construct with primers containing 40bp of homology to the human MDM2 promoter on each end (pMDM2_insert_BLAST_Fwd and pMDM2_M1300_insert_rev). We then transfected this construct together with a CAS9 plasmid expressing a gRNA (gRNA_prMDM2) targeted at the MDM2 promoter. Cells were selected with blasticidin and single clones obtained. Clones were PCR tested for integration and one clone with integration of the mMDM2 promoter in one allele was selected for subsequent experiments.
Virus production and infection
Virus was produced using 293T cells transfected with p53-YFP or RelA-CFP constructs and viral packaging vectors (3rd Gen Lenti). Viral supernatant was collected after three days. For viral infection, cells were plated at low density and infected with virus in media containing HEPES and protamine sulfate for 6hrs. Cells were allowed to recover in nonselective media for one day. Productively infected cells were selected with the appropriate antibiotic.
RNA extraction and qRT-PCR
Fifty-Thousand cells were plated in 6 well plates. Cells were cultured for 24-48hrs and then treated with the indicated compounds. RNA was extracted by treatment with Trizol and subsequent purification on a Zymo RNA column. The bulk RNA was reverse transcribed using the high capacity reverse transcription kit (applied biosystems) to produce cDNA. Transcript abundance was quantified by specific primers for MDM2 using a Sybr green (lifetechnologies) based qPCR and normalized to Actin.
Microscopy
For live cell imaging cells were plated in glass bottom 35mm dishes (Matek) 24-48hrs before imaging, 1-2hrs before imaging cells were switched to transparent media (RPMI lacking riboflavin and phenol red, Invitrogen). Live cell imaging was performed with Nikon Eclipse TE-2000 microscope equipped with a heating chamber and CO2 source, an epi-fluorescent source, either mercury arc lamp (Prior) or LED system (Lumencor), automated stage (Prior), YFP filter set (Chroma) and CCD or CMOS camera (Hamamatsu).
Microscopy data analysis
Microscopy data was processed with custom MATLAB code. Single cells were tracked manually using the phase images with a MATLAB interface (both scripts are available on request and at https://github.com/JacobStewartOrnstein/Lahav_ImageAnalysis_Matlab_Code). Single cell tracks were projected onto the fluorescent images, which were then background corrected (by Median filtering and subsequent tophat background subtraction) and nuclear signal (estimated as the average of the top 10 pixels in the nuclear area) was then computed for the applicable channels. Images displayed in the body of the paper were smoothed with a median or Gaussian filter and background subtracted. Contrast was adjusted for optimal visualization and is consistent between pre- and post-treatment for all images.
Calculation of peak shape and degradation rates
To compute the shape of p53 peaks and compare them between species we first identified the location of peaks within a trace (ignoring the first peak after DNA damage) using the MATLAB findpeaks function on smoothed data (running mean, window 3). We then computed the width for each peak (using unsmoothed data) by recording the first position that drops below 0.2* peak height-peak trough on each side of the peak. Overall peak width was then divided by the period to obtain a dimensionless measure of shape.
Degradation times from experiments described in Figure 3B was performed by treating the cell with Nutlin for 30minutes, then washing the drug out. Two hours of the subsequent single cell traces was analyzed for the T1/2 (time at which the trace drops below 0.5 × max-min). The distribution of these values was given as a boxplot. Delay times for Figure 4B was analyzed similarly. After washout of 3hrs of transcription inhibitor, the subsequent time trace (6hrs) was analyzed to identify the peak of the p53 response by smoothing the trace (running mean, window of 3) and finding the maximum value. This value was computed for all cells and shown as a boxplot.
ChIP-Seq data analysis
Human p53 (SRR575904, SRR847014, SRR941556, SRR287800) or mouse p53 ChIP-Seq datasets (SRR832846, SRR088304) were downloaded from the sequence read archive. Reads were aligned to HG18 or mm9 genome and peaks called with HOMER. Peak regions were ‘lifted-over’ to other genome builds (rheMac8, canFam3, MM9, RN4, oryCun2, susScr3, monDom5). Sequences were scanned with p53 PWM from JASPAR and fraction of peaks scored as a binding site was quantified for each species and normalized to human (or mouse) fraction.
Computational Modeling
We implemented a three species, seven parameter model of p53 signaling (Geva-Zatorsky et al., 2006 – Model IV). Parameters governing MDM2 and p53 degradation or synthesis were varied over 5 log2s around parameters that approximated the human-like oscillatory period to explore which features have the strongest effect on the p53 oscillatory period. We preformed all simulations in MATLAB (see below).
Constants
k(1)=1.7; % Mdm2 mediated p53 degradation rate
k(2)=1.1 % p53 mediated Mdm2 transcription
k(3)= 0.8; % transition between Mdm2 intermediate
k(4)=0.8; % Mdm2 degradation rate
k(5)=0.9; % p53 production rate
k(6)=0.0001; % basal (Mdm2-independent) p53 degradation rate
k(7)=0.0001; % Mdm2/p53 hill function
Starting parameters
y0(1)=0; %p53
y0(2)=0.1; %m2 intermediate
y0(3)=0.8; %Mdm2
Differential equations
dp53 = k(5) - k(6)*y(1) - k(1)*(p53/(p53+k(7)))*MDM2;
dMDM2int(2)= k(2)*p53 - k(3)*MDM2int;
dMDM2(3)=k(3)*MDM2int-k(4)*MDM2;
Replication
All experiments have a minimum of one biological replicate, N (cells or replicates) of experiments is indicated in figure legends.
Strategy for randomization and/or stratification
None
Blinding at any stage of the study
Image analysis uses only bright field to track cells, blinding the tracker to the underlying dynamic signal.
Inclusion and exclusion criteria of any data or subjects
For analysis of p53 frequency cells with no clear oscillatory behavior (defined as a detectable peak in autocorrelation) were excluded. Where applicable outliers are shown in boxplots.
Quantification and Statistical Analysis
Statistical analyses were performed using MATLAB. All t-tests were two sided and a 0.05< p value was regarded as significant. Summary statistics (mean, median, standard deviation, SEM) and number of replicates are indicated in the figure legends. Boxplots use the MATLAB default representation with the median and 25-75 percentiles, whiskers show maximum of data or up to 2.7sigmas, notch indicates 1.57(median – 75percentile)/sqrt(n) and non-overlapping notches indicate significance at the 5% level. Samples were not blinded or randomized. Distributions of data were examined for mono-modality before using t-tests.
Supplementary Material
Highlights.
-p53 oscillatory period varies across species but not cell lines within a species
-Mice and rats show more rapid p53 oscillations compared to humans
-Higher frequency p53 oscillations are driven by enhanced p53-mdm2 feedback
Acknowledgments
We thank Antonia Hafner, Caroline Mock, Jose Reyes, and the entire Lahav Lab for comments and discussion. We also thank the Nikon Imaging Center at Harvard Medical School for help with microscopy. This research was supported by The Harvard Ludwig Cancer Research Center and the National Institute of Health grants GM083303 and CA207727 (J.S-O).
Footnotes
Author Contributions: Collected data: JSO and JC. Analyzed data, preformed computational modeling: JSO. Wrote the paper: JSO, JC, GL. Supervised the study: JSO and GL.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akdemir KC, Jain AK, Allton K, Aronow B, Xu X, Cooney AJ, Li W, Barton MC. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014;42(1):205–23. doi: 10.1093/nar/gkt866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Vinikoor LC, Lee RA, Koch-Paiz CA, Ahn J, Reimers M, Chen Y, Scudiero DA, Weinstein JN, Trent JM, Bittner ML, Meltzer PS, Fornace AJJr. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008;68(2):415–24. doi: 10.1158/0008-5472.CAN-07-2120. [DOI] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD. Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci U S A. 2011;108(18):7493–8. doi: 10.1073/pnas.1019177108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Or RL, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of Oscillation by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci USA. 2000;97:11250–11255. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor E, Loewer A, Mock C, Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7(488) doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcherds W, Theillet FX, Katzer A, Finzel A, Mishall KM, Powell AT, Wu H, Manieri W, Dieterich C, Selenko P, Loewer A, Daughdrill GW. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10(12):1000–2. doi: 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- Brandt T, Petrovich M, Joerger AC, Veprintsev DB. Conservation of DNA-binding specificity and oligomerisation properties within the p53 family. BMC Genomics. 2009;10(628) doi: 10.1186/1471-2164-10-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo-Casso A, Kang HW, Lim S, Hong CI. Robustness and period sensitivity analysis of minimal models for biochemical oscillators. Sci Rep. 2015;5(13161) doi: 10.1038/srep13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeisler CA, Duffy JF, Shanahan TL, Brown EN, Mitchell JF, Rimmer DW, Ronda JM, Silva EJ, Allan JS, Emens JS, Dijk DJ, Kronauer RE. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284(5423):2177–81. doi: 10.1126/science.284.5423.2177. [DOI] [PubMed] [Google Scholar]
- Demidenko ZN, Blagosklonny MV. Flavopiridol induces p53 via initial inhibition of Mdm2 and p21 and, independently of p53, sensitizes apoptosis-reluctant cells to tumor necrosis factor. Cancer Res. 2004;64(10):3653–60. doi: 10.1158/0008-5472.CAN-04-0204. [DOI] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, Yarnitzky T, Liron Y, Polak P, Lahav G, Alon U. Oscillations and variability in the p53 system. Mol Syst Biol. 2006;2(0033) doi: 10.1038/msb4100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;(109 Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Hamstra DA, Bhojani MS, Griffin LB, Laxman B, Ross BD, Rehemtulla A. Real-time evaluation of p53 oscillatory behavior in vivo using bioluminescent imaging. Cancer Res. 2006;66(15):7482–9. doi: 10.1158/0008-5472.CAN-06-1405. [DOI] [PubMed] [Google Scholar]
- He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, Zeitlinger J. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat Genet. 2011;43:414–420. doi: 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
- Hedges SB. The origin and evolution of model organisms. Nat Rev Genet. 2002;3(11):838–49. doi: 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- Horvath MM, Wang X, Resnick MA, Bell DA. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet. 2007;3(7):e127. doi: 10.1371/journal.pgen.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298(5596):1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hunziker A, Jensen MH, Krishna S. Stress-specific response of the p53-Mdm2 feedback loop. BMC Syst Biol. 2010;4:94. doi: 10.1186/1752-0509-4-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegga AG, Inga A, Menendez D, Aronow BJ, Resnick MA. Functional evolution of the p53 regulatory network through its target response elements. Proc Natl Acad Sci U S A. 2008;105(3):944–9. doi: 10.1073/pnas.0704694105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, Huber W, Schiebel E, Knop M. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol. 2014;30(7):708–14. doi: 10.1038/nbt.2281. [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27(9):1016–31. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Lane DP, Madhumalar A, Lee AP, Tay BH, Verma C, Brenner S, Venkatesh B. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle. 2011;10(24):4272–9. doi: 10.4161/cc.10.24.18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142(1):89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Karanam K, Mock C, Lahav G. The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol. 2013;11:114. doi: 10.1186/1741-7007-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138(4):760–73. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen O, Scally M, Douady CJ, Kao DJ, DeBry RW, et al. Parallel adaptive radiations in two major clades of placental mammals. Nature. 2001(409):610–614. doi: 10.1038/35054544. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, et al. Molecular phylogenetics and the origins of placental mammals. Nature. 2001(409):614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165(3):631–42. doi: 10.1016/j.cell.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336(6087):1440–4. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells TR, Booth LN, Tuch BB, Johnson AD. Intersecting transcription networks constrain gene regulatory evolution. Nature. 2015;523(7560):361–5. doi: 10.1038/nature14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefflova K, Thybert D, Wilson MD, Streeter I, Aleksic J, Karagianni P, Brazma A, Adams DJ, Talianidis I, Marioni JC. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013;154:530–540. doi: 10.1016/j.cell.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Ornstein J, Lahav G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci Signal. 2017;10(476) doi: 10.1126/scisignal.aah6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Ornstein J, Lahav G. Dynamics of CDKN1A in Single Cells Defined by an Endogenous Fluorescent Tagging Toolkit. Cell Rep. 2016;14(7):1800–11. doi: 10.1016/j.celrep.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single-cell NFkappaB dynamics reveal digital activation and analogue information processing. Nature. 2010;466(7303):267–71. doi: 10.1038/nature09145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Tan NS, Ho B, Ding JL. Evidence for the ancient origin of the NFkappaB/IkappaB cascade: its archaic role in pathogen infection and immunity. Proc Natl Acad Sci U S A. 2006;103(11):4204–9. doi: 10.1073/pnas.0507044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.