

SUMMARY
Neurons have more extended and complex shapes than other cells and consequently face a greater challenge in distributing and maintaining mitochondria throughout their arbors. Neurons can last a lifetime, but proteins turn over rapidly. Mitochondria, therefore, need constant rejuvenation no matter how far they are from the soma. Axonal transport of mitochondria and mitochondrial fission and fusion contribute to this rejuvenation, with local protein synthesis likely also to be involved. Maintenance of a healthy mitochondrial population also requires the clearance of damaged proteins and organelles. This involves degradation of individual proteins, sequestration in mitochondria-derived vesicles, organelle degradation by mitophagy and macroautophagy, and in some cases transfer to glial cells. Both long-range transport and local processing are thus at work in achieving neuronal mitostasis – the maintenance of an appropriately distributed pool of healthy mitochondria for the duration of a neuron’s life. Accordingly, defects in the processes that support mitostasis are significant contributors to neurodegenerative disorders.
Keywords: axonal transport, homeostasis, mitochondria, mitophagy, neurons
INTRODUCTION: MITOSTASIS IN NEURONS
Neurons face unique problems because of their exceptional architecture. To transmit electric signals rapidly between regions of the CNS and to and from peripheral organs, neurons have evolved long and highly branched processes. In humans, peripheral nerves and cortico-spinal tracts can have axons a meter long. Moreover, because information processing requires extensive convergence and divergence of signals, the cytoplasmic volume of highly branched arbors easily dwarfs the volume of the neuronal cell body (Figure 1, center). In the human brain, individual neurons of the substantia nigra pars compacta are calculated to give rise to 4.5 m of axon, once all the branches are summed (Bolam and Pissadaki, 2012; Matsuda et al., 2009). A similar calculation for cholinergic basal forebrain and raphe neurons has predicted over a hundred meters of axon can arise from a single soma (Wu et al., 2014).
Figure 1. Neuronal Mitochondria in a Mouse Neuron.
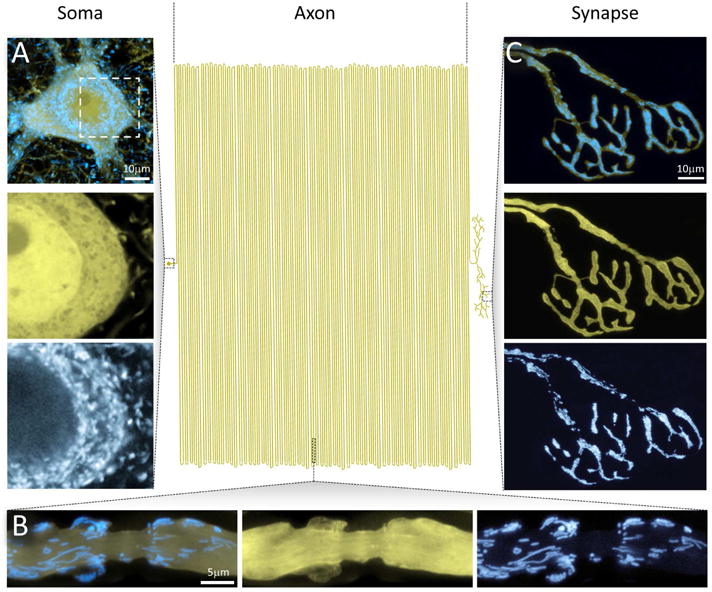
The extended geometry of neurons poses special challenges for maintaining mitostasis – here illustrated by a schematic attempt to draw a mammalian motor neuron to scale, with the soma to the left, the axon in the center, and the synapse-studded branches to the right (modified from (Devor, 1999)). A small soma (A) with a limited pool of mitochondria has to supply a vast axonal arbor that contains a far larger population of unevenly distributed mitochondria. At the same time, neuronal compartments with specific bioenergetic needs and hence accumulations of neuronal mitochondria, such as paranodes (B) or presynaptic terminals (C), need to be served.
And yet, a neuron is merely a cell and, like other cells, is constrained by the limitations and machinery of cells. Humans may live for 80 years, but most proteins survive only for days or weeks due to constant turnover by proteolytic digestion and ribosomal resynthesis (Goldberg, 2003). Because neurons are not dividing cells and need to last a human’s lifetime, the discrepancy between cellular and protein turnover is particularly extreme. Moreover, as in every cell, nearly all neuronal proteins are encoded by genes in the nucleus, no matter how far that nucleus may be from the protein’s site of action. Likewise, all cells need to transport components from one part of the cell to another by motor proteins that move along microtubule tracks, but the demands placed on those motors are more extreme in the context of highly extended and branched axons.
We focus here on one particular challenge for neuronal cell biology that we have termed neuronal mitostasis: how do neurons preserve a healthy, functioning pool of mitochondria throughout their arbor and throughout their lives? Mitostasis can be thought of as a specialized form of homeostasis, a mechanism by which mitochondrial number and quality are maintained over time in each compartment of the neuron. As mitochondrial proteins must, inevitably, be turned over and replaced, how is mitostasis accomplished? Vertebrate mitochondrial DNA (mtDNA) encodes only 13 proteins (Alston et al., 2017) and the remainder of the >1000 proteins found in mitochondria (Calvo et al., 2016) are encoded in the nucleus. How do those genes appropriately generate proteins that are required at the extremities of the cell? Because mitochondrial biogenesis involves mtDNA replication, mitochondria cannot be made de novo but must derive from other mitochondria. Where are mitochondria made and where destroyed? What are the roles of mitochondrial movements, fusions, and fissions in this process? Neuronal mitostasis, though increasingly appreciated, still has many unsolved questions.
Neurons depend on mitochondria. ATP generation by mitochondrial respiration in the brain consumes 20% of the body’s oxygen although the brain is only 2% of the mass of the body (Attwell and Laughlin, 2001). Maintaining resting potentials and firing action potentials are energetically expensive, as is neurotransmission on both the pre- and post-synaptic sides (Howarth et al., 2012). Although some of the ATP can be provided by glycolysis, mitochondrial respiration carries most of the burden (Rangaraju et al., 2014); this is why one rapidly loses consciousness if deprived of oxygen. Every part of the neuron requires ATP and therefore requires mitochondria to be present. Therefore, some mitochondria need to be retained in the soma while other mitochondria need to be trafficked to and positioned all along axons and dendrites to be concentrated at energy-hungry sites, such as presynaptic terminals and near nodes of Ranvier (Figure 1 A–C and 2) (Berthold et al., 1993; Fabricius et al., 1993; Shepherd and Harris, 1998). In this regard, the transport of mitochondria poses a complex challenge for the neuron: balancing energy supply to energy demand requires attention to the needs of each region of the neuron.
Figure 2. Neuronal Mitochondria in a Zebrafish Neuron.
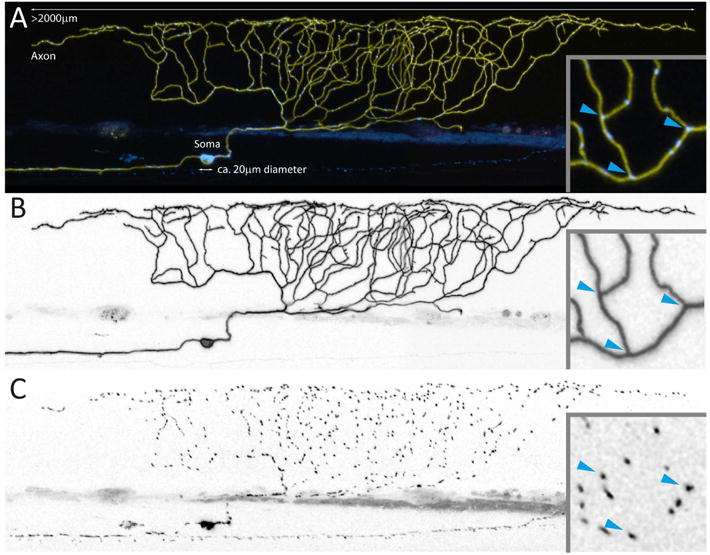
Smaller model organisms, such as flies, nematodes and zebrafish larvae (shown here), allow studying mitostatic mechanisms in a slightly less complex in vivo environment than in mammals. (A) depicts a zebrafish sensory Rohon-Beard neuron (96 hours post fertilization), which expresses a yellow fluorescent protein in the membrane and a cyan fluorescent protein in the mitochondrial matrix – giving an impression of the relative size of soma and axon (Plucinska et al., 2012). Optical accessibility allows entire axonal arbors (B) to be visualized and interrogated for mitochondrial distribution (C) and turnover. Even in these unmyelinated axons lacking synapses, mitochondria do not appear to be randomly distributed – for instance, many mitochondria reside at branch points (blue arrowheads in inset).
To understand mitostasis, therefore, it is necessary to understand the dynamics of mitochondria; their movements are crucial both for achieving an initial distribution in the cell and in accomplishing the resupply, as mitochondria and mitochondrial proteins are turned over. Mitochondria are governed by four dynamic processes: motility, anchoring, fission, and fusion (Figure 3). The movement of mitochondria is readily appreciated by video microscopy of mitochondria that have been fluorescently labeled either with a fluorescent dye, such as Tetramethylrhodamine (TMRM), or with a fluorescent protein targeted for import into mitochondria. As discussed in detail below, whether in tissue culture or in vivo, one population of mitochondria is in motion, while another is stationary (Plucinska and Misgeld, 2016; Saxton and Hollenbeck, 2012; Schwarz, 2013). The distinction between these two classes of mitochondria appears to be stable, reflecting two different pools; the motile mitochondria may temporarily pause, but tend to continue onwards and only infrequently alter their direction. Thus, some of the motile mitochondria are designated to move anterograde, down the axon, while others prefer retrograde movement; what creates this distinction is unknown. Mitochondria in the stationary pool, in contrast, remain in place often for as long as the experimenter can image (Faits et al., 2016; Lewis et al., 2016; Smit-Rigter et al., 2016). The movement of mitochondria is mediated by a motor adaptor complex that attaches the anterograde kinesin-1 motor (Kif5B in mammals) and retrograde dynein motor to the mitochondrial outer membrane (Figure 3 A). The attachment is mediated by the membrane-anchored Miro (also called RhoT1/2) and its motor-binding partner Milton (also called TRAK1/2) (Schwarz, 2013). This complex is present on all axonal mitochondria, even the stationary pool (Wang and Schwarz, 2009b). The stationary pool, at least in part, consists of mitochondria that are held in place by anchoring proteins. Syntaphilin, a protein attached to the outer mitochondrial membrane, is one such anchor and tethers the mitochondrion via a microtubule-binding domain (Kang et al., 2008). Mitochondria can also be anchored via the actin cytoskeleton (Pathak et al., 2010).
Figure 3. Mechanisms for Mitochondrial Dynamics in Neurons.
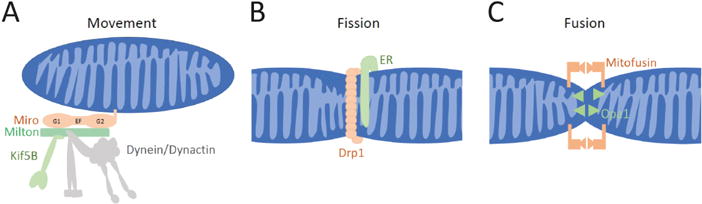
(A) To move along microtubules in both the anterograde direction (towards the + ends of microtubules) and retrograde (towards – ends), mitochondria employ a motor/adaptor complex that links Kif5b and the dynein/dynactin complex to the mitochondrial surface (Saxton and Hollenbeck, 2012; Schwarz, 2013). (B) Fission of both the inner and outer mitochondrial membranes is mediated by a ring of the dynamin-like GTPase Drp1. In many cases, fission appears to be initiated by a contact with the endoplasmic reticulum (ER). (C) Full fusion of mitochondria has two components. For the outer membranes to fuse, Mitofusins must be present on the membranes of both mitochondria and their interaction initiates the fusion. The inner membranes undergo a separate fusion that is mediated by Opa1.
Fission and fusion are also likely to play an essential role in mitostasis (Figure 3 B,C). In many cell types, mitochondria do not exist as small, distinct organelles, but rather form a highly interconnected reticulum with contiguous inner and outer membranes and multiple nucleoids containing mtDNA (Braschi and McBride, 2010). This reticulum is highly dynamic with ongoing divisions and reconnections. The balance of fission and fusion determines the length of mitochondria and is regulated by stress and nutrient availability (Wai and Langer, 2016). Interconnected mitochondria are also found in neuronal cell bodies, but in order to enter the axon a mitochondrion must undergo a fission reaction that frees it from the reticulum (Berthet et al., 2014; Fukumitsu et al., 2016; Verstreken et al., 2005). Fission is mediated by the GTPase Drp1 (Dynamin-related protein 1) and in many cases, contact with the endoplasmic reticulum and actin initiate the process together with the Drp1 receptor MFF (Mitochondrial Fission Factor). The opposing fusion reaction is particularly likely to occur when mitochondria meet end-to-end (Labbe et al., 2014; Twig et al., 2010). Full fusion requires two separate events: outer membrane fusion mediated by Mitofusins (MFN1/2) and inner membrane fusion mediated by Opa1 (optic atrophy 1). The importance of these processes for neurons is underscored by the neurological disorders caused by mutations in key components. Mitofusin2 mutations cause Charcot-Marie-Tooth disease type 2A (Kijima et al., 2005; Zuchner et al., 2004) and Opa1 mutations are responsible for autosomal dominant optic atrophy (Alexander et al., 2000; Delettre et al., 2000; McFarland et al., 2010). Mutations of Drp1, MFN, and Trak1 (Milton) are very rare in humans, but cause early lethality with broad defects that include neurological symptoms such as fetal encephalopathy and optic atrophy (Barel et al., 2017; McFarland et al., 2010).
The centrality of mitochondrial function – and consequently mitostasis – to neuronal health is further underscored by the prevalence of additional neurological disorders with a mitochondrial basis. These disorders fall in three categories – those that are directly due to defects in bioenergetics and metabolic functions, those that are quite closely linked to mitostatic processes such as mitochondrial clearance and dynamics, and those that indirectly influence mitostasis through broader defects in axonal transport or autophagy. The bioenergetic/metabolic disorders, which can arise from mutations in either mtDNA or nuclear DNA, are very heterogeneous in their presentation, and often affect many organ systems; neurological syndromes are among the most frequent and serious (McFarland et al., 2010). Examples would include many mutations in the complexes of the electron transport chain that result in encephalopathies, ataxia, and cerebellar atrophy. True mitostatic disorders, caused by mutations in the proteins that govern mitochondrial dynamics and mitophagy, result in diseases that range from severe developmental abnormalities, to optic atrophy, peripheral neuropathy, and Parkinson’s disease (Barel et al., 2017; Chen and Chan, 2006; Pickrell and Youle, 2015). Disorders that more broadly affect axonal transport, such as tauopathies and SOD1 mutations, may include phenotypes with a significant mitochondrial etiology (De Vos et al., 2008; Millecamps and Julien, 2013). More recently changes in mitostatic processes also have been described during neuroinflammation (Sadeghian et al., 2016; Sorbara et al., 2014; Witte et al., 2014) and after neurotrauma (Cartoni et al., 2016; Han et al., 2016; Sheng, 2017; Zhou et al., 2016). Such links to pathology rightly spur interest in understanding mitostasis.
THE PROBLEM OF MITOSTASIS IN NEURONS
The challenge of maintaining a healthy and demand-matched pool of mitochondria in a neuron concerns the large number of neuronal mitochondria, as well as their residence at sites far removed from the genomic information and biosynthetic hubs of the neuron’s soma. The peripheral mass of mitochondria scales with the size of the neuron, and consequently with the size and developmental state of the organism. From our own measurements, we estimate that a typical sensory neuron in larval zebrafish maintains a pool of several hundred mitochondria in its periphery (Plucinska et al., 2012). Notably in an adult mouse, a single peripheral synapse – a neuromuscular junction – likely contains a similar number of mitochondria (Misgeld et al., 2007)(Misgeld T., unpublished). This implies that the synaptic compartment of a moderately-sized murine motor unit of 50 synapses (Keller-Peck et al., 2001; Lu et al., 2009) contains several tens of thousands of mitochondria. The length of the axon, which even in axial muscles of small rodents can add to several centimeters (Lu et al., 2009) and typically contains 3 mitochondria per micrometer (Hanan S. and Misgeld T., unpublished), adds a similar mitochondrial mass on top. Finally, if one considers the numbers for diffuse projecting axons in the central nervous systems, which might have added branch lengths of several tens of centimeters even in mice, and then scale-corrections for larger organisms, such as humans, the numbers of mitochondria maintained by a <100 μm neuron soma easily grow into the millions. This suggests that the mitostatic mechanism of a neuron must have substantial capacity and are scalable across orders of magnitude. A single neuron of the human substantia nigra, for example, has been estimated to form 1 to 2 million striatal synapses (Bolam and Pissadaki, 2012). If mitochondria are present at almost half the synapses (Shepherd and Harris, 1998) and throughout several meters of axon, an estimated population of 2 million mitochondria per cell is reasonable.
Such geometrical considerations not only apply to organelle numbers, but also to the distances that a mitostatic system in neurons have to cover. The number of one meter of axon length from soma to synapse for human neurons is often cited. Even larger organisms, such as blue whales or giraffes, possess much longer projection neurons. Moreover, neurons are not only long, but branched, in a pattern that in its details at least in vertebrates is likely an imprint of the cell’s developmental history (Lu et al., 2009). Such developmental remodeling results in arbors that are highly irregular, not predicated in genomic information, and not easily captured by any idealized geometric description. The neuron needs to supply each of these branches, and the synapses that exist along their length, with an adequate number of organelles to ensure mitostatic stability. Together these geometrical features – overall size, linear extent and irregular branching – raise three conundrums that a comprehensive description of mitostatic regulation in neurons needs to account for:
The problem of “sufficient supply”: The fact that mitochondria show substantial anterograde transport, has led to the notion that most mitochondrial biosynthesis happens in the soma – after all, in vertebrates, the vast majority (~99%) of mitochondrial proteins are encoded in the nuclear genome. Are the biosynthetic capacity of the soma and the rate of mitochondrial export sufficient to maintain all peripheral mitochondrial compartments? If not, how are peripheral mitochondria supplied?
The problem of “long commutes”: The distance between the soma and the synapses of neurons in many species, together with the measured speed of mitochondrial transport, suggest that the journey of a mitochondrion from soma to synapse in the larger neurons of many species might exceed the available estimates of the lifetimes of mitochondrial proteins. This raises the question, how a mitochondrion maintains its protein composition and stoichiometry as it travels out towards the neuronal periphery?
The problem of “local demand matching”: The number and size of synapses supplied by a given axonal sub-arbor is not easily predicted and probably subject to change by synapse remodeling and long-term plasticity. How is the provision of mitochondria matched to these local and shifting demands?
The answer to these questions largely lies in the details of how mitochondrial dynamics, and especially long-distance transport of mitochondria, work in a neurons.
HOW (MUCH) DO MITOCHONDRIA MOVE IN VIVO?
In early studies, the size and shape of dynamic organelles seen by Nomarski contrast, e.g. in isolated frog axons (Cooper and Smith, 1974), suggested that many neuronal mitochondria are on the move. The specific dye (and later genetic) labeling of mitochondria then revealed in neuronal cell cultures the basic translocation behaviors of this organelle (Chang and Reynolds, 2006; Saxton and Hollenbeck, 2012), with a dominant pool of persistently stationary mitochondria and a smaller proportion that move (10–40%; Table 1 for references). Moreover, mitochondrial transport was shown to be, on the one hand, dependent on the same motors and microtubules that characterize axonal transport of other organelles (Figure 3). On the other hand, the detailed characteristics of mitochondrial transport in neurons were found to be unique: First, there are mitochondria that move in either direction (anterograde and retrograde) with few apparent reversals of directionality mid-axon. Second, there are pauses interspersed in the phases of sustained translocation, resulting in average speeds (0.25–1 μm/s; Table 1 for references) that are slower than the classical fast transport speeds of other organelles – even though substantial variability exists in the reported parameters, likely due to technical differences in the methods used to measure transport (Figure 4). Third, in addition to transport, mitochondria show another dynamic behavior, fusion and fission, which complicate analysis of mitochondrial transport, as a travelling organelle’s individuality can be abolished by interaction with other mitochondria (Owens and Walcott, 2012). Finally, the existence of pools of mitochondria associated with specific neuronal compartments, such as presynaptic boutons and around nodes of Ranvier, suggest a unique layer of regulation related to a neuron’s activity state – a notion that was confirmed by manipulations that globally or locally alter electrical activity or membrane potential (Chang and Reynolds, 2006).
Table 1. In vivo parameters of mitochondrial transport.
Compilation of in vivo values measured for basic mitochondrial transport parameter such as moving fraction (“% mov”), anterograde-to-retrograde ratio (“Flux a/r”), length (“Length a/r”) and anterograde and retrograde speed (“Speed a” and “Speed r”). Note, for comparison, corresponding average values as measured in rodent cell culture systems: motile fraction, 31 ± 8% (mean ± standard deviation); anterograde-to-retrograde ratio: 1.3 ± 0.5.
| Species/preparation | % mov |
Flux a/r |
Length a/r |
Speed a |
Speed r |
Reference |
|---|---|---|---|---|---|---|
| Homarus americanus leg nerve | >1 | 0.80 | 0.72 | 1.33 | (Forman et al., 1987) | |
| C. elegans PLML or PLMR neurons | 25% | (Fatouros et al., 2012) | ||||
| Drosophila larva intersegmental nerve | 43% | 1.5 | 0.84 | 0.26 | 0.45 | (Pilling et al., 2006) |
| Drosophila larva segmental nerve | 1.1 | 0.08 | 0.07 | (Baqri et al., 2009) | ||
| Drosophila larva segmental nerve | 26% | 1.3 | (Devireddy et al., 2015) | |||
| Drosophila larva L3 peripheral nerve | 35% | (Avery et al., 2012) | ||||
| D. rerio Rohon Beard neurons | 17% | 1.7 | 0.96 | 0.68 | 0.73 | (Plucinska et al., 2012) |
| D. rerio Rohon Beard neurons | 23% | 0.5 | 0.69 | (O’Donnell et al., 2013) | ||
| D. rerio Rohon Beard neurons | 27% | 2.3 | (O’Donnell et al., 2014) | |||
| M. musculus intercostal nerve | 13% | 1.9 | 0.87 | 1.02 | 1.41 | (Misgeld et al., 2007) |
| M. musculus dorsal column axons | 3.9 | 0.7 | 0.4 | (Sorbara et al., 2014) | ||
| M. musculus tibialis nerve | 3.2 | 0.6 | 0.5 | (Gilley et al., 2012) | ||
| M. musculus retina | 1.3 | (Takihara et al., 2015) | ||||
| M. musculus sciatic nerve | 2.7 | 0.4 | 0.4 | (Bilsland et al., 2010) | ||
| M musculus sciatic nerve | 22% | (Magrané et al., 2014) | ||||
| Summary | 26% | 2.1 | 0.9 | 0.6 | 0.7 |
A: anterograde; r: retrograde
Figure 4. Challenges in Measuring Mitochondrial Movement.
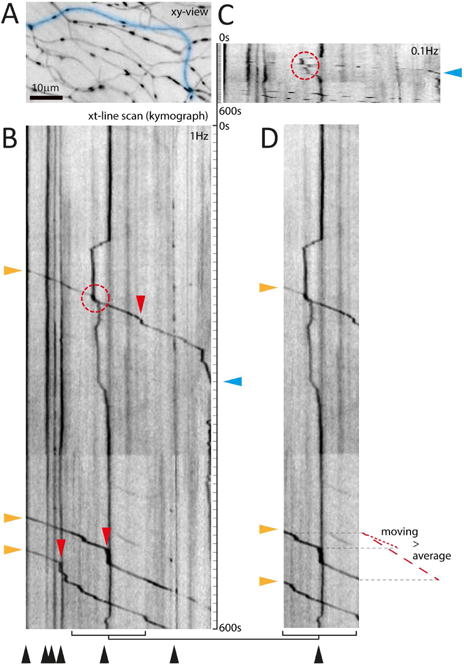
The parameters of motility are often difficult to compare, as different studies may choose to quantify the flux through a point, the percent of mitochondria that move, or the fraction of time that mitochondria spend in motion. Moreover, parameters that would appear to be fundamental properties are in fact operationally defined. (A–D) illustrate how the imaging protocol can affect analysis of kymographs from sensory axons in the skin of larval zebrafish (Plucinska et al., 2012). (A) xy-view of the region of interest imaged at 1 Hz. (B) A fully resolved kymograph along the blue line in A reveals different mitochondrial populations – stationary as vertical lines (black arrowheads) vs. moving as angled lines (three anterograde mitochondria; orange arrowheads), with short vertical segments (i.e. pauses; red arrowheads). Such pauses explain the difference between average speed (dashed red line next to D) vs. ‘moving’ speed (excluding pauses; dotted red line). (C) Temporal under-sampling (0.1 Hz was mimicked), leads to broken tracks that are difficult to follow (red circle) and obscures small pauses (compare track with cyan triangle). (D) The size of the field of view can affect the measured motile fraction (B, 3/9 mitochondria = 33% vs. D, 3/4 = 75%), as can the recording length because the stationary count will stay constant as moving mitochondria continue to pass through the field (not shown; consider the motile fraction in the upper, or lower half of B: 11% and 25%, respectively). Together these sampling discrepancies, but also signal-to-noise and movement artifacts, explain some of the reported ranges of transport parameters (Table 1). Not illustrated here are the effects of axonal branching or of fission and fusion, which further complicate such measurements.
Thus in vitro observations have been able to paint a consistent picture of how neuronal mitochondria can behave and have allowed fundamental cell biological and mechanistic analysis. However, such experiments are typically done in neurons of embryonic origin that exhibit a rather unrealistic geometry and are embedded in networks of unknown functional significance. Thus, how neuronal mitochondria behave in vivo has also required attention. The last decade has seen substantial progress to address this question, based on experimental paradigms that allow monitoring mitochondrial dynamics in vivo in the peripheral and central nervous system of a wide range of species including nematodes (Fatouros et al., 2012; Rawson et al., 2014; Williams et al., 2013), fruit flies (Babic et al., 2015; Pilling et al., 2006; Vagnoni and Bullock, 2016; Wang and Schwarz, 2009a), zebrafish (O’Donnell et al., 2013; Plucinska et al., 2012), and mice (Chandrasekaran et al., 2006; Misgeld et al., 2007). Reassuringly, the basic tenets of mitochondrial dynamics derived from in vitro work have been confirmed in vivo. Beyond this, however, the in vivo work has refined many aspects of the canonical in vitro description, has raised a number of new questions, and opened up studies in complex developmental and disease contexts that could not be modelled in vitro.
First, there have been seemingly conflicting reports on the degree to which mitochondria move in vivo. On the one hand, a substantial body of studies has shown that about 10–20% of the mitochondria in axons near the neuronal soma are moving at any given time, even under conditions in which neuronal activity is either intrinsically absent (due to surgical isolation) or suppressed by anesthesia (Bilsland et al., 2010; Gilley et al., 2012; Misgeld et al., 2007; Sorbara et al., 2014; Takihara et al., 2015). However, some other studies have suggested that baseline mitochondrial flux in fully mature neurites would be low, with a steep developmental decline (Faits et al., 2016; Lewis et al., 2016; Smit-Rigter et al., 2016). However, a full appreciation of the effects of branched neuronal geometry on local flux rates reconciles these seemingly conflicting reports: In some systems measurements are possible in proximal axonal locations, where flux reflects the totality of moving mitochondria needed to supply the arbor, while in others, due to imaging constraints, only very distal parts of the arbor can be visualized. Each time an axon bifurcates, the number of moving mitochondria per branch will be halved and therefore in each fine terminal branch, the moving pool will be a very small fraction of what once left the soma. Another methodological constraint has to do with the way in which the moving pool is determined – either as an absolute flux, or as a percentage of all mitochondria. Clearly, the latter, as a relative measurement, depends on the size of the stationary pool. In developing arbors, this percentage might be dropping as a function of an increasing denominator as mitochondria accumulate, without a major change in the absolute numbers of mitochondria entering the axon. Similarly, in different neuronal compartments – for example with a gradient along the axon – the size of the stationary pool might vary, again resulting in different values for the moving fraction despite consistent flux.
In parallel to determining the characteristics of baseline transport, a number of in vivo studies have attempted to determine the effect of in vivo activity on mitochondrial transport and positioning. Somewhat surprisingly, the majority of in vivo studies has failed to detect acute effects of activity (either spontaneous or imposed) and plasticity-inducing manipulations on bulk transport rates (Faits et al., 2016; Smit-Rigter et al., 2016). This questions the prevailing notion of neuronal activity as a major driver of baseline transport (but cf. (Sajic et al., 2013; Zhang et al., 2010) – and rather points to a model, where activity-related effects are subtle and chronic, with other factors more likely determining the predilection sites of mitochondrial residence in synapses and around nodes. The anchoring that creates the stationary pool is thus distinct from the calcium sensitivity of the Miro adaptor, which will temporarily arrest mitochondria in vitro (Macaskill et al., 2009; Wang and Schwarz, 2009b), but neither contributes to the distinction between stationary and motile pools nor appears to be activated by variations in firing rate in vivo.
Taken together, it is probably fair to say that work in intact preparations has confirmed the presence of a consistent and steady stream of mitochondria into and from the axonal arbor. Regarding the “sufficient supply” problem, for cell types where measurements near or at the axon hillock were possible, rough calculations suggest that the measured flux could sustain a turnover rate of the distal stationary mitochondria on the order of several days (Misgeld et al., 2007; Plucinska et al., 2012). Such a lifetime for mitochondria seems consistent with previous estimates based on proteome dynamics in the brain (Price et al., 2010; Vincow et al., 2013), even though the ability of mitochondria to fuse and divide, probably in a manner that does not evenly distribute molecular content, complicates the notion of a single ‘lifetime’ for the entire organelle. There remains the puzzle, however, how neurons with more extreme geometries (such as the diffusely projecting neuromodulatory neurons mentioned above) or in species larger than small experimental animals establish flux sufficient to sustain their distal mitochondrial population. Their estimated arbor size suggests flux rates in the order of several tens of mitochondria per minute, a value well above the 5–10 mitochondria that enter a murine motor neuron axon every minute. It will be informative to see such measurements from more cell types, e.g. with endoscopic imaging probes, to reveal whether the same transport rules apply across all arbor sizes, just scaled up for enormous mitochondrial flux. Alternatively, extreme neurons could have found ‘work-arounds’, e.g. by altering other parameters, such as mitochondrial lifetimes or dependence on long-distance replacement.
Similar considerations apply regarding the “long commute” problem. For small model organism the observed average transport speed (∼0.5μm/s) leads to reasonable travel times (for example, for fly segmental nerves about 1 hour from soma to synapse, while for mouse intercostal nerves around 10 hours for the same trip). For larger neurons and species, however, such calculations yield less reasonable results, as they exceed the lifetimes of short-lived nuclear-encoded mitochondrial proteins (Price et al., 2010; Vincow et al., 2013). One solution to this conundrum can be found in the ample evidence for the axonal presence of transcripts for mitochondrial proteins, not only in vitro (Aschrafi et al., 2016), but also in mature neurons in vivo (Shigeoka et al., 2016) – compatible with a model of peripheral mitochondrial maintenance by local protein synthesis. Such a model essentially shifts the cell biological problem towards the transport, lifetime and persistence of mRNAs and axonal ribosomes. Indeed, the biosynthetic machinery needed to make mitochondrial proteins is relatively simple compared to e.g. transmembrane proteins (which in addition to ribosomes require rough endoplasmic reticulum and Golgi outposts). The amplification inherent in translational outposts eases the concern that the soma alone could sustain such a vast distal organelle population. In addition, some mitochondrial proteins are imported into the organelle prior to their folding and therefore import needs to follow rapidly after their synthesis on cytoplasmic ribosomes or the unfolded state needs to be stabilized by chaperones (Ahmed and Fisher, 2009). Transport of mRNA for mitochondrial proteins may hold advantages for the cell over transport of the protein itself because of the amplification afforded, the possibility of co-translational import, and the ability to deliver proteins with half-lives too short to survive transport down axons. Local protein synthesis also raises the possibility of the generation of entire fresh mitochondria in distal axon locations, which would not only require availability and assembly of all mitochondrial proteins, but also the machinery for mitochondrial DNA replication and lipid biogenesis. Indeed, at least in vitro, metabolic labeling with nucleotide homologues confirmed the peripheral replication of mitochondrial DNA (Amiri and Hollenbeck, 2008). Moreover, the presence of smooth ER in axons (Wu et al., 2017; Yalcin et al., 2017) signifies that the capacity for lipid synthesis is also locally available.
Another pathway of supplying the neuronal periphery with mitochondria is organelle transfer from other cells. It is becoming clear that many metabolic pathways are shared between axons and glia by exchange of specific metabolites across the extracellular space (Saab et al., 2013). This concept has been expanded to the exchange of entire organelles, such as ribosomes (Court et al., 2008). There is a growing body of evidence that mitochondria can also be exchanged between cells, either via local cell fusions and “nanotubes” or via microvesicles that contain organelles (Sinha et al., 2016). While there are first reports that this might have functional implications in disease settings, such as stroke (Berridge et al., 2016), and could contribute to mitochondria clearance in specific anatomical locations (Davis et al., 2014), that mitochondrial exchange is a general feature that significantly contributes to mitostasis appears unlikely. Numerous experiments have taken advantage of cell-type specific expression of fluorescent mitochondrial proteins, and the fluorescent mitochondria are not seen spreading out from the cells in which they are expressed (Misgeld et al., 2007; Plucinska et al., 2012) – suggesting that such inter-cellular transfer is either rare, restricted to specific situations or followed by swift removal.
The supply of mitochondria to axons, therefore, can be either the canonical source (synthesis in the soma and transport) or a non-canonical source (local synthesis or transfer from an adjacent cell). How then do these mechanisms relate to the majority of axonal and synaptic mitochondria, which appear to be completely stationary and yet must somehow be turned over or rejuvenated? In vivo observations have shown that stationary mitochondria are so sparse that lateral fusion is not happening in axons on the time scale characteristic of other cells, suggesting that they are turned over individually. At least three models could explain the rejuvenation of the stationary pool: 1) The “changing of the guard” model: Stationary mitochondria may pick up and leave their guard stations, perhaps once every several weeks, to undergo mitophagy and then be replaced by a new arrival from the motile pool. This would be such an infrequent event that it would be rarely observed in most imaging protocols. 2) The “space station” model: They are undergoing rejuvenation by fusion with the motile pool in much the same way that deliveries by rockets from earth can keep the space station resupplied. This would require a low fusion probability for motile mitochondria as they move through the axon, because otherwise the most proximal mitochondria would get all the deliveries, leaving none to arrive at the periphery. In imaging experiments of axonal mitochondria, some fusion events are observed but they are indeed infrequent, as expected. 3) The “locally grown produce” model: Mitochondria undergo constant low-level rejuvenation from local sources such as axonal protein synthesis. Of course all of these mechanisms may operate in parallel and only careful – and difficult – quantification of rates of re-mobilization, fusion, and protein synthesis will sort out their contributions.
The “non-canonical” mechanisms of mitochondrial provision to axonal arbors also suggests that a straight-forward accounting approach to mitostasis by counting and measuring mitochondria as they come and go from the cell body is too simplistic a model of the life cycle of a mitochondrion and of mitostasis. Not all mitochondrial mass needs to arise in the soma, and, as reviewed in the next section, not all mitochondrial mass needs to be degraded in the soma. Nevertheless, if one focuses on transport to and from the soma, an important difference emerges. Almost all in vivo studies have noted that more mitochondria are delivered to, than are retrieved from the axon – even if size differences are taken into consideration (ratio mean 2.1; ranges 1.1–3.9 in vivo and intact preparations; notably this effect is less consistent in vitro; Table 1 for details and references). Although in some cases this could be due to axonal growth requiring a net addition of axonal mitochondria, measurements in fully-grown organisms find a similar discrepancy. If, as now seems likely, there is also substantial mitochondrial biogenesis in axons, the discrepancy between their supply and retrieval in the soma is yet greater and implies that the peripheral removal of mitochondria from axons and synapses is substantial.
HOW ARE MITOCHONDRIA CLEARED?
Though the question of mitochondrial supply to axons is critical, the “how” and “where” of mitochondrial clearance and protein turnover are no less significant. As Fred Goldberg has pointed out, cells are not a safe place for a protein; no competent biochemist would chose to store a protein at 37C instead of the freezer, nor leave it in the presence of potentially harmful cellular components (Goldberg, 2003). Proteins are prone to misfolding, denaturation, oxidation, and glycation, and therefore must be regularly degraded and replaced to preserve the quality of the cellular machinery. Mitochondrial proteins are no exception and, indeed, mitochondrial proteins may be at greater risk for damage because the electron transport chain and other mitochondrial processes are probably producing most of the reactive oxygen species in the cell (Andreyev et al., 2015). Mitochondrial proteins, therefore, need to be degraded as neuronal mitochondria are refreshed. In non-neuronal cells, many pathways have been identified for the turnover of mitochondria and their proteins. Though less work has been done in neurons, they are likely to use the same pathways. These pathways differ in their capacity: they can remove proteins individually, remove a small portion of a mitochondrion, remove a large fragment or an entire mitochondrion, or sweep up mitochondria together with other cellular components as part of a broad macroautophagic process (Figure 5).
Figure 5. Mechanisms of Mitochondrial Protein Turnover.
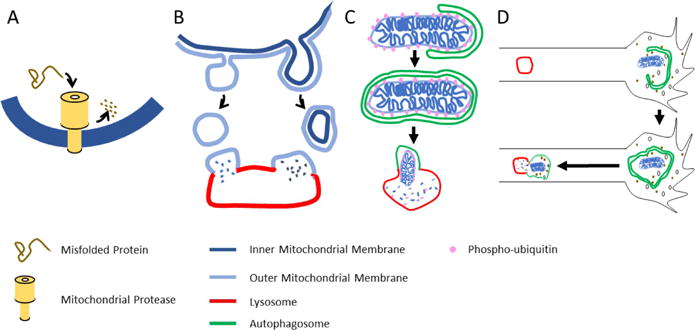
The mitochondrial quality is preserved by four mechanisms illustrated in order of their increasing scale. (A) Individual misfolded or damaged proteins are degraded by any of several mitochondrial proteases. (B) Mitochondrial components can be shed from the organelle in MDVs that bud from the organelle and can contain either only outer membrane components or also components of the inner membrane and matrix. (C) Mitophagy can engulf mitochondria, whose membrane potential or protein misfolding have triggered the PINK1/Parkin pathway to add phospho-ubiquitin to the outer membrane and thereby induce formation of autophagosomal membranes. The autophagosome will subsequently fuse with a lysosome. (D) Macroautophagy occurs in growth cones and can sweep up mitochondria along with other cytoplasmic components. Once formed, the autophagosome moves retrograde and fuses with lysosomes in the axon.
Individual mitochondrial proteins can be degraded by proteases resident within the mitochondrion. The most significant class for the removal of misfolded and denatured proteins is the mitochondrial AAA+ proteases (Glynn, 2017). These barrel-like proteases are present in the matrix and also in the inner mitochondrial membrane, facing both matrix and intermembrane space. They will recognize the hydrophobic domains that are exposed on misfolded and oxidized proteins and efficiently unfold and degrade them. The significance of these proteases for mitochondrial quality control in neurons is clear from the neurologic phenotypes associated with mutations in their genes. These phenotypes include non-syndromic mental retardation, spastic paraplegia, and spastic and spinocerebellar ataxias (Levytskyy et al., 2017). When oxidative damage has occurred, this mode of protein turnover may be considered the mitochondrion’s first line of defense, capable of removing a potentially problematic protein as soon as it forms. Indeed, the mitochondrial proteases in neurons may be commandeered for non-mitochondrial quality control: misfolded cytosolic proteins can also be imported into mitochondria for degradation (Ruan et al., 2017).
Mitochondria-derived vesicles (MDVs) represent a degradative pathway with the potential to remove large assemblies of protein and lipid rather than individual molecules (Roberts et al., 2016; Shlevkov and Schwarz, 2014) and, as such, may become particularly important when a stressor causes a local accumulation of damaged proteins. MDVs are particles that bud off from the mitochondrion and can then be targeted to either peroxisomes (Neuspiel et al., 2008), lysosomes (McLelland et al., 2014; Soubannier et al., 2012), or multivesicular bodies and exocytosis (Matheoud et al., 2016). They are a diverse population whose subtypes are still being clarified. As they form, they can select for distinct components of the outer mitochondrial membrane or include inner membrane and matrix proteins as well. Different forms of oxidative or heat stress can induce different subtypes of MDVs (Matheoud et al., 2016; McLelland et al., 2014; Soubannier et al., 2012). Much remains to be learned about the triggers of MDV formation, the mechanisms by which they form, and how they select their cargo. Formation of at least some MDVs depends on the Parkinson’s disease proteins PINK1 and Parkin, discussed further below, and also Drp1, the GTPase that mediates mitochondrial fission (Figure 3). Other MDVs are Drp1-independent but require Rab9 and the sorting nexin SNX-9. MDV production has been studied almost exclusively in cell lines, however, and its importance for neurons, though likely, remains to be demonstrated.
In contrast to the pathways mentioned above, mitophagy targets an entire mitochondrion or fragmented mitochondrion for destruction. Mitophagy is frequently studied in the context of severe damage to mitochondria (such as application of agents that dissipate the mitochondrial membrane potential) or an exceptional need to reduce mitochondrial content (such as hypoxia or erythrocyte maturation). The extent to which mitophagy figures in the normal, on-going turnover of mitochondria is less certain. Mitophagy is a highly specialized form of autophagy and has three necessary stages: recognition of the mitochondrion to be targeted, formation of an autophagic membrane around the organelle, and fusion of the resulting mitoautophagosome with a lysosome for degradation. The formation of autophagic membranes and trafficking to lysosomes employs mechanisms shared by all forms of autophagy and is reviewed elsewhere (Yin et al., 2016). Mitophagy, however, requires specialized mechanisms for the targeting stage; mechanisms to identify a mitochondrion that requires clearance and then recruit to it the autophagosomal membranes. One key pathway involves PINK1 and Parkin, proteins whose importance in mitochondrial quality control was first demonstrated in Drosophila and provided a strong indication mitochondria play a key role in the etiology of Parkinson’s disease (Clark et al., 2006; Park et al., 2006; Poole et al., 2008; Yang et al., 2008). Subsequent studies from many labs have illuminated a pathway by which PINK1, a protein kinase, serves as a sensor of mitochondrial quality and can trigger subsequent Parkin-dependent recruitment of LC-3 (Greene et al., 2012; Heo et al., 2015; Lazarou et al., 2015; McWilliams and Muqit, 2017; Pickrell and Youle, 2015; Wong and Holzbaur, 2014). In this schema, PINK1 undergoes constant synthesis, import into mitochondria, and proteolysis by mitochondrial enzymes that keeps the pathway in an “off” state. Because import across the inner mitochondrial membrane is driven by the mitochondrial membrane potential and because misfolding of proteins can also block import, the ability to import (and thereby disable) PINK1 depends on mitochondrial health. If the mitochondrion loses its membrane potential or builds up misfolded proteins, PINK1 will instead be stabilized on the outer surface of the mitochondrion and trigger mitophagy. The events downstream of PINK1 stabilization include phosphorylation of Miro and Mitofusin (Figure 3), phosphorylation (and activation) of Parkin, an E3 ubiquitin ligase, and phosphorylation of ubiquitin. Parkin binds to both phospho-miro and phospho-ubiquitin and is thereby recruited to the mitochondrial surface (Heo et al., 2015; Kane et al., 2014; Lazarou et al., 2015; McWilliams and Muqit, 2017; Shlevkov et al., 2016; Wong and Holzbaur, 2014). The resulting positive feedback loop results in extensive addition of phospho-ubiquitin to the mitochondrial surface. Phospho-ubiquitin in turn binds one of several mitophagy adaptors that recruit the autophagosomal membrane so that engulfment can proceed. It should be noted that this pathway is heavily dependent on the presence of ATP, due to the constant need for PINK1 synthesis and for phosphorylation of Parkin and ubiquitin. Experimental conditions that poison all the mitochondria in a neuron will therefore also likely shut down the PINK1/Parkin pathway to mitophagy (Cai et al., 2012; Van Laar et al., 2011). There are additional routes to mitophagy that are not dependent on either PINK1 or Parkin or both (Roberts et al., 2016), and the relative contribution and extent of redundancy of these alternatives is not clear (Devireddy et al., 2015; Sung et al., 2016). Nor are the actions of PINK1 and Parkin necessarily restricted to triggering mitophagy (Johnson et al., 2012; Morais et al., 2014; Muller-Rischart et al., 2013); in particular, as mentioned above, they can also mediate formation of MDVs and it is not known what determines the choice between MDV formation and full-fledged mitophagy.
In parallel with mitophagy, cells have an ongoing process of macroautophagy in which sections of cytoplasm are engulfed by autophagosomes. A basal rate of macroautophagy is present in all cells, but it is also subject to regulation, including by amino acid availability and the mTOR pathway (Yin et al., 2016). Macroautophagy is not an intrinsically selective process, and therefore no special receptors for mitochondria are needed, however mitochondria, or fragments of mitochondria, can be swept up alongside other cytoplasmic components. Macroautophagy is thus another process by which mitochondrial proteins can be delivered to lysosomes and degraded. Though not selective for oxidized proteins or depolarized mitochondria, degradation upon macroautophagy can nonetheless contribute to protein turnover and thereby enhance mitochondrial quality.
To what extent do mitophagy and macroautophagy figure in neurons? The relevance of the PINK1/Parkin pathway in neurons is apparent from the neurodegenerative phenotype of early-onset Parkinson’s caused by mutations in those genes. Moreover, when mitophagy is triggered, either by antimycin A applied to axons or by reactive oxygen species produced by photobleaching of mito-KillerRed, autophagosomes are recruited to the axonal mitochondria in a manner dependent on both PINK1 and Parkin (Ashrafi et al., 2014). In growth cones of cultured neurons, macroautophagy is very active and can include engulfment of mitochondria (Fu et al., 2014; Maday and Holzbaur, 2014). Less clear at present is the contribution of PINK1, Parkin, and autophagy to the overall maintenance of mitochondrial quality. In Drosophila, loss of PINK1 decreases the membrane potential of axonal mitochondria and causes structural abnormalities to the organelles (Devireddy et al., 2015), but knockout of PINK1 and Parkin in rodent models caused little or no neurodegeneration. Patients carrying these mutations generally do not have the onset of symptoms for three or four decades. Taken at face value, the relatively greater severity of neurologic phenotypes when the mitochondrial AAA-proteases are mutated suggests that mitophagy is the less important pathway, at least in mammals, and potentially triggered only by exceptional levels of mitochondrial damage. Defects in macroautophagy can produce neurological disorders (Yin et al., 2016), but to what extent these arise from altered mitostasis, rather than aggregation of misfolded proteins, remains unclear.
Further evidence that mitophagy and autophagy are not the major route of turnover for mitochondrial proteins was obtained in a study of the half-lives of mitochondrial proteins in Drosophila neurons (Vincow et al., 2013). This study found that half-lives varied widely from protein to protein and this was not consistent with a mechanism in which all the content of a mitochondrion was turned over simultaneously by engulfment and fusion with a lysosome. In addition, mutations in atg7, a key autophagy protein, and in Parkin, while altering half-lives, differed in their influence from protein to protein and for most mitochondrial proteins caused less than a 50% change in half-life. Thus at least four pathways lead to clearance of mitochondrial proteins: mitochondrial proteases, MDVs, the PINK1/Parkin mitophagic pathway, and macroautophagy. Each of these pathways is likely to contribute to neuronal mitostasis, maintaining a correctly sized pool of high-quality mitochondria.
WHERE ARE MITOCHONDRIA CLEARED?
In considering the location of mitochondrial clearance, the special problems posed by axons raise issues that small cells do not face. In particular, can mitochondria be degraded wherever they reside? It has often been presumed that they need to return to the soma for removal, like salmon returning to the stream where they were spawned. The logic behind this presumption is three-fold: 1) that mitochondria need to be degraded by lysosomes; 2) that lysosomes are most abundant in the soma; and 3) that the presence of a retrogradely moving population of mitochondria is best explained by a mitochondrial need to return to the soma at the end of their useful life. Early evidence for this model came from a study of the voltage sensitive dye JC-1 in axons, which found that those mitochondria moving retrograde had lower membrane potentials than those moving anterograde and that inhibition of the electron transport chain (ETC) with Antimycin A increased retrograde movement (Miller and Sheetz, 2004). A subsequent study, however, that used TMRM to assay mitochondrial membrane potential found no distinction between anterograde and retrograde populations (Verburg and Hollenbeck, 2008) and others have observed the arrest of mitochondrial movement in response to inhibitors of the ETC, an arrest mediated by the proteolytic degradation of Miro via the PINK1/Parkin pathway (Hsieh et al., 2016; Liu et al., 2012; Shlevkov et al., 2016; Wang et al., 2011). Most recently, a study employed exposures of dorsal root ganglion neurons for 6 hours or longer to very low levels of antimycin A to mimic a long-term mild stress, followed by an hour of recovery. In those conditions, membrane potential and directionality correlated, with better membrane potentials preferentially moving anterograde (Lin et al., 2017). One further complication need be mentioned: if retrogradely moving mitochondria are destined for degradation, once they enter the soma they should be kept apart for mitophagy. Instead, our unpublished observations suggest that mitochondria that move from the axon into the soma undergo fusion with the resident somatic pool and mix their components in with the resident and presumably younger pool of mitochondria. One set of experiments with the slowly oxidizing indicator mitoTimer (Ferree et al., 2013), found that distal axonal mitochondria were, on average, older than somatic mitochondria but one did not see intact, old mitochondria as a distinct population returning to the soma. Thus, though studies of retrograde transport, differing in their approaches, have also diverged in their findings, there seems to be little evidence to support the idea that the majority of retrogradely moving mitochondria in a normal, healthy neuron are older or of worse quality than the rest of the mitochondrial pool; they are not all salmon returning home to die.
In light of the paucity of evidence for the ‘dying salmon’ model, it is worth examining the presumptions behind it. As noted in the previous section, not all forms of mitochondrial turnover require lysosomes. Many misfolded or oxidized proteins can be degraded by the proteases in mitochondria and this may represent the primary means of protein turnover in mitochondria. MDVs that are shed by mitochondria may be transported to lysosomes independent of the mitochondrion from which they arose. Furthermore, although lysosomes are most abundant in the soma, there is evidence for lysosomes in axons as well (Ashrafi et al., 2014; Maday and Holzbaur, 2014) and for late endosomes arising in distal axons that mature into lysosomes and increase in their proteolytic capacity en route as they move retrograde (Gowrishankar et al., 2017). The axonal pool of lysosomes may be sufficient to mediate clearance, when needed, in an otherwise healthy neuron. Given the abundance of stationary mitochondria and the amount of time necessary for transport from distal regions of an axon to the soma, a different set of considerations would favor the logic of local clearance as preferable for efficiency and for preventing damaged mitochondria from releasing reactive oxygen species throughout the cell while journeying back to the soma.
The locus of mitochondrial protein turnover may therefore vary depending on the method of degradation of the components and the conditions that drive it. Mitochondrial proteases are presumably present in all neuronal mitochondria and may be continuously preserving mitochondrial quality in distal axons. The proteins removed in this fashion would, however, need to be replaced either by local protein synthesis or by fusion with the motile pool. Similarly, for MDVs there is at present no information about either their generation or fate in axons – but they can presumably be generated anywhere in the neuron. If a mitochondrion is diminished by shedding MDVs, it too will need replacement proteins from local synthesis or the motile pool. At least in cultured neurons, macroautophagy that incorporates mitochondria appears to be a very active process at axon tips and much less frequent in the soma or along the length of the axon (Maday and Holzbaur, 2014). Whether macroautophagy has a very particular role in the tip is less clear, but it may be necessary to clear the large volume of material that is shipped down growing axons (Maday and Holzbaur, 2016). What is clear is that the autophagosome once formed, recruits the retrograde dynein motor and moves retrograde in a highly processive fashion and with increasing acidity consistent with its maturation into an autophagolysosome (Maday et al., 2012). When retrograde mitochondrial transport is measured, it is unlikely that mitochondrial fragments within autophagosomes are a significant contributor to the calculated flux because the fragments are small and degradation en route will remove the markers. Macroautophagy, if it is also active at mature synapses, may thus account for some of the imbalance between delivery of mitochondrial material to the distal reaches and the amount that returns.
Mitophagy via the PINK1/Parkin pathway presents the most complex questions regarding the location of clearance and necessarily involves two issues: where are PINK1 and Parkin first activated to engage autophagosome formation, and where does the autophagosome undergo fusion with a lysosome and degradation? Ashrafi et al. observed the local recruitment of Parkin to damaged axonal mitochondria followed by the recruitment of autophagosomal markers and eventual fusion with lysosomes and loss of the mitochondrial markers (Ashrafi et al., 2014). This was consistent with the degradation of Miro by this pathway (Liu et al., 2012; Wang et al., 2011); the consequent arrest of mitochondrial movement made it unlikely that this process required damaged mitochondria to be translocated to the soma. That said, the observation of local mitophagy does not exclude the possibility that, under some circumstances (Lin et al., 2017) and particularly those where there is unlikely to be enough ATP to support the PINK1/Parkin pathway, older or slightly damaged mitochondria will not be arrested and will preferentially move retrograde.
In conclusion, local clearance of mitochondria – whether by mitochondrial proteases, MDVs, mitophagy, or macroautophagy – is likely to be a major component of mitochondrial protein turnover in neurons and may account for much of the discrepancy in the amount of mitochondria transported anterograde down axons compared to that which moves retrograde. Moreover, whereas this review has focused entirely on cell-autonomous means of mitochondrial clearance, there is evidence as well that axonal mitochondria can be shed and engulfed by glia – a pathway that provides a further alternative for turnover without retrograde transport (Bishop et al., 2004; Davis et al., 2014; Melentijevic et al., 2017). Additional clearance in the soma and dendrites will also contribute, but there is little reason to invoke an obligatory return to the soma. Moreover, because returning mitochondria fuse with other somatic mitochondria, the majority of retrogradely transported mitochondria do not appear to be sequestered and destined for degradation as isolated organelles.
MITOSTATIC DISEASES
The literature on the involvement of mitochondria in neurological diseases is exploding in volume. By now it is difficult to a name a neurological disorder in which this organelle has not been implicated. This poses the challenge to differentiate epiphenomenological damage to a vulnerable organelle from a true pathomechanistic role of mitochondria in a disease process. We believe that the concept of mitostatic neurological disorders can help to organize the plethora of roles that mitochondria play in the diseased nervous system. Such conditions would be defined as diseases, where clear-cut disturbances in aspects of mitostasis have been demonstrated – altered provision of mitochondria by biosynthesis and transport, disrupted fusion/fission kinetics or disturbed clearance. This definition would exclude conditions, where simply bioenergetic failure, depolarization, altered shape or ultrastructure of mitochondria are observed as the sole argument of mitochondrial involvement.
In some diseases, genetics provides a strong indicator for a mitostatic origin. This is certainly true for hereditary disorders with disruptions of genes with well-defined functions in mitostatic pathway. One example are neuropathies – of the optic and peripheral nerves – that emerge because of disruptions in mitochondrial fission and fusion genes. Their manifestation as degeneration of specific axonal projections underscores the special role that mitochondrial dynamics play in neurons. Indeed, the number of – individually rare – mutations discovered in the pathways that are illustrated in Figure 3 is growing. For instance, mutations in DRP1 can cause lethal encephalopathies, even though the relative role of disrupted homeostasis of mitochondria or peroxisomes is not clear here (Waterham et al., 2007). Mutations in Mitofusin2 cause the neuropathic degeneration of motor and sensory axons known as Charcot-Marie-Tooth disease type 2A (Kijima et al., 2005; Zuchner et al., 2004). Mutations in Opa1 cause autosomal-dominant optic atrophy, a disease of progressive blindness due to degeneration of retinal ganglion cells, but which can also include more widespread neuronal degeneration (Alexander et al., 2000; Chen and Chan, 2006; Delettre et al., 2000; McFarland et al., 2010). Truncations of the human milton isoform TRAK1 cause fatal encephalopathy (Barel et al., 2017). As indicated above, the most common mitostatic disease identified to date is Parkinson’s disease. Some forms of familial Parkinson’s disease are due to mutations in PINK1 and Parkin that alter mitochondrial clearance (Pickrell et al., 2015; Pickrell and Youle, 2015; Vincow et al., 2013). At the same time, the toxicology of Parkinsonism-inducing toxins, such as MPTP+ and rotenone, point to a mitochondrial origin of the disease (Sherer et al., 2001). Most recently, a study of mitophagy in neurons from patient-derived iPSCs found that slowed degradation of Miro, and consequent failure to arrest mitochondrial movement, was a characteristic of many cell lines derived from Parkinson’s disease patients, not just those with PINK1 and Parkin mutations. This included LRRK2 mutations and sporadic cases without a known genetic link (Hsieh et al., 2016). Parkinson’s illustrates the complexity of the interrelationship of mitochondrial movement and mitochondrial clearance. In healthy cells, adequate mitochondrial movement helps to keep the axonal pool healthy, but if either axonal transport is disrupted, or mitochondrial health is compromised, the movement of mitochondria will be diminished and mitochondrial health can further suffer. Persistent movement of damaged mitochondria in PD neurons, and the consequent spreading of ROS, may be as deleterious to the cell as insufficient movement. Moreover, spurious activation of the PINK1/Parkin pathway in an otherwise healthy context, by arresting mitochondrial movement, may prove as deleterious as loss of the pathway when PINK1 or Parkin are mutated. This may explain why kinetin, a PINK1 activator, failed to have the beneficial effects that were hoped for in a synuclein PD rodent model (Orr et al., 2017).
Mitochondria have also been implicated in many other diseases, such as Alzheimer’s disease, amyotrophic lateral sclerosis, and other motor neuron diseases, and of course in conditions where bioenergetic substrates are missing, such as hypoxia and stroke. In all these cases, mitochondrial pathology is a conspicuous, and even an early, pathological change, and potential roles in pathogenesis have been put forward as reviewed previously (Schon and Przedborski, 2011; Sheng and Cai, 2012). It remains unclear, however, whether there is a genuine mitostatic component in these diseases. In another category of diseases, mutations in motors or microtubule-associated proteins may impact the movement of many types of organelle. It is likely – but mostly unproven – that the disruption of mitostasis is among the serious pathogenic consequences (De Vos et al., 2008).
There are also ‘new kids on the block’, conditions in which a role for mitochondria has been suggested more recently. These include demyelinating and neuroinflammatory diseases, such as multiple sclerosis, and the response of neurons after trauma. In multiple sclerosis, mitochondrial damage is attracting increasing attention as a potential contributor to axonal damage, an important hallmark of the disease and a central driver of lasting disability. In neuroinflammatory lesions, altered mitochondria are present (Nikic et al., 2011; Witte et al., 2014). In vivo imaging in disease models has revealed reduced mitochondrial transport, which results in accumulations of dysfunctional mitochondria locally and a lack of mitochondria in the distal axonal arbor (Sadeghian et al., 2016; Sorbara et al., 2014). These processes seem largely driven by the inflammatory milieu, which appears to impair transport, as well as to prevent normal function of mitochondria, possibly via reactive oxygen or nitrogen species. Notably, demyelination, another hallmark of multiple sclerosis lesions, per se influences mitochondrial trafficking and density, in part via the anchoring protein syntaphilin. A knockout of syntaphilin, intriguingly, prolonged the survival of Purkinje cells in the shiverer mouse model of chronic demyelination (Joshi et al., 2015), but had the opposite effect in a model of acute demyelination (Ohno et al., 2014) and did not enhance motor neurons survival in a SOD1 mutant ALS model (Zhu and Sheng, 2011). The inflammation- and demyelination-mediated effects intersect in a complex fashion, in part augmenting each other (regarding local accumulations), in part by counterbalancing (transport speed). Hence, the role of mitochondria in multiple sclerosis, and in converse the effect of neuroinflammatory demyelination on mitochondrial dynamics, are complex – and from the currently available data a clear mechanistic role in the pathology cannot yet deduced. Still, this string of work by several labs illustrates how new technical approaches to mitochondrial morphology and dynamics in situ can start shedding light on the specific contributions of this neuronal organelle to nervous system disease.
Another area of recent progress is in the field of neuroregeneration. Parallel work in invertebrate and rodent model organisms has shown that, on the one hand, mitochondria are likely to influence axonal growth patterns during development (Courchet et al., 2013; Spillane et al., 2013), but also after axotomy during regeneration (Cartoni et al., 2016; Han et al., 2016; Knowlton et al., 2017; Zhou et al., 2016). In this case, most data point towards a classical bioenergetic role, where axonal mitochondria at or near the growing axon tip are simply needed for sustained axon growth. Importantly, an axotomized neuron, ranging from C. elegans to mice, seems to endogenously upregulate the export of mitochondria into the axonal stump (Han et al., 2016; Misgeld et al., 2007). These observations underscore the evolutionary persistence even of higher order regulation of mitostatic processes, and opens the opportunity to use the power of genetic screens to understand the role of neuronal mitochondria in disease.
Despite much progress in understanding mitostatic processes and their importance to neuronal survival, many open questions remain. 1) Regarding mitochondrial populations, are mobile and stationary mitochondria metabolically equivalent; do inter-organelle contacts play a role in creating mitochondrial diversity; and how different are mitochondria between cell types in the nervous system? 2) Regarding mitochondrial biogenesis, which modes of mitochondrial rejuvenation are most important, and how are they tied to transport, fusion and fission; what is the relative contribution of somatic versus peripheral biogenesis; what is the contribution of axonal and dendritic translation and is there a dedicated translational machinery in neurites that serves mitochondria? 3) Regarding mitochondrial transport, what determines the direction of movement; how far do neuronal mitochondria typically travel; how long do they maintain identity as isolated organelles; how is exchange with the stationary pool regulated; and what is the function of retrograde mitochondrial transport? 4) Regarding mitochondrial distribution, what holds mitochondria at synapses; how are specific compartments enriched in mitochondria and how is spacing ensured in areas of low density; how is flow into branches matched to axon arbor geometry; and how are axonal vs. dendritic fates specified? 5) Regarding mitochondrial clearance, do all the described pathways operate in neurons and where in the cell? 6) Regarding mitochondrial transfer, what is the extent to which mitochondria move between cells in the nervous system; how much does this movement contribute to the removal of neuronal mitochondria in health and disease; is this process important for glial support of axons? 7) Regarding axon to nucleus communication, is transcription of nuclear genes coupled to changes in energy needs or mitochondrial status in distant regions of the cell? 8) Regarding neuropathology, why do mitostatic disorders differ in their cell-type specific effects; how can mitostasis be enhanced; and will enhancing mitostasis help to preserve neurons?
In conclusion, neurons have met the complexity of their mitostatic challenge with a diverse set of mechanisms to drive biogenesis, transport, fission, fusion, and clearance. Though much remains unknown about the interplay of these mechanisms and their relative contributions, the centrality of mitostasis to neuronal survival is clear. The importance of understanding the mitostatic apparatus transcends those disorders that are most directly caused by disruption of that apparatus; in many pathological states improving mitostasis may improve mitochondrial health and benefit neuronal survival. Cell biologists have much still to explore in the fundamentals of mitochondrial dynamics and turnover in neurons.
Acknowledgments
Research in T.S.’s laboratory was supported by NIH grant R01GM069808, the Mathers Foundation, the ARSACS Foundation, and the Michael J. Fox Foundation. T.M.’s work on mitochondria is supported by the DFG through the Munich Center for Systems Neurology (SyNergy; EXC 1010), the Center for Integrated Protein Science Munich (CIPSM, EXC 114), Collaborative Research Center 870 and research grants Mi694/7-1 and 8-1. Further support came from the European Research Council under the European Union’s Seventh Framework Program (FP/2007-2013; ERC Grant Agreement n. 616791) and the German Center for Neurodegenerative Diseases (DZNE Munich). We thank Shabab Hannan with help on mitochondria counts in motor axons and compilation of transport parameters, Tatjana Kleele for the neuron drawing in Figure 1, and Angelika Harbauer and Jill Fallk for reading an earlier version of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed AU, Fisher PR. Import of nuclear-encoded mitochondrial proteins: a cotranslational perspective. Int Rev Cell Mol Biol. 2009;273:49–68. doi: 10.1016/S1937-6448(08)01802-9. [DOI] [PubMed] [Google Scholar]
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. Journal of Pathology. 2017;241:236–250. doi: 10.1002/path.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. 2008;68:1348–1361. doi: 10.1002/dneu.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015;80:517–531. doi: 10.1134/S0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Kar AN, Gale JR, Elkahloun AG, Vargas JN, Sales N, Wilson G, Tompkins M, Gioio AE, Kaplan BB. A heterogeneous population of nuclear-encoded mitochondrial mRNAs is present in the axons of primary sympathetic neurons. Mitochondrion. 2016;30:18–23. doi: 10.1016/j.mito.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. Journal of Cell Biology. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR. Wld S prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca 2+ buffering. Current Biology. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic M, Russo GJ, Wellington AJ, Sangston RM, Gonzalez M, Zinsmaier KE. Miro’s N-terminal GTPase domain is required for transport of mitochondria into axons and dendrites. J Neurosci. 2015;35:5754–5771. doi: 10.1523/JNEUROSCI.1035-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baqri RM, Turner BA, Rheuben MB, Hammond BD, Kaguni LS, Miller KE. Disruption of mitochondrial DNA replication in Drosophila increases mitochondrial fast axonal transport in vivo. PLoS One. 2009;4:e7874. doi: 10.1371/journal.pone.0007874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barel O, Christine V Malicdan M, Ben-Zeev B, Kandel J, Pri-Chen H, Stephen J, Castro IG, Metz J, Atawa O, Moshkovitz S, et al. Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain. 2017;140:568–581. doi: 10.1093/brain/awx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MV, Schneider RT, McConnell MJ. Mitochondrial Transfer from Astrocytes to Neurons following Ischemic Insult: Guilt by Association? Cell Metabolism. 2016;24:376–378. doi: 10.1016/j.cmet.2016.08.023. [DOI] [PubMed] [Google Scholar]
- Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K. Loss of Mitochondrial Fission Depletes Axonal Mitochondria in Midbrain Dopamine Neurons. Journal of Neuroscience. 2014;34:14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersen B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol. 1993;22:925–940. doi: 10.1007/BF01218351. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20523–20528. doi: 10.1073/pnas.1006869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–1483. doi: 10.1002/mds.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, McBride HM. Mitochondria and the culture of the Borg: Understanding the integration of mitochondrial function within the reticulum, the cell, and the organism. BioEssays. 2010;32:958–966. doi: 10.1002/bies.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial Parkin Translocation and Degradation of Damaged Mitochondria via Mitophagy in Live Cortical Neurons. Current biology : CB. 2012;22:545–552. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, He Z. The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron. 2016;92:1294–1307. doi: 10.1016/j.neuron.2016.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran K, Hazelton JL, Wang Y, Fiskum G, Kristian T. Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci. 2006;26:13123–13127. doi: 10.1523/JNEUROSCI.4191-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DTW, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Progress in Neurobiology. 2006;80:241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Current Opinion in Cell Biology. 2006;18:453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cooper PD, Smith RS. The movement of optically detectable organelles in myelinated axons of Xenopus laevis. J Physiol. 1974;242:77–97. doi: 10.1113/jphysiol.1974.sp010695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchet J, Lewis TL, Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Hendriks WT, MacGillavry HD, Alvarez J, van Minnen J. Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J Neurosci. 2008;28:11024–11029. doi: 10.1523/JNEUROSCI.2429-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C-HO, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, et al. Transcellular degradation of axonal mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9633–9638. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Devireddy S, Liu A, Lampe T, Hollenbeck PJ. The Organization of Mitochondrial Quality Control and Life Cycle in the Nervous System In Vivo in the Absence of PINK1. Journal of Neuroscience. 2015;35:9391–9401. doi: 10.1523/JNEUROSCI.1198-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain Suppl. 1999;6:S27–35. doi: 10.1016/S0304-3959(99)00135-9. [DOI] [PubMed] [Google Scholar]
- Fabricius C, Berthold CH, Rydmark M. Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J Neurocytol. 1993;22:941–954. doi: 10.1007/BF01218352. [DOI] [PubMed] [Google Scholar]
- Faits MC, Zhang C, Soto F, Kerschensteiner D. Dendritic mitochondria reach stable positions during circuit development. Elife. 2016;5:e11583. doi: 10.7554/eLife.11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of Tau aggregation in a novel caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Human Molecular Genetics. 2012;21:3587–3603. doi: 10.1093/hmg/dds190. [DOI] [PubMed] [Google Scholar]
- Ferree AW, Trudeau K, Zik E, Benador IY, Twig G, Gottlieb RA, Shirihai OS. MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy. 2013;9:1887–1896. doi: 10.4161/auto.26503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman DS, Lynch KJ, Smith RS. Organelle dynamics in lobster axons: anterograde, retrograde and stationary mitochondria. Brain Res. 1987;412:96–106. doi: 10.1016/0006-8993(87)91443-0. [DOI] [PubMed] [Google Scholar]
- Fu MM, Nirschl JJ, Holzbaur ELF. LC3 Binding to the scaffolding protein jip1 regulates processive dynein-driven transport of autophagosomes. Developmental Cell. 2014;29:577–590. doi: 10.1016/j.devcel.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Molecular and Cellular Neuroscience. 2016;71:56–65. doi: 10.1016/j.mcn.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, Misgeld T, Brion JP, Anderton B, Hanger DP, Coleman MP. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging. 2012;33:621 e621–621 e615. doi: 10.1016/j.neurobiolaging.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Glynn SE. Multifunctional Mitochondrial AAA Proteases. Frontiers in Molecular Biosciences. 2017;4:34–34. doi: 10.3389/fmolb.2017.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gowrishankar S, Wu Y, Ferguson SM. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J Cell Biol. 2017 doi: 10.1083/jcb.201612148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M. Mitochondria Localize to Injured Axons to Support Regeneration. Neuron. 2016;92:1308–1323. doi: 10.1016/j.neuron.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:1222–1232. doi: 10.1038/jcbfm.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schüle B, Krainc D, Palmer TD, Wang X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell. 2016;19:709–724. doi: 10.1016/j.stem.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A. 2012;109:6283–6288. doi: 10.1073/pnas.1113248109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi DC, Zhang CL, Lin TM, Gusain A, Harris MG, Tree E, Yin Y, Wu C, Sheng ZH, Dempsey RJ, et al. Deletion of mitochondrial anchoring protects dysmyelinating shiverer: implications for progressive MS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:5293–5306. doi: 10.1523/JNEUROSCI.3859-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Peck CR, Walsh MK, Gan WB, Feng G, Sanes JR, Lichtman JW. Asynchronous synapse elimination in neonatal motor units: studies using GFP transgenic mice. Neuron. 2001;31:381–394. doi: 10.1016/s0896-6273(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, Ogawa M, Ishizaki Y, Kitamura T, Shozawa Y, Hayasaka K. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005;116:23–27. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- Knowlton WM, Hubert T, Wu Z, Chisholm AD, Jin Y. A Select Subset of Electron Transport Chain Genes Associated with Optic Atrophy Link Mitochondria to Axon Regeneration in Caenorhabditis elegans. Front Neurosci. 2017;11:263. doi: 10.3389/fnins.2017.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levytskyy R, Bohovych I, Khalimonchuk O. Metalloproteases of the inner mitochondrial membrane. Biochemistry. 2017 doi: 10.1021/acs.biochem.7b00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Turi GF, Kwon SK, Losonczy A, Polleux F. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Current Biology. 2016;26:2602–2608. doi: 10.1016/j.cub.2016.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, Sheng ZH. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron. 2017;94:595–610.e596. doi: 10.1016/j.neuron.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012;8:e1002537. doi: 10.1371/journal.pgen.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tapia JC, White OL, Lichtman JW. The interscutularis muscle connectome. PLoS Biol. 2009;7:e32. doi: 10.1371/journal.pbio.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016;36:5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur ELF. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Developmental Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Human Molecular Genetics. 2014;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 2016;166:314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. The Lancet Neurology. 2010;9:829–840. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Muqit MMK. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Current Opinion in Cell Biology. 2017;45:83–91. doi: 10.1016/j.ceb.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, Taub D, Parker JA, Neri C, Gabel CV, et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367–371. doi: 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nature reviews Neuroscience. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. Journal of Cell Science. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nature methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grunewald A, Seibler P, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014;344:203–207. doi: 10.1126/science.1249161. [DOI] [PubMed] [Google Scholar]
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Bruck W, Bishop D, Misgeld T, Kerschensteiner M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- O’Donnell KC, Lulla A, Stahl MC, Wheat ND, Bronstein JM, Sagasti A. Axon degeneration and PGC-1α-mediated protection in a zebrafish model of α-synuclein toxicity. Disease models & mechanisms. 2014;7:571–582. doi: 10.1242/dmm.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KC, Vargas ME, Sagasti A. WldS and PGC-1α regulate mitochondrial transport and oxidation state after axonal injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:14778–14790. doi: 10.1523/JNEUROSCI.1331-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, Sheng ZH, Komuro H, Trapp BD. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9953–9958. doi: 10.1073/pnas.1401155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AL, Rutaganira FU, de Roulet D, Huang EJ, Hertz NT, Shokat KM, Nakamura K. Long-term oral kinetin does not protect against alpha-synuclein-induced neurodegeneration in rodent models of Parkinson’s disease. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GC, Walcott EC. Extensive fusion of mitochondria in spinal cord motor neurons. PLoS One. 2012;7:e38435. doi: 10.1371/journal.pone.0038435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J Neurosci. 2010;30:8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron. 2015;87:371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plucinska G, Misgeld T. Imaging of neuronal mitochondria in situ. Curr Opin Neurobiol. 2016;39:152–163. doi: 10.1016/j.conb.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Plucinska G, Paquet D, Hruscha A, Godinho L, Haass C, Schmid B, Misgeld T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J Neurosci. 2012;32:16203–16212. doi: 10.1523/JNEUROSCI.1327-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S. Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci U S A. 2010;107:14508–14513. doi: 10.1073/pnas.1006551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825–835. doi: 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, Jorgensen EM. Axons degenerate in the absence of mitochondria in C. elegans. Curr Biol. 2014;24:760–765. doi: 10.1016/j.cub.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RF, Tang MY, Fon EA, Durcan TM. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. International Journal of Biochemistry and Cell Biology. 2016;79:427–436. doi: 10.1016/j.biocel.2016.07.020. [DOI] [PubMed] [Google Scholar]
- Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, Florens L, Li R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543:443–446. doi: 10.1038/nature21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, Nave KA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23:1065–1072. doi: 10.1016/j.conb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Sadeghian M, Mastrolia V, Rezaei Haddad A, Mosley A, Mullali G, Schiza D, Sajic M, Hargreaves I, Heales S, Duchen MR, Smith KJ. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci Rep. 2016;6:33249. doi: 10.1038/srep33249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajic M, Mastrolia V, Lee CY, Trigo D, Sadeghian M, Mosley AJ, Gregson NA, Duchen MR, Smith KJ. Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biol. 2013;11:e1001754. doi: 10.1371/journal.pbio.1001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. Journal of cell science. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Review Mitochondria : The Next (Neurode) Generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017;27:403–416. doi: 10.1016/j.tcb.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG, Harris KM. Three-dimensional structure and composition of CA3->CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. The Journal of Neuroscience. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Greenamyre JT. Pathogenesis of Parkinson’s disease. Curr Opin Investig Drugs. 2001;2:657–662. [PubMed] [Google Scholar]
- Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin Julie Q, Amieux Paul S, Holt Christine E. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell. 2016;166:181–192. doi: 10.1016/j.cell.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A. 2016;113:E6097–E6106. doi: 10.1073/pnas.1612283113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E, Schwarz TL. Have you seen? For parkin, it’s not all or nothing. EMBO J. 2014;33:277–279. doi: 10.1002/embj.201387723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P, Islam MN, Bhattacharya S, Bhattacharya J. Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr Opin Genet Dev. 2016;38:97–101. doi: 10.1016/j.gde.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit-Rigter L, Rajendran R, Silva CAP, Spierenburg L, Groeneweg F, Ruimschotel EM, van Versendaal D, van der Togt C, Eysel UT, Heimel JA, et al. Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Current Biology. 2016;26:2609–2616. doi: 10.1016/j.cub.2016.07.033. [DOI] [PubMed] [Google Scholar]
- Sorbara CD, Wagner NE, Ladwig A, Nikic I, Merkler D, Kleele T, Marinkovic P, Naumann R, Godinho L, Bareyre FM, et al. Pervasive axonal transport deficits in multiple sclerosis models. Neuron. 2014;84:1183–1190. doi: 10.1016/j.neuron.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013;5:1564–1575. doi: 10.1016/j.celrep.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Tandarich LC, Nguyen K, Hollenbeck PJ. Compartmentalized Regulation of Parkin-Mediated Mitochondrial Quality Control in the Drosophila Nervous System In Vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:7375–7391. doi: 10.1523/JNEUROSCI.0633-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A. 2015;112:10515–10520. doi: 10.1073/pnas.1509879112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJ, Las G, Yaniv G, Hajnoczky G, Shirihai OS. Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am J Physiol Cell Physiol. 2010;299:C477–487. doi: 10.1152/ajpcell.00427.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A, Bullock SL. A simple method for imaging axonal transport in aging neurons using the adult Drosophila wing. Nat Protoc. 2016;11:1711–1723. doi: 10.1038/nprot.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial Membrane Potential in Axons Increases with Local Nerve Growth Factor or Semaphorin Signaling. Journal of Neuroscience. 2008;28:8306–8315. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proceedings of the National Academy of Sciences. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends in Endocrinology & Metabolism. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. Imaging axonal transport of mitochondria. Methods Enzymol. 2009a;457:319–333. doi: 10.1016/S0076-6879(09)05018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009b;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Williams DC, ElBejjani R, Ramirez P, Coakley S, Kim SA, Lee H, Wen Q, Samuel A, Lu H, Hilliard MA, Hammarlund M. Rapid and Permanent Neuronal Inactivation InVivo via Subcellular Generation of Reactive Oxygen with the Use of KillerRed. Cell Reports. 2013;5:553–563. doi: 10.1016/j.celrep.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends in Molecular Medicine. 2014;20:179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439–4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J. Complete morphologies of basal forebrain cholinergic neurons in the mouse. eLife. 2014;2014:1–17. doi: 10.7554/eLife.02444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A. 2017;114:E4859–E4867. doi: 10.1073/pnas.1701078114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Zhao L, Stofanko M, O’Sullivan NC, Kang ZH, Roost A, Thomas MR, Zaessinger S, Blard O, Patto AL, et al. Modeling of axonal endoplasmic reticulum network by spastic paraplegia proteins. Elife. 2017;6 doi: 10.7554/eLife.23882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Pascual C, Klionsky D. Autophagy: machinery and regulation. Microbial Cell. 2016;3:588–596. doi: 10.15698/mic2016.12.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Ho PL, Kintner DB, Sun D, Chiu SY. Activity-dependent regulation of mitochondrial motility by calcium and Na/K-ATPase at nodes of Ranvier of myelinated nerves. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:3555–3566. doi: 10.1523/JNEUROSCI.4551-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol. 2016;214:103–119. doi: 10.1083/jcb.201605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YB, Sheng ZH. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J Biol Chem. 2011;286:23432–23440. doi: 10.1074/jbc.M111.237818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]