

SUMMARY
Spinal cord injury (SCI) above cervical level 4 disrupts descending axons from the medulla that innervate phrenic motor neurons, causing permanent paralysis of the diaphragm. Using an ex vivo preparation in neonatal mice, we have identified an excitatory spinal network which can direct phrenic motor bursting in the absence of medullary input. After complete cervical SCI, blockade of fast inhibitory synaptic transmission caused spontaneous, bilaterally-coordinated phrenic bursting. Here, spinal cord glutamatergic neurons were both sufficient and necessary for induction of phrenic bursts. Direct stimulation of phrenic motor neurons was insufficient to evoke burst activity. Transection and pharmacological manipulations showed that this spinal network acts independently of medullary circuits which normally generate inspiration, suggesting a distinct non-respiratory function. We further show that this “latent” network can be harnessed to restore diaphragm function after high cervical spinal cord injury in adult mice and adult rats.
Keywords: Diaphragm, breathing, respiration, phrenic motor neuron, spinal cord injury, spasticity, rodent
eTOC Blurb
Cregg et al. uncover a spinal network that can direct diaphragm-innervating motoneurons to burst. This network is functionally independent of descending bulbospinal inspiratory circuits, pointing to a different physiologic function. Targeting this network restores diaphragm function after cervical SCI.
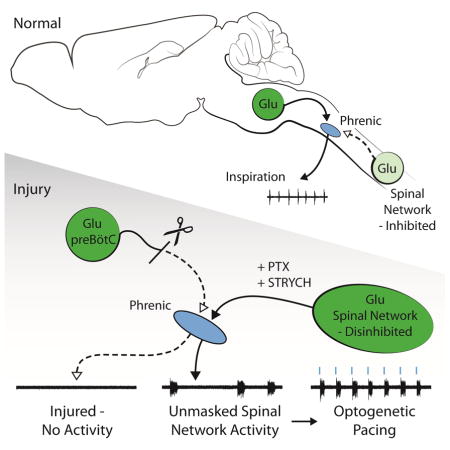
INTRODUCTION
Inspiration—the act of drawing air into the lungs—is executed primarily by the diaphragm, which contracts ~20,000 times per day and 108 times over the average human lifespan. The diaphragm is solely innervated by phrenic motor neurons (PMNs), which are anatomically positioned at spinal cord levels C3-5/6. Thus, injury to the cervical spinal cord severely compromises diaphragm function.
Excitatory circuits of the preBötzinger complex (preBötC), located in the ventrolateral medulla, generate inspiration and relay inspiratory drive to PMNs via a medullary premotor nucleus termed the rostral ventral respiratory group (rVRG; Smith et al., 1991). Neurons of the rVRG maintain bulbospinal projections that synapse directly with PMNs (Davies et al., 1985; Dobbins and Feldman, 1994; Duffin and van Alphen, 1995; Ellenberger and Feldman, 1988; Ellenberger et al., 1990) and, therefore, it is well established that rhythmic PMN bursts arise from circuits rostral to the spinomedullary junction. Evidence that rVRG axons make direct synaptic contact with PMNs comes from studies examining cross-correlation between rVRG units and phrenic nerve activity, which exhibit 1–2 ms latencies typical of monosynaptic connections (Davies et al., 1985; Duffin and van Alphen, 1995). Additional evidence comes from anatomical studies demonstrating close apposition of rVRG axons, identified by anterograde labeling, with the dendritic arbors of retrogradely labeled PMNs (Ellenberger and Feldman, 1988; Lane et al., 2008). Also, synapses between rVRG axon terminals and PMNs have been identified at the ultrastructural level (Ellenberger et al., 1990).
The simplest model, which is widely accepted, is that PMN bursts are always directly evoked by rVRG input. Nonetheless, there is also data indicating that in the absence of supraspinal (rVRG) input, PMN activity can be initiated centrally by various pharmacological manipulations (Coglianese et al., 1977; Ghali and Marchenko, 2016; Reinoso et al., 1996; Viala et al., 1979; Zimmer and Goshgarian, 2007), or by electrical stimulation (Huang et al., 2016; Kowalski et al., 2013). Although it has been suggested that this activity might be generated by a spinal analogue of the preBötC (Ghali and Marchenko, 2016), the origin of this activity has always been elusive. Indeed, it is largely unclear whether this PMN activity is caused by pharmacologic/electrical action on PMNs themselves, or whether these manipulations engage propriospinal neurons which are also known to synapse with PMNs (Dobbins and Feldman, 1994; Lane et al., 2008; Lipski et al., 1993; Lois et al., 2009). Importantly, there is no existing evidence that when isolated from the medulla, propriospinal neurons can generate spontaneous PMN burst activity.
If there is a population of propriospinal neurons that can generate PMN bursts, then it is important to determine if these interneurons simply relay inspiratory drive from the preBötC (e.g. preBötC → rVRG → interneuronal burst population → PMNs), or whether these neurons act as part of some other independent system—and what relevance this system might have. In this study we establish an ex vivo model of spinal cord injury, and combine this model with pharmacologic and optogenetic perturbations to investigate whether propriospinal circuits are capable of evoking PMN bursts. We identify a recurrent excitatory network that is both sufficient and necessary for PMN bursting in the absence of the medulla, and show that activity generated by this network is dissociable from bona fide inspiration. Furthermore, we demonstrate that this network can be used to promote diaphragm function after spinal cord injury in adult mice and rats.
RESULTS
Blockade of inhibitory synaptic transmission initiates persistent PMN bursting
We first established an ex vivo model of cervical spinal cord injury for tractable interrogation of phrenic premotor network organization. Mouse ex vivo preparations exhibit fictive inspiration (Figures 1A, 1B and S1A). Inspiratory frequency in ex vivo preparations is depressed relative to in vivo conditions due to lack of vagal feedback as well as the relatively cooler temperatures required for maintaining the preparation during recording (23–26°C). In these neonatal preparations (P2-4), we found that C1 lateral hemisection eliminates phrenic nerve inspiratory burst activity on the side ipsilateral to the lesion (Figure S1B, n = 8), and that complete transection at C1 arrests fictive inspiratory activity in both phrenic nerves (Figures 1B and S1C, n = 8). Since these data phenocopy well-characterized physiology of the adult mammal after spinal cord injury (Alilain et al., 2008), we proceeded to use this ex vivo preparation to understand the functional organization of phrenic premotor circuits.
Figure 1. After C1 transection, blockade of inhibitory synaptic transmission initiates persistent PMN bursting.

(A) Ex vivo preparation (P2-4) consists of medulla and cervical through sacral spinal cord. Suction recording electrodes were placed on the phrenic nerves as they enter the pleural cavity.
(B) Spinal cord medullary preparations exhibit fictive inspiration. Phrenic nerve inspiratory activity is abolished after a C1 transection injury. Blockade of fast inhibition (+PTX/STRYCH) initiates spontaneous PMN bursting in the absence of descending bulbospinal input.
(C) Spontaneous spinal cord derived PMN bursting initiated by blockade of fast inhibition exhibits a slower frequency as compared with fictive inspiration (n = 8 mice, **P = 0.0019, U = 62, Mann-Whitney U test).
(D) PMN bursting initiated by blockade of fast inhibition exhibits a significantly longer burst duration as compared with fictive inspiration (n = 8 mice, ***P = 0.00009, F1,7.1 = 62.1, Welch’s ANOVA).
(E) Burst duration for individual PMN bursts (n) initiated by blockade of fast inhibition does not predict timing or onset of subsequent bursts (n + 1, m = −0.06, r2 = 0.00).
(F) Poincaré analysis of variability in interburst interval (IBI). Fictive inspiration is highly rhythmic (contour plot falls on line of identity for IBIn versus IBIn+1). Spinal cord derived PMN bursting does not exhibit a stable rhythm (contours do not fall on line of identity).
See also, Figure S1 and S2.
We first examined whether spinal premotor circuits are capable of generating PMN bursts given tonic excitatory input. After a C1 transection injury, we found that bath application of NMDA/5HT elicited very modest PMN unit activity in 50% of cases (Figure S2A, n = 2 of 4). This unit activity did not exhibit any noticeable patterning between the left and right phrenic nerves, and PMNs did not exhibit any clear bursts. In contrast, application of NMDA/5HT evoked robust locomotor-like bursting from the ventral roots of the L2 segment, which exhibited right/left alternation (Figure S2B).
To further address whether an excitatory stimulus could evoke PMN bursts in the absence of medullary input, we used an optogenetic strategy to stimulate cervical Vglut2+ neurons. We found that after complete C1 transection, 30 s of continuous photostimulation of the ventral cervical spinal cord of Vglut2Cre;R26RChR2 preparations generated only modest PMN unit activity at light onset, with unit firing tapering off over the course of the photostimulation paradigm (Figure S2C). We examined PMN responses at multiple light intensities (10–100%), different light stimulus frequencies (20 Hz, 50 Hz), and different pulse durations (20 ms, 100 ms), and found that continuous light generated the strongest response consisting solely of PMN unit activity but no PMN bursts (Figure S2C). In contrast, photostimulation of the lumbar cord evoked fictive locomotion (Hägglund et al., 2010), with alternating burst activity between right and left L2 roots (Figure S2D). Together these results indicate that simply increasing excitation in the absence of medullary input does not evoke PMN burst activity.
These data suggest that either the contribution of interneurons to PMN output is diminutive, or that inhibition suppresses the action of excitatory interneurons under these conditions. To test these alternatives we examined PMN output after blockade of fast inhibitory synaptic transmission. Unexpectedly, bath application of picrotoxin (PTX) and strychnine (STRYCH) in the absence of the medulla initiated spontaneous bilaterally-coordinated PMN bursting (Figure 1B). The frequency of spontaneous PMN bursting caused by blocking inhibition was much slower than fictive inspiration (Figure 1C). Spinal cord derived PMN bursts also exhibited a significantly longer burst duration compared with fictive inspiratory bursts (Figure 1D). The duration of each individual spontaneous PMN burst did not predict the onset of subsequent bursts (Figure 1E). Finally, whereas fictive inspiration was highly rhythmic (Figure 1F, top), spontaneous PMN bursting observed in the absence of inhibition was not rhythmic (Figure 1F, bottom).
Excitatory interneurons are sufficient and necessary for spinal cord derived PMN bursting
Spinal cord derived PMN bursting initiated by blockade of inhibitory synaptic transmission may be caused via two distinct mechanisms: First, bursts could arise from PMNs themselves via synchronization of subthreshold activity in electrically coupled dendritic arbors (Tresch and Kiehn, 2000). Previous electrophysiological analyses, however, have suggested that PMNs do not exhibit electrical coupling via gap junctions (Lipski, 1984). Second, PMN bursts could arise from recurrent excitatory premotor networks. To distinguish between these possibilities we examined PMN burst generation after washing out Ca2+, which is required for synaptic transmission. Ca2+ washout completely eliminated spinal cord derived PMN bursts (Figure S3A). Moreover, we found that blockade of NMDA and non-NMDA glutamate receptors also abolished spontaneous PMN bursting (Figure S3B). These data indicate that excitatory presynaptic input is required for spontaneous PMN bursting after blockade of inhibitory synaptic transmission.
We next tested whether PMN bursts could be evoked by stimulation of cervical glutamatergic interneurons. In PTX/STRYCH disinhibited preparations from Vglut2Cre;R26RChR2 mice, a series of 200 ms photostimuli centered on the ventral cervical spinal cord was sufficient to evoke bilaterally coordinated PMN bursting (Figures 2A–2D). PMN bursting could be driven at a frequency similar to that of fictive inspiration (Figures 2D–F, ~5 bursts min−1). The duration of individual burst episodes was significantly shorter during trains of photostimulation compared with those observed spontaneously (Figure 2G). Light-evoked PMN bursts do not simply reflect direct activation of PMNs by Vglut2+ sensory or bulbospinal fibers because stimulation of Vglut2+ fibers in the absence of PTX/STRYCH did not evoke PMN bursts (Figure S2C). Additionally, the brief 200 ms photostimulus caused activity which lasted on the order of seconds (Figures 2C, 2D, 2G, and 2H). This suggests that the stimulus is recruiting recurrent excitatory premotor networks.
Figure 2. Excitatory neurons are sufficient and necessary for PMN burst activity in the absence of bulbospinal input.

(A) Experimental ex vivo paradigm for photostimulation of cervical Vglut2+ neurons.
(B) Distribution of neurons exhibiting Vglut2Cre induced expression of EYFP from a R26REYFP reporter allele in cervical spinal cord level C5. Vglut2+ neurons are situated in spinal cord laminae I–X. Motor neurons identified by their position in lamina IX also exhibit Cre-induced expression of EYFP (chevron); however a region corresponding to the location of the phrenic motor nucleus lacked any Vglut2+ soma (arrow). Scale bars: top = 100 μm, bottom = 30 μm.
(C) After complete C1 transection, spinal cord preparations exhibit no PMN activity. PTX/STRYCH application induces bilaterally coordinated bursting of PMNs, and single bursts can be evoked by ChR2-mediated stimulation of Vglut2+ glutamatergic neurons with 200 ms pulses of blue light. Light was pulsed at a frequency of 5 min−1.
(D) Inset from (C).
(E) Burst probability plot and raster plot for PTX/STRYCH induced bursts. Individual trials are aligned by light onset and grey boxes are used to highlight independent biological replicates (3 trials per animal, n = 8 mice).
(F) Burst frequency is increased during photostimulation (n = 8 mice, ***P = 8.5 × 10−11 before vs. during photostimulation, ***P = 6.2 × 10−11 during vs. after photostimulation, *P = 0.013 before vs. after photostimulation, F2,21 = 156.5, one-way ANOVA and post hoc Tukey-Kramer HSD).
(G) Burst duration is significantly reduced during photostimulation (n = 8 mice, ***P = 0.0007 before vs. during photostimulation, ***P = 0.0002 during vs. after photostimulation, F2,21 = 14.6, one-way ANOVA and post hoc Tukey-Kramer HSD).
(H) PMN bursting evoked by stimulation of Vglut2+ neurons requires glutamate transmission (n = 2 mice).
See also, Figure S3 and S4.
Blockade of NMDA and non-NMDA glutamate receptors abolished light-evoked PMN burst activity (Figure 2H), indicating a presynaptic origin of light-evoked bursts. Although it is well known that lumbar motor neurons release glutamate and express Vglut2 (Mentis et al., 2005; Nishimaru et al., 2005; Talpalar et al., 2011), we observed only sparse Vglut2Cre mediated recombination of an EYFP-reporter allele in putative PMNs (Figure 2B, arrow). To directly examine Vglut2Cre-mediated recombination in PMNs, we retrogradely traced PMNs by intrapleural injection of CTB-555 (Figures S4A–S4C). PMNs labeled by CTB-555 were positioned between the medial and lateral motor columns in spinal cord levels C3-5 (Figures 3A and 3B). 27.2% of CTB-labeled PMNs expressed LacZ in Vglut2Cre;Taulsl-LacZ mice (50/184 neurons, n = 4, Figures S4A–C).
Figure 3. Direct photostimulation of PMNs is not sufficient to evoke PMN burst activity.

(A) Anatomical coordinates of retrogradely traced PMNs, which occupy a discrete position in the ventral horn of cervical spinal cord levels C3-5 at postnatal day 10 (321 neurons in 4 animals).
(B) ChATCre;R26RChR2 mice exhibit expression of ChR2-EYFP in ChAT+ motor neurons (ventral horn) and cholinergic interneurons (arrows). CTB-555 labeled PMNs express both ChAT and ChR2-EYFP (see also, Figure S4). Scale bar = 100 μm.
(C) After removal of the medulla by transection at C1 and blockade of fast inhibitory synaptic transmission, 200 ms photostimulation of ChAT+ PMNs resulted in PMN unit activity for the duration of the stimulus. In contrast, 200 ms photostimulation of Vglut2+ neurons resulted in PMN burst activity lasting several seconds.
(D) 200 ms photostimulation of Vglut2+ neurons resulted in a significantly longer PMN response when compared with direct photostimulation of ChAT+ PMNs (4 mice per genotype, n = 22 responses for ChATCre;R26RChR2 and n = 223 responses for Vglut2Cre;R26RChR2, ***P = 1.0 × 10−14, U = 4906, Mann-Whitney U test).
(E) Direct photostimulation of ChAT+ PMNs resulted in unit activity for the duration of the stimulus: 200 ms, 500 ms, 1000 ms, 2000 ms, 4000 ms. Ca2+ washout—which disrupts synaptic vesicle release from presynaptic terminals—demonstrates that unit response is caused by direct photostimulation of PMNs rather than cholinergic interneurons.
(F) PMN response was directly proportional to photostimulus duration in ChATCre;R26RChR2 spinal cord preparations bathed in PTX/STRYCH (top, n = 3 mice) or in Ca2+-free aCSF (bottom, n = 3 mice), approaching a relationship of identity for both conditions (m = 1.006).
See also Figure S4.
We next performed in situ analysis to assess whether PMNs express Vglut2 mRNA (Figures S4D–S4F). While some motor neurons of the lateral column expressed Vglut2, most PMNs lacked expression (n = 4, 4/104 neurons, Figures S4D–S4F). This data indicates that Vglut2Cre-mediated recombination in a subset of PMNs is caused by transient rather than sustained Vglut2 expression.
After Ca2+ washout, stimulation of ChR2-expressing PMNs in Vglut2Cre;R26RChR2 mice evoked only minor PMN unit activity but no PMN bursts (Figures S4I and S4J). These results indicate that light-evoked bursts arise from excitatory premotor networks rather than from direct stimulation of PMNs.
Direct stimulation of PMNs is not sufficient to evoke PMN bursts
To further test whether stimulation of PMNs would be sufficient to evoke PMN bursts, we used a ChATCre allele to allow motor neuron-specific expression of ChR2 (Figure 3B). 99.1% of PMNs expressed ChR2-EYFP in ChATCre;R26RChR2 mice (216/218 neurons, n = 7, Figures S4G and S4H). We also observed ChR2-EYFP expression in cholinergic interneurons (Figure 3B, arrows), which represent a small fraction of cervical spinal interneurons (Barber et al., 1984).
After C1 transection and blockade of inhibitory synaptic transmission, 200 ms ChATCre;R26RChR2 photostimulation resulted in PMN unit activity for 193 ± 6 ms, but did not evoke bursts which outlasted the stimulus duration (Figure 3C). In comparison, 200 ms photostimulation of Vglut2+ interneurons resulted in a burst of PMN activity that lasted for 3635 ± 556 ms (Figures 3C and 3D). We examined whether varying the stimulus duration in ChATCre;R26RChR2 preparations would lead to burst responses lasting longer than the light stimulus. Photostimuli of 200–4000 ms evoked PMN unit activity that lasted only for the duration of the photostimulus (Figures 3E and 3F). Since light-evoked PMN unit activity persisted after Ca2+ washout, it is caused by direct stimulation of PMNs rather than being mediated by ChR2+ cholinergic interneurons (Figures 3E and 3F). Lastly, after Ca2+ washout, photostimulation of PMNs in ChATCre;R26RChR2 mice caused a much more robust unit response than photostimulation of PMNs in Vglut2Cre;R26RChR2 mice (Figures S4I and J). This is consistent with our anatomical data demonstrating that only a subset of PMNs exhibit Vglut2Cre-mediated recombination (Figure S4C). Together these data indicate that direct stimulation of PMNs is not sufficient to evoke sustained PMN bursts. We conclude that PMN bursts arise from recurrent excitatory premotor networks.
Dissociation of spinal cord derived PMN bursting from bona fide inspiration
Our results provide evidence for an excitatory spinal cord network that can cause PMN bursting. To address whether this network relays excitatory inspiratory drive from higher-order medullary nuclei (e.g., preBötC → rVRG → interneuronal burst population → PMNs), we examined PMN activity after blockade of inhibition in spinomedullary preparations. Interestingly, we observed two distinct modes of PMN bursting under these conditions: high frequency/short duration and low frequency/long duration (Figures 4A and 4B, +PTX/STRYCH). We reasoned that these distinct modes of PMN bursting may reflect the action of two distinct premotor networks: PMN bursts of shorter duration are likely driven by the preBötC because they exhibit a frequency and duration similar to that of fictive inspiration (Figure 4B), and the longer PMN bursts are likely driven by an excitatory spinal cord network that emerges following blockade of inhibitory transmission.
Figure 4. Spinal cord derived PMN bursting is independent of bona fide inspiration.

(A) In spinomedullary preparations, blockade of fast inhibitory synaptic transmission evoked two distinct modes of PMN bursting—high frequency/short duration as well as low frequency/long duration (red traces). C1 hemisection (right side, rPN) eliminated high frequency/short duration bursts, whereas low frequency/long duration bursts were retained on both the lesioned and intact sides (blue, n = 8). Application of the μ-opioid receptor agonist DAMGO selectively depressed the amplitude and duration of medullary—but not spinal cord—derived PMN bursts (orange). ◇ indicates absence of medullary derived PMN burst on the side ipsilateral to C1 hemisection.
(B) Dual modes of PMN bursting visualized in burst duration histograms (n = 8). Fictive inspiratory bursts (black) are concentrated in the range of 0–2.5 s. PTX/STRYCH application (red) causes a shift in the distribution: While a majority of bursts are 0–2.5 s in duration, a second population of longer duration bursts emerges with significantly longer duration. C1 hemisection distinguishes medullary derived PMN bursts of high frequency/short duration (open bars) from spinal cord derived PMN bursts of low frequency/long duration (shaded bars), as shorter bursts are lost on the hemisected side.
(C) Application of the μ-opioid receptor agonist DAMGO caused medullary derived bursts to become less frequent than spinal cord derived bursts (n = 7 mice, *P = 0.038, t12 = 2.3, two-tailed t-test). Opioid agonist selectively depressed the amplitude of medullary (med.)—but not spinal cord (spi.)—derived PMN bursts (n = 7 mice, *P = 0.0202 and U = 41 for med., *P = 0.0204 and U = 41 for spi., Mann-Whitney U test). Opioid agonist selectively shortened medullary—but not spinal cord—derived PMN bursts (n = 7 mice, ***P = 3.6 × 10−5 for med. and t12 = 6.4, P = 0.0783 and t12 = 1.9 for spi., two-tailed t-test).
See also, Figure S5.
To test this, we performed right C1 hemisection lesions to eliminate descending medullary input to PMNs ipsilateral to the lesion. We found that C1 hemisection eliminated short duration bursts only on the side ipsilateral to the lesion (Figure 4A, diamonds). Notably, synchronous long duration PMN bursts were retained on both sides (Figures 4A and 4B). We conclude therefore that the high frequency/short duration bursts are of medullary origin, and long duration bursts are of spinal cord origin. Importantly, these experiments demonstrate that in disinhibited preparations, excitatory medullary and spinal cord premotor networks do not interact; one type of bursting does not drive the other.
We next examined whether medullary and spinal cord premotor networks exhibit differential pharmacological sensitivity. Opioids are known to depress inspiration via direct action on preBötC circuits (Gray et al., 1999). In spinomedullary preparations, bursts of medullary origin exhibited slower frequency, smaller amplitude, and shorter duration in response to bath application of the μ-opioid agonist DAMGO (Figures 4A and 4C), whereas bursts of spinal cord origin were remarkably insensitive to DAMGO (Figures 4A and 4C). In contrast, spinal cord—but not medullary—derived PMN bursts were largely eliminated by application of riluzole, a non-selective drug that blocks persistent sodium current (INaP, Figures S5A–D). Consistent with previous data, riluzole application had no effect on medullary derived inspiratory burst frequency (Figures S5A and S5B; Thoby-Brisson et al., 2009), but shortened both the amplitude and duration of inspiratory bursts (Figures S6C and S6D; Peña et al., 2004). Together these pharmacological manipulations demonstrate that spinal cord derived PMN bursting is dissociable from bona fide inspiration.
Spontaneous spinal cord derived PMN bursts originate in the thoraco-lumbar cord
To further examine the anatomical origin of spontaneous burst initiation after blockade of inhibitory synaptic transmission, we simultaneously recorded from the phrenic nerve and the L2 ventral root (Figure 5A). Previous work in the lumbar spinal cord has demonstrated that blockade of inhibitory synaptic transmission initiates synchronous bursting across multiple ventral roots (Bracci et al., 1996). This synchronous burst activity has been attributed to excitatory premotor circuits associated with locomotion (Bracci et al., 1996; Hägglund et al., 2010; Talpalar et al., 2011). Upon application of PTX/STRYCH in C1 transected preparations, we found that PMNs exhibited long duration bursting synchronous with lumbar motor neurons in L2 (Figures 5A and 5B). Furthermore, PTX/STRYCH application caused similar bursting in the radial and musculocutaneous nerves (not shown), suggesting that long duration bursts generated by excitatory interneurons recruit motor neurons at every spinal level. Importantly, PMN bursting in the disinhibited cord was insensitive to application of the muscle AChR antagonist d-Tubocurarine (1.44 ± 0.35 min−1 before vs. 1.29 ± 0.35 min−1 after, n = 2), demonstrating that spontaneous PMN bursts are recruited directly by an excitatory premotor network rather than indirectly via intercostal muscle sensory afferents (Decima et al., 1969).
Figure 5. Spinal cord derived PMN bursts originate in the thoraco-lumbar cord.

(A) In spinal cord preparations, blockade of fast inhibitory synaptic transmission initiated PMN bursting that was tightly coupled with L2 bursting. T8 transection disrupted PN/L2 coupling, and C8 transection eliminated PMN bursting initiated by blockade of inhibitory synaptic transmission.
(B) Phase diagrams demonstrate tight PN/L2 coupling after blockade of fast inhibitory synaptic transmission. Coupling was disrupted by T8 spinal cord transection.
(C) PN burst frequency vs. L2 burst frequency. Sequential rostral spinal cord transections demonstrate that spontaneous PMN bursts originate in the thoraco-lumbar cord. PN burst frequency is significantly reduced relative to L2 after T8 transection (n = 8 mice; P = 0.0704 and t14 = 0.4 for intact cord, two-tailed t-test; ***P = 0.00093 and U = 64 for T8-Tx, Mann-Whitney U test), and virtually eliminated after C8 transection (n = 8 mice, ***P = 0.00055 and U = 64, Mann-Whitney U test).
(D) Although spontaneous PMN bursting is eliminated after C8 transection, small PMN bursts can still be evoked by ChR2 stimulation of Vglut2+ neurons in the cervical spinal cord.
(E) Bursts evoked by photostimulation of Vglut2+ neurons after C8 transection exhibited a higher failure rate (n = 4 mice, P = 0.067, U = 12, Mann-Whitney U test; n = 4 mice exhibited no failures when the spinal cord was intact, and n = 2 of 4 mice failed at a rate of 100% after C8 transection). Vglut2Cre;R26RChR2-evoked response duration was much shorter after C8 transection compared with the intact spinal cord (n = 4 mice, **P = 0.0092, F1,5.8 = 14.7, Welch’s ANOVA).
(F) Summary of findings. In the intact disinhibited spinal cord (1), low frequency/long duration bursts propagate within the rostrocaudal extent of the cord. After T8 transection (2), long duration bursts propagate independently within the C1-T8 and T9-sacral segments. Long duration bursts are generated at a higher frequency within the T9-sacral segment. After C8 transection, spontaneous long duration bursts are largely eliminated from the cervical cord but can still be evoked by photostimulation of Vglut2+ neurons. These findings indicate that spinal cord glutamatergic networks exhibit a gradient of excitability which is highest in the lumbar cord and lowest in the cervical cord. The rostrocaudal extent of burst propagation is indicated by green arrows, and the thickness of the green arrows represents excitability.
To better define the location of networks generating spontaneous bursts in the disinhibited spinal cord, we transected the spinal cord at T8. Interestingly, regions of spinal cord rostral and caudal to the transection both continued to generate bursts, but these bursts were no longer synchronous, and the frequency of the bursts rostral to T8 was reduced (Figures 5A–5C). In a purely excitatory network, such as the early embryonic spinal cord or SA-AV node coupling in the heart, burst initiation is determined by the most excitable population of cells. Our data suggests that the most excitable population of glutamatergic neurons in the disinhibited spinal cord resides caudal to T8, and that this population drives network-wide bursts. However, in their absence, other excitable populations of glutamatergic neurons rostral to T8 can take over and initiate network-wide bursts, albeit at a lower frequency (Figure 5C). We subsequently performed sequential rostral spinal cord transections and found that transection at C8 abolished spontaneous spinal cord derived PMN bursting (Figures 5A and 5C). Although C8 transection abolished spontaneous PMN bursting (Figures 5A and 5C), small PMN bursts could still be evoked by stimulation of cervical glutamatergic neurons in a subset of cases (n = 2 of 4 mice, Figures 5D and 5E). These data indicate that recurrent excitatory networks in the disinhibited spinal cord exhibit a gradient of excitability which is highest in the lumbar cord and lowest in the cervical cord (Figure 5F).
Spinal cord premotor networks can restore diaphragm function after hemiparalysis
A few previous studies have demonstrated that blockade of inhibitory synaptic transmission can initiate PMN activity in adult rodents (Ghali and Marchenko, 2016; Zimmer and Goshgarian, 2007). Although the nature of this activity was largely unclear, it was suggested that this activity might represent a spinal cord origin for respiratory rhythm (Ghali and Marchenko, 2016). Given that hemisection lesions can dissociate spinal cord burst activity from bona fide inspiration (Figures 4A and 4B; also shown in Figure 6A for comparison with the adult), we examined whether similar lesions would dissociate PTX/STRYCH initiated PMN activity from respiration in adult mice. In anesthetized mice >8 weeks old, C2 hemisection abolished diaphragm activity ipsilateral to the lesion (Figure 6B). Subsequent microinjection of PTX/STRYCH into the cervical spinal cord rapidly initiated activity in the paralyzed hemidiaphragm (Figure 6B, green arrows). This activity was largely independent of rhythmic inspiratory circuits of the preBötC because medullary driven inspiratory bursts did not evoke weak unit or burst activity ipsilateral to the lesion (Figures 6B and 6C; indicated by diamonds). However, given the fast dynamics of mouse respiration (~90 min−1), we found that long spinal cord derived bursts almost always overlapped with medullary derived respiratory bursts to some degree (Figures 6B and 6C).
Figure 6. Spinal cord derived PMN bursting can be evoked after adult spinal cord injury.

(A) In the ex vivo preparation from neonatal mice, left C1 hemisection eliminates fictive inspiration on the side ipsilateral to lesion, and application of PTX/STRYCH initiates spinal cord derived burst activity from the dennervated phrenic motor pool (green arrows distinguish spinal cord derived bursts from bursts of medullary origin). PTX/STRYCH evoked bursting is thus dissociable from fictive inspiration (see also, Figure 4A).
(B) In the adult mouse, left C2 hemisection eliminates left diaphragm activity (EKG artifact is still visible). Cervical microinjection (C3–5, 3 sites, see Experimental Procedures) of PTX/STRYCH restores some PMN bursting in the paralyzed hemidiaphragm (n = 3). Green arrows indicate spinal cord derived PMN bursts.
(C) Inset from (B), EKG artifact highlighted in gray. Black portion of trace represents EMG. Diamonds indicate medullary-derived respiratory bursts which fail to initiate PMN bursting on the side ipsilateral to the lesion.
(D) In the adult rat, left C2 hemisection eliminates left diaphragm activity (EKG artifact is still visible. Intrathecal injection of PTX/STRYCH at the cervical level restored some PMN bursting in the paralyzed hemidiaphragm in 3 of 4 rats.
(E) Inset from (D), EKG artifact highlighted in gray. Diamonds indicate medullary-derived respiratory bursts which fail to initiate PMN bursting on the side ipsilateral to the lesion. In several instances, long spinal cord derived bursts were clearly spaced in between medullary derived inspiratory bursts.
To demonstrate that long duration spinal cord derived bursts can occur independently of medullary derived bursts, we performed similar experiments in adult rats, which exhibited a slower respiratory rate than mice (~30 min−1; Figure 6D). C2 hemisection lesions abolished diaphragm activity ipsilateral to the lesion (Figure 6D), and intrathecal application of PTX/STRYCH initiated spontaneous activity in the paralyzed hemidiaphragm (Figure 6D). Again, activity observed in the paralyzed hemidiaphragm (green arrows) was independent of the preBötC because medullary driven inspiratory bursts did not evoke weak unit or burst activity ipsilateral to the lesion (Figures 6D and 6E; indicated by diamonds). Likely owing to the slower dynamics of rat respiration, we observed several long spinal cord derived bursts which were clearly spaced in between medullary derived inspiratory bursts (Figures 6D and 6E). In both adult mice and rats, we observed ipsilateral diaphragm activity after administration of PTX/STRYCH at the cervical level, suggesting that recurrent excitatory networks rostral to C8 (see evoked activity in Figures 5D and 5E) may give rise to spontaneous burst activity in vivo due to enhanced neural dynamics (37°C vs. 23–26°C ex vivo). It is also likely that PTX/STRYCH diffused far beyond the initial site of application because we also noted spastic-like movements of the hindlimbs. These data are largely consistent with our ex vivo data (Figure 6A), indicating that the spinal network we characterize in neonatal preparations has a correlate in adult animals. Together these data demonstrate that spontaneous spinal cord derived PMN bursting is generated independently of respiratory rhythm in adult animals (Figure 7), and may be harnessed to promote diaphragm activity in the absence of descending bulbospinal input in the context of SCI.
Figure 7. Summary of findings.
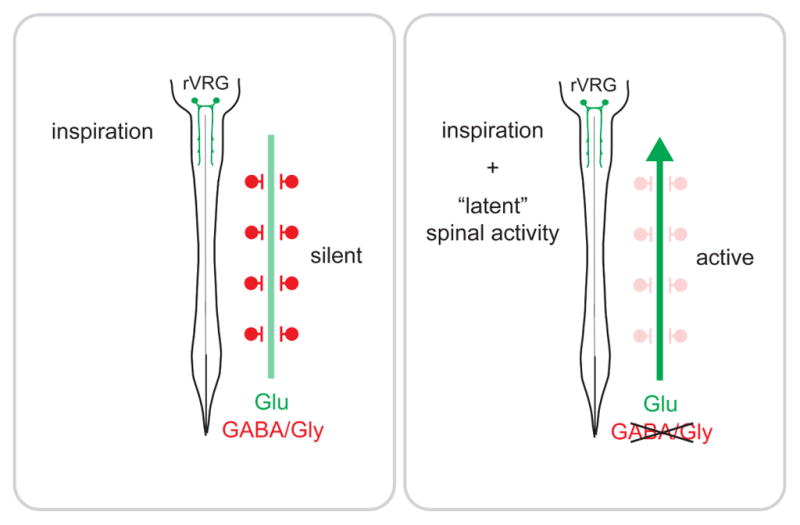
Two independent premotor networks control PMN activity postnatally: a descending, bulbospinal inspiratory network under control of the preBötC, and a second “latent” network which exhibits spontaneous activity after suppression of GABA/glycinergic synaptic transmission.
DISCUSSION
Our data puts to rest a lingering controversy—which has endured in the literature for more than a century—as to whether a “spinal respiratory rhythm generator” can initiate rhythmic inspiratory activity independent of supraspinal input (Brown-Séquard, 1860; Porter, 1895). Our data provides evidence against this hypothesis by demonstrating that while PMN bursts of spinal cord origin can occur, they 1) are not spontaneous and can only be evoked under certain pharmacological conditions, 2) are not rhythmic (Figure 1F), 3) are anatomically and pharmacologically distinguishable from bona fide inspiratory circuits under the control of the preBötC, and 4) are likely derived from more caudal regions of the spinal cord associated with circuits of the locomotor CPG. The idea that a lack of inhibition could cause spontaneous PMN bursting is perhaps not surprising given that this occurs in premotor circuits of the lumbar cord, for which propriospinal circuits act as the basis for locomotion and effects of disinhibition are well described (Bracci et al., 1996). Nevertheless, these data are surprising in the context of a vast literature on PMNs, which are thought to derive their dominant excitatory input from supraspinal neurons of the rVRG. Our data thus lead to several exciting new hypotheses concerning physiological role(s) for episodic spinal cord derived PMN bursts.
One possibility is that a caudally-derived excitatory burst module is specifically associated with PMN control under certain circumstances. This could be related to premotor circuits anatomically positioned caudal to C8, such as those associated with control of sympathetic preganglionic neurons or intercostal motor neurons. Alternatively, an excitatory circuit that coordinates bilaterally symmetric motor neuron activity throughout the rostrocaudal extent of the spinal cord—but is dissociable from rhythmic respiratory circuits of the brainstem—might represent a substrate for innate behavioral responses such as startle. In point, hyperekplexia is an exaggerated startle disorder in humans associated with dysfunction of glycinergic transmission (Rees et al., 2006). In contrast to these hypothetical roles, there is strong evidence that such spinal cord derived PMN burst activity does occur in situations of increased network excitability. Two well-described cases of increased network excitability exist physiologically: First, during embryogenesis, spinal circuits are highly excitable due to depolarizing action of GABA/glycine (Jean-Xavier et al., 2007). Second, spinal cord injury results in a variety of membrane and/or synaptic changes as well as anatomical remodeling that causes increased network excitability (Bellardita et al., 2017; Buttry and Goshgarian, 2014).
Low frequency, long duration bursts propagate within the spinal cord during embryogenesis as early as E12 (Hanson and Landmesser, 2003; Momose-Sato et al., 2012a, 2012b; Myers et al., 2005). Interestingly, PMNs exhibit activity-dependent intramuscular nerve branching and innervation of the diaphragm during this early stage of embryogenesis (E12.5–13.5; Brandon et al., 2003). Here, evoked ACh release from PMNs (rather than spontaneous vesicle fusion) regulates diaphragm innervation (Usiak and Landmesser, 1999; Washbourne et al., 2002). This evoked release is likely caused by spinal waves of propagating burst activity since activation of PMNs by the preBötC occurs much later at E15–15.5 (Thoby-Brisson et al., 2009). Although waves of propagating burst activity during embryogenesis exhibit a number of similarities to those we observe in the disinhibited spinal cord, a notable difference is that burst activity during embryogenesis is generated chiefly by excitatory cholinergic rather than glutamatergic synaptic transmission (Hanson and Landmesser, 2003; Momose-Sato et al., 2012a, 2012b). The mechanisms underlying transition from acetylcholine to glutamate as the primary excitatory neurotransmitter in the spinal cord are not understood; however, evidence that glutamatergic networks compensate for loss of acetylcholine transmission in ChAT−/− embryos, and that proper assembly of locomotor networks is disrupted in ChAT−/− embryos, suggests that downregulation of cholinergic signaling may help to instruct the development of future glutamatergic premotor networks (Myers et al., 2005). These data lend support to the hypothesis that the diffuse excitatory spinal circuit which can generate PMN activity postnatally might represent a “latent” or “holdover” network from development.
Increased excitation after spinal cord injury can lead to adverse outcomes, including muscle spasticity and/or autonomic dysreflexia (Bellardita et al., 2017; Ueno et al., 2016). An emergent view is that spasticity is caused by recruitment of disparate populations of excitatory neurons into functional circuits—leading to enhanced excitation independent of motor neuron excitability or inhibition (Bellardita et al., 2017). Interestingly, cervical spinal cord injury in human C4 tetraplegic patients is sometimes accompanied by diaphragm spasticity (Silver and Lehr, 1981). Moreover, we have observed occasional diaphragm spasticity after spinal cord injury in the adult rat in the context of experimental manipulations aimed at restoring function to the chronically denervated phrenic motor pool (P.M. Warren, J. Silver, unpublished data). It has been difficult to understand why diaphragm spasticity occurs since premotor networks controlling phrenic bursting have been thought to reside in the medulla. These medullary networks should be relatively insensitive to lesion of the cervical spinal cord, suggesting a different origin of spastic activity. Recent transynaptic tracing experiments initiated in PMNs after C2 hemisection injury have demonstrated extensive anatomical plasticity in PMN premotor networks (Buttry and Goshgarian, 2014). The origin of diaphragm spasticity after adult spinal cord injury could thus be due to heightened engagement of a pre-existing spinal network which directs PMN bursting independently of descending bulbospinal input.
Excitingly, the same mechanisms that cause network dysfunction and/or spasticity might also be harnessed toward restoration of normal circuit function. In recent years, restoring diaphragm function after SCI has become a model system for understanding functional regeneration beyond the glial scar (Alilain et al., 2011; Cregg et al., 2014), because normal function of PMNs is controlled by direct monosynaptic connection with neurons of the rVRG (Davies et al., 1985; Duffin and van Alphen, 1995; Ellenberger and Feldman, 1988; Ellenberger et al., 1990). This model presents a somewhat simpler system (vs. locomotion or micturition) for understanding how regenerating axons engage circuits caudal to a spinal cord lesion. In two examples from our previous work, we observed recovery of diaphragm function after hemiparalysis which seemed to involve the contribution of interneuronal networks (Alilain et al., 2008, 2011). These results were perplexing at the time because we did not understand that a spinal cord network could evoke PMN bursting independently of bona fide inspiration as our present study has shown (Alilain et al., 2008, 2011). Thus, those attempting to enhance regeneration of axons and restoration of a “simple” motor behavior may need to consider the dynamic interplay between intact networks and networks that undergo dramatic reorganization caudal to a spinal lesion.
Finally, future experiments aiming to identify spinal PMN excitatory burst populations in more anatomical detail could lead to a diaphragm “pacing” device (Huang et al., 2016; Kowalski et al., 2013). We demonstrate pacing of spinal cord derived PMN bursting at a rate similar to fictive inspiration, and given considerable technical advancements, this proof-of-concept could be therapeutically relevant in vivo after cervical spinal cord injury. Toward identification of PMN excitatory burst populations, our finding that spontaneous PMN burst activity is eliminated upon transection at C8 may be useful. In point, Kowalski and colleagues recently demonstrated that PMN unit activity—as opposed to burst activity demonstrated here—could be evoked by epidural stimulation at spinal cord level T2 (Kowalski et al., 2013). Altogether, our data provides new insight into PMN premotor network organization and establishes a conceptual framework for targeting treatment and recovery of diaphragm function after cervical SCI.
EXPERIMENTAL PROCEDURES
For a full description of all methods, see Supplemental Experimental Procedures.
Animals
All animal procedures were performed in accordance with Case Western Reserve University or University of Kentucky College of Medicine Institutional Animal Care and Use Committee (IACUC) guidelines. Mice were obtained from Jackson Laboratories. Vglut2Cre or ChATCre mice were crossed to R26RChR2-EYFP, R26REYFP, or Taulsl-LacZ mice. Ex vivo experiments were carried out in P2-4 male/female mice. Experiments in adult mice were performed in >8 week old male/female mice. Sprague-Dawley rats were obtained from Charles River Laboratories, Inc. Rat experiments were performed in adult females >8 weeks old.
Ex vivo Electrophysiology
After cryoanesthesia, rapid dissection was carried out in oxygenated Ringers solution. The hindbrain and spinal cord were exposed by ventral laminectomy, and the phrenic nerves were dissected free. Suction electrodes were attached to phrenic nerves and/or ventral roots from L2. A Polychrome V monochromator (Till Photonics) was used for photostimulation. The light intensity used in this study was ~0.20 mW mm−2. We used the following drugs: NMDA (7 μM), 5HT (8 μM), picrotoxin (10 μM), strychnine (0.3 μM), CNQX (20 μM), AP5 (20 μM), DAMGO (2 μM), riluzole (10 μM), and d-Tubocurarine (10 μM).
Histology
Retrograde tracing of PMNs was performed as described previously (Mantilla et al., 2009). Spinal cord tissue from was fixed in 4% paraformaldehyde for 30 min, and sectioned in the transverse plane at 20 μm. We used primary antibodies against GFP or ChAT. For X-gal staining, sections were incubated overnight at 37°C in X-gal staining buffer. In situ hybridization was performed as described (Philippidou et al., 2012). A Vglut2 probe generated from a cDNA library with the following primers: 5′-tggagaagaagcaggacaac and 5′-TAATACGACTCACTATAGGGgccagaacatgtaccagacc.
C2 Hemisection and Diaphragm EMG
After anesthesia, bipolar recording electrodes were inserted into the left and right hemidiaphragm to record baseline diaphragm activity. C2 hemisection was performed just caudal to the C2 dorsal root. In mice, 250 nl of PTX (50 mM)/STRYCH (30 mM) was injected at 3 sites in levels C3-5 on the side ipsilateral to the lesion at a depth of 1 mm. In rats, we injected 10 ul PTX/STRYCH intrathecally at the level of the C2 hemisection.
Analysis and Statistics
Representative raw traces are presented. Phase diagrams were constructed as previously described (Kjaerulff and Kiehn, 1996). Details on statistical tests used for each experiment can be found can be found in the accompanying figure legend. n represents the number of biological replicates (animals) for each group, unless stated otherwise.
Supplementary Material
Highlights.
Blockade of inhibition uncovers a spinal cord network that elicits phrenic bursting
Vglut2 interneurons were sufficient and necessary for induction of phrenic bursts
Spinal cord derived phrenic bursting is dissociable from bona fide respiration
This propriospinal network can be harnessed to allow diaphragm function after SCI
Acknowledgments
This work was supported by NSF DGE-0951783 (J.C.), University of Kentucky College of Medicine (W.J.A.), NIH NS101105 (W.J.A.), Case Western Reserve University (P.P.), Mt. Sinai Foundation (P.P.), NIH NS085037 (P.P.), NIH NS074199 (L.T.L.), and NIH NS025713 (J.S.). We thank JingQiang You for technical assistance, and Thomas (TED) Dick, Philippa Warren, and Christopher Wilson for helpful commentary and discussion.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, J.C., L.T.L., and J.S.; Methodology, J.C., W.J.A., and P.P.; Investigation, J.C., K.A.C., L.E.H., R.S.M., D.R.S., and M.E.; Writing – Original Draft, J.C.; Writing – Review & Editing, J.C., L.T.L., and J.S.; Supervision – W.J.A., P.P., L.T.L., and J.S. Funding Acquisition, J.C., W.J.A., P.P., L.T.L., and J.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alilain WJ, Li X, Horn KP, Dhingra R, Dick TE, Herlitze S, Silver J. Light-induced rescue of breathing after spinal cord injury. J Neurosci. 2008;28:11862–11870. doi: 10.1523/JNEUROSCI.3378-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber RP, Phelps PE, Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: An immunocytochemical study. J Comp Neurol. 1984;229:329–346. doi: 10.1002/cne.902290305. [DOI] [PubMed] [Google Scholar]
- Bellardita C, Caggiano V, Leiras R, Caldeira V, Fuchs A, Bouvier J, Löw P, Kiehn O. Spatiotemporal correlation of spinal network dynamics underlying spasms in chronic spinalized mice. Elife. 2017;6:e23011. doi: 10.7554/eLife.23011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracci E, Ballerini L, Nistri A. Localization of rhythmogenic networks responsible for spontaneous bursts induced by strychnine and bicuculline in the rat isolated spinal cord. J Neurosci. 1996;16:7063–7076. doi: 10.1523/JNEUROSCI.16-21-07063.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. 2003;23:539–549. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Séquard C-E. Course of lectures on the physiology and pathology of the central nervous system: delivered at the Royal College of Surgeons of England in May, 1858. Philadelphia: Collins; 1860. [PMC free article] [PubMed] [Google Scholar]
- Buttry JL, Goshgarian HG. Injection of WGA-Alexa 488 into the ipsilateral hemidiaphragm of acutely and chronically C2 hemisected rats reveals activity-dependent synaptic plasticity in the respiratory motor pathways. Exp Neurol. 2014;261:440–450. doi: 10.1016/j.expneurol.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Coglianese CJ, Peiss CN, Wurster RD. Rhythmic phrenic nerve activity and respiratory activity in spinal dogs. Respir Physiol. 1977;29:247–254. doi: 10.1016/0034-5687(77)90001-9. [DOI] [PubMed] [Google Scholar]
- Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J. Functional regeneration beyond the glial scar. Exp Neurol. 2014;253:197–207. doi: 10.1016/j.expneurol.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JG, Kirkwood PA, Sears TA. The detection of monosynaptic connexions from inspiratory bulbospinal neurones to inspiratory motoneurones in the cat. J Physiol. 1985;368:33–62. doi: 10.1113/jphysiol.1985.sp015845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decima EE, von Euler C, Thoden U. Intercostal-to-phrenic reflexes in the spinal cat. Acta Physiol Scand. 1969;75:568–579. [PubMed] [Google Scholar]
- Dobbins EG, Feldman JL. Brainstem network controlling descending drive to phrenic motoneurons in rat. J Comp Neurol. 1994;347:64–86. doi: 10.1002/cne.903470106. [DOI] [PubMed] [Google Scholar]
- Duffin J, van Alphen J. Bilateral connections from ventral group inspiratory neurons to phrenic motoneurons in the rat determined by cross-correlation. Brain Res. 1995;694:55–60. doi: 10.1016/0006-8993(95)00765-i. [DOI] [PubMed] [Google Scholar]
- Ellenberger HH, Feldman JL. Monosynaptic transmission of respiratory drive to phrenic motoneurons from brainstem bulbospinal neurons in rats. J Comp Neurol. 1988;269:47–57. doi: 10.1002/cne.902690104. [DOI] [PubMed] [Google Scholar]
- Ellenberger HH, Feldman JL, Goshgarian HG. Ventral respiratory group projections to phrenic motoneurons: electron microscopic evidence for monosynaptic connections. J Comp Neurol. 1990;302:707–714. doi: 10.1002/cne.903020403. [DOI] [PubMed] [Google Scholar]
- Ghali MGZ, Marchenko V. Patterns of phrenic nerve discharge after complete high cervical spinal cord injury in the decerebrate rat. J Neurotrauma. 2016;33:1115–1127. doi: 10.1089/neu.2015.4034. [DOI] [PubMed] [Google Scholar]
- Gray PA, Rekling JC, Bocchiaro CM, Feldman JL. Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBötzinger complex. Science. 1999;286:1566–1568. doi: 10.1126/science.286.5444.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hägglund M, Borgius L, Dougherty KJ, Kiehn O. Activation of groups of excitatory neurons in the mammalian spinal cord or hindbrain evokes locomotion. Nat Neurosci. 2010;13:246–252. doi: 10.1038/nn.2482. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23:587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Baca SM, Worrell JW, Liu X, Seo Y, Leiter JC, Lu DC. Modulation of respiratory output by cervical epidural stimulation in the anesthetized mouse. J Appl Physiol. 2016;121:1272–1281. doi: 10.1152/japplphysiol.00473.2016. [DOI] [PubMed] [Google Scholar]
- Jean-Xavier C, Mentis GZ, O’Donovan MJ, Cattaert D, Vinay L. Dual personality of GABA/glycine-mediated depolarizations in immature spinal cord. Proc Natl Acad Sci. 2007;104:11477–11482. doi: 10.1073/pnas.0704832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaerulff O, Kiehn O. Distribution of networks generating and coordinating locomotor activity in the neonatal rat spinal cord in vitro: a lesion study. J Neurosci. 1996;16:5777–5794. doi: 10.1523/JNEUROSCI.16-18-05777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski KE, Hsieh YH, Dick TE, DiMarco AF. Diaphragm activation via high frequency spinal cord stimulation in a rodent model of spinal cord injury. Exp Neurol. 2013;247:689–693. doi: 10.1016/j.expneurol.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, White TE, Coutts MA, Jones AL, Sandhu MS, Bloom DC, Bolser DC, Yates BJ, Fuller DD, Reier PJ. Cervical prephrenic interneurons in the normal and lesioned spinal cord of the adult rat. J Comp Neurol. 2008;511:692–709. doi: 10.1002/cne.21864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipski J. Is there electrical coupling between phrenic motoneurons in cats? Neurosci Lett. 1984;46:229–234. doi: 10.1016/0304-3940(84)90447-6. [DOI] [PubMed] [Google Scholar]
- Lipski J, Duffin J, Kruszewska B, Zhang X. Upper cervical inspiratory neurons in the rat: an electrophysiological and morphological study. Exp Brain Res. 1993;95:477–487. doi: 10.1007/BF00227141. [DOI] [PubMed] [Google Scholar]
- Lois JH, Rice CD, Yates BJ. Neural circuits controlling diaphragm function in the cat revealed by transneuronal tracing. J Appl Physiol. 2009;106:138–152. doi: 10.1152/japplphysiol.91125.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Alvarez FJ, Bonnot A, Richards DS, Gonzalez-Forero D, Zerda R, O’Donovan MJ. Noncholinergic excitatory actions of motoneurons in the neonatal mammalian spinal cord. Proc Natl Acad Sci U S A. 2005;102:7344–7349. doi: 10.1073/pnas.0502788102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose-Sato Y, Nakamori T, Sato K. Spontaneous depolarization wave in the mouse embryo: origin and large-scale propagation over the CNS identified with voltage-sensitive dye imaging. Eur J Neurosci. 2012a;35:1230–1241. doi: 10.1111/j.1460-9568.2012.07997.x. [DOI] [PubMed] [Google Scholar]
- Momose-Sato Y, Nakamori T, Sato K. Pharmacological mechanisms underlying switching from the large-scale depolarization wave to segregated activity in the mouse central nervous system. Eur J Neurosci. 2012b;35:1242–1252. doi: 10.1111/j.1460-9568.2012.08040.x. [DOI] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46:37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Nishimaru H, Restrepo CE, Ryge J, Yanagawa Y, Kiehn O. Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc Natl Acad Sci U S A. 2005;102:5245–5249. doi: 10.1073/pnas.0501331102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. 2004;43:105–117. doi: 10.1016/j.neuron.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Philippidou P, Walsh CM, Aubin J, Jeannotte L, Dasen JS. Sustained Hox5 gene activity is required for respiratory motor neuron development. Nat Neurosci. 2012;15:1636–1644. doi: 10.1038/nn.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter WT. The path of the respiratory impulse from the bulb to the phrenic nuclei. J Physiol. 1895;17:455–485. doi: 10.1113/jphysiol.1895.sp000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, Beatty S, Graham GE, Armstrong L, Shiang R, et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38:801–806. doi: 10.1038/ng1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinoso MA, Sieck GC, Hubmayr RD. Respiratory muscle coordination in acute spinal dogs. Respir Physiol. 1996;104:29–37. doi: 10.1016/0034-5687(95)00097-6. [DOI] [PubMed] [Google Scholar]
- Silver JR, Lehr RP. Dyspnoea during generalised spasms in tetraplegic patients. J Neurol Neurosurg Psychiatry. 1981;44:842–845. doi: 10.1136/jnnp.44.9.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Ellenberger H, Ballanyi K, Richter D, Feldman J. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpalar AE, Endo T, Löw P, Borgius L, Hägglund M, Dougherty KJ, Ryge J, Hnasko TS, Kiehn O. Identification of minimal neuronal networks Involved in flexor-extensor alternation in the mammalian spinal cord. Neuron. 2011;71:1071–1084. doi: 10.1016/j.neuron.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Karlén M, Wu N, Charnay P, Champagnat J, Fortin G. Genetic identification of an embryonic parafacial oscillator coupling to the preBötzinger complex. Nat Neurosci. 2009;12:1028–1035. doi: 10.1038/nn.2354. [DOI] [PubMed] [Google Scholar]
- Tresch MC, Kiehn O. Motor coordination without action potentials in the mammalian spinal cord. Nat Neurosci. 2000;3:593–599. doi: 10.1038/75768. [DOI] [PubMed] [Google Scholar]
- Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y. Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci. 2016;19:784–787. doi: 10.1038/nn.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usiak MF, Landmesser LT. Neuromuscular activity blockade induced by muscimol and d-tubocurarine differentially affects the survival of embryonic chick motoneurons. J Neurosci. 1999;19:7925–7939. doi: 10.1523/JNEUROSCI.19-18-07925.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala D, Vidal C, Freton E. Coordinated rhythmic bursting in respiratory and locomotor muscle nerves in the spinal rabbit. Neurosci Lett. 1979;11:155–159. doi: 10.1016/0304-3940(79)90119-8. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- Zimmer MB, Goshgarian HG. GABA, not glycine, mediates inhibition of latent respiratory motor pathways after spinal cord injury. Exp Neurol. 2007;203:493–501. doi: 10.1016/j.expneurol.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.