

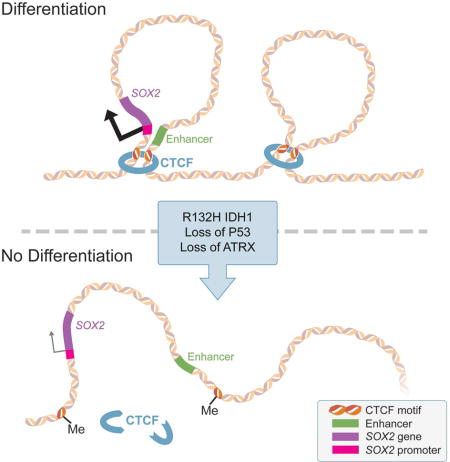
SUMMARY
Low-grade astrocytomas (LGA) carry neomorphic mutations in Isocitrate Dehydrogenase (IDH), concurrently with P53 and ATRX loss. To model LGA formation, we introduced R132H IDH1, P53 shRNA and ATRX shRNA in human neural stem cells (NSCs). These oncogenic hits blocked NSC differentiation, increased invasiveness in vivo and led to a DNA methylation and transcriptional profile resembling IDH1-mutant human LGAs. The differentiation block was caused by transcriptional silencing of transcription factor SOX2, secondary to disassociation of its promoter from a putative enhancer. This occurred due to reduced binding of the chromatin organizer CTCF to its DNA motifs and disrupted chromatin looping. Our human model of IDH-mutant LGA formation implicates impaired NSC differentiation due to repression of SOX2 as an early driver of gliomagenesis.
Keywords: Low-grade glioma, astrocytoma, IDH, P53, ATRX, neural stem cells, SOX2, chromatin looping, CTCF, DNA methylation
Graphical abstract
INTRODUCTION
Diffuse low-grade gliomas (LGGs) are slow-growing (WHO grade II) brain tumors that occur in early adult life (Cancer Genome Atlas Research et al., 2015; Ceccarelli et al., 2016) and progress to high-grade gliomas. Current treatments for LGG are ineffective and often induce hypermutated recurrent tumors (Johnson et al., 2014), highlighting the need for improved understanding of LGG biology. LGGs are classified as astrocytomas and oligodendrogliomas. The single most prevalent genetic change in either type is mutation in the Isocitrate Dehydrogenase (IDH) gene family (Parsons et al., 2008; Yan et al., 2009). Among IDH-mutated LGGs, astrocytomas carry concurrent P53 and chromatin remodeler ATRX loss-of-function mutations, while oligodendrogliomas carry 1p/19q co-deletion and TERT promoter mutations (Cancer Genome Atlas Research et al., 2015). The presence of mutant IDH in two divergent tumor lineages suggests it is a common genetic driver that occurs early in a multipotent progenitor cell (Bardella et al., 2016; Pirozzi et al., 2017; Tirosh et al., 2016; Venteicher et al., 2017).
IDH normally converts isocitrate to α-ketoglutarate (αKG). Heterozygous neomorphic mutations in the catalytic site of IDH (R132H in the cytosolic isoform IDH1) result in production of the oncometabolite 2-hydroxyglutarate (2HG) (Dang et al., 2009). 2HG competitively inhibits αKG-dependent dioxygenases responsible for demethylation of DNA and histones (Xu et al., 2011). DNA methylation and histone modifications dynamically shape the epigenome, which we define as heritable transcriptional states determined by means other than changes in the DNA sequence. Inhibition of DNA and histone demethylation by 2HG leads to a hypermethylated epigenetic state, which may cause dysregulation of oncogenes and tumor suppressors (Figueroa et al., 2010; Lu et al., 2012; Turcan et al., 2012). Flavahan et al. (2016) postulated that hypermethylation may disrupt the binding of methylation-sensitive chromatin organizer CTCF, leading to chromatin disorganization and aberrant expression of oncogenes in IDH-mutated high-grade gliomas. Other groups have linked the accumulation of 2HG and epigenetic hypermethylation to a block in differentiation, which predisposes to oncogenesis (Figueroa et al., 2010; Lu et al., 2012; Saha et al., 2014; Turcan et al., 2012). Recent mouse models have suggested that expression of mutant IDH1 in progenitors of the subventricular zone (SVZ) may induce a pre-tumorigenic state (Bardella et al., 2016; Pirozzi et al., 2017; Sasaki et al., 2012). The mechanism whereby the IDH1 mutation cooperates with loss of P53 and ATRX to promote LGA formation remains unknown.
We modeled mutant IDH1 LGA formation in neural stem cells (NSCs) derived from human embryonic stem cells (hESCs). We systematically introduced the 3 core genetic changes found in LGA via lentiviral expression of R132H mutant IDH1, and short hairpin RNA (shRNA)-mediated knockdown of P53 and ATRX, in order to study progression of gliomagenesis on an oncogenic hit-by-hit basis. We show that the combination of 3 hits blocks NSC differentiation and evokes brain invasiveness. The differentiation block is caused by transcriptional downregulation of transcription factor SOX2, the master regulator of NSC multipotency. The etiology of this transcriptional silencing is disrupted chromatin looping due to hypermethylation of DNA binding sites for chromatin insulator CTCF, leading to disassociation of the SOX2 promoter from critical enhancer elements.
RESULTS
Generation of human NSCs with astrocytoma mutations
We generated neural progenitor lineages from hESCs modified with a HES5::GFP bacterial artificial chromosome (BAC) reporter (Placantonakis et al., 2009) (Figure 1A, S1A). This reporter identifies early neuroepithelial multipotent precursors termed rosettes, in which activation of Notch signaling results in transcription of the HES5 gene. Human ESC colonies were differentiated into HES5::GFP+ rosette-NSCs (Edri et al., 2015; Elkabetz et al., 2008) (Figure 1A, S1A–C), which were mechanically picked and further differentiated into monolayers of EGF/FGF2-responsive NSCs (Figure 1A, S2). Such NSCs are thought to resemble adult SVZ neural progenitors in the adult SVZ, which we hypothesize are the cell of origin in LGA (Bardella et al., 2016). These NSCs are enriched for Nestin (~90% positive), lose HES5::GFP expression (Edri et al., 2015) and are multipotent, as demonstrated by directed differentiation to all three arms of the neuroglial lineage: neurons, oligodendrocytes and astrocytes (Figure S2A–E) (Elkabetz et al., 2008; Tabar et al., 2005).
Figure 1. Generation of human NSCs with ectopically expressed R132H IDH1, P53 knockdown and ATRX knockdown.
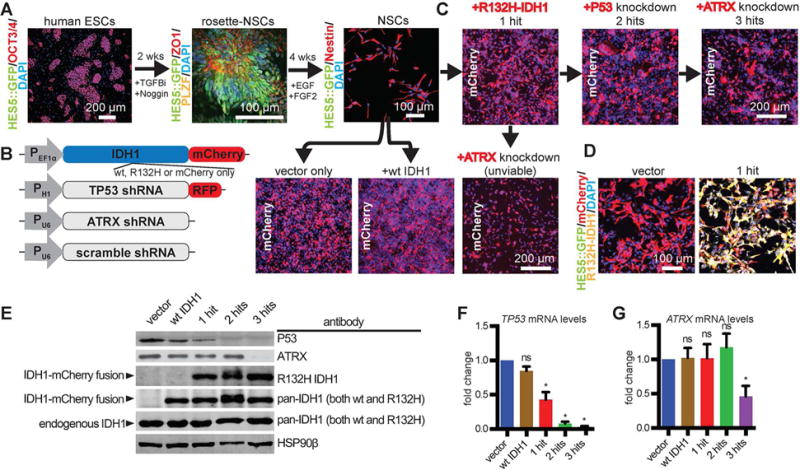
A. Human ESCs (OCT3/4+, HES5::GFP−) were progressed to rosette-NSCs (ZO1+, PLZF+, Hes5::GFP+) over two weeks with TGFβ inhibitor SB431542 (TGFBi; 10 μM) and noggin (100 ng/mL). HES5::GFP+ rosette structures were mechanically dissociated and plated at high densities in EGF and FGF2 over 4 weeks to produce NSCs growing as a monolayer (Nestin+, HES5:: GFP−).
B. Lentiviral constructs used to engineer NSCs. PEF1a, EF1a promoter; PH1; H1 promoter; PU8, U8 promoter; RFP, red fluorescent protein.
C. Wild-type NSCs were infected with lentiviruses to constitutively express either mCherry alone (vector only), wild-type IDH1-mCherry, or mutant R132H-IDH1-mCherry (1-hit). Cells were then purified for mCherry via FACS sorting. Following these transductions and sorts, cells were transduced with shRNA lentiviruses against P53 or ATRX, in either order. Cells that received ATRX shRNA as the second hit became unviable.
D. Immunofluorescence microscopy of mCherry, HES5::GFP and R132H-IDH1 in vector and 1-hit NSCs.
E. Western blot using antibodies against P53, ATRX, the R132H mutation and total IDH1. HSP90β, loading control.
F. qRT-PCR of TP53 mRNA levels across different conditions (n = 3/condition; ANOVA F(4,10)=48.49, p=0.0048). *p<0.05, post hoc Dunnett’s test; ns, not significant.
G. qRT-PCR of ATRX mRNA levels across different conditions (n = 3/condition). *p<0.05, t-test between vector and 3-hit conditions. ns, not significant.
To test how mutant IDH1 and loss of P53 and ATRX work together to promote gliomagenesis, we serially introduced an IDH1-mCherry fusion gene (R132H or wild-type or mCherry alone) into NSCs via lentivirus (Figure 1B,C), generated a pure population by FACS isolation, and added lentiviral shRNA against P53, followed by shRNA against ATRX (Figure 1B,C). We focused on conditions that are most biologically relevant: vector only (“vector”), mutant IDH1 alone (“1-hit”), mutant IDH1 with P53 knock-down (“2-hits”) and mutant IDH1 with P53 and ATRX knockdown (“3-hits”). Mutant-IDH1 NSCs with ATRX knockdown in a wild-type P53 background resulted in poor cell viability (Figure 1C), which precluded subsequent P53 knockdown. Immunofluorescent microscopy, qPCR and immunoblot verified our lines were expressing IDH1-mCherry and knocking down P53 and ATRX (Figure 1D–G).
The DNA methylome, transcriptome and karyotype of IDH-mutated NSCs resemble those of human LGA
Using mass spectrometry (MS), we found a >1000-fold increase of oncometabolite 2HG amongst all samples that contained the IDH1 mutation (Figure 2A). Elevated 2HG levels induce DNA hypermethylation in glioma (Figueroa et al., 2010; Turcan et al., 2012). To understand the roles of mutant IDH1, P53 and ATRX in our NSCs, we profiled their DNA methylomes and transcriptomes using Illumina 450k methylation arrays and RNA-seq, respectively. Global assessment of CpG sites in promoters, genes and within CpG islands showed elevated levels of methylation in all conditions that included mutant IDH1 (Figure 2B). Increased global methylation was seen across all chromosomes (Figure S3A). Interestingly, we discovered a large number of differentially methylated CpG regions among 1-, 2- and 3-hit conditions (Figure S3B).
Figure 2. Metabolic, transcriptional, epigenetic and karyotypic properties of transgenic NSCs.
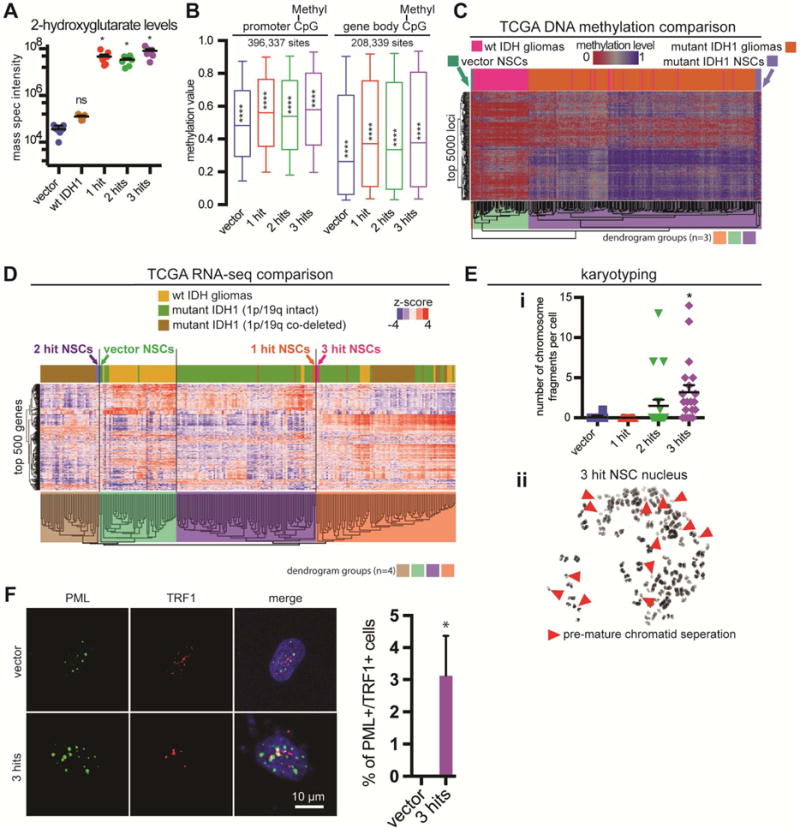
A. Quantitative MS for 2HG in genetically modified NSCs (n=6/condition, ANOVA F(4,30)=17.91, p<0.0001). *p<0.05, post hoc Dunnett’s test; ns: not significant.
B. Boxplots of beta (methylation) values of CpG sites with probes at genome-wide sites marked as promoters (−1.5kb to +0.5kb from TSS, n=396,337 CpG sites) and at gene bodies (coding and non-coding regions of a gene, n=208,339 sites). NSCs with IDH1 mutation have globally elevated levels of methylation. Statistics for promoter CpGs: n=2 arrays/condition, ANOVA F(3,122696)=475.5, p<0.0001; Statistics for gene body CpGs: n=2 arrays/condition, ANOVA F(3,123352)=283.7, p<0.0001. ****p<0.0001, post hoc Tukey’s test.
C. Hierarchical clustering of NSCs with LGGs (wild-type and mutant IDH1 demarcated) using the top 5000 most variable loci defined amongst the TCGA samples. The first three dendrogram levels are highlighted.
D. Hierarchical clustering of NSCs with LGGs (wild-type vs. mutant IDH1 and 1p/19q status demarcated) using the top 500 most variable genes defined amongst the TCGA samples. The first four dendrogram levels are highlighted.
E. i. The number of chromosome fragments per cell were significantly increased in 3-hit NSCs (n=20, ANOVA F(3,76)=11, p=0.0004). *p<0.05, post hoc Tukey’s test. ii. 3-hit NSCs had a high frequency of premature chromatid separation (red arrowheads).
F. PML and TRF1 show co-localization in ~3.1% of 3-hit NSCs, but are not detected in vector NSCs (n=3 trials, ≥3 fields per trial; n=139 total vector NSCs, n=165 total 3-hit NSCs; *p<0.05, paired t-test).
We compared our DNA methylation data to 270 human LGGs from the TCGA (Cancer Genome Atlas Research et al., 2015). Using supervised hierarchical clustering based on the top 5000 most variable methylation sites from TCGA samples, we found IDH-mutated LGGs clustered separately from wild-type IDH LGGs (Figure 2C). NSCs transduced with vector alone were the most dissimilar sample (orange dendrogram in Figure 2C). Mutant-IDH1 NSCs clustered with mutant-IDH1 gliomas (violet dendrogram in Figure 2C), suggesting epigenetic similarity.
To determine transcriptional similarities between our NSCs and human LGG, we used RNA-seq data from 239 TCGA patients (Cancer Genome Atlas Research et al., 2015). We analyzed RNA-seq data from our NSCs using the same pipeline employed by the TCGA. Supervised hierarchical clustering using the top 500 most variable TCGA genes resulted in four groups that closely resembled clusters identified by the TCGA: wild-type IDH gliomas; IDH mutant 1p/19q co-deleted gliomas (oligodendrogliomas); IDH mutant 1p/19q intact gliomas (astrocytomas); and a mixed group of IDH mutated gliomas (Figure 2D). 1-hit NSCs grouped with mutant-IDH1 1p/19q intact astrocytomas, while 3-hit NSCs clustered with the mixed group of mutant-IDH1 gliomas. Vector and 2-hit NSCs grouped with the wild-type IDH and mutant-IDH1 1p/19q co-deleted oligodendroglioma groups, respectively.
Given the known roles of P53 and ATRX in genomic integrity (Negrini et al., 2010; Ramamoorthy and Smith, 2015; Ritchie et al., 2008), we performed karyotype analysis (Figure S3C). 2-hit and 3-hit NSCs had abnormal nuclei (n=20 karyotypes/condition). We observed significantly elevated numbers of chromosomal fragments in 3-hit NSCs (Figure 2Ei), suggesting genomic instability, a known feature of LGGs (Cancer Genome Atlas Research et al., 2015; Cohen et al., 2015). The 3-hit culture had evidence of premature sister chromatid separation (Figure 2Eii), likely due to loss of ATRX, which also leads to Alternative Lengthening of Telomeres (ALT) (Ramamoorthy and Smith, 2015). To test whether ALT was utilized by our NSCs, we performed co-stains for two markers that co-localize in nuclei of ALT cells, PML and TRF1 (Yeager et al., 1999). We found that only 3-hit NSCs showed nuclear co-localization of the markers (Figure 2F). Collectively, these results show epigenetic, transcriptional and karyotypic similarity of 3-hit NSCs to IDH-mutant LGA.
Effects of the 3 hits on cell cycle, cell death and brain invasion
Using in vitro growth assays, we found that two conditions, 1-hit and mutant IDH1 with ATRX knockdown, grew more slowly than the others (Figure 3A). This finding correlated with cell cycle alterations (Figure 3B,C). To test if apoptosis contributed to growth differences, we performed TUNEL assays (Figure 3D,E). 1-hit and mutant IDH with ATRX knockdown NSCs had significantly elevated levels of TUNEL+ cells. P53 knockdown in the 2-hit and 3-hit conditions decreased TUNEL staining to control levels. A similar pattern was observed with flow cytometric assessment of cell death marker Annexin V (Figure 3F,G, S3D). These findings indicated that decreased cellular proliferation and increased cell death caused by R132H IDH1 were rescued by P53 knockdown in the 2-hit and 3-hit conditions. Furthermore, these experiments indicated that loss of ATRX as the second hit leads to non-viable cells.
Figure 3. Mutant IDH1, P53 shRNA and ATRX shRNA differentially alter NSC growth rate, cell death and invasiveness.
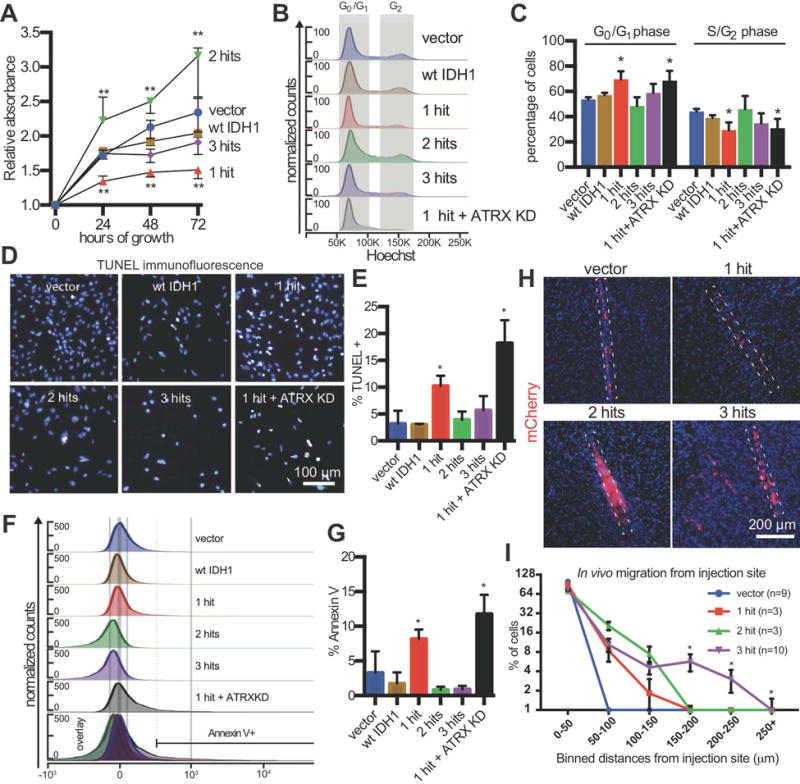
A. In vitro growth assay. NSCs were plated at equal densities and absorbance was measured by WST-1 daily to determine cellular proliferation (n=3/condition, ANOVA F(4,8)=35.81, p<0.001). **p<0.01, post hoc Dunnett’s test.
B. Representative flow cytometric cell cycle analysis.
C. Quantification of cell cycle phases (n=3/condition, ANOVA F(1,4)=7.98, p=0.0003). **p<0.01, post hoc Dunnett’s test.
D. Representative immunofluorescence microscopic image of TUNEL staining.
E. Quantification of TUNEL+ cells in (D) (n=3/condition, ANOVA F(2,10)=17.58, p= 0.0208). *p<0.05, post hoc Tukey’s test.
F. Representative histogram plots of Annexin V flow cytometry.
G. Annexin V was significantly elevated in 1-hit and R132H/ATRX KD NSCs (n=3/condition, ANOVA F(5,12)=16.5, p<0.0001). *p<0.05, post hoc Tukey’s test.
H. In vivo invasion assay. mCherry immunostaining of brain sections showing xenografts of NSCs (2.5×105 cells, 4 weeks post-injection) in the frontal lobe of NOD.SCID mice. The dotted rectangles outline the injection tracks.
I. Quantification of the percentage of mCherry+ NSCs in binned distances away from the injection site (n=3–10 mice/condition, ≥ 3 fields per mouse). Above the 150 μm cutoff, the 3-hit condition was significantly different from all others (two-way ANOVA F(3,16)=9.487, p<0.001). *q<0.05, post hoc Benjamini correction.
Diffuse brain infiltration is a hallmark of LGA. To understand whether our model replicates this aspect of the disease, we generated xenografts in NOD.SCID mice by injecting cells into the frontal lobe and quantifying cellular migration from the injection site 4 weeks later (Figure 3H,I). In vivo xenografts of 3-hit, but not 1-, 2-hit, or vector NSCs recapitulated the diffuse invasive nature of LGA, a key clinical feature.
3 hits block NSC differentiation
Expression of mutant IDH1 in progenitor cells inhibits differentiation (Figueroa et al., 2010; Lu et al., 2012; Pirozzi et al., 2017; Rosiak et al., 2016; Turcan et al., 2012; Xu et al., 2011). However, the effect of combined IDH mutation and P53/ATRX loss on NSC self-renewal and differentiation is not known. Under self-renewing conditions, immunofluorescent staining of our NSCs showed expression of Nestin, a NSC marker, in >90% of cells in all groups, with no significant staining for differentiation markers GFAP (astrocytic), TUJ1 (neuronal) or NG2 (oligodendroglial) (Figure 4A). Flow cytometric staining with CD133, a surface NSC marker (Uchida et al., 2000), and CD44, a restricted glial progenitor marker (Liu et al., 2004), revealed a different trend (Figure 4B,C, S4A,B). Vector and 2-hit NSCs were CD133high and CD44low, whereas 1-hit and 3-hit NSCs became CD133low and CD44high. These findings suggested a switching stem-like phenotype with serial introduction of the core mutations.
Figure 4. Impaired differentiation in 3-hit NSCs is associated with transcriptional downregulation of SOX2.
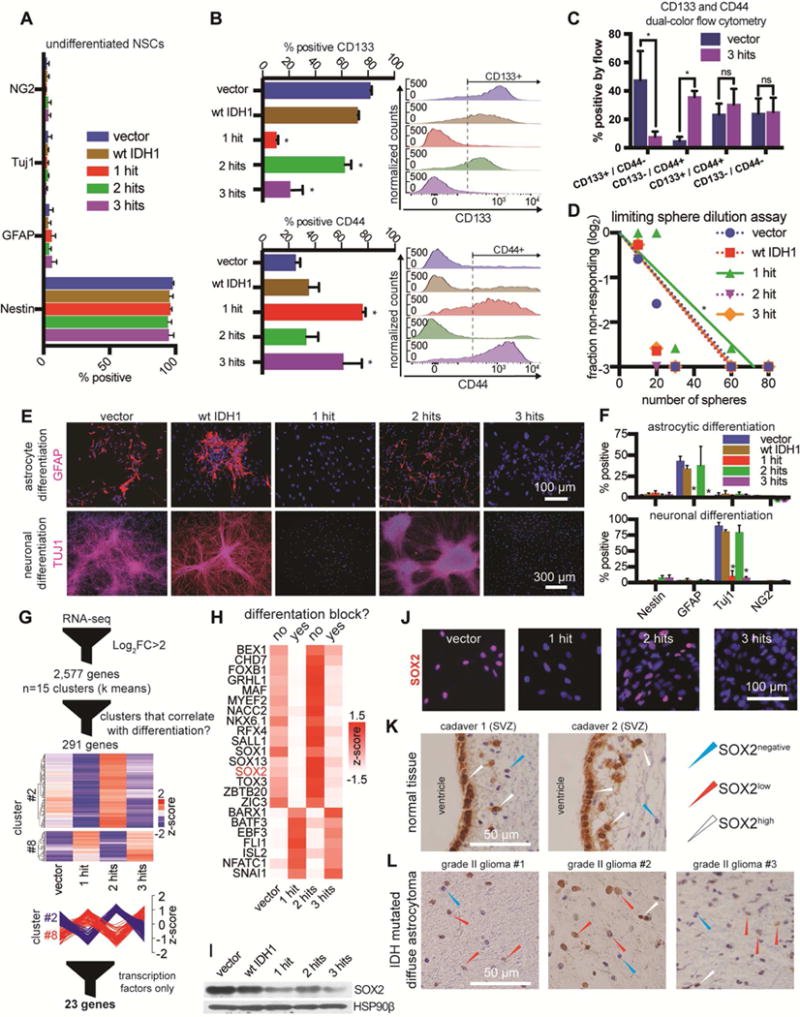
A. Immunofluorescence quantification of Nestin, GFAP, TUJ1 and Ng2 in NSCs cultured with EGF/FGF2.
B. Flow cytometric detection of CD133 (n=3/condition, ANOVA F(4,10)=53.09, p<0.0001). and CD44 (n=3/condition, ANOVA F(4,10)=6.83, p<0.001). *p<0.05, post hoc Tukey’s test.
C. CD133 and CD44 dual color flow cytometry (n=3/condition, multiple t-tests). *p<0.05; ns, not significant.
D. Sphere formation assay in limiting dilutions (n=3/condition, χ2 pairwise tests, p<0.001).
E. Representative immunofluorescence microscopic images of GFAP and TUJ1 in cultures that were differentiated to astrocytes (top) or neurons (bottom).
F. Quantification of immunofluorescence markers in astrocytic (top) or neuronal (bottom) differentiation conditions. Astrocyte differentiation: n=3/condition, ANOVA F(4,40)=10.75, p<0.0001; Neuronal differentiation: n=3/condition, ANOVA F(4,40)=86.68, p<0.0001. *p<0.05, post hoc Tukey’s test.
G. Vector, 1-hit, 2-hit and 3-hit NSCs were subjected to RNA-seq in duplicates. We found 2,577 differentially expressed genes (log2 fold-change>2 and qvalue<0.05). Genes were clustered into 15 k means groups and represented on a heatmap with no column clustering. Two of the 15 groups had genes with expression patterns that strongly correlated or anti-correlated with the ability of NSCs to differentiate (n=291 genes). Within these 2 groups we identified 23 transcription factors with IPA.
H. Transcription factors from (G) were visualized on a heatmap. The list includes SOX2, whose levels correlated with the ability of cells to differentiate.
I. Western blot of NSCs shows that SOX2 protein is downregulated most heavily in 1-hit and 3-hit NSCs, and to a lesser degree in 2-hit NSCs. HSP90β, loading control.
J. Immunofluorescence microscopy of NSCs stained for SOX2.
K. Cadaver tissue immunohistochemical staining of SOX2 reveals SOX2high ependymal cells and NSCs in the SVZ (n=2 adult patients).
L. LGA tumors (IDH mutated, P53 and ATRX loss, 1p/19q intact) showing diffuse brain infiltration contain predominantly SOX2low tumor cells (n=3 tumors).
We next subjected these cells to extreme limiting dilution assays to test their ability to clonally form spheres from single cells in self-renewing conditions (Figure 4D). The 1-hit line had mildly diminished ability to form spheres, but the addition of P53 and ATRX knockdown rescued this deficit. To look for differences in differentiation capabilities, we directed NSCs to differentiate to neurons and astrocytes (Figure 4E,F). Similar to the flow cytometry analysis, a switching phenotype emerged. While control and 2-hit NSCs were able to differentiate, 1-hit and 3-hit cells showed a near complete differentiation block.
SOX2 is transcriptionally downregulated by the 3 hits
In order to identify transcription factors (TFs) that correlated with the differentiation phenotype, we performed RNA-seq analysis. We discovered 2,577 genes that were differentially regulated amongst all four conditions (log2FC>2 and qvalue<0.05; FC, fold change) (Figure 4G). We performed k means clustering and visualized the results on a scaled heatmap (Figure S4C; Supplemental file titled RNA-seq clusters). Two of 15 clusters correlated with the differentiation phenotype and contained genes that were high (cluster 2) or low (cluster 8) in conditions capable of differentiation. Within these two clusters there were 291 genes and 23 candidate TFs (Figure 4H).
One of the candidates was SOX2 (Sex Determining Region Y-Box2), which was downregulated in conditions (1- and 3-hit) that had a differentiation block. SOX2 is a master regulator of NSC self-renewal and multipotency (Amador-Arjona et al., 2015; Ferri et al., 2004; Lodato et al., 2013). Using qRT-PCR, we confirmed SOX2 mRNA was downregulated ~10-fold in 1-hit and 3-hit NSCs and ~5-fold in 2-hit NSCs (Figure S4D). By immunoblot and immunofluorescence, SOX2 protein showed the same expression pattern as SOX2 mRNA (Figure 4I,J). We noticed a similar trend with mRNA levels of SOX1, a closely related gene, and the pro-neuronal differentiation TFs NEUROD1 and NGN2, which are maintained in a bivalent (poised) state by SOX2 (Amador-Arjona et al., 2015; Lodato et al., 2013) (Figure S4C). When compared to vector NSCs, 3-hit NSCs injected in the mouse brain showed lower SOX2 protein levels in vivo (Figure S5A).
To validate our observations regarding SOX2 levels, we performed immunohistochemistry (IHC) and immunofluorescence staining for SOX2 in human specimens (Supplemental Table 1). We assessed the level of SOX2 in the human cadaveric SVZ, which showed SOX2high NSCs and ependymal cells (Figure 4K, S5B,C) (Baer et al., 2007). When we profiled brain-infiltrating IDH1/P53/ATRX mutated LGA cells using IHC and immunofluorescence techniques for SOX2, we found low levels of SOX2 relative to NSCs in the SVZ (n=7) (Figure 4L, S5C,D). These findings support our hypothesis that SOX2 is downregulated in the gliomagenic transformation of NSCs and reproduce previous observations that SOX2 expression is relatively low in low-grade gliomas (Annovazzi et al., 2011; Guo et al., 2011; Ma et al., 2008).
The genomic region surrounding the SOX2 locus is hypermethylated and has diminished CTCF occupancy
To investigate the mechanism of SOX2 downregulation, we examined the methylation status of the SOX2 promoter using our methylation array data. We found no appreciable DNA methylation change in 3-hit relative to vector NSCs (Figure 5A). We confirmed this finding by comparing wild-type IDH (n=53) and mutant IDH (n=157) LGGs from the TCGA (Cancer Genome Atlas Research et al., 2015). When we examined a 1.2 Mb-wide genomic region surrounding the SOX2 locus, we observed increased DNA methylation in specific areas upstream and downstream of SOX2 in 3-hit vs. vector NSCs, and a similar trend when comparing mutant to wild-type IDH gliomas in the TCGA (Figure 5B). We therefore hypothesized there may be other regulatory elements sensitive to methylation in these regions.
Figure 5. Mutant IDH1-mediated hypermethylation around the SOX2 locus leads to diminished CTCF occupancy.
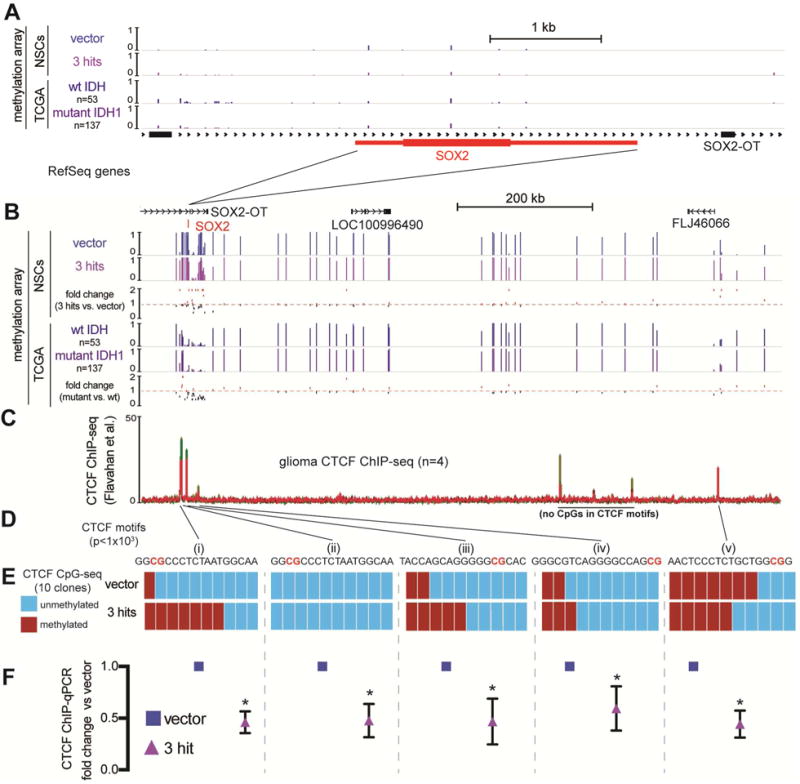
A. The SOX2 locus (~6 kb window) aligned to 450k methylation array tracks of NSCs with the indicated transgenes (n=2) and averaged methylation beta values from TCGA grade II wild-type IDH gliomas (n=53) and mutant IDH, 1p/19q intact astrocytomas (n=157). The SOX2 locus has low levels of methylation in all these conditions. RefSeq genes are shown.
B. The region (~1 Mb) containing the SOX2 locus with tracks from (A). Shown below each pair of tracks is the calculated fold change in methylation beta value. Mutant IDH astrocytomas vs. wild-type IDH gliomas share a similar pattern of changes in methylation as the comparison of 3-hit NSCs vs. vector NSCs.
C. CTCF ChIP-seq tracks from grade III and IV gliomas (Flavahan et al., 2016). Red: mutant IDH grade III gliomas; Green: wild-type IDH grade IV gliomas.
D. CTCF motif analysis of each ChIP peak reveals high confidence CTCF motif locations with CpG sites highlighted in red (MEME suite motif scanning, p<0.001)
E. Targeted bisulfite sequencing of CpG sites within indicated CTCF motifs. Ten clones of vector and 3-hit NSCs were sequenced for every site. The most robust hypermethylation in 3-hit NSCs was observed in a CTCF site upstream of SOX2 (i).
F. CTCF ChIP-qPCR of each corresponding CTCF site. CTCF occupancy was reduced approximately ~2-fold in every site assayed (p<0.01, multiple t-tests).
CTCF functions as a genomic insulator, but can also facilitate contacts between promoters and enhancers (Bell et al., 1999; Kim et al., 2007; Phillips and Corces, 2009). As an insulator, CTCF is necessary for delimiting borders between Topologically Associating Domains (TADs) (Dixon et al., 2012; Nora et al., 2012). Its binding to its DNA motifs is sensitive to methylation (Bell and Felsenfeld, 2000; Wang et al., 2012). Flavahan et al. (2016) showed that DNA hypermethylation in IDH-mutated high-grade gliomas causes the promoter of an oncogene (PDGFRA) to ectopically associate with an enhancer in a neighboring TAD due to disruption of CTCF binding and loss of inter-TAD insulation. Using available CTCF chip-seq data from wild-type and mutated IDH high-grade gliomas from Flavahan et al., we identified CTCF sites around the SOX2 gene with CpGs in their motifs (Figure 5C,D). This raised the possibility that downregulation of SOX2 in 3-hit NSCs could be due to aberrant chromatin looping brought about by reduced CTCF binding at hypermethylated motifs.
All four CTCF motifs upstream of the SOX2 promoter, and a single site ~1Mb downstream, had CpGs directly in the motif, suggesting sensitivity to methylation. To understand if direct hypermethylation of CTCF sites could lead to decreased CTCF occupancy at these CTCF sites, we performed targeted bisulfite sequencing of vector and 3-hit NSCs (n=10/motif) (Figure 5E). We found higher levels of methylation in 3 of 5 sites examined. The hypermethylation in 3-hit NSCs was particularly prominent at a CTCF motif 11 kb upstream of the SOX2 gene (Figure 5Ei). To assess whether or not CTCF occupancy at these motifs had changed, we performed ChIP-qPCR for CTCF (Figure 5F). Every site assayed had a ~2-fold decrease in CTCF occupancy, supporting the hypothesis that DNA hypermethylation in 3-hit NSCs leads to reduced CTCF occupancy around the SOX2 locus.
The SOX2 promoter has diminished contacts with putative downstream long-range enhancer regions in 3-hit conditions
Since CTCF organizes chromatin looping, we analyzed publicly available HiC plots of NSCs (Dixon et al., 2015) and found SOX2 has strong contacts with multiple areas within its TAD (Figure 6A, S6A). To understand if loss of CTCF binding affects chromatin contacts at the SOX2 locus, we performed circularized chromosome conformation capture (4C-Seq) using the SOX2 promoter as bait to identify genomic interactions that may regulate its expression (Figure 6B, S6B). We discovered strong cis interactions ~700 kb downstream of SOX2 in our control NSCs. These interactions were all apparent in NSC HiC data (Figure 6A) and significantly reduced in 3-hit NSCs.
Figure 6. The promoter of SOX2 has decreased long-range contacts with putative downstream enhancers in 3-hit NSCs.
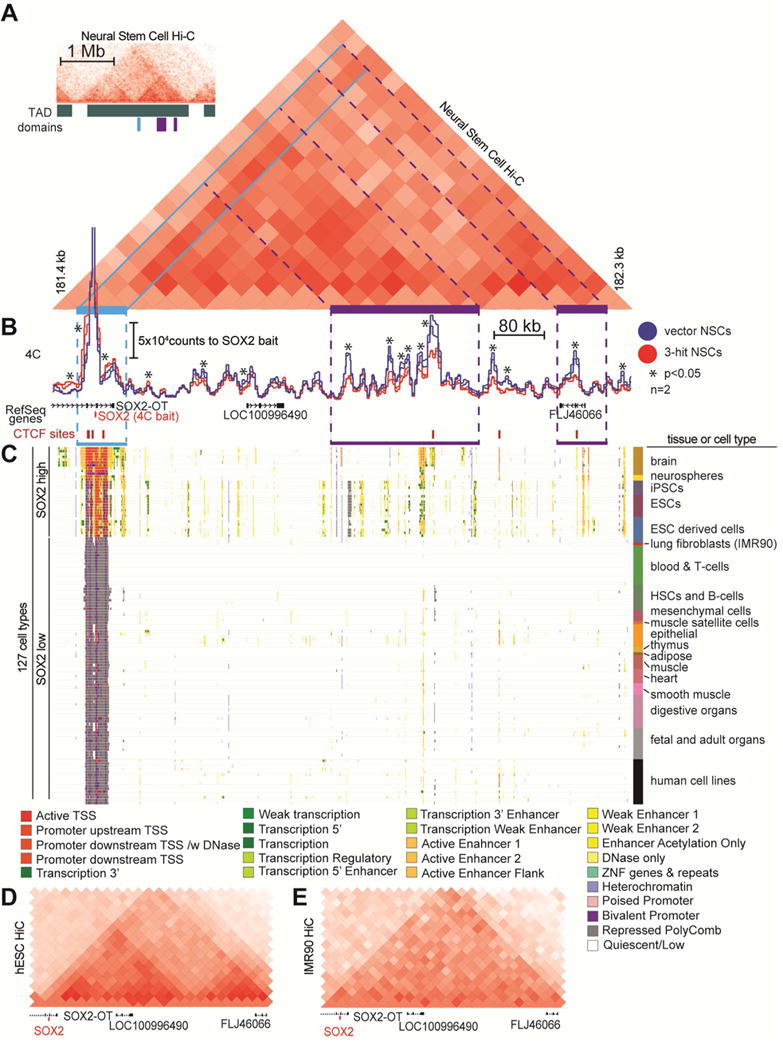
A. HiC plots in human NSCs (derived from H1 hESCs) in 40 kb bins (Dixon et al., 2015), were reprocessed. The inset on the top left shows a 2 Mb window of the HiC plot, with TADs demarcated. The large HiC plot of a 1.2Mb window shows the genomic region containing SOX2 (light blue highlight) has strong long-range contacts within the entire TAD region (purple highlights).
B. 4C using the SOX2 promoter as bait reveals strong long-range downstream contacts in vector NSCs that are diminished in 3-hit NSCs (n=2/condition, p<0.001).
C. Epigenetic states of the genomic region downstream of SOX2. Epigenetic data is from the NIH Roadmap Epigenetics Consortium (Roadmap Epigenomics et al., 2015). Each row represents a cell type (n=127) with integrated epigenome data corresponding to 25 different chromatin states (see legend). Cell types that are high for SOX2 have active TSSs and epigenetic marks in enhancer elements downstream of SOX2 that correspond to 4C and HiC contact signals in (A) and (B) (purple highlighting).
D,E. HiC plot of (D) hESCs (SOX2 high) and (E) IMR90 lung fibroblasts (SOX2 low) using the same 1.2 Mb genomic window as in (A). These plots demonstrate the genomic organization differences within the TAD that contains SOX2.
To determine if this strong long-range contact represents a promoter-enhancer interaction, we examined available epigenome data from the roadmap epigenome project (Roadmap Epigenomics et al., 2015) (Figure 6C). Remarkably, for nearly every cell type examined (n=127), a transcriptionally active TSS site at the SOX2 gene correlated with the presence of enhancers ~0.5–1 Mb downstream of the gene. To see if this same pattern of chromatin contacts is maintained in other cell types, we examined the HiC profiles of hESCs, which express high levels of SOX2, and IMR90 lung fibroblasts, which are low for SOX2 (Figures 6D,E, S6A,C,D). We found that hESCs maintained a HiC profile very similar to that of NSCs. In contrast, in IMR90 cells, TAD boundaries were similar but SOX2 did not have robust contacts with the genomic regions that corresponded to the enhancer elements. These findings suggest that in LGA SOX2 downregulation is due to aberrant chromatin looping that results in disassociation of the SOX2 promoter from a downstream enhancer.
Ectopic expression of SOX2 rescues the block in differentiation
To validate the critical role of SOX2 in gliomagenesis, we ectopically expressed SOX2 in all conditions studied and subjected them to astrocytic and neuronal differentiation. NSCs were transduced with a lentivirus constitutively expressing SOX2-P2A-GFP, or an empty vector (Figure 7A, S7A). After ectopic SOX2 expression, 3-hit cells showed differentiation levels similar to control NSCs (Figure 7B,C, S7B,C). This finding suggested that restoring SOX2 expression in 3-hit NSCs was sufficient to rescue the differentiation block and confirmed a central role for transcriptional downregulation of SOX2 in LGA formation.
Figure 7. SOX2 overexpression rescues the differentiation block in 3-hit NSCs.
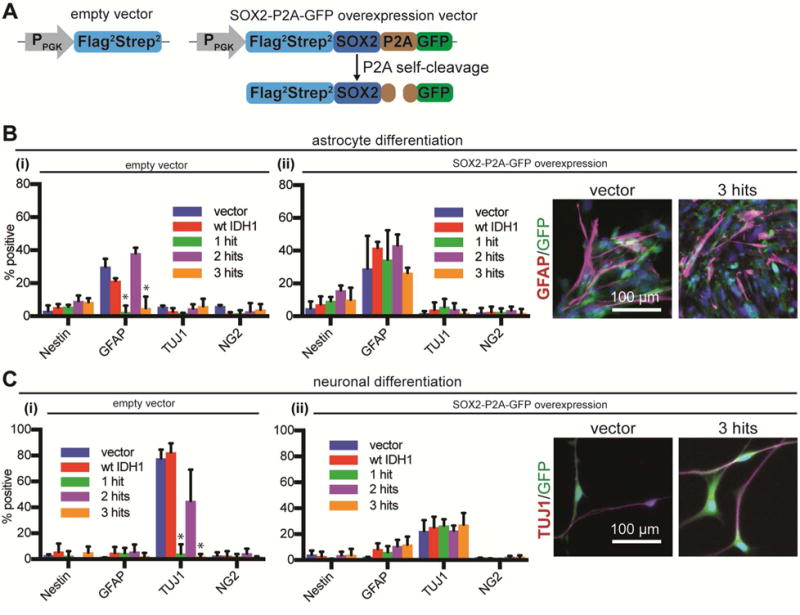
A. Lentiviral constructs used for overexpression of SOX2. PPGK, PGK promoter.
B,C. Vector and 3-hit NSCs were transduced with (i) empty vector, or (ii) SOX2-P2A-GFP lentivirus, directed to differentiate to astrocytes (B) or neurons (C) and immunostained for Nestin, GFAP, TUJ1 and NG2. There was no difference between cultures overexpressing SOX2 (n=3/condition, ANOVA F(4,40)=1.224 for astrocytic and ANOVA F(4,40)=2.1 for neuronal differentiation, both p>0.05). The expected differentiation profiles were seen with empty vector (n=3/condition, ANOVA F(4,40)=48.65 for astrocytic and ANOVA F(4,40)=113 for neuronal differentiation, both p<0.0001). *p<0.05, post hoc Tukey’s test.
DISCUSSION
The role of individual oncogenes and tumor suppressors in the initiation of LGA remains poorly understood. Mutant IDH1 is a shared core mutation in both LGA and oligodendroglioma (Cancer Genome Atlas Research et al., 2015; Parsons et al., 2008), suggesting the cell of origin is a shared neuroglial progenitor (Bardella et al., 2016; Rosiak et al., 2016).
Our model shows that the combination of the 3 oncogenic hits promotes gliomagenesis not by altering cell growth but by arresting differentiation of NSCs (Pirozzi et al., 2017; Rosiak et al., 2016; Sulkowski et al., 2017), which become locked in a self-renewing and brain-invasive state. Several lines of evidence support the notion that the IDH1 mutation is the inciting oncogenic hit in LGA. First, exome sequencing of IDH-mutated astrocytoma at initial diagnosis and recurrence showed that, while the IDH1 is preserved, distinct clonal mutations in P53 and ATRX emerge at recurrence (Johnson et al., 2014). Second, expression of mutant IDH1 alone in progenitor cells leads to pre-tumorigenic changes (Bardella et al., 2016; Figueroa et al., 2010; Lu et al., 2012; Pirozzi et al., 2017; Rosiak et al., 2016; Saha et al., 2014; Turcan et al., 2012). Third, germline mosaicism for neomorphic IDH mutations in Ollier disease can result in brain astrocytomas (Amary et al., 2011; Bonnet et al., 2016; Pansuriya et al., 2011). In contrast, loss of ATRX by itself in a wild-type P53 background does not lead to brain tumor formation (Seah et al., 2008). Similarly, TP53 loss in the brain does not result in tumor formation by itself, unless other oncogenes are introduced (Liu et al., 2011; Zheng et al., 2008). We therefore propose that the most likely order of the 3 oncogenic hits is IDH mutation, followed by loss of P53 and finally loss of ATRX.
We identified transcriptional downregulation of SOX2 as a central mechanism underlying the differentiation block. Our hypothesis that SOX2 is downregulated in the transition from NSCs to LGA tumor cells was corroborated by immunostaining of human cadaveric adult SVZ tissue and IDH1-mutant LGA. SOX2 is not only required for self-renewal, but is also the guardian of NSC multipotency, by helping maintain bivalent chromatin marks on pro-differentiation genes. Therefore, its downregulation in 3-hit NSCs provides a robust explanation for the acquired arrest in differentiation.
Despite the global DNA hypermethylation induced by mutant IDH1, we did not find hypermethylation of the SOX2 promoter. In mouse ESCs, expression of SOX2 is maintained by chromosomal looping and association with cis downstream enhancers (Li et al., 2014; Sikorska et al., 2008). Our study provides evidence that in human NSCs, SOX2 expression is dependent on contact with enhancers 700 kb downstream via CTCF-dependent chromatin looping. This chromatin loop is disrupted in 3-hit NSCs due to hypermethylation of CTCF motifs flanking the SOX2 locus, leading to decreased SOX2 expression and differentiation block. This disruption in chromatin conformation mechanistically resembles the findings of Flavahan et al. (2016) in IDH-mutated high-grade glioma. However, we show that unraveling of chromatin leads to disassociation of the SOX2 promoter from its enhancer and a differentiation block, rather than ectopic activation of an oncogene from a neighboring TAD.
Our model provides a tractable platform to study serial addition of clinically relevant oncogenic hits to human NSCs. We used this system to study the earliest stages of IDH-mutated LGA formation and discovered a fundamental gliomagenesis mechanism revolving around abnormal chromatin looping, transcriptional downregulation of SOX2 and impaired NSC differentiation. Our findings underscore the importance of chromatin conformation as a critical epigenetic mechanism of tumor initiation in LGA and likely other malignancies.
EXPERIMENTAL PROCEDURES
Detailed methods can be found in the Supplementary Experimental Procedures section. Human ESCs with a HES5::GFP reporter (Placantonakis et al., 2009) were used to derive NSCs and their progeny (astrocytes, neurons and oligodendrocytes). All hESC experiments were approved by NYU’s ESCRO (protocol #14-00267). NSCs were maintained in medium containing EGF and FGF2. Stable NSC lines were generated by delivering transgenes with lentiviruses. Standard techniques and analysis was used for flow cytometry, extreme limiting dilution sphere assays, qPCR, immunoblot, immunofluorescence and IHC. Animal procedures were performed according to IACUC protocol 160403 at NYU. For xenograft invasion assays, male NOD.SCID mice (6–8 weeks) were stereotactically injected with 2.5×105 NSCs in the frontal lobe, as previously described (Bayin et al., 2014). Xenografts were analyzed 4 weeks post injection. Images were processed wiith ImageJ software.
NSCs were harvested in self-renewing conditions for RNA-seq, DNA methylation profiling with Illumina 450K arrays, MS and 4C-seq. RNA-seq and DNA methylation data from our NSCs were compared to TCGA data (Cancer Genome Atlas Research et al., 2015). Data were visualized with R software suite (www.r-project.org). To generate a 4C library for sequencing, we adapted our recently described protocol (Raviram et al., 2016; Rocha et al., 2016) to capture interactions from the SOX2 promoter.
CTCF ChIP-seq data from 2 grade III mutant IDH and 2 grade IV wild-type IDH datasets (GSE70991), as in Flavahan et al. (2016), were used to identify CTCF binding sites near the SOX2 locus. All tracks were plotted on the same intensity scale. We visualized the 25-state imputed chromHMM model that uses integrative data from 127 cell lines that had at least five different chromatin marks used to annotate the epigenome (Roadmap Epigenomics et al., 2015). For HiC data (GSE35156 and GSE52457), publicly available (Dixon et al., 2015; Dixon et al., 2012) datasets from lung fibroblasts (IMR90), H1 hESCs and NSCs were used (Lazaris et al., 2017).
Statistical comparisons were performed with t-test, χ2 test, and ANOVA (one- and two-way). Statistical significance was set at p<0.05. Significant ANOVA comparisons were followed by post hoc Tukey’s test, Dunnett’s test, or the Benjamini false discovery rate correction. Statistical analyses were performed using Prism 7 (Graphpad) software. The n value signifies biological replicates. Statistics for RNA-seq, 450k methylation array and 4C analysis were performed using built-in statistical methods in bioinformatics packages and/or the R software suite (www.r-project.org).
Supplementary Material
HIGHLIGHTS.
R132H IDH1 and P53/ATRX knockdown block differentiation of human neural progenitors
The differentiation block is mediated by transcriptional silencing of SOX2
The SOX2 promoter disassociates from its enhancer due to disrupted chromatin looping
Reduced binding of CTCF to hypermethylated DNA motifs alters chromatin conformation
Acknowledgments
We thank these core facilities at NYU: Genome Technology, Applied Bioinformatics, High Performance Computing, Flow Cytometry, Microscopy, and Experimental Pathology. We also thank John Dankert, Michele Pagano, David Levy, Markus Schober, Susan Smith and Zharko Daniloski for reagents; and Alka Mansukhani for helpful discussions.
Funding sources: ASM, NIH T32CA009161; PPR, NIH R01GM086852; NSB, NYSTEM CO26880; LC, NIH P30CA016087, S10OD01058 and S10OD018338; GN, NIH P30CA008748; JW, NIH R01GM115384; MK and MS, Friedberg Charitable Foundation; RB, Simons Foundation; DR, Howard Hughes Medical Institute, NIH RO1GM064844; AT, ACS RSG-15-189-01-RMC, Leukemia & Lymphoma Society (8007-17), NIH P30CA016087; JAS, NIH R01GM086852, R01GM112192 and R21CA188968; TN, NIH R03NS087349, P30NS050276, 100 Women in Hedge Funds Foundation for Cancer; DGP, NIH R03NS087349, R21NS087241, R21NS088775, P30CA016087, UL1TR000038, and the Grace Jones Richardson Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
RNA-seq, methylation array and 4C-seq data can be accessed in the GEO database (www.ncbi.nlm.nih.gov/geo/) accession number GSE94962.
AUTHOR CONTRIBUTIONS
Conceptualization, ASM, DGP; Methodology, ASM, RAB, DR, AT, DZ, MS, JAS, TAN, DGP; Investigation, ASM, DG, TK, DB, JP, CB, JD, GZ, PPR, RR, CL, JMS, GL, MK, JD, NSB, JF, JS, LC, RB, GN, ASC, JGG, JW, MAK, DZ, DGP; Writing – Original Draft, AM, DGP; Writing – Review & Editing, ASM, PPR, CL, NSB, AT, MS, JAS, TAN, DGP.; Funding Acquisition, TAN, DGP; Supervision, RAB, DR, AT, DZ, MS, JAS, TAN, DGP.
Supplemental File Set: RNA-seq clusters, Related to Figure 4
References
- Amador-Arjona A, Cimadamore F, Huang CT, Wright R, Lewis S, Gage FH, Terskikh AV. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E1936–1945. doi: 10.1073/pnas.1421480112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43:1262–1265. doi: 10.1038/ng.994. [DOI] [PubMed] [Google Scholar]
- Annovazzi L, Mellai M, Caldera V, Valente G, Schiffer D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genomics Proteomics. 2011;8:139–147. [PubMed] [Google Scholar]
- Baer K, Eriksson PS, Faull RL, Rees MI, Curtis MA. Sox-2 is expressed by glial and progenitor cells and Pax-6 is expressed by neuroblasts in the human subventricular zone. Exp Neurol. 2007;204:828–831. doi: 10.1016/j.expneurol.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L, et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell. 2016;30:578–594. doi: 10.1016/j.ccell.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayin NS, Modrek AS, Dietrich A, Lebowitz J, Abel T, Song HR, Schober M, Zagzag D, Buchholz CJ, Chao MV, Placantonakis DG. Selective lentiviral gene delivery to CD133-expressing human glioblastoma stem cells. PloS one. 2014;9:e116114. doi: 10.1371/journal.pone.0116114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- Bonnet C, Thomas L, Psimaras D, Bielle F, Vauleon E, Loiseau H, Cartalat-Carel S, Meyronet D, Dehais C, Honnorat J, et al. Characteristics of gliomas in patients with somatic IDH mosaicism. Acta Neuropathol Commun. 2016;4:31. doi: 10.1186/s40478-016-0302-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. The New England journal of medicine. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Sato M, Aldape K, Mason CC, Alfaro-Munoz K, Heathcock L, South ST, Abegglen LM, Schiffman JD, Colman H. DNA copy number analysis of Grade II–III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun. 2015;3:34. doi: 10.1186/s40478-015-0213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edri R, Yaffe Y, Ziller MJ, Mutukula N, Volkman R, David E, Jacob-Hirsch J, Malcov H, Levy C, Rechavi G, et al. Analysing human neural stem cell ontogeny by consecutive isolation of Notch active neural progenitors. Nat Commun. 2015;6:6500. doi: 10.1038/ncomms7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, Zhang Y, Ling EA, Gao J, Hao A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology. 2011;59:763–775. doi: 10.1111/j.1365-2559.2011.03993.x. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaris C, Kelly S, Ntziachristos P, Aifantis I, Tsirigos A. HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC genomics. 2017;18:22. doi: 10.1186/s12864-016-3387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rivera CM, Ishii H, Jin F, Selvaraj S, Lee AY, Dixon JR, Ren B. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PloS one. 2014;9:e114485. doi: 10.1371/journal.pone.0114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Han SS, Wu Y, Tuohy TM, Xue H, Cai J, Back SA, Sherman LS, Fischer I, Rao MS. CD44 expression identifies astrocyte-restricted precursor cells. Dev Biol. 2004;276:31–46. doi: 10.1016/j.ydbio.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, Boyer LA. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet. 2013;9:e1003288. doi: 10.1371/journal.pgen.1003288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YH, Mentlein R, Knerlich F, Kruse ML, Mehdorn HM, Held-Feindt J. Expression of stem cell markers in human astrocytomas of different WHO grades. Journal of neuro-oncology. 2008;86:31–45. doi: 10.1007/s11060-007-9439-7. [DOI] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nature reviews Molecular cell biology. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk JG, Verbeke SL, Meijer D, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirozzi CJ, Carpenter AB, Waitkus MS, Wang CY, Zhu H, Hansen LJ, Chen LH, Greer PK, Feng J, Wang Y, et al. Mutant IDH1 Disrupts the Mouse Subventricular Zone and Alters Brain Tumor Progression. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-16-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placantonakis DG, Tomishima MJ, Lafaille F, Desbordes SC, Jia F, Socci ND, Viale A, Lee H, Harrison N, Tabar V, Studer L. BAC transgenesis in human embryonic stem cells as a novel tool to define the human neural lineage. Stem Cells. 2009;27:521–532. doi: 10.1634/stemcells.2008-0884. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy M, Smith S. Loss of ATRX Suppresses Resolution of Telomere Cohesion to Control Recombination in ALT Cancer Cells. Cancer Cell. 2015;28:357–369. doi: 10.1016/j.ccell.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviram R, Rocha PP, Muller CL, Miraldi ER, Badri S, Fu Y, Swanzey E, Proudhon C, Snetkova V, Bonneau R, Skok JA. 4C-ker: A Method to Reproducibly Identify Genome-Wide Interactions Captured by 4C-Seq Experiments. PLoS Comput Biol. 2016;12:e1004780. doi: 10.1371/journal.pcbi.1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Berube NG. Loss of ATRX leads to chromosome cohesion and congression defects. The Journal of cell biology. 2008;180:315–324. doi: 10.1083/jcb.200706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha PP, Raviram R, Fu Y, Kim J, Luo VM, Aljoufi A, Swanzey E, Pasquarella A, Balestrini A, Miraldi ER, et al. A Damage-Independent Role for 53BP1 that Impacts Break Order and Igh Architecture during Class Switch Recombination. Cell Rep. 2016;16:48–55. doi: 10.1016/j.celrep.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosiak K, Smolarz M, Stec WJ, Peciak J, Grzela D, Winiecka-Klimek M, Stoczynska-Fidelus E, Krynska B, Piaskowski S, Rieske P. IDH1R132H in Neural Stem Cells: Differentiation Impaired by Increased Apoptosis. PloS one. 2016;11:e0154726. doi: 10.1371/journal.pone.0154726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes & development. 2012;26:2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seah C, Levy MA, Jiang Y, Mokhtarzada S, Higgs DR, Gibbons RJ, Berube NG. Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:12570–12580. doi: 10.1523/JNEUROSCI.4048-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorska M, Sandhu JK, Deb-Rinker P, Jezierski A, Leblanc J, Charlebois C, Ribecco-Lutkiewicz M, Bani-Yaghoub M, Walker PR. Epigenetic modifications of SOX2 enhancers, SRR1 and SRR2, correlate with in vitro neural differentiation. Journal of neuroscience research. 2008;86:1680–1693. doi: 10.1002/jnr.21635. [DOI] [PubMed] [Google Scholar]
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar V, Panagiotakos G, Greenberg ED, Chan BK, Sadelain M, Gutin PH, Studer L. Migration and differentiation of neural precursors derived from human embryonic stem cells in the rat brain. Nature biotechnology. 2005;23:601–606. doi: 10.1038/nbt1088. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome research. 2012;22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59:4175–4179. [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.