

Abstract
Inflammatory pathways drive atherogenesis and link traditional risk factors to atherosclerosis and its complications. One inflammatory mediator has come to the fore as a therapeutic target in cardiovascular disease. The experimental and clinical evidence reviewed here support interleukin-1 beta (IL-1β) as both a local vascular and systemic contributor in this regard. Intrinsic vascular wall cells and lesional leukocytes alike can produce this cytokine. Local stimuli in the plaque favor the generation of active IL-1β through the action of a molecular assembly known as the inflammasome. Clinically applicable interventions that interfere with IL-1 action can improve cardiovascular outcomes, ushering in a new era of anti-inflammatory therapies for atherosclerosis. The translational path described here illustrates how advances in basic vascular biology may transform therapy. Biomarker-directed application of anti-inflammatory interventions promises to help us achieve a more precise and personalized allocation of therapy for our cardiovascular patients.
Inflammatory and Immune Mechanisms Participate in Atherothrombosis
Multiple strands of evidence, experimental and clinical, implicate inflammation in atherogenesis and its complications (1–8). Inflammatory status, monitored by C-reactive protein (CRP) concentrations measured with a high-sensitivity assay (hsCRP), can sharpen the prediction of first ever and recurrent cardiovascular events beyond that predicted by traditional risk algorithms (9,10). Targeting therapies with anti-inflammatory actions based on inflammatory status can reduce cardiovascular events (11). Reductions in events by statins may result in part from muting inflammation both by reduction in LDL and direct effects (12–14).
Despite these strong indications of the involvement of inflammation in atherosclerosis and the benefit of anti-inflammatory therapies, the use of agents such as the statins, which alter both the causal risk factor LDL and inflammatory pathways, cannot demonstrate unequivocally that a therapy that targets inflammation per se can reduce risk of atherosclerotic events. Moreover, high-dose statin treatment and other currently standard measures only prevent a fraction of recurrent events in survivors of myocardial infarction (MI) (15–17). This residual burden of events presents a pressing unmet medical need.
Cytokines Serve as Key Messengers of Inflammatory Signaling
Inflammation does not necessarily primarily trigger atherogenesis, but transduces the atherogenic effects of classical risk factors (8). Cytokines encompass a class of proteins that mediate inflammation and modulate immunity. One group of cytokines, the interleukins, bear that name because they were thought to mediate signaling between white blood cells (hence the name inter-leukin) (18). Cytokines contribute critically to atherosclerosis, among other inflammatory diseases (8,19,20).
Interleukin-1: A Primordial Pro-Inflammatory Cytokine
As indicated by its name, interleukin-1 (IL-1) figures among the first cytokines recognized. Several pathways converged on the identification of the molecules we now denote IL-1. The quest to identify the transferable sterile factor that induces fever (endogenous pyrogen activity) guided the purification of the protein we now know as IL-1 (21,22). The cloning of IL-1 cDNAs identified two related but functionally distinct isoforms: IL-1α and IL-1β(18) (Table 1). This pair of mediators has myriad effects in host defenses and in the pathogenesis of a wide variety of diseases. Notable effects of IL-1 on many cell types include the induction of prostaglandin production through the induction of cyclooxygenase-2 (COX-2); the elaboration of nitric oxide by elevation of levels of the inducible isoform of nitric oxide synthase (iNOS); induction of the expression of many cytokines, including augmenting its own gene transcription; the increased expression of leukocyte adhesion molecules and thrombogenic mediators; and the activation of cells involved in innate immunity, prominently including the mononuclear phagocytes (23) (Figure 1).
Table 1.
The IL-1 and receptor family members
| Cytokine: | Comments: |
|---|---|
| Interleukin-1 alpha (IL-1α) | IL-1α generally remains associated with the cell surface or is released by dying cells and usually acts at short distances by juxtacrine or paracrine signaling |
| Interleukin-1 beta (IL-1β) | IL-1β acts primarily extracellularly as a soluble mediator |
| Interleukin-1 receptor antagonist (IL-ra) | IL-1ra blocks the signaling receptor that binds either IL-1 α or β |
| Receptors: | |
| Interleukin-1 receptor 1 (IL-r1) | IL-1 receptor 1 binds both isoforms |
| Interleukin-1 receptor 2 (IL-r2) | IL-1 receptor 2 does not signal and thus acts as an inhibitory decoy |
Figure 1. Selected actions of IL-1 related to atherosclerosis.
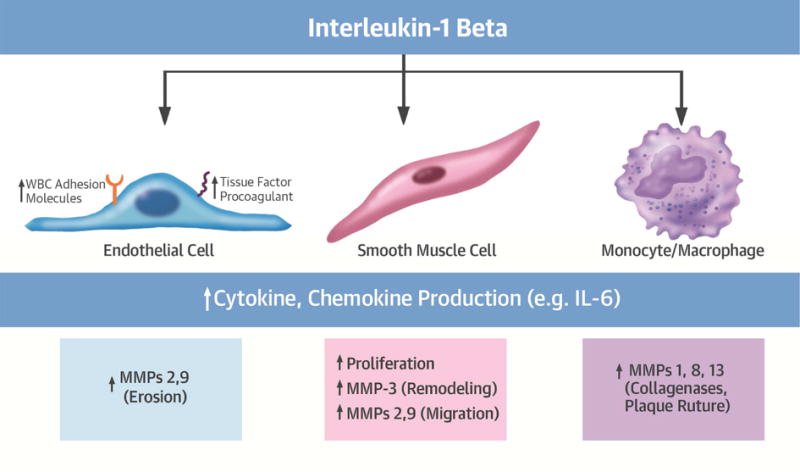
IL-1β acts on many cell types and organs, including those involved in atherogenesis such as vascular endothelial and smooth muscle cells, and macrophages. IL-1β elicits the functions shown, among many others. Blockade of IL-1 induced MMP-2 (Type IV collagenase) may contribute to the marked drop in cancer incidence and mortality seen in CANTOS by impeding tumor invasion through the collagen IV-rich basement membrane and thus limiting metastasis. MMP: matrix metalloproteinase, WBC: White blood cell.
The Interleukin-1 Family and Its Regulation
As often in the case of pluripotent biological mediators, multiple levels of regulation control IL-1 action. The IL-1 family includes the structurally related IL-1 receptor antagonist (IL-1ra) (Table 1).(18) The balance between the pro-inflammatory IL-1α and IL-1β isoforms and this endogenous inhibitor (IL-1ra) represents one important level of control. The IL-1α and IL-1β isoforms have distinct functional profiles. IL-1α typically resides on the cell surface and signals at short distances by direct contact. In contrast, IL-1β can act at a distance.
Two receptors bind the principal members of the IL-1 family. IL-1 receptor I transduces IL-1β signaling (Table 1). In contrast, the IL-1 receptor II (IL-1RII) binds the ligands but does not signal, as it lacks a cytoplasmic domain. IL-1RII thus functions as a “decoy,” providing yet another level of negative regulation of IL-1 signaling. IL-1 can induce its own gene expression in many cell types, including those implicated in atherogenesis (Figure 2A) (24,25). In the case of IL-1α or IL-1β inducing themselves or each other, a positive feedback loop pertains (Figure 2B) (26). IL-1α and IL-1β can also increase the expression of IL-1ra, enabling negative feedback inhibition—a mechanism that prevents untrammeled IL-1 signaling.
Figure 2. IL-1 activates human vascular wall cells. A: Human endothelial cells express IL-1β after exposure to bacterial endotoxin.
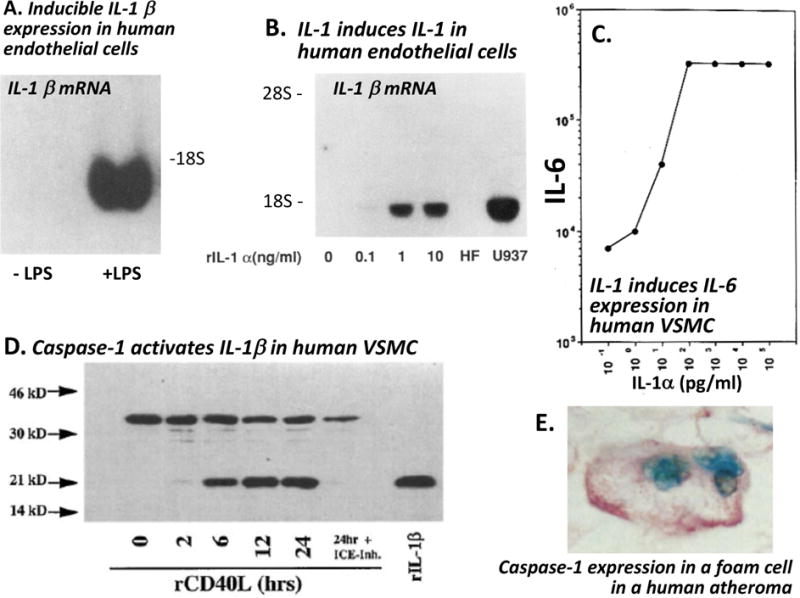
Cultured human endothelial cells were exposed to E. coli endotoxin (LPS) for 24 hours. IL-1β mRNA was evaluated by Northern blotting (42). B: IL-1 induces IL-1 in human vascular cells. Cultured human endothelial cells were exposed to recombinant IL-1α at the concentrations noted for six hours. IL-1β messenger RNA was visualized by Northern blotting.(25) Such auto induction of interleukin one isoforms occurs in human vascular smooth muscle cells and in macrophages as well (24,120). Either IL-1 isoform can induce the other or itself. C. IL-1 induces IL-6 expression in human vascular school muscle cells (VSMC.) Human the SMC were exposed to IL-1α at the concentration shown for 24 hours. IL-6 activity released by the cells is reported in arbitrary units on a logarithmic scale. IL-1 also induces IL-6 in human endothelial cells.(121). D. Caspase-1 activates IL-1β in human VSMC stimulated with recombinant CD40 ligand (rCD40L.) The 33 kD pro-IL-1β concentration declines as the active 17 kD mature form accumulates. The conversion depends on caspase-1 as shown by treatment with the caspase-1/IL-1β converting enzyme (ICE) inhibitor ZVAD (122). E. Foam cells in a human atheroma expresses caspase-1. This representative view of typical human atherosclerotic plaque shows immunohistochemical staining for caspase-1, the IL-1β converting enzyme (pink reaction product.)(27) (Republished with permission from the following sources: 1) Libby P, Ordovàs JM, Auger KR, Robbins H, Birinyi LK, Dinarello CA. Endotoxin and tumor necrosis factor induce interleukin-1 gene expression in adult human vascular endothelial cells. Am J Path 1986;124:179–186. 2) Geng Y-J, Libby P. Evidence for apoptosis in advanced human atheroma. Co-localization with interleukin-1 b-converting enzyme. Am J Pathol 1995;147:251–266. 3) Loppnow H, Libby P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol 1989;122:493–503. 4)Warner SJC, Auger KR, Libby P. Interleukin-1 induces interleukin-1. II. Recombinant human interleukin-1 induces interleukin-1 production by adult human vascular endothelial cells. J Immunol 1987;139:1911–1917. Copyright © [1987] The American Association of Immunologists, Inc. 5)Dinarello CA, Ikejima T, Warner SJC et al. Interleukin-1 induces interleukin-1. I. Induction of circulating interleukin-1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 1987;139:1902–1910. Copyright © [1987] The American Association of Immunologists, Inc. 6)Warner SJC, Auger KR, Libby P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J Exp Med 1987;165:1316–1331. 7)Schoenbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P. Ligation of CD40 activates interleukin 1beta-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1beta. Journal of Biological Chemistry 1997;272:19569–19574. © the American Society for Biochemistry and Molecular Biology.)
The Inflammasome Activates Interleukin-1β
In contrast to IL-1α, the beta isoform requires processing to exert its biological actions. Several levels of control regulate the activation of IL-1β. Pro-IL-1β requires proteolytic processing to produce mature IL-1β that possesses biological activity (Figure 3). The proteolytic enzyme responsible for producing the most mature, secreted IL-1β, first called IL-1β-converting enzyme, now bears the name caspase-1. This enzyme cleaves the 33 kD precursor of IL-1 to the active 17 kD form, analogous to processing of angiotensin I to the active angiotensin II by angiotensin-converting enzyme (Figure 3). Macrophages in human atherosclerotic plaques contain caspase-1 (Figure 2E) (27).
Figure 3. The inflammasome senses danger signals and activates IL-1 beta by proteolytic processing to the mature form by caspase 1.
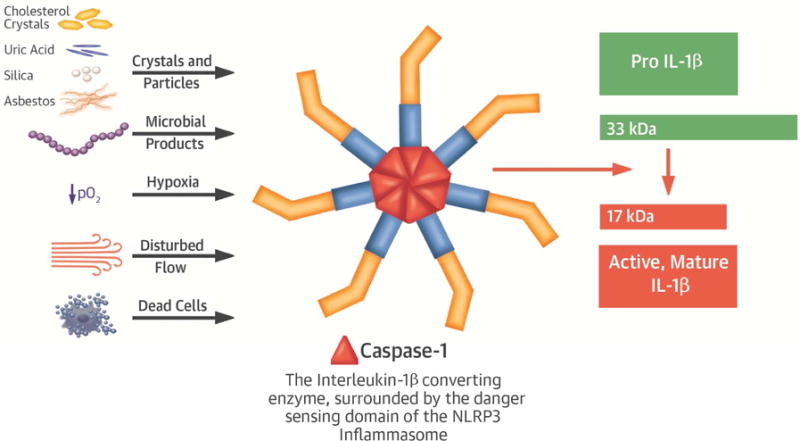
Transcription of the interleukin-1 (IL-1) beta (β) gene produces pro-IL-1β, a 33 kiloDalton (kD) protein that lacks biological activity. This precursor undergoes cleavage by an enzyme known as IL-1β converting enzyme (ICE) or caspase-1, to produce the active form of the cytokine with a molecular weight of 17 kD. The activity of caspase 1 depends on the NLRP3 inflammasome that senses danger signals such as those shown.
Caspase-1 in turn undergoes activation by a supramolecular assembly known as the inflammasome (Figure 3) (28,29). The inflammasome transduces inflammatory signals, culminating in cleavage of pro-caspase-1 to its active form, that in turn activates IL-1β. Certain rare genetic diseases demonstrate the importance of the inflammasome in control of the overwhelming pro-inflammatory potential of the active cytokine. Muckle-Wells syndrome and cryopyrin-associated periodic syndrome (CAPS) arise from gain-of-function mutations in the inflammasome (30). These mutations markedly augment IL-1β maturation and produce debilitating inflammatory diseases in children, indubitably due to overproduction of active IL-1β.
The inflammasome generally requires two signals to assemble and act (28). The co-activators or triggers of the inflammasome include components of infectious agents. Crystals can also co-stimulate the inflammasome. For example, urate crystals implicated in the pathogenesis of gout also act via the inflammasome to enhance mature IL-1β production (31). Cholesterol crystals also furnish a signal to the inflammasome (32,33). Human atheromata harbor activated inflammasomes (34). Other atherosclerosis-relevant stimuli that can augment inflammasome action include disturbed flow (such as prevails at sites of predilection to atheroma formation and complication) (35,36), and moderate hypoxia and acidosis (common in the core of plaques) (37,38).
Interleukin-1 Has Manifold Effects on the Cardiovascular System
A wealth of data implicates IL-1 in a number of cardiovascular diseases.(39,40) IL-1 induces inflammatory functions of human endothelial cells (ECs) (Figure 1).(41) IL-1 stimulates adhesion molecules that recruit leukocytes including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). IL-1 also induces chemokines such as MCP-1 (CCL-2), a chemoattractant for mononuclear phagocytes strongly implicated in inflammatory cardiovascular diseases. Cells in the atheroma produce IL-1 when exposed to inflammatory stimuli (Figure 2) (42,43). These observations led to the explicit hypothesis in the mid-1980s that IL-1 participates in atherogenesis (42).
IL-1 has multiple effects on human vascular smooth muscle cells (SMCs), a cell type intimately involved in atherogenesis (Figure 1). IL-1 can induce autocrine production of platelet-derived growth factor that can stimulate SMC proliferation.(44) IL-1 induces its own gene expression in many cell types, including ECs and SMCs, a powerful amplification loop denoted “autoinduction” (Figure 3) (24,25).
Treatment with IL-1 strongly induces SMCs and other cells to elaborate another cytokine, interleukin-6 (IL-6) (Figures 2C & 4).(45) IL-6 elicits the acute phase response: hepatocytes stimulated by IL-6 boost their synthesis of acute phase reactants, including fibrinogen and plasminogen activator inhibitor—molecules intimately involved in promoting thrombosis (fibrinogen) or limiting fibrinolysis (plasminogen activator inhibitor, PAI-1), as well as of CRP, a marker of inflammatory status (Figure 4) (46,47).
Figure 4. Amplification cascades boost IL-1 actions implicated in atherothrombosis.
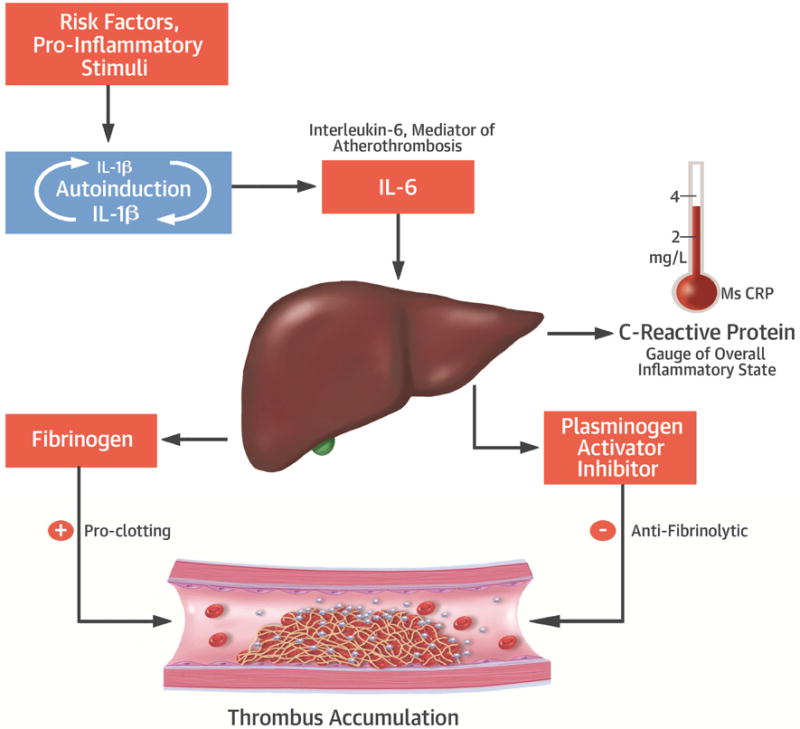
nterleukin-1 (IL-1) can induce its own gene expression in many cell types, including those implicated in atherosclerosis. This property provides a positive feedback amplification loop for boosting IL-1 levels at sites of inflammation. IL-1 also strongly augments the production of IL-6 by various cell types. IL-6 mediates the acute phase response through which the liver produces proteins that participate in host defenses, but that also augments thrombosis and inhibits fibrinolysis as explained in the text.
IL-1 therefore mediates an amplification loop whereby a single molecule of IL-1 can beget many molecules of IL-6, which in turn drive the overexpression of atherothrombosis mediators, such as those listed above (Figure 2C). Human genetic studies support causality of IL-6 in coronary heart disease (48,49). Thus, IL-1 resides upstream in the pathway that induces IL-6, a causal cytokine in atherothrombosis (Figure 4).
IL-1 alters functions of cardiac myocytes in addition to those of cells in the blood vessel wall. IL-1 impairs contractile function. This cytokine can aggravate ischemia-reperfusion injury and expansive cardiac remodeling after experimental MI (50,51). Experimental and small human studies suggest that antagonism of IL-1 can also benefit expansive remodeling after human MI, and limit the release of CRP following acute coronary syndromes (ACS) (52,53,54).
Numerous experiments involving genetically induced loss-of-function or gain-of-function of IL-1, manipulation of IL-1ra, and pharmacologic inhibition of IL-1 strongly implicate this cytokine in atherogenesis. Peri-adventitial application of IL-1 aggravates intimal thickening in pig arteries, implicating IL-1 in arterial hyperplasia, and IL-1 inhibition limits this response to injury (55,56). In hyperlipidemic mice, IL-1 generally promotes lesion formation, and interruption of IL-1 signaling limits atherogenesis (57–60). Deficiency of IL-1ra in mice augments arterial inflammation (61) and aneurysm formation (62), and hemizygous deficiency of IL-1ra limits early atherosclerosis and reduces macrophage content in hyperlipidemic mice.(63) IL-1 receptor 1–deficient hyperlipidemic mice have impaired expansive remodeling during lesion formation, attributable to reduced expression of the matrix metalloproteinase MMP-3 (stromelysin) (Figure 1) (64). Activated platelets can express IL-1α and elaborate microparticles that bear functional IL-1β, another link between this cytokine and atherothrombosis (65,66). The preponderance of data suggest a prominent contribution of IL-1 to the pathogenesis of multiple cardiovascular diseases (67).
Therapeutic Targeting of Interleukin-1β
Given the wealth of findings that implicates inflammation in atherogenesis, and the residual burden of recurrent cardiovascular events in survivors of acute coronary syndromes—even when they receive contemporary standard medical care including high-dose statins, potent anti-platelet combinations, and late generation stenting—the proposition that direct anti-inflammatory therapy could improve outcomes in such patients takes on considerable clinical urgency, beyond its theoretical interest. IL-1β’s convincing links to pro-inflammatory diseases such as atherosclerosis highlighted this cytokine as a potential therapeutic target (68,69).
Several strategies can combat IL-1 action. Administration of the decoy receptor has not undergone substantive clinical development. The IL-1ra (anakinra) has conferred clinical benefit in some cases of rheumatoid arthritis, acute gouty arthritis, and diabetes mellitus. Anakinra requires daily injections, and blocks the actions of both IL-1 isoforms, potentially impairing host defenses and hence susceptibility to infection to a greater extent than selective isoform inhibition. In contrast to the IL-1ra, canakinumab, a human monoclonal antibody, selectively neutralizes IL-1β but not IL-1α, whereas IL-1ra blocks both. An anti-IL-1α antibody has also entered clinical evaluation (70). Canakinumab benefits inflammatory diseases mediated by IL-1β, including Muckle-Wells syndrome (71), rarer forms of juvenile inflammatory arthritis (72), and common diseases such as acute gouty arthritis (73). Canakinumab has a prolonged biological half-life that permits subcutaneous dosing every 3 months. Both anakinra and canakinumab appear well tolerated in most patients, although studies of the IL-1ra have not evaluated infection risk with long-term exposure of large populations. Moreover, as atherosclerosis-related stimuli activate the inflammasome that generates mature IL-1β but not IL-1α, the β isoform was chosen as the most disease-relevant target. In a phase II study of diabetic patients, canakinumab dose-dependently reduced plasma levels of fibrinogen, IL-6, and hsCRP.(74).
The Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS): Affirmation of the Role of Inflammation in Atherothrombosis
The strong biological basis presented above provided the rationale for a large-scale clinical trial that tested the hypothesis that administration of canakinumab to neutralize IL-1β activity could improve outcomes in individuals who have sustained a prior MI.(75) CANTOS enrolled over 10,000 individuals at least one month post-acute MI in almost 40 countries. The protocol mandated aggressive use of all standard secondary prevention therapies for MI, including treatment with high-dose statins, aspirin and other anti-platelet agents, beta-adrenergic blocking agents, and agents that interrupt renin-angiotensin signaling, according to prevailing guidelines. The patients enrolled had residual inflammation despite these guideline-mandated secondary prevention measures, as indicated by levels of hsCRP above 2.0 mg/L. Stable post-MI patients eligible for CANTOS randomly received placebo or one of three doses of canakinumab, administered quarterly subcutaneously. The primary endpoint included non-fatal MI, non-fatal stroke, or cardiovascular death. The trial ended when approximately 1,400 events accrued in early 2017.
Canakinumab 150 mg subcutaneously every 3 months met CANTOS’ primary endpoint with a 15 % reduction of non-fatal MI, non-fatal stroke, or cardiovascular death (76). The 150 mg and 300 mg doses had similar effects on this endpoint, yielding p=0.007 when combined. A pre-specified expanded endpoint that included unstable angina requiring urgent revascularization showed a significant 17% reduction for the combined 150 mg and 300 mg groups. The 50 mg dose did not significantly lower events. The anti-IL-1β treatment did not alter LDL, but the two higher doses lowered hsCRP by about 60%.
These results affirm the clinical importance and therapeutic relevance of the biological aspects of IL-1β reviewed herein. Moreover, as the first large-scale blinded and placebo-controlled, randomized clinical trial that targeted inflammation but not lipids, CANTOS affirms the inflammatory hypothesis of atherosclerosis and sets the stage for a new era of cardiovascular therapeutics. Furthermore, the CANTOS results highlight the utility of assessing both LDL and hsCRP post ACS, as targeting either of these biomarkers can reduce recurrent events.
Indeed, we have entered an exciting era of “precision medicine,” which permits the allocation of therapies rationally based on readily measured biomarkers. Patients following ACS with LDL levels that remain above those desired despite statin therapy should receive consideration for adding a cholesterol absorption inhibitor (as validated in IMPROVE-IT (77)) or a PCSK9 inhibitor (as validated in FOURIER (78)). These patients represent residual LDL risk. Those who have persistently elevated inflammation despite a standard of care regimen, as gauged by an hsCRP greater than 2 mg/L, could receive canakinumab. These patients could be categorized as having residual inflammatory risk (79,80). Of note, FOURIER and CANTOS achieved a similar magnitude of event reduction on top of statins by targeting distinct aspects of residual risk.
Beyond CANTOS: Other Targets, Other Indications
IL-1β represents only one of several potential avenues for targeting inflammation in atherosclerosis directly. As inflammatory signaling pathways have considerable redundancy, targeting any one pathway (such as IL-1β signaling) may not block all inflammatory pathways implicated in atherogenesis. Following the success of CANTOS, a suite of studies should evaluate the role of other measures that manipulate inflammation and immunity in clinical scenarios beyond stable patients with prior myocardial infarction.
In this regard, some confusion has prevailed regarding interventions that target inflammation per se versus oxidative stress (81). The enzyme lipoprotein-associated phospholipase A2 (LP-PLA2) indeed arises primarily from inflammatory cells (e.g. macrophages) but acts to generate pro-oxidant lipid species. Two large clinical trials targeted LP-PLA2 with a small molecule inhibitor (losmapimod) but failed to reduce cardiovascular events (82,83). Thus, these trials did not vitiate the inflammatory hypothesis as they focused primarily on reducing oxidative stimuli rather than inflammation itself. An inhibitor of p38 MAP kinase (losmapimod) likewise failed to reduce cardiovascular events in secondary prevention. Oxidative stress also activates p38 MAP kinase, and its inhibition does not interfere selectively with the IL-6 pathway causally implicated in cardiovascular events (84,85).
Clinical trials currently in progress target inflammation in ways that are less well defined. Colchicine, long used to mitigate certain inflammatory conditions, reduced cardiovascular events in an unblinded, relatively small clinical trial (LoDoCo) (86). Two larger studies currently underway are following up this exciting and intriguing preliminary observation: the COLCOT and LoDoCo2 studies (87). The use of low-dose methotrexate has revolutionized the treatment of rheumatoid arthritis. The ongoing Cardiovascular Inflammation Reduction Trial (CIRT) evaluates whether this anti-inflammatory agent can improve cardiovascular outcomes in an at-risk population (88).
A wealth of experimental work over the last several decades has identified a number of other inflammatory targets with well-characterized mechanisms of action that merit consideration for future clinical investigation. Although IL-1α has distinct cell biological and biochemical characteristics from the β isoform, it shares many pro-inflammatory actions including induction of IL-6. Antibody reagents that neutralize IL-1 alpha have entered clinical trials and hold considerable interest for cardiovascular indications (70,89). Tocilizumab, an antibody that selectively neutralizes IL-6, can treat several inflammatory diseases, but adversely perturbs the lipid profile. Other strategies for neutralizing IL-6 that lack such unwanted actions definitely merit evaluation for cardiovascular indications. A small trial in patients with acute coronary syndromes showed that tocilizumab reduced the excursion of CRP post primary coronary artery intervention (90).
Interleukin 18, a member of the IL-1 family, depends like IL-1β on the inflammasome for activation. Several lines of evidence suggest that IL-18 participates in atherogenesis (91–93) This cytokine also deserves consideration for therapeutic targeting in atherothrombosis. As the inflammasome pathway activates the immature precursors of both IL-1β and IL-18, inhibition of the NLRP3 inflammasome could offer an approach to simultaneous limitation of the action of this pair of pro-inflammatory cytokines.(94–96) Yet, given the potential risks of interfering with host defenses, such dual effects might have more liability than inhibition of a single isoform of a single cytokine.
Considerable data implicate CD40 and its ligand CD154 in cardiovascular and metabolic diseases (97–101). While monoclonal antibody strategies encountered thrombotic complications, a late-generation antisense oligonucleotide approach has promise as an approach to limiting the action of this signaling dyad (102). A small molecule inhibitor of CD40 interaction with its signal transducing partner TRAF also holds interest in this regard (103).
Many studies have implicated tumor necrosis factor (TNF) in inflammatory diseases including atherosclerosis (104,105). Like methotrexate, anti-TNF strategies have proven highly successful in certain rheumatologic and inflammatory diseases. Yet, anti-TNF strategies can adversely affect lipid profiles. Moreover, despite preclinical promise, trials that targeted TNF in patients with heart failure showed no benefit, rather a possible hazard (105). These considerations have limited interest in targeting TNF in cardiovascular patients.
Experimental evidence implicated the monocyte chemoattractant MCP-1 (CCL2) and its receptor CCR2 in atherogenesis (106–108). Currently, research regarding monocyte heterogeneity in cardiovascular diseases has flourished (109–112). In particular, a pro-inflammatory subset of monocytes recruited via the chemokine receptor CCR2 has proven functionally important in a number of cardiovascular conditions. As monoclonal antibodies that can neutralize CCR2 have entered clinical use, they also merit clinical evaluation to quell cardiovascular inflammation.
Any intervention that interferes with innate immunity, such as those delineated above, might increase susceptibility to infections and/or impair tumor surveillance. Therefore, the clinical applications of anti-inflammatory therapies require careful consideration of potential adverse effects. Indeed, CANTOS revealed a small but significant increase in fatal infections (an excess of about 1 per 1000 patient-years) (76). Analysis of infection liability in the over 6,700 patients exposed to canakinumab will inform regarding risk factors for infection and help to mitigate this risk. The decades of experience with anti-inflammatory interventions in rheumatologic and other diseases provide guidance in this regard, as we move towards clinical application in the cardiovascular arena of biological and other strategies to modulate the inflammatory response.
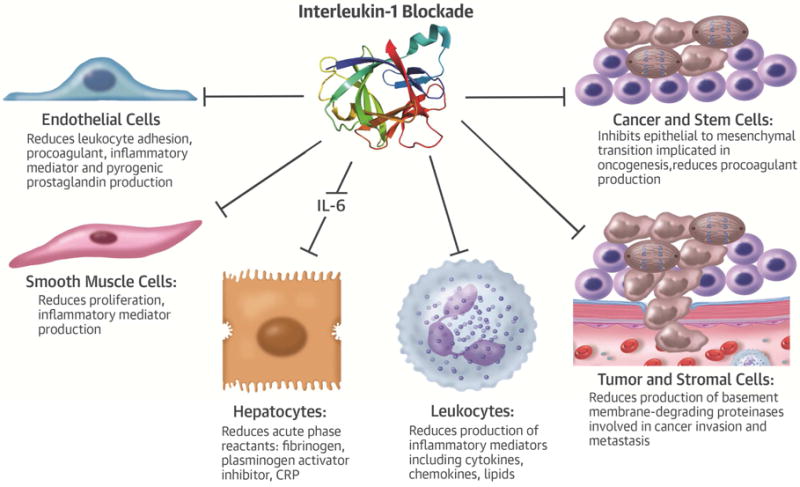
One might have expected an increase in cancer due to impaired tumor surveillance due to treatment with canakinumab, as anti-TNF strategies do entail a risk for lymphoma. Yet, in CANTOS actually showed a substantial and canakinumab-dose-dependent reduction in fatal malignancy (p=0.0007) (113). Canakinumab (300 mg/3 months) produced a 67% fall in incident lung cancer, and a 77% decrease in fatal lung cancer (p=0.0002.) The cancer benefit in this trial unlikely represents an effect on cancer initiation, although IL-1 may contribute to oncogenesis in some circumstances (114). Rather, IL-1 blockade may impede the invasion and metastasis of existing cancers. IL-1 can induce a key proteinase involved in cancer spread, matrix metalloproteinase-2 (MMP-2), implicated in breaching the basement membrane by cancers (Central Illustration) (115). IL-1 can also promote epithelial-to-mesenchymal transition implicated in tumorigenesis (116). Most cancer treatments entail cardiovascular risk. Canakinumab appears capable of reducing risk of cancer fatalities in the inflamed population studied while producing cardiovascular benefit rather than hazard.
Central Illustration. Some Effects of Interleukin-1 Blockade on Cellular Functions.
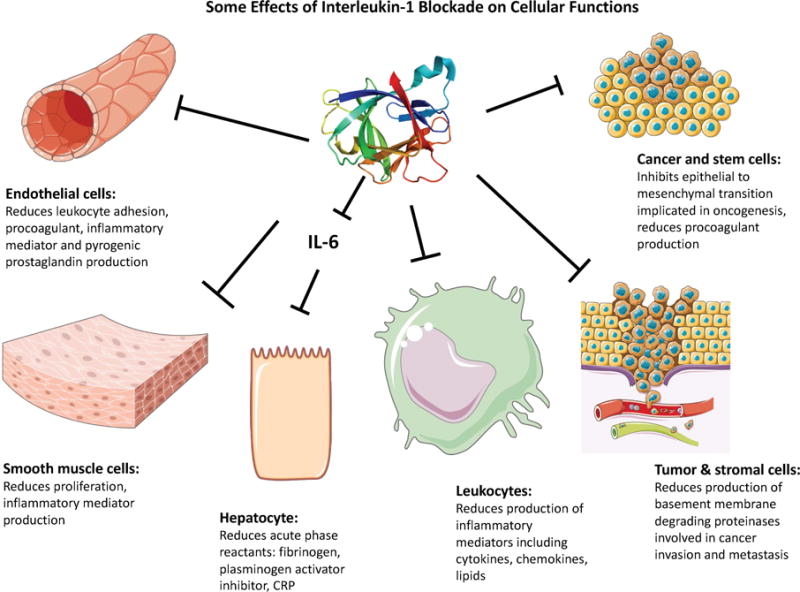
Interleukin-1 exerts many actions on cells that can contribute to pathogenesis diseases including atherosclerosis, thrombosis, oncogenesis, and invasion and metastasis of tumors. Many of the effects of interleukin-1 on hepatocytes, including the induction of the acute phase response, depend on the intermediary of interleukin 6 (IL-6), a cytokine potently induced by interleukin 1. Strong human genetic evidence implicates IL-6 as a causal factor for atherothrombosis.
Beyond the chronic phase of atherosclerosis in secondary prevention, inflammation participates pivotally in other clinical scenarios. Cytokines modulate the healing of acute ischemic myocardial injury experimentally (53). Therefore, some of the anti-cytokine strategies summarized above might foster beneficial healing after an ischemic insult in patients with ACS. For example, treatment with an anti-IL-1 beta antibody limits mobilization of pro-inflammatory monocytes to the infarcted myocardium and improves left ventricular function following coronary artery ligation in mice (117). The data available from the over 1,400 primary endpoint events in CANTOS, many of them ACS, will provide safety data to inform the use of canakinumab administration during ACS, a highly inflamed patient population excluded from CANTOS which enrolled only individuals at least 30 days following an ACS. Interventions that target CCR2 also promote healthier healing of the ischemic myocardium (118).
Conclusion - Entering an Era of Targeting Inflammation in Cardiovascular Diseases
In conclusion, we have entered an exciting era in which we can reap in the clinic the benefits of decades of research into the participation of immune and inflammatory pathways in cardiovascular disease (8). While extrapolation of preclinical studies to humans requires considerable caution (119), the burden of unmet need and residual risk in our patients mandates careful consideration of novel approaches to targeting inflammatory pathways.
Table 2.
Selected targets for limiting cardiovascular inflammation beyond IL-1β
| Inflammasomes |
| IL-1α |
| IL-6 |
| IL-18 |
| IL-33 |
| TNF |
| CD40/CD40L (CD164) |
| TRAF-6 |
| Lipid mediators of resolution |
Condensed Abstract.
Inflammatory pathways drive atherogenesis and link traditional risk factors to atherosclerosis and its complications. Interleukin-1 beta (IL-1β) has emerged as an actionable mediator in prevention of recurrent cardiovascular events. Intrinsic vascular wall cells and lesional leukocytes alike can produce this pro-inflammatory cytokine. Local stimuli in the plaque boost the generation of active IL-1β through the action of a molecular assembly known as the inflammasome. Therapies that interfere with IL-1 action can improve cardiovascular outcomes, ushering in a new era of anti-inflammatory therapies for atherosclerosis. Biomarker-directed application of anti-inflammatory interventions promise to personalize allocation of therapy for our cardiovascular patients.
Acknowledgments
Dr. Libby’s laboratory has received research support from Novartis. Dr. Libby serves as an unpaid member of the scientific advisory board of Olatec. He declines all personal remuneration from pharma and device concerns.
Abbreviations
- hsCRP
high-sensitivity C-eactive protein
- IL
interleukin-1
- MI
myocardial infarction
- ACS
acute coronary syndromes –
- cDNA
complementary DNA
- ICAM-1
intercellular adhesion molecule-1
- VCAM-1
vascular cell adhesion molecule-1
- SMC
smooth muscle cells
- EC
endothelial cells
- MMP
matrix metalloproteinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Libby P. Inflammatory and immune mechanisms in atherogenesis. In: Leaf A, Weber P, editors. Atheroclerosis Reviews. New York: Raven Press; 1990. pp. 79–89. [Google Scholar]
- 2.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–38. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lichtman AH, Binder CJ, Tsimikas S, Witztum JL. Adaptive immunity in atherogenesis: new insights and therapeutic approaches. J Clin Invest. 2013;123:27–36. doi: 10.1172/JCI63108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–93. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby P, Hansson GK. Inflammation and Immunity in Diseases of the Arterial Tree: Players and Layers. Circulation Research. 2015;116:307–311. doi: 10.1161/CIRCRESAHA.116.301313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nus M, Mallat Z. Immune-mediated mechanisms of atherosclerosis and implications for the clinic. Expert Rev Clin Immunol. 2016;12:1217–1237. doi: 10.1080/1744666X.2016.1195686. [DOI] [PubMed] [Google Scholar]
- 7.Weber C, Shantsila E, Hristov M, et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thrombosis And Haemostasis. 2016;116 doi: 10.1160/TH16-02-0091. [DOI] [PubMed] [Google Scholar]
- 8.Libby P. History of discovery: inflammation in atherosclerosis. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridker PM, Baker MT, Hennekens CH, Stampfer MJ, Vaughan DE. Alu-repeat polymorphism in the gene coding for tissue-type plasminogen activator (t-PA) and risks of myocardial infarction among middle-aged men. Arteriosclerosis, Thrombosis & Vascular Biology. 1997;17:1687–90. [PubMed] [Google Scholar]
- 10.Ridker PM. A Test in Context: High-Sensitivity C-Reactive Protein. J Am Coll Cardiol. 2016;67:712–23. doi: 10.1016/j.jacc.2015.11.037. [DOI] [PubMed] [Google Scholar]
- 11.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. New England Journal of Medicine. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 12.Schonbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004;109:II18–26. doi: 10.1161/01.CIR.0000129505.34151.23. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. New England Journal of Medicine. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 14.Jain MK, Sangwung P, Hamik A. Regulation of an inflammatory disease: Kruppel-like factors and atherosclerosis. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2014;34:499–508. doi: 10.1161/ATVBAHA.113.301925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 16.Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. European Heart Journal. 2015;36:1163–70. doi: 10.1093/eurheartj/ehu505. [DOI] [PubMed] [Google Scholar]
- 17.Masoudi FA, Ponirakis A, de Lemos JA, et al. Trends in U.S. Cardiovascular Care: 2016 Report From 4 ACC National Cardiovascular Data Registries. J Am Coll Cardiol. 2017;69:1427–1450. doi: 10.1016/j.jacc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–32. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldmann M. Many cytokines are very useful therapeutic targets in disease. J Clin Invest. 2008;118:3533–6. doi: 10.1172/JCI37346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 21.Dinarello CA. Demonstration of a human pyrogen-inducing factor during mixed leukocyte reactions. J Exp Med. 1981;153:1215–1224. doi: 10.1084/jem.153.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auron PE, Webb AC, Rosenwasser LJ, et al. Nucleotide sequence of human monocyte interleukin-1 precursor cDNA. Proc Natl Acad Sci U S A. 1984;81:7907–7911. doi: 10.1073/pnas.81.24.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annual Review Of Immunology. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 24.Dinarello CA, Ikejima T, Warner SJC, et al. Interleukin-1 induces interleukin-1. I. Induction of circulating interleukin-1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–1910. [PubMed] [Google Scholar]
- 25.Warner SJC, Auger KR, Libby P. Interleukin-1 induces interleukin-1. II. Recombinant human interleukin-1 induces interleukin-1 production by adult human vascular endothelial cells. J Immunol. 1987;139:1911–1917. [PubMed] [Google Scholar]
- 26.Beltrami-Moreira M, Vromman A, Sukhova GK, Folco EJ, Libby P. Redundancy of IL-1 Isoform Signaling and Its Implications for Arterial Remodeling. PLoS ONE. 2016;11:e0152474. doi: 10.1371/journal.pone.0152474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng Y-J, Libby P. Evidence for apoptosis in advanced human atheroma. Co-localization with interleukin-1 b-converting enzyme. Am J Pathol. 1995;147:251–266. [PMC free article] [PubMed] [Google Scholar]
- 28.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel MN, Carroll RG, Galvan-Pena S, et al. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol Med. 2017 doi: 10.1016/j.molmed.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 31.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 32.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paramel Varghese G, Folkersen L, Strawbridge RJ, et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. Journal of the American Heart Association. 2016;5 doi: 10.1161/JAHA.115.003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao H, Lu M, Lin TY, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128:632–42. doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe J, Berk BC. Atheroprone flow activation of the sterol regulatory element binding protein 2 and nod-like receptor protein 3 inflammasome mediates focal atherosclerosis. Circulation. 2013;128:579–82. doi: 10.1161/CIRCULATIONAHA.113.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ Res. 2014;115:875–83. doi: 10.1161/CIRCRESAHA.115.304437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajamaki K, Nordstrom T, Nurmi K, et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem. 2013;288:13410–9. doi: 10.1074/jbc.M112.426254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fearon WF, Fearon DT. Inflammation and cardiovascular disease: role of the interleukin-1 receptor antagonist. Circulation. 2008;117:2577–2579. doi: 10.1161/CIRCULATIONAHA.108.772491. [DOI] [PubMed] [Google Scholar]
- 40.Abbate A, Van Tassell BW, Biondi-Zoccai GG. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. BioDrugs. 2012;26:217–33. doi: 10.1007/BF03261881. [DOI] [PubMed] [Google Scholar]
- 41.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MAJ. Interleukin-1 activation of vascular endothelium effects on procoagulant activity and leukocyte adhesion. Am J Path. 1985;121:393–403. [PMC free article] [PubMed] [Google Scholar]
- 42.Libby P, Ordovàs JM, Auger KR, Robbins H, Birinyi LK, Dinarello CA. Endotoxin and tumor necrosis factor induce interleukin-1 gene expression in adult human vascular endothelial cells. Am J Path. 1986;124:179–186. [PMC free article] [PubMed] [Google Scholar]
- 43.Libby P, Ordovas JM, Birinyi LK, Auger KR, Dinarello CA. Inducible interleukin-1 expression in human vascular smooth muscle cells. J Clin Invest. 1986;78:1432–1438. doi: 10.1172/JCI112732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Libby P, Warner SJC, Friedman GB. Interleukin-1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J Clin Invest. 1988;88:487–498. doi: 10.1172/JCI113346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin-6. J Clin Invest. 1990;85:731–738. doi: 10.1172/JCI114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le JM, Vilcek J. Interleukin 6: a multifunctional cytokine regulating immune reactions and the acute phase protein response. Lab Invest. 1989;61:588–602. [PubMed] [Google Scholar]
- 47.Castell JV, Gomez-Lechon MJ, David M, Fabra R, Trullenque R, Heinrich PC. Acute-phase response of human hepatocytes: regulation of acute-phase protein synthesis by interleukin-6. Hepatology. 1990;12:1179–86. doi: 10.1002/hep.1840120517. [DOI] [PubMed] [Google Scholar]
- 48.Sarwar N, Butterworth AS, Freitag DF, et al. IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379:1205–13. doi: 10.1016/S0140-6736(11)61931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hingorani AD, Casas JP. Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214–24. doi: 10.1016/S0140-6736(12)60110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki K, Murtuza B, Smolenski RT, et al. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104:I308–I3. doi: 10.1161/hc37t1.094871. [DOI] [PubMed] [Google Scholar]
- 51.Abbate A, Salloum FN, Van Tassell BW, et al. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS One. 2011;6:e27923. doi: 10.1371/journal.pone.0027923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbate A, Van Tassell BW, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] American Journal of Cardiology. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abbate A, Dinarello CA. Anti-inflammatory therapies in acute coronary syndromes: is IL-1 blockade a solution? European Heart Journal. 2015;36:337–9. doi: 10.1093/eurheartj/ehu369. [DOI] [PubMed] [Google Scholar]
- 54.Morton AC, Rothman AM, Greenwood JP, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. European Heart Journal. 2015;36:377–84. doi: 10.1093/eurheartj/ehu272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimokawa H, Ito A, Fukumoto Y, et al. Chronic treatment with interleukin-1 beta induces coronary intimal lesions and vasospastic responses in pigs in vivo. The role of platelet-derived growth factor. J Clin Invest. 1996;97:769–76. doi: 10.1172/JCI118476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morton AC, Arnold ND, Gunn J, et al. Interleukin-1 receptor antagonist alters the response to vessel wall injury in a porcine coronary artery model. Cardiovasc Res. 2005;68:493–501. doi: 10.1016/j.cardiores.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 57.Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–4. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 58.Kirii H, Niwa T, Yamada Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2003;23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 59.Chi H, Messas E, Levine RA, Graves DT, Amar S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation. 2004;110:1678–85. doi: 10.1161/01.CIR.0000142085.39015.31. [DOI] [PubMed] [Google Scholar]
- 60.Merhi-Soussi F, Kwak BR, Magne D, et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res. 2005;66:583–93. doi: 10.1016/j.cardiores.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 61.Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191:303–12. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isoda K, Kitagaki M, Niida T, et al. Deficiency of interleukin-1 receptor antagonist promotes spontaneous femoral artery aneurysm formation in mice. Am J Pathol. 2012;180:1254–63. doi: 10.1016/j.ajpath.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 63.Isoda K, Sawada S, Ishigami N, et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2004;24:1068–73. doi: 10.1161/01.ATV.0000127025.48140.a3. [DOI] [PubMed] [Google Scholar]
- 64.Alexander MR, Moehle CW, Johnson JL, et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. 2012;122:70–79. doi: 10.1172/JCI43713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hawrylowicz CM, Santoro SA, Platt FM, Unanue ER. Activated platelets express IL-1 activity. Journal of Immunology. 1989;143:4015–8. [PubMed] [Google Scholar]
- 66.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol. 2011;186:5489–96. doi: 10.4049/jimmunol.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rader DJ. IL-1 and atherosclerosis: a murine twist to an evolving human story. J Clin Invest. 2012;122:27–30. doi: 10.1172/JCI61163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–23. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong DS, Janku F, Naing A, et al. Xilonix, a novel true human antibody targeting the inflammatory cytokine interleukin-1 alpha, in non-small cell lung cancer. Invest New Drugs. 2015;33:621–31. doi: 10.1007/s10637-015-0226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. New England Journal of Medicine. 2009;360:2416–25. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 72.Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. New England Journal of Medicine. 2012;367:2396–406. doi: 10.1056/NEJMoa1205099. [DOI] [PubMed] [Google Scholar]
- 73.Schlesinger N, Alten RE, Bardin T, et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis. 2012;71:1839–48. doi: 10.1136/annrheumdis-2011-200908. [DOI] [PubMed] [Google Scholar]
- 74.Ridker PM, Howard CP, Walter V, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–48. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- 75.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 76.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New England Journal of Medicine. 2017 doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 77.Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. New England Journal of Medicine. 2015 doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 78.Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine. 2017 doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 79.Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. European Heart Journal. 2016 doi: 10.1093/eurheartj/ehw024. [DOI] [PubMed] [Google Scholar]
- 80.Ridker PM. How Common Is Residual Inflammatory Risk? Circulation Research. 2017;120:617. doi: 10.1161/CIRCRESAHA.116.310527. [DOI] [PubMed] [Google Scholar]
- 81.Ridker P. Informative neutral studies matter—why the targeting inflammation with salsalate in cardiovascular disease (tinsal-cvd) trial deserves our attention. JAMA Cardiology. 2016;1:423–424. doi: 10.1001/jamacardio.2016.0604. [DOI] [PubMed] [Google Scholar]
- 82.White HD, Held C, Stewart R, et al. Darapladib for preventing ischemic events in stable coronary heart disease. New England Journal of Medicine. 2014;370:1702–11. doi: 10.1056/NEJMoa1315878. [DOI] [PubMed] [Google Scholar]
- 83.O’Donoghue ML, Braunwald E, White HD, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. Jama. 2014;312:1006–15. doi: 10.1001/jama.2014.11061. [DOI] [PubMed] [Google Scholar]
- 84.O’Donoghue ML, Glaser R, Aylward PE, et al. Rationale and design of the LosmApimod To Inhibit p38 MAP kinase as a TherapeUtic target and moDify outcomes after an acute coronary syndromE trial. Am Heart J. 2015;169:622–630 e6. doi: 10.1016/j.ahj.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 85.O’Donoghue ML, Glaser R, Cavender MA, et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA. 2016 doi: 10.1001/jama.2016.3609. [DOI] [PubMed] [Google Scholar]
- 86.Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–10. doi: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 87.Tardif J-C, L’Allier P. Colchicine Cardiovascular Outcomes Trial (COLCOT) 2017 [Google Scholar]
- 88.Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the cardiovascular inflammation reduction trial: A test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hong DS, Hui D, Bruera E, et al. MABp1, a first-in-class true human antibody targeting interleukin-1alpha in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 2014;15:656–66. doi: 10.1016/S1470-2045(14)70155-X. [DOI] [PubMed] [Google Scholar]
- 90.Kleveland O, Kunszt G, Bratlie M, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. European Heart Journal. 2016 doi: 10.1093/eurheartj/ehw171. [DOI] [PubMed] [Google Scholar]
- 91.Mallat Z, Corbaz A, Scoazec A, et al. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598–603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- 92.Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195:245–57. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang J, Sun C, Gerdes N, et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat Med. 2015;21:820–6. doi: 10.1038/nm.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxidants & redox signaling. 2015;22:1146–61. doi: 10.1089/ars.2014.5989. [DOI] [PubMed] [Google Scholar]
- 95.van Hout GP, Bosch L, Ellenbroek GH, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. European Heart Journal. 2016 doi: 10.1093/eurheartj/ehw247. [DOI] [PubMed] [Google Scholar]
- 96.van der Heijden T, Kritikou E, Venema W, et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice. Arteriosclerosis and Thrombosis: a journal of vascular biology. 2017 doi: 10.1161/ATVBAHA.117.309575. [DOI] [PubMed] [Google Scholar]
- 97.Mach F, Schönbeck U, Sukhova GK, et al. Functional CD 40 Ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for CD40-CD40 ligand signaling in atherosclerosis. Procedings of the National Academy of Sciences (USA) 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 99.Schonbeck U, Mach F, Libby P. CD154 (CD40 ligand) Int J Biochem Cell Biol. 2000;32:687–693. doi: 10.1016/s1357-2725(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 100.Lutgens E, Gorelik L, Daemen MJ, et al. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–6. doi: 10.1038/15271. [DOI] [PubMed] [Google Scholar]
- 101.Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype [see comments] Proc Natl Acad Sci U S A. 2000;97:7464–9. doi: 10.1073/pnas.97.13.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Donner AJ, Yeh ST, Hung G, Graham MJ, Crooke RM, Mullick AE. CD40 Generation 2.5 Antisense Oligonucleotide Treatment Attenuates Doxorubicin-induced Nephropathy and Kidney Inflammation. Mol Ther Nucleic Acids. 2015;4:e265. doi: 10.1038/mtna.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zarzycka B, Seijkens T, Nabuurs SB, et al. Discovery of small molecule CD40-TRAF6 inhibitors. J Chem Inf Model. 2015;55:294–307. doi: 10.1021/ci500631e. [DOI] [PubMed] [Google Scholar]
- 104.Warner SJ, Libby P. Human vascular smooth muscle cells. Target for and source of tumor necrosis factor. J Immunol. 1989;142:100–9. [PubMed] [Google Scholar]
- 105.Silva LC, Ortigosa LC, Benard G. Anti-TNF-alpha agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy. 2010;2:817–33. doi: 10.2217/imt.10.67. [DOI] [PubMed] [Google Scholar]
- 106.Gu L, Okada Y, Clinton S, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low-density lipoprotein-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 107.Gosling J, Slaymaker S, Gu L, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. Journal of Clinical Investigation. 1999;103:773–8. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 109.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 110.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Libby P, Nahrendorf M, Swirski FK. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease. Journal of the American College of Cardiology. 2016;67:1091–1103. doi: 10.1016/j.jacc.2015.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Swirski FK, Nahrendorf M, Libby P. Mechanisms of Myeloid Cell Modulation of Atherosclerosis. Microbiol Spectr. 2016;4 doi: 10.1128/microbiolspec.MCHD-0026-2015. [DOI] [PubMed] [Google Scholar]
- 113.Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. The Lancet. 2017 doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 114.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–29. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galis Z, Muszynski M, Sukhova G, et al. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–189. doi: 10.1161/01.res.75.1.181. [DOI] [PubMed] [Google Scholar]
- 116.Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2:840–55. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sager HB, Heidt T, Hulsmans M, et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation. 2015;132:1880–90. doi: 10.1161/CIRCULATIONAHA.115.016160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sager HB, Dutta P, Dahlman JE, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8:342ra80. doi: 10.1126/scitranslmed.aaf1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Libby P. Murine “Model” Monotheism: An Iconoclast at the Altar of Mouse. Circ Res. 2015;117:921–5. doi: 10.1161/CIRCRESAHA.115.307523. [DOI] [PubMed] [Google Scholar]
- 120.S JC, Auger KR, Libby P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J Exp Med. 1987;165:1316–1331. doi: 10.1084/jem.165.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Loppnow H, Libby P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol. 1989;122:493–503. doi: 10.1016/0008-8749(89)90095-6. [DOI] [PubMed] [Google Scholar]
- 122.Schoenbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P. Ligation of CD40 activates interleukin 1beta-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1beta. Journal of Biological Chemistry. 1997;272:19569–19574. doi: 10.1074/jbc.272.31.19569. [DOI] [PubMed] [Google Scholar]