

Summary
Insulin resistance is a hallmark of diabetes and an unmet clinical need. Insulin inhibits hepatic glucose production and promotes lipogenesis by suppressing FOXO1-dependent activation of G6pase and inhibition of Glucokinase, respectively. The tight coupling of these events poses a dual conundrum: mechanistically, as the FOXO1 corepressor of Glucokinase is unknown; and clinically, as inhibition of glucose production is predicted to increase lipogenesis. Here we report that SIN3A is the insulin-sensitive FOXO1 corepressor of Glucokinase. Genetic ablation of SIN3A abolishes nutrient regulation of Glucokinase, without affecting other FOXO1 target genes, and lowers glycemia without concurrent steatosis. To extend this work, we executed a small molecule screen and discovered selective inhibitors of FOXO-dependent glucose production devoid of lipogenic activity in hepatocytes. In addition to identifying a novel mode of insulin action, these data raise the possibility of developing selective modulators of unliganded transcription factors to dial out adverse effects of insulin sensitizers.
ETOC
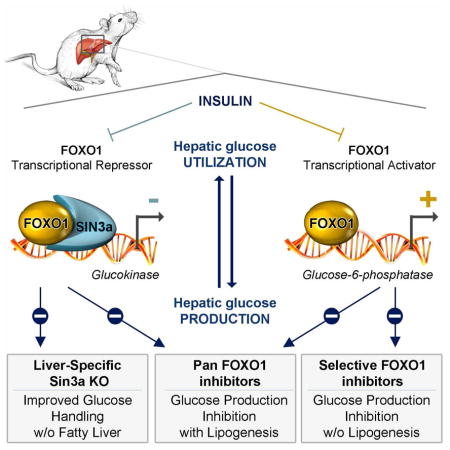
The transcriptional output of FOXO1 can be selectively modulated in a way that might reduce adverse effects of insulin sensitizers
Introduction
Insulin resistance predisposes to diabetes and metabolic diseases. Restoring insulin sensitivity is an effective approach to prevent and treat diabetes (Berkowitz et al., 1996), and to reduce its macrovascular complications (Kernan et al., 2016). However, currently available insulin sensitizers have significant adverse effects, such as weight gain due to triglyceride accumulation, fractures (possibly related to increased bone marrow adipogenesis), and hemodynamic changes (Cariou et al., 2012).
These “adverse” effects are part and parcel of increased insulin sensitivity, and cannot be effectively separated from it. This conundrum is best illustrated by the role of insulin in the liver. Insulin has pleiotropic hepatic effects mediated by diverse signaling mechanisms, including the PI3K/AKT/FOXO pathway (Dong et al., 2008; Lu et al., 2012; Matsumoto et al., 2007). Others and we have shown that insulin inhibits FOXO (1, 3a, and 4), resulting in decreased glucose production and increased glucose utilization through glycolysis, glycogen synthesis, and de novo lipogenesis (Altomonte et al., 2003; Haeusler et al., 2010a, 2010b; Matsumoto et al., 2007; Samuel et al., 2006; Zhang et al., 2006). This is achieved through an elegant, if unexplained mechanism, whereby FOXO inhibit expression of the rate-limiting enzyme of glucose utilization, glucokinase (Gck) (Dong et al., 2008; Haeusler et al., 2014; Zhang et al., 2006), while stimulating the rate-limiting enzyme of glucose production, glucose-6-phosphatase (G6pc) (Haeusler et al., 2014; Nakae et al., 2001a). When FOXO are inhibited, glucose production decreases, potentially benefiting diabetes treatment, but hepatic lipid synthesis increases, predisposing to steatosis (Pajvani and Accili, 2015).
While the activating functions of FOXO can be explained by binding to DNA via the forkhead domain (Cook et al., 2015a), the mechanism of its repressor functions in the liver is unknown. In this study, we sought to discover how FOXO suppress hepatic Gck, then leveraged this knowledge to identify selective FOXO inhibitors with the ability to inhibit G6pc, but bereft of Gck-stimulating activity.
Results
Insulin induction of Gck requires glucocorticoid-induced Foxo1 expression
Gck expression is induced within 1h of refeeding, suppressed after a 4h-fast (Haeusler et al., 2014), and inversely correlated with Foxo1 expression (Figure S1A). This regulation is abolished in liver-specific triple FOXO (1, 3a, and 4) knockout mice (L–Foxo1,3,4) (Haeusler et al., 2014). To identify the corepressor(s) required for FOXO inhibition of Gck, we established an in vitro system that recapitulated hormonal control of Gck expression. We incubated primary murine hepatocytes in the presence of cAMP, dexamethasone (dex), and insulin in various combinations. Neither insulin nor cAMP alone affected Gck (Figure 1A–B), but cAMP/dex (Figure 1C) or dex alone (Figure 1D) resulted in a 4-fold induction of Gck mRNA that was further increased by insulin. In contrast, G6pc expression was induced by cAMP and suppressed by insulin (Figure S1B–C). Dex had a small effect on G6pc expression (Figure S1D), but greatly potentiated the effect of cAMP (Figure S1E). Time course experiments revealed that Gck induction by cAMP/dex peaked at 2h and returned to basal within 12h, whereas Gck induction by insulin peaked at 4h (Figure 1E). In hepatocytes pre-treated with dex/cAMP, insulin began to induce Gck after 1h (Figure 1F). In primary hepatocytes from liver-specific FOXO1 knockout (L-Foxo1) (Matsumoto et al., 2007) or L-Foxo1,3,4 (Haeusler et al., 2010a) mice, Gck was elevated in the presence of cAMP/dex compared to WT mice and insulin failed to induce it further (Figure 1G and Figure S1F, respectively). In contrast, cAMP/dex failed to induce G6pc (Figure S1G–H). The identical pattern of Gck expression in L-Foxo1 and L-Foxo1,3,4 mice suggests that FOXO1 accounts for the bulk of the effect on Gck expression. Time-course experiments in L-Foxo1 hepatocytes revealed that the ability of cAMP/dex to promote Gck was preserved for up to 12hr, and insulin had no ability to induce it further (Figure S1I). Moreover, transfection of WT hepatocytes with adenovirus (Figure S1J–K) encoding constitutively active ADA-FOXO1 confirmed that FOXO1 time- and dose-dependently represses Gck expression.
Figure 1. Insulin induction of Gck requires glucocorticoid-stimulated FOXO1 expression.

A–D, Gck expression in primary hepatocytes after 7h treatment with vehicle or insulin (A, n=9 from 3 mice), cAMP and/or insulin (B, n=8 from 3 mice), dexamethasone (dex), cAMP and/or insulin (C, n=9 from 3 mice), and in dex or dex/insulin (D, n=8 from 2 mice). E–F, Time-course of Gck expression in primary hepatocytes treated with vehicle, cAMP/dex, or cAMP/dex/insulin for the indicated times (E, n=4 from 1 mouse; h=hours), and 6h cAMP/dex followed by insulin (F, n=6 from 2 mice, min=minutes). G. Gck expression in primary hepatocytes from WT (n=12 from 4 mice) vs. L-Foxo1 (n=12 from 4 mice) mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions. See also Figure S1, S2, S3.
As no glucocorticoid-response element is present in the hepatic Gck promoter, the permissive effect of dex on insulin induction of Gck (Katz et al., 1979; Spence and Pitot, 1979) likely requires synthesis of new factors. Consistent with this hypothesis, pre-treatment with the protein synthesis inhibitor, cycloheximide, abolished insulin induction of Gck (Figure S1L). Among transcription factors known to regulate Gck (Bae et al., 2010; Massa et al., 2011) (Figure S2A), Foxo1, Foxo3 and Hnf4α mRNA showed a time-dependent increase after treatment with cAMP/dex (Figure S2B, C, and E), whereas Pparγ and Hnf6 expression decreased (Figure S2G and I), and Foxo4, Hif1a and Srebf1 expression did not change (Figure S2D, F and H). The increase in Foxo1 and Hnf4α mRNA was due to dex (Figure S2J–L), and was associated with increased FOXO1 protein after 4h-treatment (Figure S2M–N). Primary hepatocytes from L-Foxo1 mice treated with cAMP/dex in the presence or absence of insulin did not display changes in the expression of factors modulating Gck expression (Figure S3A–H), consistent with the possibility that FOXO1 induction mediates the permissive effect of dex in insulin-induced Gck expression in vitro. To examine this in more detail in vivo, we studied mice with an induced deletion of hepatic glucocorticoid receptors, to circumvent the developmental effects of glucocorticoid receptor deficiency (C.B. and L.W., manuscript in preparation). Absence of the hepatic glucocorticoid receptor resulted in a 4-fold decrease in Gck expression (Figure S3I) that was not reversed by 5 weeks of corticosterone treatment (Figure S3I). Corticosterone failed to induce Gck in WT mice as well (Figure S3I). This treatment was associated with no changes to Hnf4α and Foxo1 expression (Figure S3J–K), suggesting that mechanisms other than glucocorticoid, or cell-autonomous factors can compensate in vivo.
Insulin induction of Gck requires FOXO1 phosphorylation or acetylation
Insulin induces Gck within 1h in vivo (Haeusler et al., 2014) or in dex/cAMP pre-treated hepatocytes (Figure 1F), consistent with the post-translational modification, rather than de novo synthesis, of an existing factor. FOXO1 is regulated by phosphorylation and acetylation (Frescas et al., 2005; Nakae et al., 2001a; Qiang et al., 2010). Transduction of primary hepatocytes from L-Foxo1 mice with adenoviruses or plasmids encoding WT and mutant FOXO1 showed that FOXO1 inhibited Gck and that insulin was able to override the suppressive effect of WT, but not of phosphorylation-defective (ADA-FOXO1) or acetylation-defective (KR-FOXO1) FOXO1 on Gck (Figure 2A–B), demonstrating that FOXO1 inhibition by phosphorylation and/or acetylation is required for insulin-induced Gck expression.
Figure 2. Insulin removes FOXO1 inhibition on Gck promoter.

A–B, Gck expression in L-Foxo1 primary hepatocytes transfected with plasmids (A, n=8 from 2 mice) or adenoviruses (B, n=4–7 from 2 mice) encoding WT and mutant FOXO1 in the presence or absence of insulin. ADA-FOXO1 = phosphorylation-defective FOXO1 at T24, S253 and S316; KR-FOXO1 = acetylation-defective FOXO1; T24A-FOXO1 = phosphorylation-defective FOXO1 at T24; S253A = phosphorylation-defective FOXO1 at S253) C, Gck expression in primary hepatocytes isolated from WT (n=7 from 2 mice) vs. KR/KR (n=8 from 2 mice) mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. D, Rat Gck promoter activity in insulin-treated primary hepatocytes transfected with control (CTL) and FOXO1 plasmids (n=6 from 2 mice). E–F, FOXO1 ChIP-qPCR in primary hepatocytes treated with cAMP/dex on Gck promoter (−1545 to +52) using overlapping primer sets (E, n=3), and on P5 (−1187 to −1040) and P22 (−93 to +52) following treatment with cAMP/dex or cAMP/dex/insulin (F, N=7 and 6, respectively). G. Gck expression in L-Foxo1 primary hepatocytes transfected with WT-FOXO1- and DBD-FOXO1- (DNA binding deficient) expressing adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel B and G, * or # are used to compare, respectively, solid and empty bars to each other). See also Figure S4.
To identify FOXO1 phosphorylation sites required for insulin-induced Gck expression, we transduced L-Foxo1 primary hepatocytes with adenoviruses encoding phosphorylation-defective FOXO1 mutants (T24A and S253A). Gck repression by these two mutants was not reversed by insulin, demonstrating that T24 or S253 phosphorylation are necessary for insulin-induced Gck expression (Figure 2B). We next examined primary hepatocytes isolated from mice carrying acetylation-defective Foxo1 alleles (KR/KR) (Banks et al., 2011). These mice display a modest gain-of-function, due to reduced sensitivity of the KR mutant to insulin-dependent phosphorylation (Banks et al., 2011). Gck expression was comparable to cAMP/dex-treated WT hepatocytes, but was not further induced by insulin, confirming that FOXO1 acetylation is required for insulin-induced Gck expression (Figure 2C). As a control, insulin inhibited G6pc expression induced by WT-FOXO1 (Figure S4A–B). Interestingly, the T24A and KR/KR mutants did not affect insulin inhibition of G6pc, unlike Gck, while the S253A mutant did (Figure S4B–C). These data suggest that the repressive function of FOXO1 is regulated through a mechanism distinct from its activating function, and that FOXO1 deacetylation and T24 dephosphorylation are required for repression, but not for activation of FOXO1-mediated transcription.
FOXO1 is recruited to the hepatic glucokinase promoter
Luciferase assays in primary hepatocytes from WT mice co-transfected with FOXO1 and a Gck reporter showed that FOXO1 inhibits Gck promoter activity (Figure 2D). To determine whether FOXO1 binds the hepatic Gck promoter, we scanned the sequence from −1500 to +50 by chromatin immunoprecipitation followed by qPCR (ChIP-qPCR). In cAMP/dex-treated primary hepatocytes, we detected an enrichment of regions P5 (−1187 to −1053 from the transcription start site) and P22 (−93 to +52) in the FOXO1 ChIP (Figure 2E). Insulin inhibited FOXO1 binding to the promoter (Figure 2F). As control, we did not detect FOXO1 in P5 and P22 in L-Foxo1 mice (data not shown).
Interestingly, neither P5 nor P22 contain a FOXO1 DNA-binding sequence (5′-TT(G/A)TTT(T/A)(G/C)-3′), suggesting that FOXO1 does not need to bind DNA in order to inhibit Gck. To test this point, we transduced L-Foxo1 primary hepatocytes with adenovirus expressing a DNA-binding and phosphorylation-deficient mutant FOXO1 (ADA/DBD-FOXO1). This mutant retained the ability to inhibit Gck (Figure 2G) but was unable to induce G6pc (Figure S4D). However, we detected DBD-FOXO1 in P5 and P22 by ChIP, suggesting that FOXO1 binds to these sequences indirectly (Figure S4E). Consistent with this notion, cAMP/dex/insulin-regulated Gck expression in primary hepatocytes from Dbd-Foxo1 knock-in mice (Cook et al., 2015a) (DBD) was similar to WT hepatocytes (Figure S4F), whereas cAMP/Dex failed to induce G6pc (Figure S4G). These data suggest that DNA binding is required for the activating, but not for the inhibitory function of FOXO1.
To identify proteins that interact with FOXO1 and bind to the Gck promoter, we inspected P5 and P22 to find known consensus transcription factor binding sites using the Jaspar database, and identified a HNF4A binding site in P22. Interestingly, this segment is necessary for insulin-induced Gck expression (Roth et al., 2004), and HNF4A is known to interact with FOXO1 (Ganjam et al., 2009; Hirota et al., 2003, 2008) and regulate Gck expression (Ganjam et al., 2009; Hirota et al., 2008; Roth et al., 2004). We confirmed this interaction by co-immunoprecipitation (Figure S4H). Luciferase assays in primary hepatocytes from WT mice transfected with either FOXO1, HNF4A, or both showed that FOXO1 inhibited Gck promoter activity, whereas HNF4A increased it. Interestingly, FOXO1 inhibited HNF4A-induced Gck promoter activity (Figure S4I). However, we could not detect HNF4A on P5 and P22 by ChIP. Rather, HNF4A bound to P20 and P21 following cAMP/dex treatment, and its binding was further increased by insulin (Figure S4J). As P21 and P22 segments overlap, these data can be construed to indicate that HNF4 is the FOXO1 binding partner, as described previously (Ganjam et al., 2009). However, the failure of FOXO1 to bind to P21 indicated that there must be additional FOXO1 binding partners.
A FOXO1/SIN3A/HDAC complex represses Gck
To identify the corepressor(s) required for FOXO1 inhibition of Gck, we first used deletion mapping to identify a corepressor-interacting domain on FOXO1. A dominant-negative FOXO1 mutant lacking the transactivation domain at the C-terminus (Δ256-FOXO1) (Altomonte et al., 2003; Nakae et al., 2001a) was unable to induce G6pc (Figure S4K), but retained the ability to suppress Gck and to be inhibited by insulin (Figure 3A). These data are consistent with the observation that T24 and S253 phosphorylation are necessary for insulin-induced Gck expression, since both sites are contained within this mutant (Figure 2B), and indicate that the alleged corepressor binds to the FOXO1 NH2-terminal half. This region contains three alanine-rich putative repression domains, the third of which is unique to FOXO1 compared to FOXO3a and 4 (Figure S4L). We hypothesized that this unique 18-amino acid segment (AA126-143) contained the corepressor binding site. To test the hypothesis, we generated a plasmid expressing a deletion mutant lacking amino acids 126-144 (Δ19-FOXO1). When transfected in WT primary hepatocytes, this mutant induced G6pc and underwent normal insulin-induced phosphorylation as WT-FOXO1 did (Figure S4M), but failed to inhibit Gck (Figure 3B) or a Gck promoter-driven luciferase reporter (Figure S4N).
Figure 3. FOXO1 interacts with SIN3A through its NH2-terminus.

A, Gck expression in L-Foxo1 primary hepatocytes transfected with WT-FOXO1 and Δ256-FOXO1 (AA1-256) adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). B, Gck expression in WT primary hepatocytes transfected with FOXO1 and Δ19-FOXO1 (without AA126-144) plasmids in the presence or absence of insulin (n=8 from 2 mice). C, Schematic representation of SIN3A interacting domain (SID) locations and mutations of the FOXO1 N-terminal domain. D, Gck expression in WT primary hepatocytes transfected with FOXO1 and SID mutant FOXO1 plasmids (n=8 from 3 mice). E, Co-immunoprecipitation of SIN3A and FOXO1 in primary hepatocytes. F, SIN3A ChIP-qPCR on P5 (−1187 to −1040), P20 (−219 to −77), P21 (−154 to −9) and P22 (−93 to +52) in primary hepatocytes treated with cAMP/dex or cAMP/dex/insulin (n=6). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions. See also Figure S4.
SIN3A and B are pleiotropic corepressors involved in neoplastic transformation (Kadamb et al., 2013). As the Δ19 deleted region contains one of two putative SIN3-interacting domains (SID1 and 2) (AAXXL) found in the FOXO1 NH2-terminal half (Pang et al., 2003), we sought to determine whether SIN3 is the FOXO1 corepressor of Gck. To this end, we generated single SID1 (SID1-FOXO1), SID2 (SID2-FOXO1) or double SID1/2 mutants (SID1/2-FOXO1) (Figure 3C). Mutation of SID1 had no effect on FOXO1-dependent Gck inhibition, whereas the SID2 and SID1/2 mutations largely prevented it (Figure 3D). Moreover, SID2-FOXO1 was unable to inhibit Gck promoter activity in luciferase assays (Figure S4N). These data are consistent with the hypothesis that SIN3A is the FOXO1 corepressor of the Gck promoter. Interestingly, this hypothesis is also supported by the protein interaction predictive algorhytm, PrePPI (Zhang et al., 2012).
We asked whether FOXO1 and SIN3A interact by performing co-immunoprecipitation experiments. Consistent with the data above, WT, but not Δ19 or SID2-FOXO1 could be detected in reciprocal co-immunoprecipitations (Figure 3E). In ChIP experiments performed with SIN3A antibodies in primary hepatocytes treated with cAMP/dex, we detected an enrichment of P20, P21 and P22 (−93 to +52) that was inhibited by insulin (Figure 3F). These data are consistent with the hypothesis that FOXO1 recruits SIN3A, and the latter binds to the Gck promoter to inhibit Gck expression.
mRNA studies showed that Sin3a expression is regulated by cAMP, dex, and insulin in a FOXO1-independent manner. Indeed, Sin3a expression decreased in response to dex, cAMP, or insulin alone and combined cAMP/dex/insulin in vitro (Figure S5A–D), and upon refeeding in vivo (refed/fast=0.84 AU, P=0.03). The pattern of Sin3a expression in response to cAMP/dex with and without insulin was similar in primary hepatocytes isolated from WT, L-Foxo1, and KR/KR mice (Figure S5E–F), demonstrating that FOXO1 does not regulate Sin3a. Moreover, there were no changes of Sin3a mRNA in L-Foxo1,3,4 mice (KO/WT=1.02 AU, P=0.75) or following FOXO1 overexpression (Figure S5G).
Next, we examined the role of SIN3A in the regulation of Gck by gain- and loss-of-function experiments in primary hepatocytes. SIN3A overexpression lowered Gck (Figure 4A), whereas Foxo1 expression was unaffected (Figure S5H). When co-transfected with FOXO1, SIN3A potentiated Gck inhibition by FOXO1 (Figure 4A). In the same cells, Sin3a knockdown with siRNA increased Gck expression (Figure 4B), leaving Foxo1 expression unaffected (Figure S5I). Co-transfection of Sin3a siRNA and FOXO1 showed that SIN3A deletion prevents FOXO1 inhibition of Gck (Figure 4B).
Figure 4. SIN3A regulates hepatic lipid and glucose metabolism.

A–B, Effect of FOXO1 expression on Gck expression in WT primary hepatocytes co-transfected with SIN3A and control (CTL) plasmids (A, n=8 from 2 mice), or Sin3a siRNA and control siRNA (siCTL) (B, n=8 from 2 mice). C–D, Glucose production (c, n=8 from 2 mice) and de novo lipogenesis (D, n=9 from 3 mice) in primary hepatocytes transfected with SIN3A plasmid. E, Histone 3 acetylation on Gck promoter in WT primary hepatocytes treated with cAMP/dex or cAMP/dex/insulin (n=4). F–G, Gck expression in WT primary hepatocytes transduced with FOXO1 and KR-FOXO1 adenoviruses in the presence or absence of trichostatin A (TSA) (F, n=8 from 2 mice), or transfected with FOXO1 plasmid and treated with TMP269 or TC-H106 (G, n=6 from 2 mice). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel A, B, C, D and G, * or # are used to compare, respectively, solid and empty bars to each other). See also Figure S5.
FOXO1 controls glucose production and triglyceride synthesis through its dual regulation of G6pc and Gck (Haeusler et al., 2014). Thus, the critical test of the hypothesis that SIN3A is the FOXO1 corepressor was to interrogate its ability to uncouple glucose production from lipogenesis. To assess this point, we overexpressed SIN3A in primary hepatocytes and measured glucose release into the medium in response to cAMP/dex/insulin, as well as C14 incorporation into triglycerides. We found that SIN3A had no effect on the ability of insulin to suppress cAMP/dex-induced glucose production (Figure 4C), but blunted the effect of insulin to induce lipogenesis by nearly 50% (Figure 4D). These data demonstrate a physiologic correlate to the FOXO1/SIN3A interaction, and provide a potential mechanism to uncouple the stimulatory and inhibitory effects of FOXO1 by selective inhibition of the SIN3A-independent functions of FOXO1.
SIN3A recruits histone deacetylases (HDAC) to inhibit gene expression (Hassig et al., 1997; Kadamb et al., 2013). To investigate whether HDAC are required for its FOXO1-dependent effect on Gck, we first examined histone acetylation at the hepatic Gck promoter by ChIP-qPCR. Treatment with cAMP/dex/insulin increased histone3 (H3) acetylation on the Gck promoter, especially on P11, P15, and P18 (Figure 4E). In primary hepatocytes transduced with WT-FOXO1, the class I/II HDAC inhibitor, Trichostatin A (TSA), prevented FOXO1 inhibition of Gck, suggesting that class I/II HDAC are involved in this effect (Figure 4F). The same effect occurred in hepatocytes transduced with KR-FOXO1 (deacetylated FOXO1 mutant) (Figure 4F), indicating that Gck induction in the presence of TSA does not result from FOXO1 acetylation (Banks et al., 2011; Mihaylova et al., 2011). This effect appeared to be mediated by class I HDACs, as the class II-specific inhibitor (TMP 269) did not prevent FOXO1 inhibition of Gck, whereas two separate class I-specific inhibitors, TC-H106 (Figure 4G) and FK228 (Figure S5J), did.
Impaired Gck regulation in Sin3a/b knockout mice
To investigate the metabolic functions of SIN3A in hepatocytes, we generated liver-specific single and double knockouts Sin3alox/lox:Sin3blox/lox (Dannenberg et al., 2005; David et al., 2008) using Albumin-cre (Postic et al., 1999) transgenic mice to drive Cre recombination-mediated deletion of the two genes. Single knockouts haploinsufficient for the other isoform, L-Sin3a−/−:Sin3b+/− (L-Sin3a) or L-Sin3a+/−:Sin3b−/− (L-Sin3b), as well as double knockouts L-Sin3a/b (L-Sin3a/b) were born at term in Mendelian ratios. At post-natal day 2 (P2), L-Sin3a/b and L-Sin3a mutants became hypoglycemic compared to L-Sin3b, double heterozygotes, or WT littermates (Figure 5A), while maintaining normal weight (Figure 5B). Interestingly, this is the same time point at which L-Foxo1,3,4 mice develop hypoglycemia due to inappropriate activation of Gck (Haeusler et al., 2014). L-Sin3a/b and L-Sin3a mice developed jaundice at P5 and became growth-retarded starting at P15. At weaning, L-Sin3a/b and L-Sin3a were ~50% smaller, hypoglycemic, hypoactive, and jaundiced, but all parameters normalized after P35 (Figure 5A–B). L-Sin3b also showed lower glycemia and weight compared to WT or double heterozygous littermates (Figure 5A–B).
Figure 5. Impaired hepatic development and metabolism in L-Sin3a/b−/− mice.

A–B, Time course of glycemia (A) and body weight (B) in WT, heterozygous, L-Sin3a, L-Sin3b and L-Sin3a/b mice (n=5–9). C–E, Weight (C), fat mass (D), and lean mass (E) of adult male WT and L-Sin3a/b mice on chow diet (n = 11/10). F, Glucose levels in ad libitum-fed (n=32/22), overnight-fasted (n=32/22), 30min- (n=20/11) and 4h-refed (n=20/11) WT and L-Sin3a/b mice. G–H, GTT (G) and PTT (H) carried out after an overnight fast in WT (n=7 for GTT, 8 for PTT) and L-Sin3a/b mice (n=7 for GTT, 8 for PTT). I, Liver weight in 12h-fasted (n=12/11) and 4h-refed (n=20/11) WT and L-Sin3a/b mice. J, Liver H&E trichrome and PAS staining in WT and L-Sin3a/b mice (arrowhead = fibrotic tissue; asterisk = necrosis). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions. Scale bar = 100 μm. See also Figure S5 and Table S1.
Adult male L-Sin3a/b mice showed normal body weight, but lower fat mass and higher lean mass (Figure 5C–E). They showed lower ad libitum glucose levels (Figure 5F), and lower glucose excursions during glucose and pyruvate tolerance tests compared to WT littermate controls (Figure 5G–H). Ketone levels after a 12h-fast were lower, whereas cholesterol levels were higher compared to littermate controls. After 4h-refeeding, insulin levels were higher in L-Sin3a/b mice (Table S1). L-Sin3a/b mice also showed evidence of cholestasis, with elevated plasma alkaline phosphatase, aspartate transaminase (AST), alanine transaminase (ALT), cholesterol, triglycerides, bile acids and bilirubin (Table S1). These findings were associated with hepatomegaly and histological abnormalities that included fibrosis and biliary infarcts (Figure 5I–J, Figure S5K), whereas glycogen content, as assessed histochemically by Periodic Acid Schiff staining, was similar to WT mice. L-Sin3a/b mice also displayed splenomegaly and enlarged kidneys (Figure S5L–M).
Although the metabolic phenotype of L-Sin3a/b and L-Sin3a mice is consistent with altered regulation of Gck, their developmental and histological abnormalities confounded the analysis. To circumvent this problem, we deleted Sin3a and Sin3b in adult mouse liver by injecting AAV8-TBG-CRE in Sin3a lox/lox:Sin3blox/lox mice to achieve liver-restricted recombination (iL-Sin3a/b mice). AAV8-TBG-GFP injection in WT mice served as control (iWT mice). Two weeks after injection, there were no histological abnormalities or hepatomegaly in iL-Sin3a/b compared to iWT mice (Figure S6A–B). Ad libitum, fasting, and 30min-refeeding glucose levels were lower in iL-Sin3a/b mice compared to controls (Figure 6A), while insulin levels, body weight, and body composition remained normal (Figure S6C–F). Moreover, glucose excursions during GTT and PTT were reduced (Figure 6B–C). Three weeks after injection, we killed mice that had been fasted for 12h followed or not by 4h-refeeding. Blood tests revealed that plasma cholesterol and glucose levels were lower, whereas AST and ALT were higher in iL-Sin3a/b mice compared to iWT mice (Table S2). Similarly to L-Sin3a/b KO mice, iL-Sin3a/b displayed enlarged kidneys in the fasted state (Figure S6G). Moreover, hepatic triglyceride and cholesterol content in the fasted state were lower (Figure 6D–E), while glycogen content was normal. We observed a slight decreased of glycogen content in the refed state compared to iWT mice (Figure 6F). As a critical test of our hypothesis, we measured Gck expression in the fasted and refed state. Fasting levels were higher, and there was a complete lack of induction by refeeding (Figure 6G). GCK protein levels followed the same pattern of Gck mRNA expression (Figure S6H–I). In contrast, regulation of other FOXO1 targets, such as G6pc and Pck1, was preserved (Figure 6H–I). To determine the functional consequences of the impaired regulation of Gck, we measured glycogen content, glucose production, glycolysis, and lipogenesis in primary hepatocytes from iL-Sin3a/b mice two weeks after induction of recombination. Confirming in vivo studies, we saw no changes in glycogen content (Figure S6J). Insulin inhibited cAMP/dex-dependent glucose production (Figure 6J), but was unable to induce lipogenesis (Figure 6K) and glycolysis (Figure S6K). These data indicate that Gck can no longer be regulated in response to the feeding cycle. Finally, chromatin immunoprecipitation revealed that Sin3a/b deletion did not alter FOXO1 binding to the Gck promoter (Figure S6L).
Figure 6. Impaired Gck regulation in iL-Sin3a/b−/− mice.

A, Glucose levels in ad libitum-fed (n=15/16 each genotype), overnight-fasted (n=15/16), or 30min- (n=9/9) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. B–C, Glucose (GTT) (B) and pyruvate tolerance tests (PTT) (C) carried out after an overnight fast in WT (before virus injection, n=13 for GTT, 14 for PTT), iWT (n=6 for GTT, 7 for PTT) and iL-Sin3a/b mice (n=7 for GTT and PTT). D–E, Hepatic triglyceride (D) and cholesterol (E) content in 12h-fasted (n=6/7) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. f, Hepatic glycogen content in 12h-fasted (n=5/5) and 4h-refed (n=5/5) iWT and iL-Sin3a/b mice. g–i, Hepatic Gck (G), G6pc (H) and Pck1 (I) expression in 12h-fasted (n=8/8) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. J–K, Glucose production (J, n=7 from 2 mice) and de novo lipogenesis (K, n=5 from 2 mice) in iWT and iL-Sin3a/b primary hepatocytes. Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel H, I, J and K, * or # are used to compare, respectively, solid and empty bars to each other). See also Figure S6 and Table S2.
Small molecule FOXO inhibitors uncouple regulation of glucose production from lipogenesis
In type 2 diabetes, hepatic insulin resistance increases glucose and triglyceride production leading to the two main abnormalities of this disease, hyperglycemia and hypertriglyceridemia. A bona fide insulin sensitizer acting as a FOXO inhibitor would be expected to decrease glucose production by diverting carbons into increased triglyceride synthesis. In contrast, an ideal hepatic insulin sensitizer should reduce the excessive hepatic glucose production characteristic of diabetes, without increasing hepatic lipid deposition. The identification of SIN3A as the FOXO1 corepressor of Gck raised the possibility that the two functions of FOXO1 could be independently modulated by selective FOXO inhibitors. We executed a high throughput screen of a small molecule library to identify FOXO inhibitors after establishing a FOXO1 reporter gene assay in HEK293s cells as described in the Methods section. The related transcription factor FOXA2, which is not inhibited by insulin (Haeusler et al., 2010a) but shares common regulatory elements and binds to the same DNA sequence as FOXO1 was used as a counterscreen. We also evaluated inhibition of the closely related family member FOXO3. In a 106 compound library, ~1.4% of the compounds showed human FOXO1 inhibitory activity. Using computational analysis for diversity, we pared down the number to 6,000 compounds that were tested in potency determinations in human FOXO1 vs. human FOXA2 reporter gene assays at concentrations between 50 μM and 1.6 nM. 170 hits with at least 10-fold selectivity for FOXO1 over FOXA2 were further characterized for cytotoxicity using an Alamar Blue Assay and for potency against human FOXO3 and mouse FOXO1 in reporter gene assays (Figure S7, Table S3). Selected compounds were then tested in primary hepatocytes for their ability to modulate G6pc and Gck expression. The expectation was that a pan-FOXO1 inhibitor would decrease the former and increase the latter. We identified three distinct classes of compounds (Figure 7A–D): compounds 1, 3, 4, 6, 7, 10, 11, and 12 behaved as non-specific inhibitors that decreased both G6pc and Gck expression, or had no discernible effects; compounds 2, 5, and 13 behaved as pan-inhibitors, decreasing G6pc and altering basal or/and insulin-induced regulation of Gck. Compounds 5, despite clearing the first selection, showed poor separation between FOXO1 and FOXA2 activity (<5-fold, Table S3), and was not further considered. A third group of compounds, including 8 and 9, demonstrated selective inhibition by decreasing G6pc without affecting Gck (Figure 7A–D, Table S4). To assess the functional consequences of these selective inhibitors, we tested compounds 8, 9, and 13 in glucose production and de novo lipogenesis assays in primary hepatocytes (Figure 7E–F). Compounds 8 and 13 curtailed cAMP/dex-induced glucose production, whereas 9 had no effect (Figure 7E). In addition, 13 increased lipogenesis, as expected of a full FOXO inhibitor, whereas 8 decreased it (Figure 7F). The data are consistent with selective regulation of FOXO-dependent gene expression by these compounds.
Figure 7. Effect of small molecule FOXO inhibitors on G6pc and Gck expression.

A–D, G6pc (A–B) and Gck (C–D) expression in primary hepatocytes treated for 7h with vehicle, cAMP/dex, or cAMP/dex/insulin in the presence or absence of FOXO inhibitors (Cpd). Cpd 1–7 were applied to a final concentration of 50μM; Cpd 8–13 at 10μM. (A,C, n=3 from 1 mouse; B,D, n=4 from 1 mouse). E–F, Glucose production (E, n=4 for # 8, 13, n=8 for DMSO and # 9, from 2 mice) and de novo lipogenesis (F, n=3 for DMSO, # 8, 13, n=6 for # 9, from 2 mice) in WT primary hepatocytes in the presence or absence of FOXO inhibitors # 8, 9 and 13. G, FOXO1 ChIP-qPCR on Gck (P22 = −93 to +52) and G6pc promoter (−230 to −31) in primary hepatocytes treated with cAMP/dex in the presence or absence of FOXO1 inhibtors (n=5). H, SIN3A ChIP-qPCR on Gck (P22 = −93 to +52) in primary hepatocytes treated with cAMP/dex in the presence or absence of FOXO1 inhibtors (n=5). Data are means ± s.e.m. In panel A–D: a = P<0.05 compared to vehicle. b = P<0.05 compared to cAMP/dex. c = P<0.05 compared to DMSO in cAMP/Dex condition. d = P<0.05 compared to DMSO in cAMP/Dex/Insulin condition. In panel F–G: *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel E–F, # is used to compare empty bars to each other). See also Figure S7 and Table S3–4.
To establish a mechanism for the differential actions of compounds 8 and 13 on Gck vs. G6pc expression, we tested their effects in chromatin immunoprecipitation assays in cAMP/Dex hepatocytes. Compounds 8 and 13 cleared FOXO1 from G6pc promoter (Figure 7G), explaining its inhibition. In contrast, compound 8 failed to clear FOXO1 and SIN3a from the Gck promoter, whereas 13 was able to induce a substantial clearance (Figure 7G–H). These data indicate that the differential actions of these compounds are associated with different effects on the FOXO1 transcriptional complex.
Discussion
FOXO1 can activate and repress gene expression, but the molecular mechanism underlying its ability to inhibit hepatic gene transcription was unknown. The key conclusions of this work are that: (i) FOXO1 functions in a corepressor complex with SIN3A via a discrete NH2-terminal motif; (ii) SIN3A plays a heretofore overlooked role in hepatic development and metabolism; and (iii) pharmacological modulation of the activator and repressor balance of FOXO can be achieved by small molecules that fulfill the criteria for a new class of insulin sensitizers.
The balance between the activating and repressive functions of FOXO1 is crucial in the liver since, by inducing G6pc and by inhibiting Gck, FOXO1 regulates the G6pc/Gck ratio, intracellular glucose-6-phosphate levels, and hepatic glucose handling (Haeusler et al., 2014; Pajvani and Accili, 2015). Unlike previous studies showing that insulin induction of Gck expression requires the recruitment of transcriptional activators on Gck promoter (Foretz et al., 1999; Kim et al., 2004, 2009; Roth et al., 2004), the present data show that it requires clearance of a FOXO1 corepressor. Interestingly, the repressive function of FOXO1 seems to be differentially regulated from its activating function, as clearing the Gck corepressor requires phosphorylation of two sites, T24 and S253, whereas inhibition G6pc requires phosphorylation of one site, S253. This finding is consistent with two previous observations: (i) T24 phosphorylation is differentially regulated by insulin and IGF1 receptors (Nakae et al., 2000), providing a potential explanation for the different biological actions of the two receptors; and (ii) T24 and S253 kinases are distinct (Jacinto et al., 2006; Nakae et al., 2001b). We also show that DNA binding is required for the activating, but not for the inhibitory function of FOXO1 (Cook et al., 2015a).
The repressive and activating functions of FOXO1 are achieved through the recruitment of cofactors (van der Vos and Coffer, 2008). PGC-1α (Matsumoto et al., 2007; Puigserver et al., 2003) and PRMTs (Choi et al., 2012) are known transcriptional co-activators of FOXO1-dependent gluconeogenesis. FCOR inhibits FOXO1 function in adipose tissue, but does not act by inhibiting gene transcription (Nakae et al., 2012). SIN3A was not known to be a FOXO1 corepressor or to regulate metabolism. Besides a carboxy-terminal acidic serine/threonine-rich region that serves as a transactivation domain, FOXO1 also contains amino-terminal proline- and alanine-rich domains that allow for the recruitment of the SIN3A/HDAC I complex. This complex is found on the Gck promoter in fasting conditions, and is cleared by insulin. The mechanism is probably multi-faceted. First, it can result from FOXO1 inhibition; second, since SIN3A is phosphorylated on S431 through the PI3K/Akt pathway (Humphrey et al., 2013, 2015), it too can be a direct target of insulin. Deletion of Sin3a in the liver causes hypoglycemia, mimicking L-Foxo1 mice (Haeusler et al., 2014; Matsumoto et al., 2007). The hypoglycemia probably reflects the inability to fully suppress Gck, with increased glucose vs. lipid oxidation during fasting, and increased glucose clearance following a meal. However, unlike L-Foxo mice, iL-Sin3a/b mice have reduced hepatic triglycerides, consistent with the inability to induce Gck in response to nutrients and thus with an inability to prime lipogenesis. Interestingly, cholesterol content is also decreased in iL-Sin3a/b mice. This can reflect GCK- or/and FOXO1-independent functions of SIN3A in liver metabolism, for example through its role to regulate mitochondrial activity (Barnes et al., 2014; Pile et al., 2003), or cholesterol biosynthesis (Solaimani et al., 2013). There are likely to be additional functions to hepatic SIN3A, in particular in the development of bile ducts, that account for the abnormal liver function tests, histopathologic abnormalities, and splenomegaly (likely secondary to portal hypertension) seen in mice with constitutive liver deletion. Finally, while our results indicate that SIN3A is crucial for nutrient regulation of Gck, they do not exclude the presence of other components in the repressor complex, such as SHP, which is known to interact with SIN3a (Farhana et al., 2007) and to decrease Gck expression (Kim et al., 2009).
On the therapeutic side, this study demonstrates the selective pharmacological targeting of FOXO1. Studies have shown beneficial effects of FOXO1 inhibition on diabetic hyperglycemia by reducing hepatic glucose production. However, these benefits can be offset by an increase in hepatic fat content (Pajvani and Accili, 2015). We have discovered small molecules with the ability to fine-tune the FOXO1 activator/repressor balance and alter the ratio of G6pc to Gck. In a clinical setting, this can reduce hepatic glucose production without increasing triglyceride accumulation. Interestingly, compound 8 has even the ability to suppress lipogenesis. Unfortunately, we have not been able to test the compounds in vivo due to their pharmacokinetic properties. Nevertheless, through structure refinement it should prove possible to modify the lead compounds and achieve the necessary distribution and coverage to engage the target in vivo. The findings should be viewed as providing proof of principle that the transcriptional output of unliganded transcription factors can be selectively modulated for therapeutic purposes, in a way that is conducive to dialing out potential adverse effects of insulin sensitizers.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Domenico Accili (da230@columbia.edu).
Experimental Model and Subject Details
Animals
Mice were maintained under standard laboratory conditions with water and standard chow diet (PicoLab rodent diet 20, 5053; Purina Mills) freely available. Mice were housed on a 12 h light/12 h dark cycle with lights on at 07:00 and off at 19:00. All animal experiments were in accordance with NIH guidelines for Animal Care and Use, approved and overseen by Columbia University Institutional Animal Care and Use Committee (IACUC). Male mice aged 9–24 weeks were used for most of experiment. Male and female mice aged 2–60 days were used for developmental studies.
L-Foxo1, L-Foxo1,3,4, KR/KR and Dbd-Foxo1 knock-in mice have been previously described (Banks et al., 2011; Cook et al., 2015a; Haeusler et al., 2010a; Matsumoto et al., 2007). To generate L-Sin3a/b mice, we crossed Sin3alox/lox:Sin3blox/lox (Dannenberg et al., 2005; David et al., 2008) and Albumin-cre (Postic et al., 1999) transgenic mice. The genotyping primers were Sin3A-1: 5′-GTCCTCAGGGAAGACGTTGA-3′, Sin3A-4: 5′-GCCCTGTCCTATCTTGACCA-3′, Sin3A-5: 5′-AGGACCACCAAAGTTCAGGA-3′, Sin3B FO-2: 5′-TACAACGGCTTCCTGGAGAT-3′, Sin3B FO-5: 5′-ACACCCAACACTCCCTGTTC-3′, Sin3B KO-D: 5′-CCCTCGAGGTCGACCCCGGGAAGC-3′, Sin3B KO B2: 5′-CCCAACACTCCCTGTTCAGGCCTC-3′. Peripheral GR knockout mice were generated by crossing mice that were floxed for the GR allele with a tamoxifen-inducible Rosa26CreERT2 line (Taconic #6466, Hudson, NY) to generate GRlox/lox,Rosa26CreERT2+/− (GRper KO) and GRlox/lox,Rosa26CreERT2−/− (control) mice.
For corticosterone treatment, animals were provided with corticosterone-supplemented water. Corticosterone (CORT; Sigma, St. Louis MO) was dissolved in 100% ethanol, at a concentration of 10 mg/mL. This stock solution was then diluted with regular tap water to obtain a final ethanol concentration of 1%, which yielded a final concentration of 100 μg/mL CORT. The GR gene is inactivated after daily ip injection of tamoxifen only in the periphery, but not the central nervous system. Deletion of GR protein in the liver was confirmed by Western Blot (data not shown). Livers from these mice were examined 2 weeks after the GR inactivation at which point animals had unaltered body composition and normal glucose tolerance (data not shown). For in vivo studies on Sin3alox/lox:Sin3blox/lox mice, we injected 1×1011 purified viral particles (AAV8.TBG.eGFP or AAV8.TBG.Cre) per mice via tail vein; we performed metabolic analysis on day 14 and killed the animals at day 21 after injection. The deletion efficiency was confirmed by genotyping (data not shown).
Primary Hepatocyte Culture and transfection
Primary hepatocytes were isolated from male mice via collagenase perfusion, as previously described (Cook et al., 2015b). We anesthetized mice with ketamine and xylazine (ketamine 100 mg/kg IP, xylazine 10 mg/kg, i.p. injection). We clamped the supradiaphragmatic inferior vena cava (IVC), catheterized the inferior vena cava with a 24-gauge catheter (Exel international) and infused 50 cc EGTA-based perfusion solution followed by 100 cc type I collagenase solution (Worthington Biochemicals). Following cell dissociation, we filtered cells with 100-μm mesh cell strainers, and gradient centrifugation steps to purify cell suspension. Then, we suspended hepatocytes at 5×105 cells per ml in Medium 199 (Sigma), 10% FBS (Life Technologies), antibiotics (called Plating medium) and plated them on collagen-coated cultureware for 2h. Following attachment, cells were incubated for 4h with Plating medium. For some experiments, hepatocytes have been transfected during this step. Plasmids transfection (500ng/5×105 cells, 48h) was performed using Lipofectamine2000, Opti-MEM and serum-free, antibiotics-free Medium 199 following manufacturer’s instructions. Adenoviruses (107 pfu/5×105 cells, 24h) were directly added to the plating medium. For RNA interference, Sin3a silencing was achieved using Stealth RNAi™ siRNA (40pmol/5×105 cells, 48h) directed against murine Sin3a and lipofectamine2000 reagent according to the manufacturer’s instructions. A scrambled siRNA was used as negative control. After 4h, transfection medium was removed. Prior to the experimental assays described below, cells were washed twice with PBS and incubated overnight in Medium 199, 1% BSA [w/v], antibiotics (called serum-free medium). For most of experiment, cells were incubated with 100 uM 8-CPT-cAMP, 1 uM dex, 100 nM insulin or vehicle for 7 h. For timing experiments, cells were incubated with the same concentration for 1h, 2h, 4h and 12h. For timing-reverse experiments, cells were incubated with 100 uM 8-CPT-cAMP, 1 uM dex for 6h after what insulin has been added for 30min, 1h and 2h.
HEK293s cell culture and generation of cells for the reporter gene assay
HEK293s cells (female, ATCC) were maintained in DMEM (Invitrogen 41965-039) containing 4.5 g/l glucose supplemented with Glutamax (Invitrogen 35050-038), non-essential amino acids (Invitrogen 11140-035), 25 mM HEPES (Invitrogen 15630-056), and 10% FBS (Sigma-Aldrich F2442) at 37°C and 5% CO2.
To generate transiently transfected cells for each of the four reporter gene assays and for the cytotoxicity assay, HEK293s cells were grown in 10-layer cell factories (Corning) and transiently co-transfected with 150 μg total DNA/150×106 cells, using a 1:4 ratio of the respective FOXO-expressing construct (human or mouse pIRESneo3-FOXO1 vector, the pIRESneo3-FOXA2 vector, or the pIRESneo3-FoxO3 vector respectively) to luciferase reporter construct. The MaxCyte® STX™ Scalable Transfection System was used to transfect in total 15×109 cells, which were cryopreserved following a 20 minute recovery and revived before running the reporter gene assays as described in the Method details section. The cells were confirmed by IDEXX BioResearch (Columbia, MO, USA) to be Mycoplasma-free and of the expected identity (based on short tandem repeat analysis of 9 alleles).
Method Details
Chemicals and Antibodies
Ketamine is from KetaSet® and Xylazine from AnaSed®. Medium 199, HBSS, EGTA, HEPES, PenStrep and Gentamycin are from Life Technology. Collagen type 4 is pursached from Worthington. Insulin (Humulin® R U-100) was purchased from Eli Lilly. 8-(4-chlorophenylthio) (CPT)-cAMP, dexamethasone, cycloheximide, Bovine Serum Albumin (BSA), D-glucose and sodium pyruvate were from Sigma-Aldrich. Lipofectamine2000 and Stealth RNAi™ siRNA against Sin3a were from Thermo Fisher. Trichostatin A (TSA), TMP269 and FK228 were from Selleckchem. TC-H 106 was from Cayman chemical. Insulin was diluted in sterile water. 8-(4-chlorophenylthio) (CPT)-cAMP, D-glucose, sodium pyruvate were dissolved in sterile water. Dexamethasone and cycloheximide were dissolved in ethanol (100%). TSA, TMP195, FK228, TC-H 106 were dissolved in DMSO. Anti-FOXO1 (for Western Blot and co-immunoprecipitation, C29H4), anti-SIN3A (for Western Blot, D1B7), and anti-HNF4A (for Western Blot, C11F12) antibodies were from Cell Signaling. Anti-HNF4A ChIP Grade (ab41898), anti-FOXO1A ChIP Grade (ab39670), anti-SIN3A ChIP Grade (ab3479), anti-H3 (ab1791), anti-Ac H3 (ab47915), anti-actin (ab8227) and control IgG antibodies were from Abcam. GCK antibody was from Dr. Magnuson (Vanderbilt University School of Medicine, Nashville, TN).
Plasmids and Viruses
Plasmids encoding RFP (CTL plasmid) and FOXO1 have been described before (Frescas et al., 2005). Plasmids encoding HNF4A (#33006) and SIN3A (#30454) were purchased from Addgene. Δ19-FOXO1 mutant was made by introducing PstI restriction site in WT-FOXO1. Mutations were introduced by site-directed mutagenesis using the QuickChange II (Stratagene). To generate FoxO1Δ19AA mutant, FoxO1-WT was used as a PCR template. Two pairs of primers were designed: PstI1,5′-CCACCGACCGGGCCGCTGCAGCAGCCCCCACCCGTGCCTC-3′ (sense) and 3′-GAGGCACGGGTGGGGGCTGCTGCAGCGGCCCGGTCGGTGG-5′(antisense); PstI2, 5′-CGCCGCGGGGCCACTCGCGCTGCAGCCGCGCAAGACCAGC-3′ (sense) and 3′-GCTGGTCTTGCGCGGCTGCAGCGCGAGTGGCCCCGCGGCG-5′ (antisense). Both amplified final PCR fragments bear PstI restriction sites, have been then digested and ligated to eliminate 19 amino acids sequence. For the Gck promoter reporter construct, we cloned the region lying between −1470 and +28 from rat genomic DNA into pGL3-basic vector. Plasmids encoding WT-FOXO1, SID1-FOXO1, SID2-FOXO1 and SID1/2-FOXO1 were purchased from Origene. WT-FOXO1, ADA-FOXO1 (T24A-S253D,S316A mutations), ADA/DBD-FOXO1 (T24A, S253D and S316A mutations (ADA) + N208A and H212R mutations (DBD)), KR-FOXO1 (Lys(219,242,245,259,262,271,291)Arg mutations), Δ256-FOXO1 (AA1-256, truncated form), T24A-FOXO1 (T24A mutation) and S256A-FOXO1 (S253D mutation) adenoviruses have been described before (Altomonte et al., 2003; Nakae et al., 2001a; Qiang et al., 2010). AAV8.TBG.PI.eGFP.WPRE.bGH and AAV8.TBG.PI.Cre.rBG were purchased from Penn Vector Core.
To prepare constructs used to generate transiently transfected cells for the HEK293s reporter gene assays, four copies of a 21-bp insulin-responsive element identical to that of human IGFBP-1 (GCAAAACAAACTTATTTTGAA) were inserted into the pGL4.26-luc2/minP/hygro vector (Promega) containing firefly luciferase cDNA, building the luciferase reporter construct pGL4.26-4xIRE-luc2. In parallel, full-length cDNA of human and mouse FOXO1, human FOXA2 or human FOXO3 were inserted into the mammalian expression vector pIRESneo3 (Clontech Laboratories) to generate the human and mouse pIRESneo3-FOXO1 vector, the pIRESneo3-FOXA2 vector, and the pIRESneo3-FoxO3 vector respectively. Endotoxin-free plasmid preparations were made and the sequence of all constructs confirmed (GeneArt).
mRNA Studies
We isolated RNA with RNeasy mini-kit (Qiagen) from frozen liver (~30mg) or from hepatocytes treated as above (~5×105 cells) and 1 ug of RNA was reverse-transcribed using the GoScript reverse transcription system (Promega) following manufacturer’s instructions. cDNAs were diluted (1:10 for in vivo studies, 1:5 for in vitro studies), and qPCR was performed using GoTaq® qPCR Master Mix (Promega). Primer sequences are available upon request. Gene expression levels were normalized to TATA-binding protein (TBP) using the 2−ΔΔCt method and are presented as relative transcript levels. For RNA profiling, adult L-Foxo1,3,4 and littermate control mice were fasted for 22 hours and then chow for 4 hours. RNA was prepared with Trizol and RNeasy (Qiagen). Three samples per group were analyzed using GeneChip Mouse Exon arrays (Affymetrix) and Partek Genomics Suite software.
Protein analysis
For protein extraction, hepatocytes treated as above (~5×105 cells) were washed twice with ice-cold PBS and lysed in ice-cold lysis buffer (20 mM Tris-HCl (pH=7.4), 150 mM NaCl, 10% glycerol, 2% NP-40, 1 mM EDTA, 20 mM NaF, 30 mM Na4P2O7, 0.2% SDS, 0.5% sodium deoxycholate) supplemented with Protease/Phosphatase Inhibitor Cocktail (1X, Cell Signaling) and centrifugated for 30min (14,000rpm). For co-immunoprecipitation, hepatocytes (~5×106 cells) were washed twice with ice-cold PBS and lysed in in light ice-cold lysis buffer (20 mM Tris–HCl (pH 7.4), 10 mM KCl, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 1% NP-40, 20 mM NaF) supplemented with Protease/Phosphatase Inhibitor Cocktail (1X, Cell Signaling). Lysate was sonicated for 1min40 (5X, output 70%, 20sec/20sec) and NaCl has been added to a final concentration of 420mM. The second lysate was sonicated for 1min40 (5X, output 70%, 20sec/20sec) and centrifugated for 30min (14,000rpm). Protein concentration was assessed by Pierce BCA protein assay (Thermo scientific) and 1mg was used for IP. Primary antibody has been added to lysis buffer and rock overnight. Next morning, Protein A Agarose Beads (Cell Signaling, #9863) were used to immunoprecipitate the complexes. The Ag-Ab complex was eluted from the beads by heating samples in SDS-loading buffer. For Western Blotting, 15ug of proteins extraction or the elution obtained from IP were loaded per well. Densitometric analysis was performed using ImageJ software (National Institutes of Health). Co-immunoprecipitations were repeated at least three times.
Chromatin Immunoprecipitation Assays
We isolated intact chromatin from primary hepatocytes (~5×106 cells) by using ChIP-IT Express Kit (Active Motif) following the manufacturer’s instructions; cells were sonicated using the 550 Sonic Dismembrator (Fisher Scientific). Chromatin immunoprecipitation was followed by qPCR using using GoTaq® qPCR Master Mix (Promega). Multiple overlapping pairs of primers were designed using Primer3 to cover hepatic Gck promoter from −1447 to +52 from the start point. P5, P20, P21 and P22 sequences were as follow: P5 (forward) 5′-ATCCGCTCCGTTTGTCTCT-3′ and (reverse) 5′-ATCTCCTGGGCAAGTCACAG-3′ (−1187 to −1040); P20 (forward) 5′-GAAGGGGGCATGTGAGTG-3′ and (reverse) 5′-AAAGAACCACGTGGGATCAG-3′ (−219 to −77); P21 (forward) 5′-GTGTTCAGAGAACATGGTAGCC-3′ and (reverse) 5′-TCTGAGAGGTGGCTCCTAAAA-3′ (−154 to −9); P22 (forward) 5′-ATCCCACGTGGTTCTTTGTC-3′ and (reverse) 5′-ACTGTCTGGCTGAGTGTTGC-3′ (−93 to +52). Other primer sequences are available upon request. Fold enrichment was calculated by a modified ΔC(t) method and normalized to DNA immunoprecipitated with negative IgG control (Sigma) antibody according to the formula (Δ(C(t)IP – C(t)input)100.
Luciferase Assays in primary hepatocytes
We transfected primary hepatocytes with a luciferase construct containing the hepatic Gck promoter sequence (−1470 to +28) (1μg/5×105 cells), as well as with plasmids encoding FOXO1, Δ19-FOXO1, SID2-Foxo1, RFP (in variant combination, 0.5μg total/5×105 cells) and pRL vectors (3ng/5×105 cells), using Lipofectamine 2000 (Invitrogen) as described above. Forty hours after the transfection of plasmids, hepatocytes were lysed and luciferase assay was performed using the Dual Luciferase Reporter Assay System (Promega) following the manufacturer’s instructions and assayed using an Orion L Microplate Luminometer (Berthold).
In vitro metabolic assay
For glucose production assay, serum-free medium was replaced with glucose production medium (glucose-free and phenol red–free DMEM supplemented with 1% BSA, 3.3 g/L NaHCO3, 20 mmol/L calcium lactate, and 2 mmol/L sodium pyruvate, antobiotics). Cells were incubated with dex + 8-CPT-cAMP, dex + 8-CPT-cAMP + insulin, or vehicle for 6 h. Glucose released into the culture medium was measured via peroxidase-glucose oxidase assay (Sigma) and normalized to protein content. For de novo lipogenesis assay, following overnight incubation in Medium 199 supplemented with 0.25% fatty acid-free BSA (Fisher), we replaced the medium with insulin- (10 nM) or vehicle-containing serum-free medium. After 2 hr, cells were washed twice with PBS and radiolabeling was carried out in Medium 199 + 0.25% fatty acid-free BSA containing 0.6 μCi/ml [1,2-14C]acetic acid (PerkinElmer Life Sciences) with or without insulin (10nM) over 3 h. Lipids were extracted using 3:2 hexane:isopropyl alcohol, dried in scintillation vials under N2 gas, and resuspended in 2:1 chloroform:methanol. Radiocarbon labeling of resuspended lipids was determined by liquid scintillation counting (PerkinElmer) and normalized to total cellular protein content. For the glycolysis assay, we detected extracellular L-lactate using the Cayman’s Glycolysis Cell-Based Assay Kit following the manufacturer’s instructions. For the glycolysis assay, we used Glycogen assay kit from Abcam (ab65620) following the manufacturer’s instructions.
In vivo Time course
Male, 9-week old C57BL/6J mice were purchased from Jackson labs. Feeding was synchronized by removing food during daytime (10:00–18:00 hours) and then food was replaced at 18:00. Mice were euthanized at time 0 (20:00) as well as 1, 2, 4, 6, 12, 18, and 24 hours of fasting. For refed mice, food was replaced at 18:00 and mice were sacrificed 15 minutes later, as well as 1, 2, 4, 6, 12, 18, and 24 hours after refeeding. 5 mice were analyzed per time point. Lights were off from 18:00–8:00. Liver RNA was extracted using Trizol, cDNA was synthesized with qScript (Quanta), and quantitative PCR was performed using goTaq (Promega). Primer sequences are available upon request. Gene expression levels were normalized to m36B4 or 18S using the 2−ΔΔCt method and are presented as relative transcript levels.
In vivo metabolic studies
Only male mice aged 9–15 weeks were studied, except in developemental studies (from 2-day-old to 40-day-old), where pups of both genders were used. We performed glucose and pyruvate tolerance tests after a 16-h (6 p.m. to 10 a.m.) fast using intraperitoneal injection of 2 g per kg body weight glucose. Blood glucose measurements were made from tail vein blood using OneTouch glucose monitor (One Touch Ultra, Bayer). Prior to sacrifice, mice were overnight fasted for 13 h, from 1900 to 0800 h. Mice to be refed were then given ad libitum access to chow from 0800 to 1200 h. Insulin levels were measured by ELISA (#10-1247-01, Mercodia); triglyceride (Infinity, #TR22421, ThermoFisher), total cholesterol (Cholesterol E, #439-17501, Wako Pure chemicals), ketone bodies (Total Ketone Bodies, #415-73301, #411-73401, Wako Pure chemicals), nonesterified fatty acids (HR Series NEFA-HR(2), #999-34691, #995-34791, #991-34891, #993-35191, Wako Pure chemicals), and total bile acid (STA-631, Cell Biolabs) by colorimetric assays. Blood chemistry analysis was performed by the Institute of Comparative Medicine (Columbia University).
Liver analyses
We used paraffin-embedded sections for Hematoxylin and eosin (H&E), Periodic acid–Schiff (PAS) and Trichrome staining. To measure hepatic lipid content, hepatic lipids were extracted from ~100 mg snap-frozen tissue samples using the method of Folch (Folch et al., 1957). TG and cholesterol contents were assayed colorimetrically and normalized to sample weight. To measure hepatic glycogen content, we homogenized frozen liver in 6% (vol/vol) perchloric acid, adjusted to pH 6–7 with KOH, then incubated it with 1 mg ml−1 amyloglucosidase (Sigma) in 0.2 M acetate (pH 4.8) and quantified glucose released (glycogen breakdown value minus PCA value), as described (Haeusler et al., 2014).
High throughput screen using FOXO reporter gene assays
Compound plates for the high-throughput screen were prepared using an Echo 525 Acoustic liquid handler (Labcyte) to dispense 100nl compound or DMSO from Echo-compatible 384-well source plates (Labcyte P5525) to luminescence-compatible 384-well plates (Greiner 481080).
To test compounds in the reporter gene assay, cryopreserved transfected cells were thawed and diluted in low-glucose (1 g/l) DMEM (Invitrogen 11880-028) supplemented with GlutaMax (Invitrogen 35050-038), non-essential amino acids (Invitrogen 11140-035), 25 mM HEPES (Invitrogen 15630-056), 1% charcoal/dextran-treated FBS (Nordic Biolabs SH30068). A Multidrop Combi liquid handler (ThermoFisher Scientific) was used to dispense 16,000 cells transfected with hFoxo1+IRE-luc2-reporter constructs into each well of the compound plates. After 24 h incubation at 37°C and 5% CO2, the plates were transferred to room temperature for 30 min, and 10 μl Steady-Glo (Promega) was added using the Multidrop Combi liquid handler. Following another 30 min incubation at room temperature, the luminescence signal was measured using an EnVision® multilabel system (PerkinElmer Life Sciences) equipped with an ultra-sensitive luminescence filter.
Compound-mediated cell toxicity was determined in hFOXO1/pGL4.26-IRE_luc2-expressing cells using Alamar Blue (Invitrogen DAL1100). Briefly, the cells were revived and seeded as in the reporter gene assay. After 20 hours exposure to compound, a background measurement was performed in an EnVision® multilabel system (PerkinElmer Life Sciences), followed by incubation for 4 hours with Alamar Blue prior to a 2nd measurement.
In the high-throughput screen, we tested 1 million compounds at 10 μM for their inhibitory effect on FOXO1, using a 0.5 μM solution of 3-chloro-N-ethyl-4-(5-isoquinolyloxymethyl)-N-methyl-benzamide (AZ4337) as the control compound and the reporter gene assay protocol described above. Based on a robust z-score (Malo et al., 2006), cut-off of ≤3.0, 1.4% of the compounds inhibited FOXO1-mediated reporter gene expression. After computational analysis for diversity, 6,000 compounds were pursued for potency determination in FOXO1 and FOXA2 reporter gene assays (testing 10 concentrations from 50 μM to 1.6 nM, using the reporter gene assay protocol described above with the exception that 25,000 FOXA2-expressing cells were seeded per well). The170 hits with at least 10-fold selectivity of FOXO1 over FOXA2 and a few control compounds with lower selectivity were further characterized for cell toxicity in an Alamar Blue assay using a 10 μM solution of (Z)-4-(1-methyltetrazol-5-yl)sulfanylbut-3-en-2-one (AZ7514) as the control compound, and for potency against human FOXO3 and mouse FOXO1 using reporter gene assays and 0.5 μM AZ4337 as control compound, following the reporter gene assay protocol described above (seeding 16,000 cells/well). For potency and cell toxicity, the percent effect was calculated using the GeneData Screener software as follows: 100 * [(X-DMSO)/(DMSO-Min)], where: X is the raw signal (CPS) for the well; Min is the median raw signal (CPS) obtained from the inhibitor control wells (0.5 μM AZ4337 for potency, 10μM AZ7514 for cell toxicity) on the same plate; DMSO is the median raw signal (CPS) obtained from the neutral control wells (0.5% DMSO) on the same plate.
Characterization of FOXO inhibitors
All compounds were from the AstraZeneca compound collection. They were characterized by 1H NMR spectroscopy and high-resolution masspectrometry of solid material from the collection inventory. 1H NMR spectra were recorded at 300 or 400 MHz. Chemical shifts (ppm) were determined relative to internal solvent (1H, δ 2.50 ppm; DMSO-d6). Analytical HPLC/MS was conducted on a QTOF mass spectrometer using a UV detector monitoring either at (a) 210 nm with a BEH C18 column (2.1mm × 100 mm, 1.7 μm, 0.7 mL/min flow rate), using a gradient of 2% v/v CH3CN in H2O (ammonium carbonate buffer, pH 10) to 98% v/v CH3CN in H2O, or at (b) 230 nm with an HSS C18 column (2.1 × 100 mm, 1.8 μm, 0.7 mL/min flow rate), using a gradient of 2% v/v CH3CN in H2O (ammonium formate buffer, pH 3) to 98% v/v CH3CN in H2O. All tested compounds were determined to be ≥95% pure using the analytical method (a) or (b) described above based on the peak area percentage. High-resolution mass spectra were carried out using high-resolution electrospray ionization mass spectrometry (HRESIMS) where the spectrometer was linked together with an Aquity®UPLC system. High resolution MS and 1H NMR data are presented below for each compound (Figure S7C).
Compound 1
1H NMR (400 MHz, DMSO-d6, ppm): δ 6.78 (broad s, 2H), 3.69 (m, 4H), 2.88 (m, 2H), 2.67 (m, 2H), 1.95 (2H, m), 1.63 (2H, m), 1.54 (4H, m). HRESIMS: calcd for C12H18N4[M + H]+, 219.1609; found 219.1612
Compound 2
1H NMR (400 MHz, DMSO-d6, ppm): δ 8.86 (dd, J=1.5, J=4.4, 1H), 8.62 (dd, J=1.5, J=8.4, 1H), 8.24 (d, J=2.5, 1H), 7.92 (d, J=2.5, 1H), 7.73 (dd, J=4.4, J=8.4, 1H), 7.17 (s, 2H). HRESIMS: calcd for C9H7N7[M + H]+, 214.0841; found 214.0831
Compound 3
1H NMR (400 MHz, DMSO-d6, ppm): δ 8.82 (s, 2H), 7.89 (d, J=7.8 Hz, 2H), 7.79 (d, J=7.8 Hz, 2H), 7.48 (broad s, 1H), 3.80 (m, 4H), 3.43 (m, 4H), 2.43 (broad s, 3H), 1.43 (s, 9H). HRESIMS: calcd for C20H27N5O4S[M + H]+, 434.1862; found 434.1866.
Compound 4
1H NMR (400 MHz, DMSO-d6, ppm): δ 9.65 (broad s, 1H), 8.33 (s, 1H), 8.17 (m, 2H), 7.69 (d, J=9.0 Hz, 1H), 7.53 (d, J=8.0 Hz, 2H), 7.39 (m, 1H), 7.36 (d, J=8.0 Hz, 2H), 7.01 (broad s, 1H), 6.44 (broad s, 1H), 4.67 (broad s, 2H.). HRESIMS: calcd for C19H14F3N5O[M + H]+, 386.1229; found 386.1218
Compound 5
1H NMR (400 MHz, DMSO-d6, ppm) δ 9.64 (s, 1H), 8.82 (m, 1H), 8.73 (m, 2H), 8.41 (d, J=6.3 Hz, 1H), 8.36 (d, J=8.3 Hz, 1H), 8.26 (m, 1H), 8.08 (d, J=6.5 Hz, 1H), 7.91 (s, 2H), 7.86 (m, 1H), 7.76 (d, J=7.9 Hz, 1H), 7.71 (m, 1H), 5.60 (s, 2H), 3.68 (m, 2H), 3.17 (m, 2H). HRESIMS: calcd for C24H19Cl2N3O2[M+H]+, 452.0932; found 452.0943
Compound 6
1H NMR (400 MHz, DMSO-d6, ppm): δ 8.73 (broad s, 1H), 8.60 (broad s, 1H), 7.92 (m, 1H), 7.65 (s, 1H), 7.22 (m, 1H), 7.17 (m, 1H), 6.98 (m, 1H), 6.92 (m, 1H), 6.34 (m, 1H), 3.39 (m, 2H), 3.29 (m, 2H). HRESIMS: calcd for C14H13ClF3N5O[M + H]+, 360.0839; found 360.0836.
Compound 7
1H NMR (400 MHz, DMSO-d6, ppm) δ 8.30 (s, 1H), 8.13 (d, J=8.3 Hz, 1H), 8.03 (s, 1H), 7.66 (d, J=8.9 Hz, 1H), 7.54 (broad s, 1H), 7.39 (d, J=8.0 Hz, 1H), 7.23 (m, 1H), 7.14 (m, 1H), 6.95 (m, 1H), 6.64 (d, J=8.9 Hz, 1H), 3.35 (m, 2H), 3.30 (m, 2H). HRESIMS: calcd for C15H14ClF3N4O[M + H]+, 359.0886; found 359.0889.
Compound 8
1H NMR (300 MHz, DMSO-d6, ppm): δ 11.69 (s, 1H), 8.16 (d, J = 5.10 Hz, 1H), 7.06 (s, 1H), 7.04 (s, 1H), 6.87 (d, J = 5.10 Hz, 1H), 6.77 (broad s, 1H), 3.37 (m, 2H), 3.34 (s, 3H), 2.87 (m, 2H). HRESIMS: calcd for C12H13N5O[M + H]+, 244.1198; found 244.1190.
Compound 9
1H NMR (400 MHz, DMSO-d6, ppm): δ 13.57 (br s, 1H), 11.15 (broad s, 1H), 8.16 (d, J = 2.2 Hz, 1H), 8.08 (dd, J = 2.2, 8.7 Hz, 1H), 7.63 (m, 2H), 7.31 (m, 3H), 3.96 (s, 3H). HRESIMS: calcd for C18H14ClN5O2[M + H]+, 368.0914; found 368.0912.
Compound 10
1H NMR (400 MHz, DMSO): δ 12.94 (broad, 1H), 10.81 (broad, 1H), 8.05-7.88 (m, 2H), 7.70-7.45 (m, 2H), 7.30-6.93 (m, 5H), 3.32 - 3.26 (m, 4H, on the slope of DMSO-d5), 2.90 (m, 1H), 2.47 - 2.41 (m, 4H), 2.22 (s, 3H). HRESIMS: calcd for C22H23N7O[M + H]+, 402.2042; found 402.2057.
Compound 11
1H NMR (400 MHz, CDCl3): δ 8.38 (d, J=6.5 Hz, 1H), 7.86 (m, 1H), 7.46(m, 1H), 7.39 (s, 1H), 7.23 (m, 1H), 7.19 (s, 1H), 6.98 (d, J=6.5 Hz, 1H), 5.57 (m, 1H), 4.02-3.76 (m, 5H), 3.65-3.37 (m, 2H), 2.60 (s, 3H), 2.14 - 2.03 (m, 1H), 1.93 - 1.61 (m, 2H), 1.55 (d, J=9.6 Hz, 6H). HRESIMS: calcd for C23H27ClN6O2[M + H]+, 455.1962; found 455.1948.
Compound 12
1H NMR (400 MHz, DMSO, 24°C): δ 10.04 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 7.97 (d, J = 9.0 Hz, 2H), 7.80 (d, J = 9.0 Hz, 2H), 7.49 (s, 1H), 7.20 (d, J = 5.2 Hz, 1H), 5.65 - 5.72 (m, 1H), 4.51 (t, J = 5.4 Hz, 1H), 3.69 (t, J = 6.0 Hz, 2H), 3.53 (t, J = 6.0 Hz, 2H), 3.29 - 3.37 (m, 4H), 2.55 (s, 3H), 1.50 (d, J = 6.8 Hz, 6H). HRESIMS: calcd for C21H27N5O4S[M + H]+, 446.1862; found 446.1849.
Compound 13
1H NMR (400 MHz, DMSO): δ 10.51 (broad s, 1H), 8.41 (s, 1H), 8.20 (d, J = 4.8 Hz, 1H), 8.10 (m, 1H), 7.72 (d, J = 4.8 Hz, 1H), 7.48 (m, 1H), 7.38 (m, 1H), 6.62 (m, 1H), 4.02 (s, 3H). HRESIMS: calcd for C13H11N3O2[M + H]+, 242.0929; found 242.0926.
Quantification and Statistical Analysis
Statistical analyses were performed by use of Prism 5.0 software (Graph Pad). We calculate p values for unpaired comparisons between two groups by two-tailed Student’s t-test. One-way ANOVA followed by Tukey’s multiple comparisons test (compare all pairs of columns) was used for comparisons between three or more groups. Two-way ANOVA followed by Bonferroni post-tests was used to examine the influence of two different variables. We used the customary threshold of p < 0.05 to declare statistical significance. * means p<0.05, ** p<0.01, and *** p<0.001. All results are presented as mean ± SEM. Sample size and statistical details can be found in the figures and legends.
Supplementary Material
Table S1. Related to Figure 5. Blood analysis in WT vs. L-Sin3a/b (constitutive KO) in fasting vs. refeeding conditions. FFA=Free Fatty Acid; TG=Triglyceride; GGT= Gamma-Glutamyl Transferase; AST= Aspartate transaminase; ALT= Alanine transaminase; BUN= Blood Urea Nitrogen; CPK= Creatine phosphokinase.
Table S2. Related to Figure 6. Blood analysis in WT vs. iL-Sin3a/b (induced KO) in fasting vs. refeeding conditions. FFA=Free Fatty Acid; TG=Triglyceride; GGT= Gamma-Glutamyl Transferase; AST= Aspartate transaminase; ALT= Alanine transaminase; BUN= Blood Urea Nitrogen; CPK= Creatine phosphokinase.
Table S3. Related to Figure 7. Reporter gene assay IC50 values (μM) for compound 1–13. h = human; m = mouse; n.a. = Not analyzed; n.d. = Not determined due to irregular curve shape at the highest concentrations tested.
Table S4. Related to Figure 7. Pharmacokinetics of compound 8, 9 and 13 in vivo in mice. Data are average means (n=3).
A, Time course of hepatic Foxo1, Foxo3 and Foxo4 expression during fasting and refeeding in mice. B–E, G6pc expression in primary hepatocytes after 7h treatment with vehicle or insulin (B, n=6 from 2 mice), cAMP or cAMP/insulin (C, n=8 from 2 mice), in dex or dex/insulin (D, n=8 from 2 mice), and cAMP/dex or cAMP/dex/insulin (E, n=6 from 2 mice). F, Gck expression in primary hepatocytes from WT (n=6 from 2 mice) vs. L-Foxo1,3,4 (n=8 from 2 mice) mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. G–H, G6pc expression in primary hepatocytes from WT (n=7–10 from 3 mice) vs. L-Foxo1 (n=7–10 from 3 mice) (G), and WT (n=6 from 2 mice) vs. L-Foxo1,3,4 mice (n=7 from 2 mice) (H) after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. i, Time course of Gck expression in L-Foxo1 primary hepatocytes treated with vehicle, cAMP/dex, or cAMP/dex/insulin (n=4 from 1 mouse, h=hours). J–K, Time- (K, n=3 from 1 mouse) and dose-dependence (J, n=3 from 1 mouse) of FOXO1-induced Gck expression in primary hepatocytes. L, Gck expression in primary hepatocytes from WT vs. L-Foxo1,3,4 mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin in the presence or absence of cycloheximide (n=3 from 1 mouse). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A, Schematic representation of transcription factors regulating Gck promoter activity (HNF4, hepatic nuclear factor 4 alpha; HNF6, hepatic nuclear factor 6; SREBF1, sterol regulatory element binding transcription factor 1c; PPARγ, peroxisome proliferator-activated receptor gamma; HIF1, hypoxia induced factor 1 alpha subunit). B–I, Time course of Foxo1 (B), Foxo3 (C), Foxo4 (D), Hnf4α (E), Hif1α (F), Pparγ (G), Srebf1 (H), and Hnf6 (I) expression in primary hepatocytes treated with vehicle, cAMP/dex, or cAMP/dex/insulin (n=3 from 1 mouse, h=hours). J–L, Foxo1 (J, n=12 from 3 mice), Foxo3 (K, n=4 from 1 mouse) and Hnf4α (L, n=8 from 2 mice) expression in primary hepatocytes treated with vehicle, dex, or dex/insulin. M–N, Representative immunoblot (M) and quantification (N) of FOXO1 time-dependent induction in primary hepatocytes treated with vehicle or dex (n=3). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A–H, Foxo1 (A), Foxo3 (B), Foxo4 (C), Hnf4α (D), Hif1α (E), PPARγ (F), Srebf1 (G), Hnf6 (H) expression in primary hepatocytes from WT (n=7 from 2 mice) or L-Foxo1 (n=7 from 2 mice) animals, treated with vehicle, cAMP/dex, or cAMP/dex/insulin. I–K, Hepatic Gck (I), Foxo1 (J) and Hnf4α (K) expression in WT mice vs. mice lacking hepatic glucocorticoid receptors treated or not with corticosterone for 5 weeks (n=4–5). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A–B, G6pc expression in L-Foxo1 primary hepatocytes transfected with plasmid (A, n=4 from 1 mouse) or adenoviruses (B, n=4–6 from 2 mice) encoding WT and mutant FOXO1 in the presence or absence of insulin. C, G6pc expression in primary hepatocytes from WT (n=4 from 1 mouse) vs. KR/KR (n=4 from 1 mouse) animals after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. D, G6pc expression in L- Foxo1 primary hepatocytes transfected with ADA-FOXO1 and DBD-FOXO1 adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). E, FOXO1 ChIP-qPCR on P5 (−1187 to −1040) and P22 (−93 to +52) in primary hepatocytes transduced with ADA-FOXO1 and DBD-FOXO1 adenoviruses (n=3). F–G, Gck (F, n=10 from 3 mice) and G6pc (G, n=4 from 1 mouse) expression in primary hepatocytes from WT vs. DBD mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. H, Co-immunoprecipitation of HNF4A and FOXO1. I, Rat Gck promoter activity in primary hepatocytes following transfection of FOXO1 or/and HNF4A (n=9 from 3 mice). J, HNF4A ChIP-qPCR on P20 (−219 to −77), P21 (−154 to −9) and P22 (−93 to +52) in primary hepatocytes treated with cAMP/dex, or cAMP/dex/insulin (n=4). K, Gck expression in L-Foxo1 primary hepatocytes transduced with ADA-FOXO1 and Δ256-FOXO1 adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). L, Multiple sequence alignment of FOXO1, O3 and O4, and location of Δ19 regions. M, G6pc expression in primary hepatocytes transfected with FOXO1 and Δ19-FOXO1 plasmids in the presence or absence of insulin (n=4 from 1 mouse). N, Rat Gck promoter activity in primary hepatocytes transfected with FOXO1, Δ19-FOXO1 and SID2-FOXO1 plasmids (n=6 from 2 mice). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel B, D and K, * or # are used to compare, respectively, solid and empty bars to each other).
A–D, Sin3a expression in WT primary hepatocytes after 7h-treatment with vehicle, cAMP/dex, or cAMP/dex/insulin (A, n=15 from 4 mice), vehicle, dex, or dex/insulin (B, n=8 from 2 mice), vehicle, cAMP, or cAMP/insulin (C, n=7 from 2 mice), and vehicle or insulin (D, n=3 from 1 mouse). E–F, Sin3a expression in primary hepatocytes from WT vs. L-Foxo1 (E, n=3 from 1 mouse), and WT vs. KR/KR (F, n=4 from 1 mouse) mice after 7h treatment vehicle, cAMP/dex, or cAMP/dex/insulin. G, Sin3a expression in primary hepatocytes transfected with FOXO1 plamids (n=29 from 8 mice). H–I, Foxo1 expression in primary hepatocytes transfected with SIN3A plasmid (H, n=21 from 6 mice), or Sin3a siRNA (I, n=11 from 3 mice). J, Gck expression in WT primary hepatocytes transfected with FOXO1 plasmid in the presence or absence of FK228 (n=8 from 2 mice). K, Liver trichrome staining in WT and L-Sin3a/b mice (arrow = bile duct; asterisk = necrosis). L–M, Kidney (L) and spleen (M) weight in 12h-fasted (n=12/11 in L, 7/7 in M) and 4h-refed (n=20/11 in L, 11/7 in M WT and L-Sin3a/b mice. Data are means ± s.e.m. Scale bar = 100 μm. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel J, * or # are used to compare, respectively, solid and empty bars to each other).
A, Liver H&E, trichrome and PAS staining in iL-Sin3a/b mice. B, Liver weight in 12h-fasted (n=6/7) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. C, Insulin levels in ad libitum-fed and overnight-fasted iWT and iL-Sin3a/b mice (n=6). D–F, Weight (D, n=15), fat mass (E, n=7), and lean mass (F, n=7) of adult male iWT and iL-Sin3a/b mice on chow diet. G, Kidney weight in 12h-fasted (n=6/7) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. H–I, Representative immunoblot (H, from 2 blot simultaneously processed) and quantification (I) of hepatic GCK protein levels in 12h-fasted and 4h-refed iWT and iL-Sin3a/b mice (n=5). J, Glycogen content in primary hepatocytes obtained from iWT and iL-Sin3a/b mice (n=6 from 1 mouse). K, Glycolysis in iWT and iL-Sin3a/b primary hepatocytes (n=9 from 1 mouse). L, FOXO1 ChIP-qPCR on P22 (−93 to +52) in primary hepatocytes from iWT and iL-Sin3a/b mice in cAMP/Dex conditions (n=3). Data are means ± s.e.m. Scale bar = 100 μm. *P<0.05, **P<0.01, ***P<0.001 compared to control condition (in panel K, *, # and $ are used to compare, respectively, solid, empty and grey bars to each other).
A–B, Selectivity of compounds (Cpd) 8 (A) and 9 (B) measured in HEK293 cells by luciferase reporter gene assays. Data are average response means (percent effect, see Methods; n≥3) ± s.e.m. C, Chemical structures of the reported FOXO inhibitors.
Highlights.
Discovery of SIN3a as the FOXO corepressor of hepatic glucokinase
SIN3a regulates hepatic insulin sensitivity
Co-repressor clearance as a novel mechanism of gene induction by insulin
Selective targeting of the activator and repressor functions of FOXO1
Acknowledgments
This work was supported by NIH grants DK57539 and DK63608 (Columbia Diabetes Research Center) and by the Fondation Bettencourt Schueller (F.L.). We would like to thank Frank Jansen, Jennifer Hicks, Yantao Chen, Marléne Fredenwall, Olle Karlsson, Maria Petersson, Johan Ulander, Catherine Bardelle, Marian Preston, Brett Litten, Lena Svensson, Dorota Kakol-Palm and Eva Lundborg at AstraZeneca for their contributions; Dr. Ronald A. DePinho for providing the floxed Sin3a/b animals, and Dr. Mark Magnuson for the anti-GCK antibody. The authors declare no competing financial interests. D.L., E.E., T.N., and A.J. are employed by AstraZeneca.
Footnotes
Author Contributions. F.L. designed and performed experiments, analyzed data, and wrote the manuscript. R.A.H performed experiments, provided mouse strains, and edited the manuscript. D.L., E.E., T.N., A.J. designed and performed (E.E) experiments, analyzed data, and wrote the manuscript. K.A. performed experiments. C.B. provided tissue samples from experimental animals. D.A. designed experiments, oversaw research, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altomonte J, Richter A, Harbaran S, Suriawinata J, Nakae J, Thung SN, Meseck M, Accili D, Dong H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol - Endocrinol Metab. 2003;285:E718–E728. doi: 10.1152/ajpendo.00156.2003. [DOI] [PubMed] [Google Scholar]
- Bae JS, Kim TH, Kim MY, Park JM, Ahn YH. Transcriptional Regulation of Glucose Sensors in Pancreatic β-Cells and Liver: An Update. Sensors. 2010;10:5031–5053. doi: 10.3390/s100505031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Kim-Muller JY, Mastracci TL, Kofler NM, Qiang L, Haeusler RA, Jurczak MJ, Laznik D, Heinrich G, Samuel VT, et al. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metab. 2011;14:587–597. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes VL, Bhat A, Unnikrishnan A, Heydari AR, Arking R, Pile LA. SIN3 is critical for stress resistance and modulates adult lifespan. Aging. 2014;6:645–660. doi: 10.18632/aging.100684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz K, Peters R, Kjos SL, Goico J, Marroquin A, Dunn ME, Xiang A, Azen S, Buchanan TA. Effect of troglitazone on insulin sensitivity and pancreatic beta-cell function in women at high risk for NIDDM. Diabetes. 1996;45:1572–1579. doi: 10.2337/diab.45.11.1572. [DOI] [PubMed] [Google Scholar]
- Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab TEM. 2012;23:205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Choi D, Oh KJ, Han HS, Yoon YS, Jung CY, Kim ST, Koo SH. Protein arginine methyltransferase 1 regulates hepatic glucose production in a FoxO1-dependent manner. Hepatol Baltim Md. 2012;56:1546–1556. doi: 10.1002/hep.25809. [DOI] [PubMed] [Google Scholar]
- Cook JR, Matsumoto M, Banks AS, Kitamura T, Tsuchiya K, Accili D. A mutant allele encoding DNA binding-deficient FoxO1 differentially regulates hepatic glucose and lipid metabolism. Diabetes. 2015a;64:1951–1965. doi: 10.2337/db14-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JR, Langlet F, Kido Y, Accili D. Pathogenesis of selective insulin resistance in isolated hepatocytes. J Biol Chem. 2015b;290:13972–13980. doi: 10.1074/jbc.M115.638197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, DePinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–1595. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Grandinetti KB, Finnerty PM, Simpson N, Chu GC, DePinho RA. Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proc Natl Acad Sci. 2008;105:4168–4172. doi: 10.1073/pnas.0710285105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhana L, Dawson MI, Leid M, Wang L, Moore DD, Liu G, Xia Z, Fontana JA. Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor. Cancer Res. 2007;67:318–325. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Stanley GHS. A SIMPLE METHOD FOR THE ISOLATION AND PURIFICATION OF TOTAL LIPIDES FROM ANIMAL TISSUES. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Foretz M, Guichard C, Ferré P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci. 1999;96:12737–12742. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Ganjam GK, Dimova EY, Unterman TG, Kietzmann T. FoxO1 and HNF-4 are involved in regulation of hepatic glucokinase gene expression by resveratrol. J Biol Chem. 2009;284:30783–30797. doi: 10.1074/jbc.M109.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem. 2010a;285:35245–35248. doi: 10.1074/jbc.C110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Han S, Accili D. Hepatic FoxO1 ablation exacerbates lipid abnormalities during hyperglycemia. J Biol Chem. 2010b;285:26861–26868. doi: 10.1074/jbc.M110.134023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun. 2014;5:5190. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- Hirota K, Daitoku H, Matsuzaki H, Araya N, Yamagata K, Asada S, Sugaya T, Fukamizu A. Hepatocyte Nuclear Factor-4 Is a Novel Downstream Target of Insulin via FKHR as a Signal-regulated Transcriptional Inhibitor. J Biol Chem. 2003;278:13056–13060. doi: 10.1074/jbc.C200553200. [DOI] [PubMed] [Google Scholar]
- Hirota K, Sakamaki J, Ishida J, Shimamoto Y, Nishihara S, Kodama N, Ohta K, Yamamoto M, Tanimoto K, Fukamizu A. A Combination of HNF-4 and Foxo1 Is Required for Reciprocal Transcriptional Regulation of Glucokinase and Glucose-6-phosphatase Genes in Response to Fasting and Feeding. J Biol Chem. 2008;283:32432–32441. doi: 10.1074/jbc.M806179200. [DOI] [PubMed] [Google Scholar]
- Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stöckli J, Yang JY, James DE. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013;17:1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey SJ, Azimifar SB, Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol. 2015;33:990–995. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kadamb R, Mittal S, Bansal N, Batra H, Saluja D. Sin3: Insight into its transcription regulatory functions. Eur J Cell Biol. 2013;92:237–246. doi: 10.1016/j.ejcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Katz NR, Nauck MA, Wilson PT. Induction of glucokinase by insulin under the permissive action of dexamethasone in primary rat hepatocyte cultures. Biochem Biophys Res Commun. 1979;88:23–29. doi: 10.1016/0006-291x(79)91691-7. [DOI] [PubMed] [Google Scholar]
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R, et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N Engl J Med. 2016;374:1321–1331. doi: 10.1056/NEJMoa1506930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kim H, Kim TH, Im SS, Park SK, Lee IK, Kim KS, Ahn YH. SREBP-1c mediates the insulin-dependent hepatic glucokinase expression. J Biol Chem. 2004;279:30823–30829. doi: 10.1074/jbc.M313223200. [DOI] [PubMed] [Google Scholar]
- Kim TH, Kim H, Park JM, Im SS, Bae JS, Kim MY, Yoon HG, Cha JY, Kim KS, Ahn YH. Interrelationship between liver X receptor alpha, sterol regulatory element-binding protein-1c, peroxisome proliferator-activated receptor gamma, and small heterodimer partner in the transcriptional regulation of glucokinase gene expression in liver. J Biol Chem. 2009;284:15071–15083. doi: 10.1074/jbc.M109.006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. Statistical practice in high-throughput screening data analysis. Nat Biotechnol. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- Massa ML, Gagliardino JJ, Francini F. Liver glucokinase: An overview on the regulatorymechanisms of its activity. IUBMB Life. 2011;63:1–6. doi: 10.1002/iub.411. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Barr V, Accili D. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000;19:989–996. doi: 10.1093/emboj/19.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001a;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Ogawa W, Kasuga M, Accili D. Insulin regulation of gene expression through the forkhead transcription factor Foxo1 (Fkhr) requires kinases distinct from Akt. Biochemistry (Mosc) 2001b;40:11768–11776. doi: 10.1021/bi015532m. [DOI] [PubMed] [Google Scholar]
- Nakae J, Cao Y, Hakuno F, Takemori H, Kawano Y, Sekioka R, Abe T, Kiyonari H, Tanaka T, Sakai J, et al. Novel repressor regulates insulin sensitivity through interaction with Foxo1. EMBO J. 2012;31:2275–2295. doi: 10.1038/emboj.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Accili D. The new biology of diabetes. Diabetologia. 2015;58:2459–2468. doi: 10.1007/s00125-015-3722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y-P, Kumar GA, Zhang J-S, Urrutia R. Differential binding of Sin3 interacting repressor domains to the PAH2 domain of Sin3A. FEBS Lett. 2003;548:108–112. doi: 10.1016/s0014-5793(03)00749-x. [DOI] [PubMed] [Google Scholar]
- Pile LA, Spellman PT, Katzenberger RJ, Wassarman DA. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: implications for the regulation of energy metabolism. J Biol Chem. 2003;278:37840–37848. doi: 10.1074/jbc.M305996200. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual Roles for Glucokinase in Glucose Homeostasis as Determined by Liver and Pancreatic β Cell-specific Gene Knock-outs Using Cre Recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. 2010;285:27396–27401. doi: 10.1074/jbc.M110.140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth U, Curth K, Unterman TG, Kietzmann T. The Transcription Factors HIF-1 and HNF-4 and the Coactivator p300 Are Involved in Insulin-regulated Glucokinase Gene Expression via the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. J Biol Chem. 2004;279:2623–2631. doi: 10.1074/jbc.M308391200. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S, McKay R, Monia B, Shutter JR, Lindberg RA, et al. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes. 2006;55:2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–393. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- Solaimani P, Damoiseaux R, Hankinson O. Genome-Wide RNAi High-Throughput Screen Identifies Proteins Necessary for the AHR-Dependent Induction of CYP1A1 by 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2013;136:107–119. doi: 10.1093/toxsci/kft191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JT, Pitot HC. Hormonal regulation of glucokinase in primary cultures of adult rat hepatocytes. J Biol Chem. 1979;254:12331–12336. [PubMed] [Google Scholar]
- van der Vos KE, Coffer PJ. FOXO-binding partners: it takes two to tango. Oncogene. 2008;27:2289–2299. doi: 10.1038/onc.2008.22. [DOI] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 5. Blood analysis in WT vs. L-Sin3a/b (constitutive KO) in fasting vs. refeeding conditions. FFA=Free Fatty Acid; TG=Triglyceride; GGT= Gamma-Glutamyl Transferase; AST= Aspartate transaminase; ALT= Alanine transaminase; BUN= Blood Urea Nitrogen; CPK= Creatine phosphokinase.
Table S2. Related to Figure 6. Blood analysis in WT vs. iL-Sin3a/b (induced KO) in fasting vs. refeeding conditions. FFA=Free Fatty Acid; TG=Triglyceride; GGT= Gamma-Glutamyl Transferase; AST= Aspartate transaminase; ALT= Alanine transaminase; BUN= Blood Urea Nitrogen; CPK= Creatine phosphokinase.
Table S3. Related to Figure 7. Reporter gene assay IC50 values (μM) for compound 1–13. h = human; m = mouse; n.a. = Not analyzed; n.d. = Not determined due to irregular curve shape at the highest concentrations tested.
Table S4. Related to Figure 7. Pharmacokinetics of compound 8, 9 and 13 in vivo in mice. Data are average means (n=3).
A, Time course of hepatic Foxo1, Foxo3 and Foxo4 expression during fasting and refeeding in mice. B–E, G6pc expression in primary hepatocytes after 7h treatment with vehicle or insulin (B, n=6 from 2 mice), cAMP or cAMP/insulin (C, n=8 from 2 mice), in dex or dex/insulin (D, n=8 from 2 mice), and cAMP/dex or cAMP/dex/insulin (E, n=6 from 2 mice). F, Gck expression in primary hepatocytes from WT (n=6 from 2 mice) vs. L-Foxo1,3,4 (n=8 from 2 mice) mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. G–H, G6pc expression in primary hepatocytes from WT (n=7–10 from 3 mice) vs. L-Foxo1 (n=7–10 from 3 mice) (G), and WT (n=6 from 2 mice) vs. L-Foxo1,3,4 mice (n=7 from 2 mice) (H) after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. i, Time course of Gck expression in L-Foxo1 primary hepatocytes treated with vehicle, cAMP/dex, or cAMP/dex/insulin (n=4 from 1 mouse, h=hours). J–K, Time- (K, n=3 from 1 mouse) and dose-dependence (J, n=3 from 1 mouse) of FOXO1-induced Gck expression in primary hepatocytes. L, Gck expression in primary hepatocytes from WT vs. L-Foxo1,3,4 mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin in the presence or absence of cycloheximide (n=3 from 1 mouse). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A, Schematic representation of transcription factors regulating Gck promoter activity (HNF4, hepatic nuclear factor 4 alpha; HNF6, hepatic nuclear factor 6; SREBF1, sterol regulatory element binding transcription factor 1c; PPARγ, peroxisome proliferator-activated receptor gamma; HIF1, hypoxia induced factor 1 alpha subunit). B–I, Time course of Foxo1 (B), Foxo3 (C), Foxo4 (D), Hnf4α (E), Hif1α (F), Pparγ (G), Srebf1 (H), and Hnf6 (I) expression in primary hepatocytes treated with vehicle, cAMP/dex, or cAMP/dex/insulin (n=3 from 1 mouse, h=hours). J–L, Foxo1 (J, n=12 from 3 mice), Foxo3 (K, n=4 from 1 mouse) and Hnf4α (L, n=8 from 2 mice) expression in primary hepatocytes treated with vehicle, dex, or dex/insulin. M–N, Representative immunoblot (M) and quantification (N) of FOXO1 time-dependent induction in primary hepatocytes treated with vehicle or dex (n=3). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A–H, Foxo1 (A), Foxo3 (B), Foxo4 (C), Hnf4α (D), Hif1α (E), PPARγ (F), Srebf1 (G), Hnf6 (H) expression in primary hepatocytes from WT (n=7 from 2 mice) or L-Foxo1 (n=7 from 2 mice) animals, treated with vehicle, cAMP/dex, or cAMP/dex/insulin. I–K, Hepatic Gck (I), Foxo1 (J) and Hnf4α (K) expression in WT mice vs. mice lacking hepatic glucocorticoid receptors treated or not with corticosterone for 5 weeks (n=4–5). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions.
A–B, G6pc expression in L-Foxo1 primary hepatocytes transfected with plasmid (A, n=4 from 1 mouse) or adenoviruses (B, n=4–6 from 2 mice) encoding WT and mutant FOXO1 in the presence or absence of insulin. C, G6pc expression in primary hepatocytes from WT (n=4 from 1 mouse) vs. KR/KR (n=4 from 1 mouse) animals after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. D, G6pc expression in L- Foxo1 primary hepatocytes transfected with ADA-FOXO1 and DBD-FOXO1 adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). E, FOXO1 ChIP-qPCR on P5 (−1187 to −1040) and P22 (−93 to +52) in primary hepatocytes transduced with ADA-FOXO1 and DBD-FOXO1 adenoviruses (n=3). F–G, Gck (F, n=10 from 3 mice) and G6pc (G, n=4 from 1 mouse) expression in primary hepatocytes from WT vs. DBD mice after 7h treatment with vehicle, cAMP/dex, or cAMP/dex/insulin. H, Co-immunoprecipitation of HNF4A and FOXO1. I, Rat Gck promoter activity in primary hepatocytes following transfection of FOXO1 or/and HNF4A (n=9 from 3 mice). J, HNF4A ChIP-qPCR on P20 (−219 to −77), P21 (−154 to −9) and P22 (−93 to +52) in primary hepatocytes treated with cAMP/dex, or cAMP/dex/insulin (n=4). K, Gck expression in L-Foxo1 primary hepatocytes transduced with ADA-FOXO1 and Δ256-FOXO1 adenoviruses in the presence or absence of insulin (n=4 from 1 mouse). L, Multiple sequence alignment of FOXO1, O3 and O4, and location of Δ19 regions. M, G6pc expression in primary hepatocytes transfected with FOXO1 and Δ19-FOXO1 plasmids in the presence or absence of insulin (n=4 from 1 mouse). N, Rat Gck promoter activity in primary hepatocytes transfected with FOXO1, Δ19-FOXO1 and SID2-FOXO1 plasmids (n=6 from 2 mice). Data are means ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel B, D and K, * or # are used to compare, respectively, solid and empty bars to each other).
A–D, Sin3a expression in WT primary hepatocytes after 7h-treatment with vehicle, cAMP/dex, or cAMP/dex/insulin (A, n=15 from 4 mice), vehicle, dex, or dex/insulin (B, n=8 from 2 mice), vehicle, cAMP, or cAMP/insulin (C, n=7 from 2 mice), and vehicle or insulin (D, n=3 from 1 mouse). E–F, Sin3a expression in primary hepatocytes from WT vs. L-Foxo1 (E, n=3 from 1 mouse), and WT vs. KR/KR (F, n=4 from 1 mouse) mice after 7h treatment vehicle, cAMP/dex, or cAMP/dex/insulin. G, Sin3a expression in primary hepatocytes transfected with FOXO1 plamids (n=29 from 8 mice). H–I, Foxo1 expression in primary hepatocytes transfected with SIN3A plasmid (H, n=21 from 6 mice), or Sin3a siRNA (I, n=11 from 3 mice). J, Gck expression in WT primary hepatocytes transfected with FOXO1 plasmid in the presence or absence of FK228 (n=8 from 2 mice). K, Liver trichrome staining in WT and L-Sin3a/b mice (arrow = bile duct; asterisk = necrosis). L–M, Kidney (L) and spleen (M) weight in 12h-fasted (n=12/11 in L, 7/7 in M) and 4h-refed (n=20/11 in L, 11/7 in M WT and L-Sin3a/b mice. Data are means ± s.e.m. Scale bar = 100 μm. *P<0.05, **P<0.01, ***P<0.001 compared to control conditions (in panel J, * or # are used to compare, respectively, solid and empty bars to each other).
A, Liver H&E, trichrome and PAS staining in iL-Sin3a/b mice. B, Liver weight in 12h-fasted (n=6/7) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. C, Insulin levels in ad libitum-fed and overnight-fasted iWT and iL-Sin3a/b mice (n=6). D–F, Weight (D, n=15), fat mass (E, n=7), and lean mass (F, n=7) of adult male iWT and iL-Sin3a/b mice on chow diet. G, Kidney weight in 12h-fasted (n=6/7) and 4h-refed (n=9/9) iWT and iL-Sin3a/b mice. H–I, Representative immunoblot (H, from 2 blot simultaneously processed) and quantification (I) of hepatic GCK protein levels in 12h-fasted and 4h-refed iWT and iL-Sin3a/b mice (n=5). J, Glycogen content in primary hepatocytes obtained from iWT and iL-Sin3a/b mice (n=6 from 1 mouse). K, Glycolysis in iWT and iL-Sin3a/b primary hepatocytes (n=9 from 1 mouse). L, FOXO1 ChIP-qPCR on P22 (−93 to +52) in primary hepatocytes from iWT and iL-Sin3a/b mice in cAMP/Dex conditions (n=3). Data are means ± s.e.m. Scale bar = 100 μm. *P<0.05, **P<0.01, ***P<0.001 compared to control condition (in panel K, *, # and $ are used to compare, respectively, solid, empty and grey bars to each other).
A–B, Selectivity of compounds (Cpd) 8 (A) and 9 (B) measured in HEK293 cells by luciferase reporter gene assays. Data are average response means (percent effect, see Methods; n≥3) ± s.e.m. C, Chemical structures of the reported FOXO inhibitors.