

Humans with a rare gene mutation in SERPINE1 live longer and show evidence of protection from aging-related morbidity.
Abstract
Plasminogen activator inhibitor–1 (PAI-1) has been shown to be a key component of the senescence-related secretome and a direct mediator of cellular senescence. In murine models of accelerated aging, genetic deficiency and targeted inhibition of PAI-1 protect against aging-like pathology and prolong life span. However, the role of PAI-1 in human longevity remains unclear. We hypothesized that a rare loss-of-function mutation in SERPINE1 (c.699_700dupTA), which encodes PAI-1, could play a role in longevity and metabolism in humans. We studied 177 members of the Berne Amish community, which included 43 carriers of the null SERPINE1 mutation. Heterozygosity was associated with significantly longer leukocyte telomere length, lower fasting insulin levels, and lower prevalence of diabetes mellitus. In the extended Amish kindred, carriers of the null SERPINE1 allele had a longer life span. Our study indicates a causal effect of PAI-1 on human longevity, which may be mediated by alterations in metabolism. Our findings demonstrate the utility of studying loss-of-function mutations in populations with geographic and genetic isolation and shed light on a novel therapeutic target for aging.
INTRODUCTION
Age is the primary risk factor for most of the chronic diseases, including type 2 diabetes mellitus (DM), metabolic syndrome, and cardiovascular diseases (CVD) (1). The prevalence of age-related diseases has increased concordantly with the growing proportion of elderly individuals in the population. Aging remains one of the most challenging biological processes to unravel, with coordinated and interrelated molecular and cellular changes (2). Humans exhibit clear differential trajectories of age-related decline on a cellular level with telomere attrition across various somatic tissues and on a physiological level across multiple organ systems (3). In addition to telomere length, López-Otín and colleagues (4) proposed several molecular drivers of aging, including genomic instability, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication. Despite knowledge of these potential molecular causes of aging, no targeted interventions currently exist to delay the aging process and to promote healthy longevity (5, 6).
In the United States, cardiometabolic disease influences life span as a leading cause of death and disability in adult men and women (7). Cardiometabolic disease is associated with a shorter leukocyte telomere length (LTL) (8, 9). Telomere shortening, which results from replication of somatic cells in vitro and in vivo, may cause replicative senescence. Senescent cells and tissues exhibit a distinctive pattern of protein expression, including increased plasminogen activator inhibitor–1 (PAI-1) as a part of the senescence-associated secretory phenotype (SASP) (10, 11). PAI-1, which is encoded by the SERPINE1 gene, is the primary inhibitor of endogenous plasminogen activators and is synthesized in the liver and fat tissue. In addition to its role in regulating fibrinolysis, PAI-1 also contributes directly to cellular senescence in vitro (12). Genetic absence or pharmacologic inhibition of PAI-1 in murine models of accelerated aging provides protection from aging-like pathology, prevents telomere shortening, and prolongs life span (13). Cross-sectional human studies have demonstrated an association of plasma levels of PAI-1 with insulin resistance (14, 15). Mendelian randomization analyses from large genome-wide association studies (GWAS) provide an additional supportive evidence for a casual effect of PAI-1 on insulin resistance and coronary heart disease (16).
The role of the SASP, in general, and specifically PAI-1 in longevity in humans is uncertain. We have previously reported the identification of a rare frameshift mutation (c.699_700dupTA) in the SERPINE1 gene in the Old Order Amish (OOA), living in relative geographic and genetic isolation in the vicinity of Berne, Indiana; this mutation results in a lifelong reduction in PAI-1 levels (17, 18). Therefore, we tested the association of carrier status for the null SERPINE1 mutation with LTL as the prespecified primary end point in the only known cohort with a SERPINE1 null mutation. The secondary end points of the study included fasting insulin level, prevalence of DM, and life span. The findings of this study were extended by assessing the novel aging scores derived in the OOA cohort in a U.S. population–based cohort study, the Coronary Artery Risk Development in Young Adults Study (CARDIA).
RESULTS
Baseline characteristics of the study participants
The clinical characteristics of the study participants by SERPINE1 genotype status are shown in Table 1 and table S1. A total of 177 participants was enrolled. Forty-three participants were identified as carriers of the null SERPINE1 mutation, and seven participants were identified as homozygous for the null SERPINE1 mutation with an overall minor (null) allele frequency of 16% in the study population. The observed SERPINE1 genotype frequencies were in Hardy-Weinberg equilibrium. Heterozygous carriers of the null SERPINE1 mutation had 50% lower mean circulating plasma PAI-1 levels (5.9 ± 6.6 ng/ml versus 12.7 ± 9.8 ng/ml; P < 0.0001) compared with unaffected participants. Homozygous individuals for the null SERPINE1 mutation had no evidence of detectable PAI-1 antigen in the plasma, consistent with the loss-of-function mutation. Unaffected Amish participants had lower body mass index, fasting glucose, and triglyceride levels than the CARDIA participants sampled from the U.S. urban communities (Table 2). Rates of clinically overt disease with hypertension and type 2 DM were similar in both unaffected Amish participants and the CARDIA participants.
Table 1. Clinical characteristics of the Berne Amish kindred by SERPINE1 genotype status.
Continuous values are listed as mean ± SD unless otherwise specified. HDL, high-density lipoprotein; LDL, low-density lipoprotein.
|
SERPINE1+/+ (n = 127) |
SERPINE1+/− (n = 43) |
P* | |
| Age (years) | 46 ± 20 | 44 ± 17 | 0.97 |
| Female, n (%) | 70 (55) | 28 (65) | 0.43 |
| Hypertension, n (%) | 41 (33) | 14 (33) | 0.85 |
| Systolic blood pressure (mmHg) | 130 ±18 | 128 ± 20 | 0.69 |
| Diastolic blood pressure (mmHg) | 77 ± 10 | 78 ± 12 | 0.60 |
| Obesity, n (%) | 38 (30) | 12 (28) | 0.60 |
| Body mass index (kg/m2) | 27.7 ± 5.9 | 26.9 ± 6.3 | 0.29 |
| Diabetes, n (%) | 8 (7) | 0 (0) | <0.01 |
| Fasting insulin (uIU/ml)† | 4.9 (3.3–6.7) | 4.0 (2.9–5.1) | 0.04 |
| Total cholesterol (mg/dl) | 188 ± 40 | 188 ± 55 | 0.92 |
| HDL cholesterol (mg/dl) | 60 ± 15 | 63 ± 12 | 0.25 |
| Triglyceride (mg/dl)† | 81 (57–113) | 76 (51–110) | 0.54 |
| LDL cholesterol (mg/dl) | 109 ± 32 | 107 ± 44 | 0.67 |
| Serum creatinine (mg/dl) | 0.80 ± 0.16 | 0.75 ± 0.15 | 0.13 |
| Ever smoked, n (%) | 48 (38) | 12 (28) | 0.11 |
*Polygenic model adjusted for carrier status and family structure in SOLAR.
†Non-normally distributed, reported as median (25th to 75th percentile).
Table 2. Comparison of clinical characteristics of the Berne Amish kindred SERPINE1+/+ and CARDIA.
|
SERPINE1+/+ (n = 127) |
CARDIA (n = 2793) |
P | |
| Age (years) | 46 ± 20 | 50 ± 4 | 0.03 |
| Female, n (%) | 70 (55) | 1605 (57) | 0.60 |
| Hypertension, n (%) | 41 (33) | 1023 (37) | 0.42 |
| Systolic blood pressure (mmHg) | 130 ± 18 | 119 ± 16 | <0.01 |
| Diastolic blood pressure (mmHg) | 77 ± 10 | 75 ± 11 | 0.02 |
| Obesity, n (%) | 38 (30) | 1181 (42) | <0.01 |
| Body mass index (kg/m2) | 28 ± 6 | 30 ± 7 | <0.01 |
| Diabetes, n (%) | 8 (7) | 296 (11) | 0.17 |
| Fasting glucose (mg/dl)* | 88 (82–98) | 94 (87–102) | <0.01 |
| Total cholesterol (mg/dl) | 188 ± 40 | 192 ± 36 | 0.23 |
| HDL cholesterol (mg/dl) | 60 ± 15 | 58 ± 18 | 0.18 |
| Triglyceride (mg/dl)* | 81 (57–113) | 91 (68–132) | <0.01 |
| LDL cholesterol (mg/dl) | 109 ± 32 | 112 ± 32 | 0.28 |
| Serum creatinine (mg/dl) | 0.80 ± 0.16 | 0.88 ± 0.44 | <0.01 |
| Ever smoked, n (%) | 48 (38) | 1007 (37) | 0.77 |
*Non-normally distributed, reported as median (25th to 75th percentile).
Association of null SERPINE1 heterozygosity with telomere length and life span
Overall, the mean LTL was highly correlated with chronological age (R = −0.70, P < 0.0001) and 9% shorter per decade of age (P < 0.0001) in unaffected Amish participants. Carriers of the SERPINE1 mutation had a 10% longer mean LTL, the prespecified primary end point, after adjustment for age, sex, and familial relatedness compared with noncarriers (P = 0.007) (Fig. 1). Interaction between age and carrier status of the SERPINE1 null mutation in association with LTL was not significant. Secondary analyses, including participants who were homozygous for the SERPINE1 null mutation (n = 7), demonstrated similar results (P = 0.020; table S2). The overall heritability (h2) estimate of LTL was 0.55 (P < 0.0001), suggesting that the additive effects of genetic variation, including the SERPINE1 mutation, accounted for 55% of the total variation.
Fig. 1. Association of SERPINE1 genotype status and leukocyte telomere length as a function of age in the Berne Amish kindred.
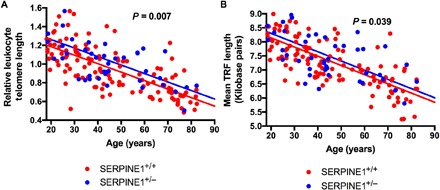
(A and B) LTL in SERPINE1 null allele carriers and noncarriers in the Berne Amish kindred as quantified by (A) polymerase chain reaction (PCR) and (B) Southern Blot. Relative LTL is shown in (A), and mean terminal restriction fragment (TRF) length is shown in (B) as a function of age stratified by SERPINE1 mutation status. P value represents difference in mean LTL and TRF by SERPINE1 mutation status (carriers versus noncarriers) after adjustment for age, sex, and family structure in Sequential Oligogenic Linkage Analysis Routines (SOLAR) (P = 0.007 and P = 0.039, respectively). Every 1-year increase in age of study participant was associated with a 0.0087 lower relative LTL (P < 0.0001) and a 30–base pair lower mean TRF (P < 0.0001).
Vital status was available on 221 individuals identified as directly related to our study participants. Genotype status for SERPINE1 was ascertained by direct genotyping or obligate ascertainment from ancestral data in deceased individuals with known dates of birth and death (n = 56). The median survival in SERPINE1 null carriers compared with unaffected individuals was longer [85 (73 to 88) versus 75 (70 to 83); P = 0.037], as shown in Fig. 2.
Fig. 2. Age at death in the extended Berne Amish kindred by genotype status for SERPINE1.
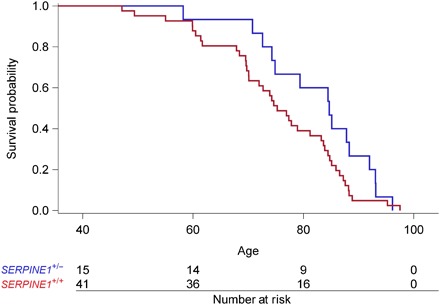
On the basis of ancestral data obtained from the extended pedigree and extensive fieldwork, we identified 221 individuals with known dates of birth and death. Genotype status for SERPINE1 was ascertained by direct genotyping or obligate ascertainment in 56 family members. The mean ± SD age at death was delayed by 7 years in SERPINE1 null carriers compared with unaffected individuals (82 ± 10 versus 75 ± 12; P = 0.037 by Wilcoxon rank sum test).
Association of null SERPINE1 heterozygosity and metabolic traits
Carriers of the SERPINE1 mutation had similar mean body mass index compared with noncarriers (P = 0.46). The prevalence of type 2 DM, one of the prespecified secondary end points, was significantly lower in carriers of the SERPINE1 null mutation compared to noncarriers, as shown in Table 1 (0% versus 7%; P = 0.001). Fasting insulin levels were 28% lower in SERPINE1 heterozygotes (P = 0.035) after adjustment for age, sex, and familial relatedness in comparison to unaffected participants. Secondary analysis including Amish participants (n = 7) who were homozygous for the null SERPINE1 mutation demonstrated similar results (P = 0.026). Sensitivity analysis performed with exclusion of Amish participants with diabetes did not alter the results. In addition, we observed a significant positive correlation between PAI-1 and fasting insulin levels in the Amish participants (R = 0.55, P < 0.0001) and the CARDIA participants (R = 0.48, P < 0.05).
Association of null SERPINE1 heterozygosity and cardiovascular parameters
Tissue Doppler e′ velocity was slightly higher in carriers than controls but did not achieve significance (12.8 ± 4.0 versus 12.3 ± 4.1; P = not significant). Brachial pulse pressure [47 (42 to 53) versus 51 (44 to 57)] and carotid intima-media thickness (cIMT) [0.60 (0.51 to 0.72) versus 0.61 (0.52 to 0.79)] were lower in carriers than controls, but differences were not significant.
Several cardiovascular measures that correlated significantly with chronological age in unaffected individuals (|R| > 0.60, P < 0.0001; Fig. 3) were identified. A composite score of cardiovascular aging that represents vascular and myocardial health (e′ velocity, brachial pulse pressure, and carotid IMT) was 0.6 units lower, on average, in carriers of the SERPINE1 mutation than noncarriers, but this difference did not achieve statistical significance (P = 0.09). A second composite score representative of cardiometabolic aging that included cardiovascular parameters and fasting insulin was 1.03 units lower, on average, in carriers (P = 0.03). Composite score 3, which was representative of comprehensive biological aging, including score 2 parameters and relative LTL, was 0.53 units lower, on average, in carriers (P = 0.005).
Fig. 3. Association of SERPINE1 genotype status and cardiovascular measures of aging as a function of age in the Berne Amish kindred.
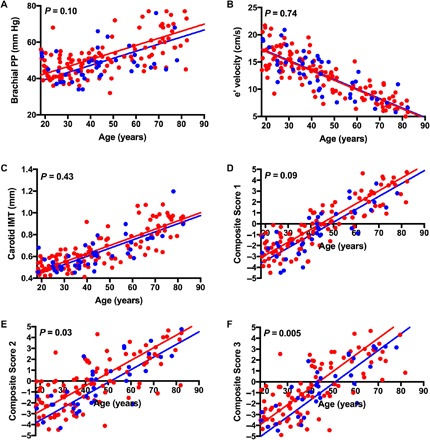
(A to C) Components of the aging composite scores, including brachial pulse pressure (A), e′ velocity (B), and carotid IMT (C) as a function age in Amish participants by genotype status for SERPINE1 null allele. (D to F) Composite scores of cardiovascular, cardiometabolic, and comprehensive biological aging in SERPINE1 null allele carriers and noncarriers in the Berne Amish kindred. Several cardiovascular measures that correlated strongly with chronological age with absolute correlation coefficients (|R|) greater than 0.60 (P < 0.0001) were identified. Pulse pressure [PP = 34.7 + 0.39 (age in years)] and carotid IMT [0.32 + 0.01 (age in years)] increased with age. e′ velocity [e′ = 20.3 − 0.2 (age in years)] decreased with age, as expected. These measures of vascular structure and stiffness and myocardial health were then standardized to have mean = 0 and SD = 1 (z scores) and were integrated into composite score 1 (cardiovascular age composed of brachial pulse pressure, e′ velocity, and carotid IMT), composite score 2 (cardiometabolic age composed of score 1 plus fasting insulin), and composite score 3 (comprehensive biological age composed of score 2 and LTL). Z scores were coded so that higher values corresponded to older levels of the measures of biological aging; that is, The z scores for LTL and e′ velocity, which decline with age, were reverse-coded so that higher z scores corresponded to lower levels [**P value represents difference in components and composite scores of aging by SERPINE1 mutation status (carriers versus noncarriers) after adjustment for age, sex, and family structure in SOLAR].
These three composite scores of aging were assessed in the CARDIA cohort to determine their associations with cardiovascular outcomes. Each score was significantly associated with the 5-year outcomes of cardiovascular morbidity and mortality (tables S3 and S4). Similar results were obtained when restricting the analysis to white participants in CARDIA. In addition, plasma PAI-1 levels in the CARDIA participants correlated significantly with all three composite scores (R = 0.27, 0.42, and 0.39 for composite scores 1, 2, and 3, respectively; P < 0.05).
Founder identification in family structure pedigree
A simplified diagram of the extended Berne Amish pedigree highlighting individuals with heterozygosity and homozygosity for the null SERPINE1 gene is shown in fig. S1. We identified either individual I-1 or I-2 as highly likely to have introduced a single copy of the mutated allele into the population six generations ago. This husband and wife couple is a common ancestor to all individuals newly and previously identified in the study as heterozygous or homozygous for the null SERPINE1 allele. Although the genetically restricted nature of this cohort has resulted in a relatively high prevalence of the SERPINE1 null allele, the mean kinship coefficient among carriers of the null SERPINE1 allele was 7%. The oldest known homozygous individual for the SERPINE1 mutation was born in 1981 as a result of the first marriage to occur between two heterozygotes. Although these individuals provide a unique opportunity to examine the biology of PAI-1 in humans, the rarity of this condition in the world and their young age limit comparisons at this time, but baseline characteristics are described (table S1).
DISCUSSION
The central findings of our study are that heterozygosity for the null SERPINE1 gene encoding PAI-1, which is associated with a lifelong reduction in PAI-1, is associated with longer LTL, a healthier metabolic profile with lower prevalence of diabetes, and a longer life span. The Berne Amish kindred provide an unprecedented opportunity to study the biological effects of a private loss-of-function mutation with a large effect on circulating PAI-1 on longevity in humans (17, 18). We confirm previous reports that suggest that LTL is inversely correlated with chronological age and is substantially heritable (19). In epidemiologic studies, LTL has been validated as a potent predictor of diabetes, CVD, and all-cause mortality (8, 9, 20–22). In addition, inherited deficiencies in telomere maintenance in mice and humans manifest with an accelerated aging phenotype, supporting the critical role of telomere attrition in the pathophysiology of aging (23, 24). These perspectives informed both our study design and the selection of LTL as the prespecified primary end point. Although age-adjusted LTL is significantly longer in the carriers of the null SERPINE1 allele, the age-related phenomenon of LTL attrition across the cohort is the same. This supports previous data that absolute LTL may be largely determined at birth, and individuals with a longer LTL may shift the onset of insulin resistance and CVD to an older age, promoting longer life span (25–27).
The current study builds upon the available cellular and animal evidence supporting the role of PAI-1, the product of SERPINE1, as an important contributor to aging (28). PAI-1 expression is increased in senescent cells and tissues and is a fundamental component of the SASP (10). There is a compelling evidence that senescent cells accumulate in the tissues and contribute to the aging process (29, 30). In addition to contributing to the molecular fingerprint of senescence, PAI-1 is necessary and sufficient for the induction of replicative senescence in vitro and is a critical downstream target of the tumor suppressor p53 (12, 31). The contribution of PAI-1 to cellular senescence is broadly relevant in the organism as a whole, and age-dependent increases in plasma PAI-1 levels have been identified in wild-type mice as they age, murine models of accelerated aging (Klotho and BubR1H/H), and humans (29, 32, 33). Partial PAI-1 deficiency in Klotho-deficient animals extended median survival nearly threefold with preserved telomere length and substantial protection against aging-like pathology in various organs (13). Both genetic and pharmacological inhibition of PAI-1 protected against the development of hypertension and arteriosclerosis and was associated with longer telomere length in mice treated with long-term nitric oxide synthase inhibition (34). These preclinical data are consistent with our findings of a longer LTL and a greater life span in carriers of the null SERPINE1 allele in humans.
Metabolism plays a fundamental role in the biology of aging, and insulin and insulin-like growth factor 1 (IGF1) are widely endorsed as critical contributors to senescence and aging in several experimental models (for example, flies, worms, and mammals) (35). Potential antiaging interventions have focused on caloric restriction and drugs with metabolic effects, including metformin and resveratrol, all of which mechanistically intersect in reducing PAI-1 expression (36–38). Conversely, PAI-1 production is enhanced by insulin, free fatty acids, and glucose (39–41). In addition, PAI-1 impairs the proteolytic degradation of IGF-binding protein–3 (IGFBP3) and IGF1, both of which are capable of initiating cellular senescence (42, 43). In observational human studies, PAI-1 levels were higher in obesity and insulin resistance and independently predicted the future development of type 2 DM (15, 44). In the only GWAS of circulating PAI-1 in 19,599 participants, a genotype-phenotype analysis identified a significant association between single-nucleotide polymorphisms (SNPs) that regulate PAI-1 and type 2 DM (45). Further, Mendelian randomization analyses from large GWAS studies support a casual role for PAI-1 and hyperglycemia (16). Here, we demonstrate that SERPINE1 null carriers have lower fasting insulin levels and lower prevalence of type 2 DM, and in a diverse U.S. population–based study (CARDIA), we demonstrate that circulating PAI-1 levels are strongly correlated with fasting insulin levels.
Our findings from the only known PAI-1–deficient kindred provide the first example of a private gene mutation on age-dependent molecular and metabolic changes in humans. Whole-genome sequencing and GWAS for healthy aging and life span have had limited success in identifying associated genetic loci, supporting the value of rare genetic variants (such as the one described here) in understanding biological mechanisms that regulate aging and life span (46–48). The genetic mechanism of PAI-1 deficiency in the Berne Amish kindred is explained by the presence of a dinucleotide insertion (TA) within the coding sequence of the PAI-1 gene (exon 4), resulting in a frameshift mutation and formation of a premature stop codon and the synthesis of a truncated, nonfunctional PAI-1 protein. This frameshift mutation has not been identified in the general population based on the sequence data publicly available for review from 60,706 unrelated individuals in the Exome Aggregation Consortium (ExAC). Ten distinct loss-of-function mutations, including three frameshift mutations, are reported in the ExAC database in SERPINE1 and have exceedingly rare mutated allele prevalence ranging from 0.000008239 to 0.0005850 (49). Further, in GWAS, SNPs account for no more than 3.7% of the total variation in plasma PAI-1 levels beyond that of age and sex (45). Therefore, discovery and validation of SERPINE1 SNPs in GWAS of longevity may be difficult, given the limited contribution of these variants in the general population to circulating PAI-1 levels, and highlight the strength of our study with a large effect size on circulating PAI-1 levels as a result of a loss-of-function mutation.
Additional longitudinal studies in this cohort will help inform potential links between the null SERPINE1 mutation, metabolism, and life span. Although there is a possibility that the SERPINE1 mutation segregates with other mutations that may contribute to the phenotype observed, the qualitative and quantitative effects we observed with SERPINE1 heterozygosity in humans are fully concordant with the predicted effects observed in preclinical experimental models (13, 34). We acknowledge that the generalizability of the findings identified here may be limited, given that this cohort carries a rare private mutation, is functionally isolated from the “modern world” in several ways, and appears to be relatively healthier when compared to a general population sample as represented by CARDIA. Average plasma PAI-1 levels even in unaffected Amish participants without the null SERPINE1 allele were 50% lower than participants enrolled in U.S. population–based cohorts such as CARDIA and the Framingham Heart Study (50).
Use of small-molecule selective inhibitors of human PAI-1 or other components of the SASP may be of interest in preventing or treating age-related morbidities. Orally active PAI-1 inhibitors have already completed extensive preclinical evaluation and are currently in early-phase clinical testing in humans in Japan (34, 51). The absence of bleeding complications in heterozygotes for the SERPINE1 mutation in over 20 years of clinical observation suggests that therapeutic agents that selectively reduce but incompletely inhibit PAI-1 activity may be safe from a hemostatic perspective (17).
Our study has several limitations. First, our results are based on an observational study and include limited assessments of LTL, metabolic parameters and disease, and measures of cardiovascular structure and function. Given the current lack of consensus regarding the quantitative assessment of biological age, we developed novel composite scores of aging that integrate age-dependent physiological measures of cardiovascular health including vascular structure and stiffness, subclinical atherosclerosis, myocardial relaxation, and LTL (52, 53). Although we demonstrate that our composite scores are significantly associated with the 5-year cardiovascular outcomes in CARDIA, events are limited, and these quantitative assessments should be replicated in additional population-based cohorts with longer follow-up times. Second, although the aging phenotype of homozygous individuals is certainly of interest, these individuals are extremely rare in the kindred and relatively young (all less than 34 years old), limiting any meaningful comparisons at the present time. Therefore, we focused on the potential impact of SERPINE1 heterozygosity and LTL in this study. Third, all of the heterozygous participants share a common ancestor in generation I from the late 1800s, and this lends a possibility that other unidentified genetic or epigenetic factors contribute to the present findings. To mitigate this risk, we adjusted our analyses for relatedness in SOLAR (54).
In conclusion, our data indicate that PAI-1 is associated with a number of parameters that may reflect biological aging, including LTL, metabolism, and life span. Overall, our findings are the first to identify the physiological association of a null mutation in PAI-1 with LTL and life span in humans and suggest that PAI-1, a component of the senescence-related secretome, may influence the aging process. Future studies will provide the opportunity to investigate the contribution of PAI-1 to individual telomere attrition over time, the development of incident diabetes and other age-related diseases, and perhaps ultimately differences in health and life span in humans.
MATERIALS AND METHODS
Amish study participants
This cross-sectional observational study was conducted in the OOA—a founder population who originally settled in Berne, Indiana, which is characterized by a homogeneous diet and lifestyle—and included participants aged 18 years and older. The Institutional Review Board at the Indiana Hemophilia and Thrombosis Center and the Northwestern University approved the study protocol. A total of 450 individuals was invited to participate in the study, and 177 individuals were enrolled in the study. All study participants provided written informed consent and were genotyped for the c.699_700dupTA frameshift mutation in SERPINE1. For the primary analyses, participants homozygous for the null SERPINE1 allele were excluded because of small numbers (n = 7) and their relatively young age (18 to 34 years). The primary end point of telomere length and the secondary end points of fasting insulin, diabetes status, and life span were prospectively selected.
Molecular markers and laboratory testing
Genotyping for SERPINE1 null mutation status was performed at the University of Michigan with a preidentified primer for the presence of the c.699_700dupTA frameshift mutation in the PAI-1 (SERPINE1) gene. Genomic DNA isolated from peripheral blood leukocytes (DNeasy Blood and Tissue kit, Qiagen) was used to measure relative LTL by quantitative PCR and TRF length by Southern blot by blinded technicians using previously described methods (55–57). Quantitative PCR samples were run in triplicate, and Southern blot samples were run in duplicate. The inter-assay coefficient of variation (CV) was 9.6% for relative LTL and 2.5% for mean TRF.
Plasma samples preserved in citrate were assayed for human PAI-1 total antigen with the Molecular Innovations PAI-1 ELISA (Molecular Innovations) using BioTek Synergy HT Gen5 software (BioTek). All samples were run in duplicate, and the mean of two measurements was used. The intra-assay CV was 6.15%, and the inter-assay CV was 5.98%. Metabolites (glucose and insulin) and cholesterol (total cholesterol, HDL cholesterol, LDL cholesterol, and triglyceride) were measured in morning fasting blood samples according to a standardized protocol in the main laboratory at the Northwestern Memorial Hospital.
Cardiovascular assessment
All study participants underwent a comprehensive two-dimensional echocardiographic examination with Doppler and tissue Doppler imaging using a commercially available ultrasound system with harmonic imaging (Vivid 7 or Vivid i, GE Healthcare) and a 3.5-MHz transducer (GE Healthcare) by blinded sonographers. Cardiac structure and function were quantified as recommended by the American Society of Echocardiography (ASE) (58). Deidentified images were copied in RAW format and analyzed offline using GE EchoPAC software version 7.0.0 (GE Healthcare). All measurements were made by a single experienced research sonographer and verified by an experienced investigator with expertise in echocardiography (both blinded to clinical data and genotype status) according to the guidelines of the ASE.
Carotid imaging was performed by an experienced sonographer blinded to genotype status using a commercially available ultrasound machine (Vivid 7 or Vivid i, GE Healthcare) and an 8-MHz transducer (GE Healthcare). Deidentified images were copied in RAW format for offline analysis and analyzed using GE EchoPAC software version 7.0.0 (GE Healthcare). All measurements were performed by a single experienced research sonographer and verified by an experienced investigator (both blinded to clinical data and genotype status). IMT measurements were performed using GE’s semiautomated edge detection method according to the ASE recommendations (59).
Genealogical information and life span
Detailed genealogic data spanning three generations were obtained from each participant. The pedigree was supplemented by an extensive fieldwork to fill in available birth and death dates and was used to adjust for family relatedness by calculation of kinship matrix for each of our enrolled participants in the SOLAR polygenic models. We additionally used ancestral data from the extended pedigree for life-span analysis. We restricted our life-span analysis to individuals known to have died at the age of ≥45 years to minimize the potential bias from premature cause of death, including infection, accidental trauma, and childbirth, as previously described in similar analyses (60).
Validation of composite aging scores
We sought to validate the biological aging composite scores in a generalized U.S. population using the CARDIA study. The CARDIA study is a multicenter, community-based longitudinal cohort study designed to investigate the risk factors for the development of CVD. In 1985–1986, 5115 black and white men and women aged 18 to 30 years were recruited from four urban sites across the United States (Birmingham, AL; Chicago, IL; Minneapolis, MN; and Oakland, CA). Recruitment was balanced within each center by sex, age, race, and education. The CARDIA participants have been followed for more than 30 years with collection of detailed demographic and clinical data. All participants provided written informed consent at each examination, and institutional review boards from each field center and the coordinating center approved the study annually. Detailed descriptions of the study design and conduct had been previously published (61). For this analysis, all participants who attended the year-25 examination and had available data for components of composite scores 1 and 2 were included (N = 2793). A subset of participants who also had data for composite score 3 (n = 892) were also examined separately.
Statistical analysis
We compared baseline demographics between heterozygous and unaffected participants with adjustment for family structure. We used polygenic models adjusted for age, sex, and family structure to determine the association of heterozygosity in SERPINE1 with LTL as the primary end point, as measured by quantitative PCR and Southern blot. We estimated heritability (h2) defined as the proportion of total variation explained by additive genetic effects for LTL. We also investigated the association of SERPINE1 status with parameters of metabolism, including fasting insulin and the prevalence of type 2 DM and life span. Composite scores of aging were created by the summation of standardized z scores among aging-related characteristics that had the highest values for Pearson correlation with chronological age (|R| > 0.5). Composite score 1 included log-transformed brachial pulse pressure, tissue Doppler e′ velocity (a marker of left ventricular relaxation), and log-transformed average carotid IMT. Composite score 2 included components in score 1 and fasting insulin, and composite score 3 included components of score 2 and LTL. The three composite aging scores were validated in a generalized U.S. population using the CARDIA study.
Supplemental descriptive analysis was performed including the homozygous participants (n = 7) with limited power given their small sample size and young age. Polygenic analyses were conducted using SOLAR (version 8.1.1; Southwest Foundation for Biomedical Research). All other analyses were conducted using SAS software version 9.4 (SAS Institute). Two-sided P values <0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank our Amish liaisons and the Indiana Hemophilia and Thrombosis Center’s Research clinic staff, as well as the Feinberg Cardiovascular Research Institute volunteers. In addition, we extend our deepest gratitude to the Berne Amish community for their time, participation, and support, without which these studies would not have been possible. A pedigree is also included in the Supplementary Materials and is abbreviated to provide anonymity. We thank the participants of the CARDIA study for their long-term commitment and important contributions to the study. Funding: This work was supported by grants from the NIH (R01 HL051387 to D.E.V.; F32 HL129695 to S.S.K.; R01 HL127028 and HL107577 to S.J.S.; and R01HD071180, R01HL116446, and R01HL134840 to A.A.). The CARDIA is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the University of Alabama at Birmingham (HHSN268201300025C and HHSN268201300026C), Northwestern University (HHSN268201300027C), University of Minnesota (HHSN268201300028C), Kaiser Foundation Research Institute (HHSN268201300029C), and Johns Hopkins University School of Medicine (HHSN268200900041C). CARDIA is also supported, in part, by the Intramural Research Program of the National Institute on Aging (NIA) and an intra-agency agreement between NIA and NHLBI (AG0005). This manuscript has been reviewed by CARDIA for scientific content. Author contributions: D.E.V., S.S.K., and S.J.S. provided substantial contribution to the conception and design of the study, data acquisition and analysis, and drafting and critical revision of the manuscript. A.S.B. contributed to data analysis and critical manuscript revisions. E.K., M.E., A.T.P., D.M.L.-J., E.P., A.A., T.M., and M.H. contributed to data acquisition and critical revision of the manuscript. S.G. and A.D.S. contributed substantially to the conception and design of the study, data acquisition, and critical revision of the manuscript. All coauthors approved the final version of the manuscript to be published and agreed to be accountable for the accuracy and integrity of all aspects of the work. D.E.V. claims responsibility for all figures in the manuscript and the Supplementary Materials. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/11/eaao1617/DC1
Supplementary Materials and Methods
table S1. Clinical characteristics of homozygous participants for the null SERPINE1 mutation.
table S2. Association of null SERPINE1 mutant allele status with PAI-1, telomere length, and fasting insulin: secondary analyses.
table S3. Association of cardiometabolic aging composite scores with 5-year CVD morbidity and mortality in the CARDIA cohort (n = 2793).
table S4. Association of biological aging composite scores including telomere length with 5-year CVD morbidity and mortality in the CARDIA cohort (n = 872).
fig. S1. Abbreviated pedigree of the Berne Amish kindred.
REFERENCES AND NOTES
- 1.Kaeberlein M., Rabinovitch P. S., Martin G. M., Healthy aging: The ultimate preventative medicine. Science 350, 1191–1193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fontana L., Partridge L., Longo V. D., Extending healthy life span—From yeast to humans. Science 328, 321–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daniali L., Benetos A., Susser E., Kark J. D., Labat C., Kimura M., Desai K. K., Granick M., Aviv A., Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 4, 1597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo V. D., Antebi A., Bartke A., Barzilai N., Brown-Borg H. M., Caruso C., Curiel T. J., de Cabo R., Franceschi C., Gems D., Ingram D. K., Johnson T. E., Kennedy B. K., Kenyon C., Klein S., Kopchick J. J., Lepperdinger G., Madeo F., Mirisola M. G., Mitchell J. R., Passarino G., Rudolph K. L., Sedivy J. M., Shadel G. S., Sinclair D. A., Spindler S. R., Suh Y., Vijg J., Vinciguerra M., Fontana L., Interventions to slow aging in humans: Are we ready? Aging Cell 14, 497–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.López-Otín C., Galluzzi L., Freije J. M. P., Madeo F., Kroemer G., Metabolic control of longevity. Cell 166, 802–821 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Kochanek K. D., Xu J., Murphy S. L., Minino A. M., Kung H.-C., Deaths: Preliminary data for 2009. Natl. Vital Stat. Rep. 59, 1–51 (2011). [PubMed] [Google Scholar]
- 8.D’Mello M. J. J., Ross S. A., Briel M., Anand S. S., Gerstein H., Paré G., Association between shortened leukocyte telomere length and cardiometabolic outcomes: Systematic review and meta-analysis. Circ. Cardiovasc. Genet. 8, 82–90 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Haycock P. C., Heydon E. E., Kaptoge S., Butterworth A. S., Thompson A., Willeit P., Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 349, g4227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchkonia T., Zhu Y., van Deursen J., Campisi J., Kirkland J. L., Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Özcan S., Alessio N., Acar M. B., Mert E., Omerli F., Peluso G., Galderisi U., Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 8, 1316–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kortlever R. M., Higgins P. J., Bernards R., Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 877–884 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eren M., Boe A. E., Murphy S. B., Place A. T., Nagpal V., Morales-Nebreda L., Urich D., Quaggin S. E., Budinger G. R. S., Mutlu G. M., Miyata T., Vaughan D. E., PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. U.S.A. 111, 7090–7095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alessi M. C., Juhan-Vague I., PAI-1 and the metabolic syndrome: Links, causes, and consequences. Arterioscler. Thromb. Vasc. Biol. 26, 2200–2207 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Festa A., D’Agostino R. Jr, Tracy R. P., Haffner S. M.; Insulin Resistance Atherosclerosis Study, Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetes 51, 1131–1137 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Song C., Burgess S., Eicher J. D., O’Donnell C. J., Johnson A. D., Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J. Am. Heart Assoc. 6, e004918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fay W. P., Parker A. C., Condrey L. R., Shapiro A. D., Human plasminogen activator inhibitor-1 (PAI-1) deficiency: Characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 90, 204–208 (1997). [PubMed] [Google Scholar]
- 18.Fay W. P., Shapiro A. D., Shih J. L., Schleef R. R., Ginsburg D., Brief report: Complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. N. Engl. J. Med. 327, 1729–1733 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Njajou O. T., Cawthon R. M., Damcott C. M., Wu S.-H., Ott S., Garant M. J., Blackburn E. H., Mitchell B. D., Shuldiner A. R., Hsueh W.-C., Telomere length is paternally inherited and is associated with parental lifespan. Proc. Natl. Acad. Sci. U.S.A. 104, 12135–12139 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cawthon R. M., Smith K. R., O’Brien E., Sivatchenko A., Kerber R. A., Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361, 393–395 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Codd V., Nelson C. P., Albrecht E., Mangino M., Deelen J., Buxton J. L., Jan Hottenga J., Fischer K., Esko T., Surakka I., Broer L., Nyholt D. R., Mateo Leach I., Salo P., Hägg S., Matthews M. K., Palmen J., Norata G. D., O’Reilly P. F., Saleheen D., Amin N., Balmforth A. J., Beekman M., de Boer R. A., Böhringer S., Braund P. S., Burton P. R., de Craen A. J. M., Denniff M., Dong Y., Douroudis K., Dubinina E., Eriksson J. G., Garlaschelli K., Guo D., Hartikainen A.-L., Henders A. K., Houwing-Duistermaat J. J., Kananen L., Karssen L. C., Kettunen J., Klopp N., Lagou V., van Leeuwen E. M., Madden P. A., Mägi R., Magnusson P. K. E., Männistö S., McCarthy M. I., Medland S. E., Mihailov E., Montgomery G. W., Oostra B. A., Palotie A., Peters A., Pollard H., Pouta A., Prokopenko I., Ripatti S., Salomaa V., Suchiman H. E. D., Valdes A. M., Verweij N., Viñuela A., Wang X., Wichmann H.-E., Widen E., Willemsen G., Wright M. J., Xia K., Xiao X., van Veldhuisen D. J., Catapano A. L., Tobin M. D., Hall A. S., Blakemore A. I. F., van Gilst W. H., Zhu H.; CARDIoGRAM consortium; Erdmann J., Reilly M. P., Kathiresan S., Schunkert H., Talmud P. J., Pedersen N. L., Perola M., Ouwehand W., Kaprio J., Martin N. G., van Duijn C. M., Hovatta I., Gieger C., Metspalu A., Boomsma D. I., Jarvelin M.-R., Slagboom P. E., Thompson J. R., Spector T. D., van der Harst P., Samani N. J., Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 45, 422–427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao J., Miao K., Wang H., Ding H., Wang D. W., Association between telomere length and type 2 diabetes mellitus: A meta-analysis. PLOS ONE 8, e79993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armanios M., Blackburn E. H., The telomere syndromes. Nat. Rev. Genet. 13, 693–704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blasco M. A., Lee H.-W., Hande M. P., Samper E., Lansdorp P. M., DePinho R. A., Greider C. W., Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Benetos A., Kark J. D., Susser E., Kimura M., Sinnreich R., Chen W., Steenstrup T., Christensen K., Herbig U., von Bornemann Hjelmborg J., Srinivasan S. R., Berenson G. S., Labat C., Aviv A., Tracking and fixed ranking of leukocyte telomere length across the adult life course. Aging Cell 12, 615–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Factor-Litvak P., Susser E., Kezios K., McKeague I., Kark J. D., Hoffman M., Kimura M., Wapner R., Aviv A., Leukocyte telomere length in newborns: Implications for the role of telomeres in human disease. Pediatrics 137, e20153927 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunt S. C., Kark J. D., Aviv A., Association between shortened leukocyte telomere length and cardio-metabolic outcomes. Circ. Cardiovasc. Genet. 8, 4–7 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Cesari M., Pahor M., Incalzi R. A., Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 28, e72–e91 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker D. J., Childs B. G., Durik M., Wijers M. E., Sieben C. J., Zhong J., Saltness R. A., Jeganathan K. B., Verzosa G. C., Pezeshki A., Khazaie K., Miller J. D., van Deursen J. M., Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demaria M., O’Leary M. N., Chang J., Shao L., Liu S., Alimirah F., Koenig K., Le C., Mitin N., Deal A. M., Alston S., Academia E. C., Kilmarx S., Valdovinos A., Wang B., de Bruin A., Kennedy B. K., Melov S., Zhou D., Sharpless N. E., Muss H., Campisi J., Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W., Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Takeshita K., Yamamoto K., Ito M., Kondo T., Matsushita T., Hirai M., Kojima T., Nishimura M., Nabeshima Y., Loskutoff D. J., Saito H., Murohara T., Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin. Thromb. Hemost. 28, 545–554 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Aillaud M. F., Pignol F., Alessi M. C., Harle J. R., Escande M., Mongin M., Juhan-Vague I., Increase in plasma concentration of plasminogen activator inhibitor, fibrinogen, von Willebrand factor, factor VIII:C and in erythrocyte sedimentation rate with age. Thromb. Haemost. 55, 330–332 (1986). [PubMed] [Google Scholar]
- 34.Boe A. E., Eren M., Murphy S. B., Kamide C. E., Ichimura A., Terry D., McAnally D., Smith L. H., Miyata T., Vaughan D. E., Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-l-arginine methyl ester–induced hypertension and vascular senescence. Circulation 128, 2318–2324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatar M., Bartke A., Antebi A., The endocrine regulation of aging by insulin-like signals. Science 299, 1346–1351 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Ølholm J., Paulsen S. K., Cullberg K. B., Richelsen B., Pedersen S. B., Anti-inflammatory effect of resveratrol on adipokine expression and secretion in human adipose tissue explants. Int. J. Obes. 34, 1546–1553 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Nagi D. K., Yudkin J. S., Effects of metformin on insulin resistance, risk factors for cardiovascular disease, and plasminogen activator inhibitor in NIDDM subjects: A study of two ethnic groups. Diabetes Care 16, 621–629 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Colman R. J., Beasley T. M., Kemnitz J. W., Johnson S. C., Weindruch R., Anderson R. M., Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 5, 3557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilsson L., Banfi C., Diczfalusy U., Tremoli E., Hamsten A., Eriksson P., Unsaturated fatty acids increase plasminogen activator inhibitor-1 expression in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 18, 1679–1685 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Alessi M. C., Juhan-Vague I., Kooistra T., Declerck P. J., Collen D., Insulin stimulates the synthesis of plasminogen activator inhibitor 1 by the human hepatocellular cell line Hep G2. Thromb. Haemost. 60, 491–494 (1988). [PubMed] [Google Scholar]
- 41.Chen Y.-Q., Su M., Walia R. R., Hao Q., Covington J. W., Vaughan D. E., Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J. Biol. Chem. 273, 8225–8231 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Deelen J., Uh H.-W., Monajemi R., van Heemst D., Thijssen P. E., Böhringer S., van den Akker E. B., de Craen A. J. M., Rivadeneira F., Uitterlinden A. G., Westendorp R. G. J., Goeman J. J., Slagboom P. E., Houwing-Duistermaat J. J., Beekman M., Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age 35, 235–249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elzi D. J., Lai Y., Song M., Hakala K., Weintraub S. T., Shiio Y., Plasminogen activator inhibitor 1—Insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc. Natl. Acad. Sci. U.S.A. 109, 12052–12057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosito G. A., D’Agostino R. B., Massaro J., Lipinska I., Mittleman M. A., Sutherland P., Wilson P. W. F., Levy D., Muller J. E., Tofler G. H., Association between obesity and a prothrombotic state: The Framingham Offspring Study. Thromb. Haemost. 91, 683–689 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Huang J., Sabater-Lleal M., Asselbergs F. W., Tregouet D., Shin S.-Y., Ding J., Baumert J., Oudot-Mellakh T., Folkersen L., Johnson A. D., Smith N. L., Williams S. M., Ikram M. A., Kleber M. E., Becker D. M., Truong V., Mychaleckyj J. C., Tang W., Yang Q., Sennblad B., Moore J. H., Williams F. M. K., Dehghan A., Silbernagel G., Schrijvers E. M. C., Smith S., Karakas M., Tofler G. H., Silveira A., Navis G. J., Lohman K., Chen M.-H., Peters A., Goel A., Hopewell J. C., Chambers J. C., Saleheen D., Lundmark P., Psaty B. M., Strawbridge R. J., Boehm B. O., Carter A. M., Meisinger C., Peden J. F., Bis J. C., McKnight B., Öhrvik J., Taylor K., Franzosi M. G., Seedorf U., Collins R., Franco-Cereceda A., Syvänen A.-C., Goodall A. H., Yanek L. R., Cushman M., Müller-Nurasyid M., Folsom A. R., Basu S., Matijevic N., van Gilst W. H., Kooner J. S., Hofman A., Danesh J., Clarke R., Meigs J. B.; DIAGRAM Consortium; Kathiresan S., Reilly M. P.; CARDIoGRAM Consortium; Klopp N., Harris T. B., Winkelmann B. R., Grant P. J., Hillege H. L., Watkins H.; C4D Consortium; Spector T. D., Becker L. C., Tracy R. P., März W., Uitterlinden A. G., Eriksson P., Cambien F.; CARDIOGENICS Consortium; Morange P.-E., Koenig W., Soranzo N., van der Harst P., Liu Y., O’Donnell C. J., Hamsten A., Genome-wide association study for circulating levels of PAI-1 provides novel insights into its regulation. Blood 120, 4873–4881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erikson G. A., Bodian D. L., Rueda M., Molparia B., Scott E. R., Scott-Van Zeeland A. A., Topol S. E., Wineinger N. E., Niederhuber J. E., Topol E. J., Torkamani A., Whole-genome sequencing of a healthy aging cohort. Cell 165, 1002–1011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christensen K., Johnson T. E., Vaupel J. W., The quest for genetic determinants of human longevity: Challenges and insights. Nat. Rev. Genet. 7, 436–448 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deelen J., Beekman M., Uh H.-W., Helmer Q., Kuningas M., Christiansen L., Kremer D., van der Breggen R., Suchiman H. E. D., Lakenberg N., van den Akker E. B., Passtoors W. M., Tiemeier H., van Heemst D., de Craen A. J., Rivadeneira F., de Geus E. J., Perola M., van der Ouderaa F. J., Gunn D. A., Boomsma D. I., Uitterlinden A. G., Christensen K., van Duijn C. M., Heijmans B. T., Houwing-Duistermaat J. J., Westendorp R. G. J., Slagboom P. E., Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell 10, 686–698 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Exome Aggregation Consortium, ExAC Browser (Beta) (ExAC, 2016); http://exac.broadinstitute.org.
- 50.Lieb W., Larson M. G., Benjamin E. J., Yin X., Tofler G. H., Selhub J., Jacques P. F., Wang T. J., Vita J. A., Levy D., Vasan R. S., Mitchell G. F., Multimarker approach to evaluate correlates of vascular stiffness: The Framingham Heart Study. Circulation 119, 37–43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izuhara Y., Yamaoka N., Kodama H., Dan T., Takizawa S., Hirayama N., Meguro K., van Ypersele de Strihou C., Miyata T., A novel inhibitor of plasminogen activator inhibitor-1 provides antithrombotic benefits devoid of bleeding effect in nonhuman primates. J. Cereb. Blood Flow Metab. 30, 904–912 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belsky D. W., Caspi A., Houts R., Cohen H. J., Corcoran D. L., Danese A., Harrington H., Israel S., Levine M. E., Schaefer J. D., Sugden K., Williams B., Yashin A. I., Poulton R., Moffitt T. E., Quantification of biological aging in young adults. Proc. Natl. Acad. Sci. U.S.A. 112, E4104–E4110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levine M. E., Modeling the rate of senescence: Can estimated biological age predict mortality more accurately than chronological age? J. Gerontol. A Biol. Sci. Med. Sci. 68, 667–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khoury M. J., Cohen B. H., Diamond E. L., Chase G. A., McKusick V. A., Inbreeding and prereproductive mortality in the Old Order Amish. III. Direct and indirect effects of inbreeding. Am. J. Epidemiol. 125, 473–483 (1987). [DOI] [PubMed] [Google Scholar]
- 55.Cawthon R. M., Telomere measurement by quantitative PCR. Nucleic Acids Res. 30, e47 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pooley K. A., Sandhu M. S., Tyrer J., Shah M., Driver K. E., Luben R. N., Bingham S. A., Ponder B. A. J., Pharoah P. D. P., Khaw K.-T., Easton D. F., Dunning A. M., Telomere length in prospective and retrospective cancer case-control studies. Cancer Res. 70, 3170–3176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kimura M., Stone R. C., Hunt S. C., Skurnick J., Lu X., Cao X., Harley C. B., Aviv A., Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat. Protoc. 5, 1596–1607 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Lang R. M., Bierig M., Devereux R. B., Flachskampf F. A., Foster E., Pellikka P. A., Picard M. H., Roman M. J., Seward J., Shanewise J. S., Solomon S. D., Spencer K. T., St John Sutton M., Stewart W. J., Recommendations for chamber quantification: A report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. 18, 1440–1463 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Stein J. H., Korcarz C. E., Hurst R. T., Lonn E., Kendall C. B., Mohler E. R., Najjar S. S., Rembold C. M., Post W. S., Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: A consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force Endorsed by the Society for Vascular Medicine. J. Am. Soc. Echocardiogr. 21, 93–111 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Mitchell B. D., Hsueh W.-C., King T. M., Pollin T. I., Sorkin J., Agarwala R., Schäffer A. A., Shuldiner A. R., Heritability of life span in the Old Order Amish. Am. J. Med. Genet. 102, 346–352 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Friedman G. D., Cutter G. R., Donahue R. P., Hughes G. H., Hulley S. B., Jacobs D. R. Jr, Liu K., Savage P. J., CARDIA: Study design, recruitment, and some characteristics of the examined subjects. J. Clin. Epidemiol. 41, 1105–1116 (1988). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/11/eaao1617/DC1
Supplementary Materials and Methods
table S1. Clinical characteristics of homozygous participants for the null SERPINE1 mutation.
table S2. Association of null SERPINE1 mutant allele status with PAI-1, telomere length, and fasting insulin: secondary analyses.
table S3. Association of cardiometabolic aging composite scores with 5-year CVD morbidity and mortality in the CARDIA cohort (n = 2793).
table S4. Association of biological aging composite scores including telomere length with 5-year CVD morbidity and mortality in the CARDIA cohort (n = 872).
fig. S1. Abbreviated pedigree of the Berne Amish kindred.