

Summary
Disruptions in lipid homeostasis have been observed in many neurodevelopmental disorders that are associated with dendrite morphogenesis defects. However, the molecular mechanisms of how lipid homeostasis affects dendrite morphogenesis are unclear. We find that easily shocked (eas), which encodes a kinase with a critical role in phospholipid phosphatidylethanolamine (PE) synthesis, and two other enzymes in this synthesis pathway are required cell-autonomously in sensory neurons for dendrite growth and stability. Further, we show that the level of Sterol Regulatory Element Binding Protein (SREBP) activity is important for dendrite development. SREBP activity increases in eas mutants, and decreasing the level of SREBP and its transcriptional targets in eas mutants largely suppresses the dendrite growth defects. Furthermore, reducing Ca2+ influx in neurons of eas mutants ameliorates the dendrite morphogenesis defects. Our study uncovers a role for EAS kinase and reveals the in vivo function of phospholipid homeostasis in dendrite morphogenesis.
eTOC Blurb
Meltzer et al. show that EAS, a conserved kinase in the phospholipid phosphatidylethanolamine synthesis pathway, regulates dendrite growth via SREBP signaling and Ca2+ influx. Their study reveals the role of phospholipid homeostasis in dendrite morphogenesis in vivo.
Introduction
How neurons achieve the proper wiring pattern during development is an important question, as the dendrite arborization pattern of each neuron is critical for the function of the nervous system (Jan and Jan, 2010; Lefebvre et al., 2015). Defects in dendrite morphogenesis are common anatomical features of many neurodevelopmental disorders (Kaufmann and Moser, 2000). Although abnormal lipid metabolism has been observed in mouse models as well as patients with neurodevelopmental disorders (Buchovecky et al., 2013; Tamiji and Crawford, 2010; Tint et al., 1994) and 60 percent of the human brain dry mass is lipids (Wong and Crawford, 2014), relatively little is known about the importance of lipid homeostasis for dendrite morphogenesis during neural development.
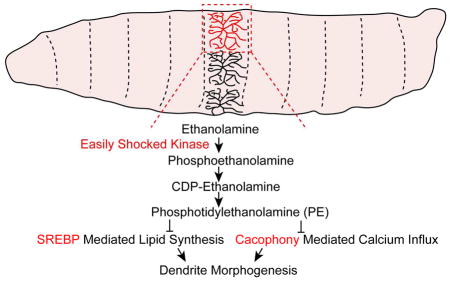
We have used the dendritic arborization (da) sensory neurons of the Drosophila larval peripheral nervous system (PNS) to identify the molecular mechanisms that regulate dendrite morphogenesis of these neurons, which fall into four distinct classes (I–IV) based on their dendrite morphologies and axon projections (Grueber et al., 2002; 2007). From a genetic screen for genes that affect dendritic growth in class IV da neurons, we identified easily shocked (eas) as an important gene that regulates dendrite morphogenesis in sensory neurons. The eas gene encodes a conserved ethanolamine kinase, the first enzyme in the cytidine 5′-diphosphate (CDP)-ethanolamine pathway for the synthesis of the membrane phospholipid phosphotidylethanolamine (PE) (Kennedy, 1957). PE is the predominant phospholipid in membranes in Drosophila (Jones et al., 1992), and the second most abundant phospholipid in mammals (Vance, 2014). In Drosophila, PE regulates the processing and activity of sterol regulatory element-binding protein (SREBP), a highly conserved basic helix-loop-helix leucine zipper transcription factor that is crucial for lipid homeostasis (Dobrosotskaya et al., 2002). Reduction of the PE level causes SREBP precursor proteins to be transported from the endoplasmic reticulum to the Golgi apparatus, where they are cleaved to release the transcriptionally active domain that translocates to the nucleus to activate the expression of lipogenic genes (Rawson, 2003). It was unknown how this phospholipid homeostasis process affects dendrite development.
Originally identified by Seymour Benzer, eas mutants respond to a mechanical jolt by exhibiting transient paralysis and then recover, whereas wild type flies are unaffected (Benzer, 1971). eas mutant flies were subsequently used as a Drosophila model of seizure; their seizure-like phenotype could be suppressed by mutations that reduced the hyperexcitability of eas mutants (Parker et al., 2011; Pavlidis et al., 1994). It is unknown whether these mutations that reduce hyperexcitability also affect neural morphogenesis in eas mutants. Reducing the level of cacophony (cac), the Drosophila homolog of the Cav2 voltage-gated calcium channel genes and a major mediator of neuronal Ca2+ influx (Peng and Wu, 2007; Saras and Tanouye, 2016), suppresses the seizure phenotype present in eas mutants. We found that reducing cac gene activity ameliorated the dendrite morphogenesis defects of eas mutants.
Here, we show that the dendrite morphogenesis defects of eas mutants are attributable to increased lipogenesis and altered Ca2+ influx. Our results uncover a role for the conserved ethanolamine kinase and for SREBP signaling in dendrite morphogenesis and highlight an important role of phospholipid and lipid homeostasis during neuronal development.
Results
EAS Kinase Acts Cell-Autonomously to Regulate Dendrite Morphogenesis in da Neurons
From an RNA interference (RNAi) screen for regulators of dendrite development in Drosophila, we identified eas, which encodes ethanolamine kinase (Pavlidis et al., 1994), as a candidate. To corroborate the RNAi results, we first examined the existing eas allele, eas1, which produces a truncated protein lacking kinase activity (Pavlidis et al., 1994). We further generated knockout mutants, easKO (Figures S1A and S1B), in which the entire coding region is removed via CRISPR/Cas9 (Port et al., 2014). Both alleles showed similar dendrite outgrowth defects at 120 hours (hrs) after egg laying (AEL), with dramatic decreases in the number of branches and the total dendrite length in class IV da neurons (Figures 1A–1C, 1E and 1F). Sholl analysis revealed that reductions in dendrite branching occurred uniformly throughout the dendritic arbor in the eas1 and easKO mutants (Figure 1G). In addition, we found decreases in the number of branches and the total dendrite length in class I and class III da neurons in the eas1 and easKO mutants (Figures S2A–S2H). We further examined the morphology of class IV da neurons at 48, 72, 96, and 120 hrs AEL in wildtype and easKO mutants. Dendrites of class IV da neurons normally establish their dendritic territories and completely tile the body wall by 48 hrs AEL, and then continue to grow throughout larval development (Parrish et al., 2007). Although the patterning of class IV da neurons in easKO mutants examined at 48 hrs AEL initially proceeded normally, dendrite growth defects became apparent at 72 hrs AEL (Figures 1E and 1F), suggesting that eas is required for dendrite outgrowth and/or stability after the initial dendrite territory is established. The overall pattern of axon projections in easKO animals appears to be similar to but much fainter than that of wildtype animals (Figures S2I and S2J), raising the possibility that axon morphogenesis may also be affected in easKO mutants.
Figure 1. EAS Is Required in Class IV da Neurons for Dendrite Morphogenesis.

(A–D) Sample images showing dendrite morphology for the indicated genotypes.
(E and F) Quantification of number of branches (E) and total dendritic length (F) at 48 hrs, 72 hrs, 96 hrs, and 120 hrs AEL in wildtype (n = 6, 6, 6 and 10 respectively), easKO (n = 7, 6, 6 and 9 respectively) and eas1 mutants (n = 6). Student’s t tests were used to compare between wildtype and easKO mutants at 48 hrs, 72 hrs, and 96 hrs AEL. one-way ANOVA analysis was used for wildtype, eas1, and easKO mutants at 120 hrs AEL.
(G) Sholl analysis showing decreased complexity of the dendritic arbor for the indicated genotypes. n = 5 per genotype.
(H and I) Quantification of number of branches (H) and total dendritic length (I) for the indicated genotypes. n = 7 and 5 for wildtype and easKO+ppk>eas, respectively. n = 6 for all of the other genotypes.
(J–L) Sample images showing dendrite morphology for the indicated genotypes.
(M and N) Quantification of total dendritic length (M) and number of branches (N) for neuroal pect and bbc RNAi, two other critical genes that act together with eas in the CDP–ethanolamine pathway for PE synthesis. n= 6, 6, and 5 for control, pect RNAi, and bbc RNAi, respectively.
See also Figures S1 and S2. Scale bars represent 30 μm. ns, not significant; *p < 0.05; **p < 0.01; and ***p < 0.001 for all figures.
We next asked whether eas was required cell-autonomously in class IV da neurons to regulate dendrite morphogenesis. We were able to rescue the easKO and eas1 dendrite morphogenesis defects by expressing eas in class IV da neurons (Figures 1H and 1I). Furthermore, knocking down eas only in da neurons led to a similar reduction in number of branches and total dendrite length (Figures 1D, 1H and 1I), suggesting that eas is required cell-autonomously in class IV da neurons for proper dendrite morphogenesis. Interestingly, overexpressing eas in class IV da neurons did not strongly affect dendrite growth, suggesting that an increase in easily shocked kinase activity does not affect dendrite morphogenesis (Figures 1H and 1I).
Dendrites of class IV da neurons undergo complete pruning and regrowth during the pupal stage to reestablish dendrite territory (Kuo et al., 2005; Williams and Truman, 2005). We also found reductions in the number of dendrite branches and total dendritic length in 1 day old easKO adult flies (Figures S1C, S1D, S1F and S1G). Knocking down eas in the neurons also led to similar dendrite morphogenesis defects, suggesting that eas cell-autonomously regulates dendrite morphogenesis during the pupal stage (Figures S1E–S1G).
The eas gene encodes an ethanolamine kinase that catalyzes the phosphorylation of ethanolamine to phosphoethanolamine, which is modified by phosphoethanolamine cytidylytransferase (encoded by pect) to produce CDP–ethanolamine. CDP-ethanolamine donates phosphoethanolamine to diacylglycerol to generate PE, which is mediated by CDP-ethanolamine phosphotransferase (encoded by bbc) (Dobrosotskaya et al., 2002). In addition, the level of PE is reduced in eas whole flies and heads (Kliman et al., 2010; Nyako et al., 2001; Pavlidis et al., 1994). Therefore, we asked whether the dendrite morphogenesis defects were due to insufficient PE synthesis through the CDP–ethanolamine pathway. Compared with control animals, knocking down pect and bbc in neurons both led to easKO-like decreases in the number of branches and dendrite length (Figures 1J–1N), suggesting that PE synthesis through the CDP–ethanolamine pathway is important for dendrite growth.
EAS Kinase Regulates Terminal Dendrite Dynamics
Dendrites in wildtype animals grow and branch extensively starting from 48 hrs AEL, while displaying dynamic growth and retraction of the terminal dendrites (Parrish et al., 2007). Since the dendrite defects of easKO mutant neurons became apparent during this period, we asked whether eas is required for normal terminal dendrite dynamics (Figures 2A–2F). By performing in vivo time-lapse analysis during larval development at 72 hrs and 76 hrs AEL, we detected terminal dendrite growths and retractions while these dendrites formed complete field coverage in wildtype animals (Figures 2A–2C). The fraction of retracting terminal dendrites in easKO mutant class IV da neurons was increased (Figure 2H), while the fraction of growing terminal dendrites was unaltered (Figure 2G). As a result, 77.5% of the terminal dendrites were dynamic in easKO mutants (n = 10 neurons), whereas 70.1% of the terminal dendrites were dynamic in wildtype animals (n = 10 neurons, p<0.05), suggesting that eas is required for regulating terminal dendrite stability. As a result, the growth/retraction ratio of terminal dendrites was decreased in class IV da neurons in easKO mutant (Figure 2I). Our findings suggest that eas is important for terminal dendrite stability and supporting dendrite growth after the initial dendrite patterning is established.
Figure 2. EAS Regulates Terminal Dendrite Growth Dynamics.

(A and B) Sample images showing dendrite morphology for wildtype animals at 72 hrs (A) and 76 hrs (B) AEL.
(D and E) Sample images showing dendrite morphology for easKO mutants at 72 hrs (D) and 76 hrs (E) AEL.
(C and F) Branch dynamics are depicted in traces. Increased and decreased terminal branch region are marked in green and magenta, respectively.
(G–I) Quantification of fractions of growing (G) and retracting (I) terminal dendrites, as well as dendrite growth/retraction ratios for the indicated genotypes, showing decreases of dendrite stability and overall dendrite growth in easKO mutants.
Scale bars represent 5 μm. Student’s t test was used. n = 10 neurons per genotype.
Level of SREBP Activity is Critical for Dendrite Morphogenesis in the Class IV da Neurons
Previous studies have established a link between low PE levels and increased SREBP activity (Dobrosotskaya et al., 2002; Lim et al., 2011), which is critical for turning on the transcription of downstream genes for lipogenesis (Rawson, 2003). However, it is unknown whether SREBP, a highly conserved transcription factor that is essential for lipid homeostasis, regulates nervous system development.
To test whether the level of SREBP transcriptional activity is important for dendrite morphogenesis, we tested the consequence of increased SREBP activity in class IV da neurons by expressing constitutively active forms of SREBP. Indeed, expressing the N-terminal domain containing the first 452 amino acids of SREBP, which mimics the cleaved, active form of SREBP (SREBP.1-452), led to decreases in the number of branches and the total dendrite length of class IV da neurons (Figures 3A, 3B, 3D and 3E).
Figure 3. Aberrantly High Level of SREBP Transcriptional Activity Contributes to the Dendrite Morphogenesis Defects in easKO Mutants.

(A–C) Sample images showing dendrite morphology for the indicated genotypes.
(D and E) Quantification of total dendritic length (D) and number of branches (E) for neurons expressing constitutively active forms of SREBP (srebp.1-452), full-length SREBP, and RNAi construct targeting SREBP. n = 5 for ppk>srebp. n = 6 for all of the other genotypes.
(F and G) Western blot of larval brains showing the full-length (Fl-SREBP) and constitutively active, mature (m-SREBP) forms of SREBP protein in wildtype and easKO mutants. Tubulin was used as a loading control.
(H–N) Sample images showing dendrite morphology for the indicated genotypes.
(O and P) Quantification of total dendritic length (O) and number of branches (P) for the indicated genotypes. One-way ANOVA tests were used. Results of comparison between easKO mutants and each genotype are labeled on top of each column. n = 7 for wildtype and n = 6 for easKO mutants.
Scale bars represent 30 μm.
Conversely, knocking down srebp by expressing a previously-characterized SREBP RNAi in class IV da neurons (Figure 3C) (Song et al., 2014) also led to decreases in the number of branches (Figure 3E) and the total dendrite length (Figure 3D). Interestingly, both knocking down and expressing constitutively active SREBP in class IV da neurons led to many missing longitudinal and commissural projections (Figures S2K and S2L), suggesting that SREBP is also involved in regulating axon morphology. Taken together, our results demonstrate that SREBP signaling is critical for dendrite outgrowth during larval development.
Aberrant Lipid Homeostasis Mediated by SREBP Signaling Contributes to Dendrite Morphogenesis Deficits in easKO Mutants
Given that the level of SREBP activity is critical for dendrite growth, we tested the hypothesis that SREBP acts downstream of a deficiency in PE synthesis to alter dendrite morphogenesis in easKO mutants. First, we determined whether SREBP activity is altered in easKO mutants by examining the processing of endogenous SREBP proteins by Western blots using a previously characterized SREBP antibody (Lim et al., 2011). Most of the SREBP proteins were full-length, membrane-bound precursors (Fl-SREBP), and only a trace amount of the cleaved and mature form (m-SREBP) was detected in wildtype larval brains (Figure 3F). Compared to wild-type, activated SREBP protein level was increased by 3.6 ± 1.4 fold in easKO mutant brains (Figure 3G; n=3), suggesting that loss of eas results in greater activation of SREBP signaling.
To determine whether the increased transcriptional activity of SREBP contributes to dendrite morphogenesis defects in easKO mutants, we reduced the level of SREBP in easKO mutants to see whether the severity of dendrite growth defects would be ameliorated. Indeed, removing one copy of the SREBP gene in easKO mutants largely restored dendrite growth (Figures 3H and 3I) (n = 7). In Drosophila, activated SREBP protein turns on the expression of known lipogenic genes, including acetyl-coA carboxylase (acc), fatty acid synthase (fas), acetyl CoA synthase (acs) and fatty acyl CoA synthetase (acsl) (Rawson, 2003). Given that the SREBP processing is increased in easKO mutants, we reasoned that the dendrite morphogenesis defects could arise from increased expression of SREBP target genes. Indeed, expressing RNAi constructs targeting acc and fas in class IV da neurons of easKO mutants (Figures 3J and 3K) (n = 5 per genotype), and introduced one copy of the acsl or acs mutant alleles in easKO mutants (Figures 3L and 3M) (n = 6 per genotype) improved dendrite morphogenesis in easKO mutants (Figures 3O and 3P). While knocking down fas in class IV da neurons in easKO mutants also increased the number of branches, it did not increase the dendrite length, possibly due to a low RNAi efficiency or the redundancy of the three fas genes in the Drosophila genome. Further, increasing SREBP levels in class IV da neurons of easKO mutants does not enhance their dendrite growth defects (Figures 3N–3P) (n = 5). Similarly, increasing SREBP levels of class IV da neurons in wildtype animals did not lead to any defects in dendrite morphogenesis (Figures 3D and 3E) (n = 5), suggesting that the level of activated SREBP is not further increased by increasing the total amount of full-length SREBP in the neurons.
Together, these results show that hyperactivation of SREBP contributes to the dendrite growth defects in easKO mutants.
Reducing the Level of cacophony Partially Suppresses Dendrite Morphogenesis Deficits in easKO Mutants
Adult eas flies with characteristic seizure-like phenotypes have been used as a model for screening for mutations that could suppress neuronal hyperexcitability and hence bang sensitivity (Parker et al., 2011). To determine whether the seizure-like phenotype was associated with defects in dendrite development, we asked whether the altered lipid homeostasis mediated by SREBP also contributes to the seizure-like phenotype in easKO mutants (Figure S3A). Interestingly, mutations in the SREBP and its transcriptional targets, acs and acsl did not suppress the bang-sensitive seizure-like behavior in easKO mutants (Figure S3B), suggesting that dendrite morphogenesis defects mediated by the altered lipid homeostasis pathway likely do not contribute to the physiological defects that lead to seizure susceptibility in easKO mutants.
We then asked whether genes that suppressed the seizure-like phenotype could ameliorate the dendrite morphogenesis defects in easKO mutants. Mutations in cacophony (cac), a gene encoding the α1 subunit of the voltage-gated Ca2+ channel, potently suppress the seizure-like behavior in eas mutants (Saras and Tanouye, 2016). Interestingly, cac is also required to mediate dendrite Ca2+ influx and pruning of class IV da neurons during metamorphosis (Kanamori et al., 2013). We found that knocking down cac by expressing a previously characterized RNAi construct (Saras and Tanouye, 2016) in class IV da neurons ameliorated the dendrite outgrowth defects in easKO mutants (Figures 4A, 4B, 4D and 4E). Meanwhile, knocking down cac in class IV da neurons in wildtype animals did not cause any significant dendrite growth defects, suggesting that low Ca2+ influx does not impair dendrite morphogenesis (Figure 4C). It thus appears that calcium signaling is abnormally enhanced in easKO mutants. We then checked the localization of Cacophony by expressing a previously-characterized GFP-tagged Cacophony (Cac-GFP) in class IV da neurons (Kawasaki et al., 2004). We found that the distribution of Cac-GFP remains unchanged in wildtype and easKO mutants (Figures S3G–S3L), suggesting that eas does not regulate the localization of Cacophony. Although overexpressing escargot (esg, a member of the snail family of transcription factors) (Hekmat-Scafe et al., 2005) or kazachoc (kcc, K+-Cl− cotransporter) (Hekmat-Scafe et al., 2010) was able to suppress seizure-like phenotype in eas mutants, their overexpression in class IV da neurons did not suppress the dendrite phenotype in easKO mutants (Figures S3C–S3F). Therefore, the increased Ca2+ influx, rather than hyperexcitability per se, likely contribute to dendrite morphogenesis defects in easKO mutants.
Figure 4. Reducing the level of cacophony partially suppresses dendrite morphogenesis defects in easKO mutants.

(A and C) Sample images showing dendrite morphology for easKO mutants (A) (n = 9), easKO mutants with neuronal specific cacophony knocked down (B) (n = 6), and wildtype animals with neuronal specific cacophony knocked down (C) (n = 6). n = 10 for wildtype.
(D and E) Quantification of total dendritic length (D) and number of branches (E), showing that reducing the level of cacophony partially suppresses the dendrite growth defects in easKO mutants. One-way ANOVA tests were used.
See also Figure S3. Scale bars represent 30 μm.
Discussion
A growing body of evidence suggests that lipid metabolism and homeostasis is important for nervous system development (Zhang and Liu, 2015). Here, we provide evidence that EAS kinase and SREBP signaling pathway are required for dendrite growth during development. Our results do not rule out the possibility that a reduction in PE levels could also independently contribute to the eas dendritic phenotype, due to a lack of PE supply for expanding the plasma membrane during dendrite growth. Interestingly, although both reduced and increased SREBP activity led to the same dendrite reduction phenotype, the underlying mechanism may be different. Reduced lipid production caused by low SREBP activity might impair the lipid supply for growing dendrites, while aberrantly increased lipid production by SREBP might lead to lipotoxicity (Liu et al., 2015). Since the homologues of both genes are highly conserved, it will be interesting to determine their roles in dendrite morphogenesis within the developing mammalian nervous system.
We observed that terminal dendrite dynamics, but not initial establishment of the dendritic territory, is perturbed in eas mutants. It is possible that terminal dendrite growth and stability rely more on PE, while the primary and secondary branches do not. It is also possible that although PE synthesis is impaired in eas mutants, neurons still have enough PE to support initial dendrite growth. As the animal grows, neurons cannot supply enough PE for the exuberant terminal dendrite growth. It would be of interest to identify how impairment in PE specifically affects terminal dendrite dynamics in future studies.
We find that reducing the level of cacophony, a subunit of the Cav2 voltage-gated Ca2+ channel, ameliorates the dendrite morphogenesis defects in easKO mutants. In contrast, reducing SREBP signaling largely rescues the dendrite phenotypes but does not suppress the adult seizure-like phenotype. It is conceivable that the part of the neural circuit that heavily relies on SREBP signaling for dendrite morphogenesis during development does not contribute to the seizure-like behavior. Consistent with this hypothesis, expressing eas acutely in neurons in adult eas flies suppresses the seizure-like phenotype (Kroll and Tanouye, 2013). It thus appears that alterations in neuronal Ca2+ influx contribute to not only seizure-like phenotypes but also dendrite morphogenesis defects during larval development.
In summary, our study uncovers an important role for phospholipid PE synthesis in neural development, and they further reveal a molecular pathway involving conserved molecules for phospholipid homeostasis as being important in the regulation of dendrite morphogenesis.
Experimental Procedures
Live Imaging
Animals were reared at 25°C in density-controlled v ials or at 29°C for bbc and pect RNAi experiments. Larvae at appropriate stages were mounted in glycerol and dendrites of da neurons were imaged using a Leica SP5 laser scanning confocal microscope as previously described (Meltzer et al., 2016). Using water as a mounting media, v’ada neuron of the 1-day old adult ventral abdomen was imaged with a Leica SP5 laser scanning confocal microscope. Dendritic length and branch number were calculated using the Simple Neurite Tracer plugin in ImageJ.
Statistical Analysis
Data were analyzed and plotted with Prism 6.0c software. Student’s t test was to compare statistical significance of two independent groups. One-way ANOVA tests were used to compare statistical significance of more than two independent groups. The Tukey’s multiple comparison test was used along with one-way ANOVA tests when multiple comparisons was required.
See Supplemental Experimental Procedures for details on fly stocks, Western blotting, generating eas knockout flies, immunohistochemistry, and bang-sensitive analysis.
Supplementary Material
Highlights.
A conserved ethanolamine kinase, EAS, regulates dendrite growth and stability
Multiple enzymes involved in phospholipid PE synthesis affect dendrite growth
EAS regulates dendrite morphogenesis via SREBP signaling
Reduction of cacophony calcium channel partially rescues eas dendrite phenotype
Acknowledgments
We thank Drs. Mark Tanouye and Rolf Bodmer for reagents. Supported by National Science Foundation Graduate Research Fellowship, AAUW American Dissertation Fellowship (to S.M.), NIH grant number R37NS040929 and R35NS097227 (to Y.-N.J.). L.Y.J. and Y.-N.J. are investigators of the Howard Hughes Medical Institute.
Footnotes
Author Contributions
S.M., J.A.B., G.L.P., Y.G., L. D., and C.E.O., conducted experiments and analyses. S.M., L.Y.J. and Y.-N.J. designed experiments and wrote the paper. J.A.B. conducted the genetic screen.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benzer S. From the gene to behavior. Jama. 1971;218:1015–1022. [PubMed] [Google Scholar]
- Buchovecky CM, Turley SD, Brown HM, Kyle SM, McDonald JG, Liu B, Pieper AA, Huang W, Katz DM, Russell DW, et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nature Publishing Group. 2013;45:1013–1020. doi: 10.1038/ng.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science. 2002;296:879–883. doi: 10.1126/science.1071124. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development. 2002;129:2867–2878. doi: 10.1242/dev.129.12.2867. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Ye B, Yang CH, Younger S, Borden K, Jan LY, Jan YN. Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development. 2007;134:55–64. doi: 10.1242/dev.02666. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Dang KN, Tanouye MA. Seizure suppression by gain-of-function escargot mutations. Genetics. 2005;169:1477–1493. doi: 10.1534/genetics.104.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Mercado A, Fajilan AA, Lee AW, Hsu R, Mount DB, Tanouye MA. Seizure sensitivity is ameliorated by targeted expression of K+-Cl− cotransporter function in the mushroom body of the Drosophila brain. Genetics. 2010;184:171–183. doi: 10.1534/genetics.109.109074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci. 2010;11:316–328. doi: 10.1038/nrn2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HE, Harwood JL, Bowen ID, Griffiths G. Lipid composition of subcellular membranes from larvae and prepupae of Drosophila melanogaster. Lipids. 1992;27:984–987. doi: 10.1007/BF02535576. [DOI] [PubMed] [Google Scholar]
- Kanamori T, Kanai MI, Dairyo Y, Yasunaga KI, Morikawa RK, Emoto K. Compartmentalized calcium transients trigger dendrite pruning in Drosophila sensory neurons. Science. 2013;340:1475–1478. doi: 10.1126/science.1234879. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Zou B, Xu X, Ordway RW. Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J Neurosci. 2004;24:282–285. doi: 10.1523/JNEUROSCI.3553-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNEDY EP. Metabolism of lipides. Annu Rev Biochem. 1957;26:119–148. doi: 10.1146/annurev.bi.26.070157.001003. [DOI] [PubMed] [Google Scholar]
- Kliman M, Vijayakrishnan N, Wang L, Tapp JT, Broadie K, McLean JA. Structural mass spectrometry analysis of lipid changes in a Drosophila epilepsy model brain. Mol BioSyst. 2010;6:958–18. doi: 10.1039/b927494d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll JR, Tanouye MA. Rescue of easily shocked mutant seizure sensitivity in Drosophila adults. J Comp Neurol. 2013;521:3500–3507. doi: 10.1002/cne.23364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CT, Jan LY, Jan YN. Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. Proceedings of the National Academy of Sciences. 2005;102:15230–15235. doi: 10.1073/pnas.0507393102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Sanes JR, Kay JN. Development of Dendritic Form and Function. Annu Rev Cell Dev Biol. 2015;31:741–777. doi: 10.1146/annurev-cellbio-100913-013020. [DOI] [PubMed] [Google Scholar]
- Lim HY, Wang W, Wessells RJ, Ocorr K, Bodmer R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes & Development. 2011;25:189–200. doi: 10.1101/gad.1992411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer S, Yadav S, Lee J, Soba P, Younger SH, Jin P, Zhang W, Parrish J, Jan LY, Jan YN. Epidermis-Derived Semaphorin Promotes Dendrite Self-Avoidance by Regulating Dendrite-Substrate Adhesion in Drosophila Sensory Neurons. Neuron. 2016;89:741–755. doi: 10.1016/j.neuron.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyako M, Marks C, Sherma J, Reynolds ER. Tissue-specific and developmental effects of the easily shocked mutation on ethanolamine kinase activity and phospholipid composition in Drosophila melanogaster. Biochem Genet. 2001;39:339–349. doi: 10.1023/a:1012209030803. [DOI] [PubMed] [Google Scholar]
- Parker L, Howlett IC, Rusan ZM, Tanouye MA. Seizure and epilepsy: studies of seizure disorders in Drosophila. Int Rev Neurobiol. 2011;99:1–21. doi: 10.1016/B978-0-12-387003-2.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Emoto K, Jan LY, Jan YN. Polycomb genes interact with the tumor suppressor genes hippo and warts in the maintenance of Drosophila sensory neuron dendrites. Genes & Development. 2007;21:956–972. doi: 10.1101/gad.1514507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Peng IF, Wu CF. Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J Neurosci. 2007;27:1072–1081. doi: 10.1523/JNEUROSCI.4746-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Chen HM, Lee T, Bullock SL. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci USa. 2014;111:E2967–E2976. doi: 10.1073/pnas.1405500111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RB. The SREBP pathway — insights from Insigs and insects. Nat Rev Mol Cell Biol. 2003;4:631–640. doi: 10.1038/nrm1174. [DOI] [PubMed] [Google Scholar]
- Saras A, Tanouye MA. Mutations of the Calcium Channel Gene cacophony Suppress Seizures in Drosophila. PLoS Genet. 2016;12:e1005784. doi: 10.1371/journal.pgen.1005784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Veenstra JA, Perrimon N. Control of Lipid Metabolism by Tachykinin in Drosophila. CellReports. 2014;9:40–47. doi: 10.1016/j.celrep.2014.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiji J, Crawford DA. The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals. 2010;18:98–112. doi: 10.1159/000323189. [DOI] [PubMed] [Google Scholar]
- Tint GS, Irons M, Elias ER, Batta AK, Frieden R, Chen TS, Salen G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- Vance JE. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic. 2014;16:1–18. doi: 10.1111/tra.12230. [DOI] [PubMed] [Google Scholar]
- Williams DW, Truman JW. Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development. 2005;132:3631–3642. doi: 10.1242/dev.01928. [DOI] [PubMed] [Google Scholar]
- Wong C, Crawford DA. Comprehensive Guide to Autism. New York, NY: Springer New York; 2014. Lipid Signalling in the Pathology of Autism Spectrum Disorders; pp. 1259–1283. [Google Scholar]
- Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6:254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.