

Summary
Alternative splicing contributes to gene expression dynamics in many tissues, yet its role in auditory development remains unclear. We performed whole exome sequencing in individuals with sensorineural hearing loss (SNHL) and identified pathogenic mutations in Epithelial Splicing Regulatory Protein 1 (ESRP1). Patient derived iPSCs showed alternative splicing defects that were restored upon repair of an ESRP1 mutant allele. To determine how ESRP1 mutations cause hearing loss we evaluated Esrp1−/− mouse embryos and uncovered alterations in cochlear morphogenesis, auditory hair cell differentiation and cell fate specification. Transcriptome analysis revealed impaired expression and splicing of genes with essential roles in cochlea development and auditory function. Aberrant splicing of Fgfr2 blocked stria vascularis formation due to erroneous ligand usage, which was corrected by reducing Fgf9 gene dosage. These findings implicate mutations in ESRP1 as a cause of SNHL and demonstrate the complex interplay between alternative splicing, inner ear development, and auditory function.
Keywords: ESRP1, alternative splicing, hearing loss, cochlear epithelium, Fgf signaling, stria vascularis, inner ear development
eTOC Blurb
Rohacek et al. identify mutations in an alternative splice regulator, ESRP1, in a family with hearing loss. Loss of Esrp1 in mice leads to morphological defects in inner ear development and cell fate switches in the lateral cochlear wall caused by altered Fgfr2 splicing patterns and Fgf ligand usage.
Introduction
Hearing loss is the most prevalent sensory deficit in humans affecting 1 in 500 newborns (Morton and Nance, 2006). Approximately half of all cases of early onset hearing loss in developed countries have a genetic etiology, with single gene mutations in over 100 different loci identified thus far (http://hereditaryhearingloss.org). Mutations in the majority of these genes result in nonsyndromic sensorineural hearing loss (SNHL), where abnormal inner ear function is the only diagnostic feature. Identification of hearing loss genes has increased our awareness of the vast clinical and genetic heterogeneity underlying this condition and has contributed greatly to our understanding of inner ear development and function through studies in animal models (Lenz and Avraham, 2011; Kazmierczak and Muller, 2012; Ohlemiller et al., 2016). Despite the large number of identified hearing loss genes, the cause of inherited SNHL still remains uncertain in many children (Sloan-Heggen et al., 2016; Mehta et al., 2016). The advent of whole exome sequencing has accelerated the pace of disease gene discovery, and its application to individuals with hearing loss is improving the rate of diagnosis (Atik et al., 2015; Sloan-Heggen et al., 2016).
The inner ear is a dual-purpose organ that senses sound and balance through the activation of mechanosensory hair cells in distinct auditory and vestibular structures (Wu and Kelley, 2012). The organ of Corti is a specialized sensory apparatus for hearing in mammals that lines the medial wall of the cochlear duct and consists of a single row of inner hair cells, three rows of outer hair cells and a variety of support cells. Hair cells convert sound induced vibrations into electrochemical signals that are transmitted to the brain for processing along auditory nerve fibers (Kazmierczak and Muller, 2012; Yu and Goodrich, 2014). Interference with the sound transduction pathway at any point, especially within hair cells, results in hearing loss (Dror et el., 2010; Schwander et al., 2010).
Cell types on the lateral side of the cochlear duct comprising Reissner’s membrane, the stria vascularis, and the outer sulcus function in the production, secretion, recycling and maintenance of endolymph, a specialized fluid of high ionic concentration that supports hair cell mechanotransduction (Patuzzi et al., 2011). Mutations in genes that disrupt the flow of potassium ions through the multilayered stria vascularis, including gap junctions, channels, pumps and transporters, are common causes of hearing loss (Locher et al., 2015). Despite their importance for auditory function, relatively little is known about the development of lateral cochlear duct cells, compared to their sensory counterparts.
Gene regulatory networks synchronize cochlear morphogenesis with the specification and differentiation of sensory and nonsensory cell types that form along its length (Groves and Fekete, 2012). While many of the key transcriptional regulators of cochlear development are known, the role that posttranscriptional events play in the formation of inner ear structures and cell types is less clear. A more comprehensive approach to the study of gene expression dynamics that integrates transcriptional and posttranscriptional mechanisms is likely to improve our fundamental understanding of inner ear biology and hearing loss.
Alternative splicing is a posttranscriptional process that increases the complexity of mRNA transcripts and proteins encoded by a finite genome (Chen and Manley, 2009; Fu and Ares, 2014). Most human multi-exon genes are alternatively spliced in a regulated manner that relies on the pairing of different combinations of 5′ and 3′ splice sites to generate mature mRNAs that differ by one or more exons (Pan et al., 2008; Wang et al., 2008). Alternatively spliced transcripts frequently produce protein isoforms with divergent properties, although the biological significance of the vast majority of these events remains unexplored (Wang et al., 2008; Pan et al., 2008; Keleman et al., 2013; Yang et al., 2016). Many mammalian tissues show cell type and stage specific expression of alternatively spliced transcripts that often fit into biologically coherent pathways (Ule et al., 2005; Fu and Ares, 2014; Yang et al., 2014; Raj and Blencowe, 2015; Bebee et al., 2015; Vuong et al., 2016; Traunmüller et al., 2016; Zhang et al., 2016). The coordinated expression of these splicing-regulatory networks is due in large part to the selective activity of specialized RNA binding proteins that mediate the inclusion or exclusion of alternatively spliced exons based on their recruitment to cis acting elements in target transcripts (Chen and Manley, 2009; Fu and Ares, 2014). Consequently, mutations that disrupt either the cis or trans regulators of alternative splicing contribute significantly to human disease (Cieply and Carstens, 2015; Xiong et al., 2015; Li et al., 2016), as well as hearing loss in mice (Nakano et al., 2012; Moayedi et al., 2014).
Epithelial Splicing Regulatory Proteins (Esrp) are a highly conserved family of RNA binding proteins that promote alternative splicing exclusively in epithelial tissues (Warzecha et al., 2012). Mammals possess two highly homologous Esrp genes, Esrp1 and Esrp2, which were originally characterized in a cell-based screen for regulators of an epithelial specific Fgfr2 splicing event (Warzecha et al., 2009). Subsequent studies using genome wide approaches in epithelial cell lines and mouse knockout models demonstrated that Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development (Dittmar et al., 2012; Bebee et al., 2015; Yang et al., 2016; Bebee et al., 2016).
In the current study, we performed whole exome sequencing in individuals with profound bilateral SNHL and identified biallelic pathogenic mutations in ESRP1. Patient derived induced pluripotent stem cells (iPSCs) showed alterations in alternative splicing, consistent with a loss of ESRP1 function. To understand how mutations in ESRP1 might cause hearing loss we evaluated Esrp1−/− mouse embryos and uncovered defects in inner ear morphogenesis, auditory hair cell differentiation and cell fate specification along the lateral wall of the cochlear epithelium. RNA-seq analysis revealed impaired expression and splicing of genes associated with cochlear development and auditory function that explain several aspects of the inner ear phenotypes in Esrp1−/− embryos. In particular, aberrant splicing of Fgfr2 from the IIIb (epithelial) to IIIc (mesenchymal) isoforms compromised the identity of cells along the cochlear lateral wall due to improper Fgf9 ligand usage. Surprisingly, ectopic Fgf9/Fgfr2-IIIc signaling also compensated for the loss of Fgfr2-IIIB to promote cochlear duct morphogenesis in Esrp1−/− mutants. These findings implicate mutations in ESRP1 as a cause of SNHL and demonstrate the complex interplay between alternative splicing, inner ear development, and auditory function.
Results
Exome sequencing reveals ESRP1 mutations in a family with SNHL
An eight-year-old female with congenital profound bilateral SNHL, born to healthy non-consanguineous parents, was evaluated at the Division of Otolaryngology at the Children’s Hospital of Philadelphia (Fig. 1A). A temporal bone CT scan of the proband showed no abnormalities in cochlear morphology. However, an unusual vestibular dysplasia was revealed, consisting of a rudimentary lateral semicircular canal deficient in its central bony island that took on a cystic appearance (Fig. S1A–D). A 14-year-old brother was also diagnosed with severe to profound bilateral SNHL from birth, and showed the same vestibular dysplasia as his sister (Fig. S1E–F). Despite the abnormal vestibular findings, neither child presented with balance or movement disorders. Four other healthy siblings, including the proband’s twin brother, have normal hearing (Fig. 1A). No other family history of hearing loss was reported.
Figure 1. ESRP1 mutations segregate with SNHL and disrupt alternative splicing.

(A) Pedigree with SNHL showing segregation of maternal (blue) and paternal (red) ESRP1 mutations to affected (compound heterozygous) and unaffected offspring. The position of the ESRP1 frameshift (p.Asp222Glyfs*32) and missense (p.Leu259Val) mutations is indicated. (B) Quantitative RT-PCR of ESRP1 and ESRP2 mRNAs from iPSCs derived from parents and affected children. (C) Schematic and sequencing tracks for CRISPR-Cas9 gene editing strategy to repair the paternal ESRP1 (c.665_683 del) mutation. (D) Bar graph and gel image of western blot for ESRP1 from patient derived iPSCs and corrected proband (cP). (E) Quantitative RT-PCR of ESRP1 mRNA from iPSCs comparing corrected to uncorrected proband and mother. (F–I) Bar graphs and gel images of radioactive RT-PCR results showing PSI values for ESRP1 dependent alternative splicing events in patient-derived iPSCs and corrected proband. (J) Graph of RT-PCR dependent alternative splicing events in mouse ESCs (Bar represents average for two independent clones). (K,L) Bar graphs and gel images of radioactive RT-PCR results showing PSI values for ESRP1 dependent splicing events in MDA-MB-231 cells transduced with ESRP1 wild type (WT), ESRP1 mutant (p.Leu259Val) and EGFP cDNAs at three different titers. Abbreviations: Mother (M), Father (F), Proband (P), Affected brother (AB), CRISPR-Cas9 corrected Proband (cP), Percent Spliced In (PSI), epi (epithelial isoform), mes (mesenchymal isoform), ns (not significant). Graphs for B and E represent mean ± SE, D and F–I,K,L represent mean ± SD (* P<0.05, **P<0.001; ***P<0.0001, (B,D–I) one-way ANOVA with Tukey’s test, (K,L) 2-way ANOVA with Dunnet’s test; n=3). See also Figure S1.
To identify damaging mutations associated with the inner ear phenotypes in this pedigree we performed whole exome sequencing on DNA isolated from peripheral blood of the parents, proband, affected brother and unaffected siblings. Coding region and splice site variants that segregated in an autosomal recessive inheritance pattern and that occurred with a population frequency less than 3% in the 1000 Genomes Project Database and Exome Sequencing Project Exome Variant Server were selected for further analysis. No pathogenic mutations in known SNHL genes were found using this approach. However, compound heterozygous mutations were identified in a novel candidate hearing loss gene, ESRP1, that segregated with SNHL in this family (Fig. 1A and Fig. S1G). The two children with SNHL inherited different ESRP1 mutations from each parent, while the unaffected siblings had either one or no mutations (Fig. 1A, Fig. S1G). The paternal ESRP1 mutation harbors a 19 bp deletion in exon 7 (c.665_683 del) that is predicted to cause a frame shift at codon Asp222 resulting in premature termination of translation 31 amino acids downstream (p.Asp222Glyfs*32). The maternal ESRP1 mutation contains a missense variant in exon 8 (c.775C>G) that results in the substitution of leucine for valine (p.Leu259Val) at a highly conserved residue within the principle RNA recognition motif (Fig. S1G,H). An additional cohort of 144 probands with bilateral SNHL was screened for mutations in ESRP1 and ESRP2 by Sanger sequencing. While no homozygous or compound heterozygous variants were identified in these genes, rare heterozygous missense substitutions in ESRP1 were found in three samples (Fig. S1I).
To determine the impact of ESRP1 mutations on alternative splicing we derived three independent iPSC lines from lymphoblastoid cells obtained from the parents and affected children in the pedigree (Fig. 1A). We first assessed the abundance of ESRP1 and found it to be reduced by 50% at both the transcript and protein level in the father and affected children, compared to the mother (Fig. 1B,D). No differences in ESRP2 expression were observed across individuals (Fig. 1B). The ESRP1 mRNA encoded by the mutant paternal allele is likely subject to nonsense-mediated decay due to the presence of a premature stop codon, accounting for its reduced expression. The paternal ESRP1 mutation is also predicted to be non-functional since it would lead to out-of-frame translation prior to the essential RRM domains (Fig. 1A).
We interrogated the effects of the paternal ESRP1 (c.665_683 del) allele in iPSCs from the proband by genetically repairing the mutation with the CRISPR-Cas9 system. A guide RNA specific to the mutant paternal ESRP1 allele was cloned into the pX330 expression vector and electroporated into iPSCs from the proband along with a single-stranded oligodeoxynucleotide (ssODN) template that was used to correct the paternal mutation by homology directed repair (Fig. 1C). Three independent iPSC clones were identified with the corrected ESRP1 paternal allele as assessed by Sanger sequencing (Fig. 1C). ESRP1 mRNA and protein levels were significantly increased in the genetically repaired iPSCs from the proband compared to the uncorrected iPSCs, and were similar to the unaffected mother (Fig. 1D,E). These results confirm the damaging effect of the paternal ESRP1 (c.665_683 del) mutation on its expression in iPSCs.
The alternative splicing patterns of several known ESRP dependent events were evaluated in patient derived iPSCs by RT-PCR. The level of exon inclusion, quantified as percent spliced in (PSI), was significantly reduced for ENAH, NF2 and RALGPS2 and increased for ARHGEF11 in iPSCs from both affected children, compared to either parent (Fig. 1F–I). These alterations in alternative splicing were restored in the genetically repaired iPSCs from the proband, indicating their dependency on ESRP1. Not all ESRP dependent exons showed significant differences in splicing (e.g. SCRIB, MACF1, GRHL1), implying that some events are less sensitive to the loss of ESRP1 than others (data not shown). The presence of ESRP2 in iPSCs is likely to explain the partial, and in some cases nonexistent, splicing switches in the affected children, consistent with the recent finding in mice that Esrp2 is able to compensate for the loss of Esrp1 in the splicing of some, but not all, regulated transcripts (Bebee et al., 2015). In support of this premise, we observed similar splicing switches in mouse embryonic stem cells with a targeted disruption in Esrp1 compared to iPSCs from the affected children (Fig. 1J).
To investigate the nature of the maternal ESRP1 (c.775C>G) mutation, we performed retroviral transduction of wild type and mutant cDNAs into the MDA-MB 231 breast cancer cell line that does not express endogenous ESRP1 or ESRP2. Inclusion of the epithelial ENAH exon and skipping of the OSBPL3 exon was significantly increased in the presence of wild type ESRP1 compared to the maternal missense allele (Fig. 1K,L). These findings further support the hypomorphic nature of the ESRP1 (c.775C>G) mutation.
Defects in inner ear morphogenesis and auditory hair cell differentiation in Esrp1−/− mouse mutants
Our finding that human mutations in ESRP1 segregate with congenital hearing loss prompted us to investigate the role of Esrp1 during mouse inner ear development. Esrp1 is broadly expressed throughout the epithelium of the otic vesicle (E10.5) and is notably absent from the surrounding mesenchyme (Fig. S2A). Esrp1 continues to show unrestricted expression in sensory and nonsensory epithelial progenitors during cochlear and vestibular morphogenesis, although the level is weaker at later stages of inner ear development (Fig. S2B–E).
We evaluated Esrp1−/− embryos for defects in inner ear morphology using the paint fill technique. Alterations in the formation of cochlear and vestibular structures were observed in Esrp1−/− embryos compared to control littermates at E14.5, including a significant shortening and widening of the cochlear duct, as well as dysgenesis of the lateral semicircular canal and common crus due to incomplete resorption of the vestibular epithelium (Fig. 2A–D). This vestibular defect is similar to that described in children with ESRP1 mutations (Fig. S1A–F). A more severe inner ear phenotype consisting of fluid filled cysts with no vestibular or cochlear outgrowth was also observed at low penetrance in Esrp1−/− embryos (16%, 23/138) and will be discussed further below (Fig. S7A,B).
Figure 2. Inner ear morphogenesis and auditory hair cell differentiation are disrupted in Esrp1−/− mouse embryos.

(A–B) Inner ear paint fills of control and Esrp1−/− embryos at E14.5. The lateral semicircular canal and apex of the cochlear duct are magnified in adjacent panels to reveal their dysmorphic features. Scale bar = 100μm. (C–D) Quantification of cochlear length and width from control and Esrp1−/− mutant embryos represented as mean ± SD (***P<0.001, unpaired t-test; n=8). (E–J, N–S) Whole mount preparations of control and Esrp1−/− cochlear sensory epithelium stained with markers of hair cells (Myosin VIIa) and actin/stereocilia (Phalloidin) at E18.5 (E–J) and P0 (N–S) at defined regions (base, mid and apex) of the cochlear duct. Scale bar = 10μm. (K–M, T–V) Quantification of inner and outer hair cells in control and Esrp1−/− embryos at E18.5 (K–M) and P0 (T–V) within a 150μm area from defined regions (base, mid and apex) of the cochlear duct represented as mean ± SD (*P<0.0001, one-way ANOVA with Tukey’s test; n=10). Abbreviations: anterior, posterior and lateral semicircular canals (asc, psc, lsc, respectively), cochlear duct (cd), common crus (cc), endolymphatic duct (ed), inner hair cell (ihc), outer hair cell (ohc), saccule (s), utricle (u). See also Figures S3 and S4.
We next determined whether the loss of Esrp1 affects sensory development within the inner ear. The cochlear sensory epithelium was isolated from Esrp1−/− and control embryos at E18.5 and co-stained by whole mount for actin (phalloidin) to visualize stereocilliary bundles, and Myosin VIIa (Myo7a), a hair cell marker. Esrp1−/− embryos displayed a 25% decrease in total hair cell number that was primarily accounted for by a near complete absence of hair cells at the apex of the cochlear duct (Fig. 2E–M and Fig. S3A,B,E–G). Prosensory progenitors marked by Sox2 were present in the requisite number at the apex of the cochlear duct of Esrp1−/− embryos at E18.5, indicating that the missing hair cells in this region are not explained by an absence of sensory progenitors (Fig. S4A–C).
Hair cells at the base and middle turn of Esrp1−/− embryos showed an immature morphology compared to control littermates as seen by the reduced complexity of stereociliary bundles (Fig. 2E–H). Support cells marked by Sox2 and Prox1 revealed a normal compliment of pillar and Dieter cells at the basal turn of Esrp1−/− embryos (Fig. S4D–I). Moreover, vestibular sensory structures (maculae and cristae) were less affected by the loss of Esrp1 with only slight alterations in size, organization and innervation at E18.5 (Fig. S4J,K). These data reveal an intriguing cochlear phenotype in Esrp1−/− mutants consisting of a truncated cochlear duct with immature or absent hair cells.
We evaluated Esrp1−/− embryos one day later at postnatal day 0 (P0) to determine whether the hair cell phenotype was due to an arrest or delay in sensory development (Fig. 2N–V and Fig. S3C,D). Hair cell morphology at the base and middle turns of Esrp1−/− mutants appeared more advanced at P0 than at E18.5, with a notable improvement in stereocilia bundle morphology that was comparable to control pups at this stage (Fig. 2N–Q). Whereas the total number of hair cells was reduced in Esrp1−/− compared to control embryos after normalizing for cochlear duct length at E18.5, this difference was no longer apparent at P0 (Fig. S3H). This finding may be explained by the continued differentiation of Myosin VIIa positive hair cells at the apex of the cochlear duct of Esrp1−/− mutants between E18.5 and P0 (Fig. 3I,J,M,R,S,V), which occurred in the absence of further elongation of the cochlear duct (Fig. S3C–H). Unfortunately, Esrp1−/− mutants die soon after birth due to complications from cleft lip and palate defects, preventing their analysis at later postnatal ages (Bebee et al., 2015). These data suggest that the sensory phenotype in Esrp1−/− mutants is due, in part, to a temporal delay in auditory hair cell development.
Figure 3. Sensory and nonsensory gene expression profiles are disrupted in the cochlear epithelium of Esrp1−/− embryos.

(A) Plot of differentially expressed genes between wild type and Esrp1−/− cochlear epithelium at E16.5 with a fold change (FC) > 0.4 (n=3 replicates, P<0.05). (B) Gene Ontology term enrichment for differentially expressed genes between wild type and Esrp1−/− cochlear epithelium. (C) Heatmap of differentially expressed genes separated into sensory and nonsensory categories. (D) Hearing loss genes are significantly enriched in the set of differentially expressed transcripts between control and Esrp1−/− mutants. Genes expressed in the stria vascularis are marked with an asterisk.
Esrp1 regulates the timing of hair cell differentiation
To identify gene expression networks that are disrupted in Esrp1−/− mutants we performed RNA-seq from purified cochlear epithelium at E16.5. Three biological replicates from Esrp1−/− and control littermates were sequenced to a depth of 40–60 million reads and mapped to the mouse genome. Our analysis revealed 751 genes (341 upregulated, 410 downregulated) that were differentially expressed between control and Esrp1−/− mutants (fold change ≥0.4, p<0.05, Fig. 3A). Gene ontology terms associated with inner ear morphogenesis, ion transport, sensory perception of sound, and auditory receptor cell differentiation showed a significant enrichment of differentially expressed genes as revealed by the DAVID 6.8 analysis tool (Fig. 3B; Huang et al., 2009).
We further grouped the list of differentially expressed genes into sensory and nonsensory categories based on their published expression patterns (Fig. 3C). Two of the sensory genes that were downregulated in Esrp1−/− embryos, Atoh1 and Pou4f3, stood out because of their essential roles in regulating hair cell differentiation and maturation (Bermingham et al., 1999; Erkman et al., 1996; Xiang et al., 1997). Atoh1 transcription initiated properly at the base and mid-base levels of the cochlea in Esrp1−/− embryos, but was delayed by approximately 48 hours at the apex (Fig. 4A–F; Fig. S5A–X; Table S2). On the other hand, Pou4f3 expression was delayed at all levels of the cochlear duct in Esrp1−/− embryos by approximately 24 hours (Fig. 4G–L; Fig. S5Y-AP; and Table S2). No differences were observed in the expression of Sox2 or P27kip1 in prosensory progenitors along the cochlear duct of Esrp1−/− and control embryos, suggesting that the delay in hair cell differentiation was not associated with alterations in the specification or cell-cycle exit of sensory progenitors at earlier stages (Fig. S4L–O).
Figure 4. Hair cell differentiation is delayed in Esrp1−/− embryos.

(A–X) Transverse sections through defined regions of the cochlear duct (Base, Mid, Apex) from control and Esrp1−/− embryos stained for Atoh1 mRNA (A–F), Pou4F3 protein (G–L) and Hey2 mRNA (M–R) at E16.5 (n=5 or 6). Staining in prosensory domain is marked with a bracket. Weak Atoh1 expression at the apex of the cochlear duct in control embryos (C) is consistently absent in Esrp1−/− embryos at this stage. (S–X) Transverse sections through the organ of Corti of control and Esrp1−/− newborn pups (P0) stained for MyoVIIa and Sox2. Sox2 staining persists in hair cell nuclei of Esrp1−/− embryos (arrow heads in W). Scale bar = 100μm (A–F, M–R), 10μm (G–L) and 5μm (S–X). (Y) Schematic of the gene regulatory network controlling hair cell differentiation. Abbreviations: inner hair cell (ihc), outer hair cells (ohc), medial (M), lateral (L). See also Figures S4 and S5 and Table S2.
A previous study implicated the Hey family of bHLH transcription factors as negative regulators of Atoh1 (Benito-Gonzalez and Doetzlhofer, 2014). We examined Hey1 and Hey2 in sensory progenitors by in situ hybridization and observed a consistent upregulation in Hey2 at the mid and apex regions of the cochlear duct of Esrp1−/− embryos at E16.5, a stage when Hey2 transcription is typically downregulated (Fig. 4M–R). No obvious change in Hey1 expression was detected (Fig. S5AQ and data not shown). Attempts to validate these results by qRT-PCR revealed a trend for increased expression of Hey2 that did not reach statistical significance (Fig. S5AQ, Table S2). The discrepancy in these results might be explained by the dynamic and graded nature of Hey2 expression along the developing cochlear duct at this stage. Hey2 has also been shown to be modulated by Shh signaling (Benito-Gonzalez and Doetzlhofer, 2014). However, we found no changes in Gli1 expression between Esrp1−/− and control embryos by in situ hybridization or qRT-PCR at E14.5 or E16.5, suggesting that elevated Shh signaling is unlikely to account for the increase in Hey2 expression in Esrp1−/− embryos (Fig. S5AQ, Table S2, and data not shown).
We also observed a failure to downregulate Sox2 in Esrp1−/− hair cells at P0 (Fig. 4S–X), which is known to be associated with delays in sensory maturation (Puligilla and Kelley, 2016). These data suggest that an imbalance in the expression of negative regulators of sensory development (Hey2 and Sox2) may underlie the delay in hair cell differentiation in Esrp1−/− embryos (Fig. 4Y). Although, the possibility also exists that the upregulation of Sox2 and Hey2 is a consequence rather than a cause of the sensory delay.
Esrp1 regulates the fate of nonsensory cells along the lateral cochlear wall
Genes in the nonsensory category displayed greater fold changes in expression between Esrp1−/− and control embryos than did the sensory genes (Fig. 3C). The most downregulated transcripts in the cochlea of Esrp1−/− mutants encode for ion channel subunits (Bsnd, Kcnq1) and the Estrogen related receptor beta (Nr3b2/Esrrβ) transcription factor that regulates their expression (Fig. 3C and Table S2). Each of these genes is expressed in marginal cells of the stria vascularis and cause hearing loss when mutated in humans and mice due to altered ion homeostasis in the endolymph (Neyroud et al., 1997; Lee et al., 2000; Schlingmann et al., 2004; Chen and Nathans, 2007; Rickheit et al., 2008; Collin et al., 2008). Of note, hearing loss genes were significantly enriched in the overall set of differentially expressed transcripts between Esrp1−/− and control embryos (Fig. 3D). These findings prompted us to investigate the integrity of the stria vascularis in Esrp1−/− embryos.
We stained the lateral wall of the cochlear epithelium with E-cadherin and observed a pronounced expansion of cells with a flattened epithelial morphology characteristic of Reissner’s membrane, and a complete absence of cuboidal shaped marginal cells in Esrp1−/− compared to control embryos at E16.5 (Fig. 5A,D). Consistent with this change in epithelial morphology, we observed a three-fold increase in the number of cells expressing Otx2, a marker of Reissner’s membrane, and a dramatic reduction of Nr3b2 positive marginal cells in Esrp1−/− compared to control embryos (Fig. 5B,C,E–G). This phenotype resulted in a two-fold net gain in the number of cells within the lateral wall of the cochlear epithelium and likely explains the widened cochlear duct in Esrp1−/− embryos.
Figure 5. Esrp1 regulates the identity of nonsensory cells along the lateral cochlear wall.

(A–F) Transverse sections through the cochlear duct of control (n=8) and Esrp1−/− (n=6) embryos at E16.5 immunostained for E-cadherin (A,D), Otx2 (B,E) and Nr3b2 (C,F). (G) Quantification of cells expressing Otx2, Nr3b2, as well as the total number of cells in the lateral cochlear epithelium represented as mean ± SD (***P<0.0001, Student’s t-test). (H–U) Transverse sections through the cochlear duct of control (n=5) and Esrp1−/− (n=5) embryos at E18.5 immunostained for E-cadherin (H,O), and indicated cell type specific markers of the stria vascularis (I–N, P–U). Scale bar = 50μm (A–F, H,O) and 25μm (I–N, P–U). (V) Schematic of the stria vascularis displaying cell types and markers analyzed in (H–U). Abbreviations: blood vessel (bv), outer hair cells (ohc), outer sulcus (os), Reissner’s membrane (Rm) and stria vascularis (sv). See also Figure S6 and Table S2.
At E18.5, the lateral cochlear epithelium of Esrp1−/− embryos was highly dysmorphic compared to controls with an expanded and partially collapsed Reissner’s membrane and no morphological evidence of marginal cells (Fig. 5H,O). Several key proteins marking distinct cell layers of the stria vascularis, including marginal cells (Kcnq1, Barttin), intermediate cells (Atp1a1, CD44), basal cells and fibrocytes (Atp1a1, Claudin-11, Connexin-26/GJB2) were reduced or absent in Esrp1−/− embryos (Fig. 5I–N,P–V). Taken together, these data suggest that the identity of cells along the lateral wall of the cochlear epithelium has been altered in Esrp1−/− mutants resulting in a gain in Reissner’s membrane at the expense of marginal cells.
Altered splicing of Fgfr2 is responsible for the lateral cochlear wall defects in Esrp1 mutants
To define and quantify the differential splicing events between Esrp1−/− and control embryos that might explain unique aspects of the cochlear phenotype we analyzed the RNA-seq dataset from E16.5 cochlear epithelium using the MAJIQ algorithm (Vaquero-Garcia et al., 2016). This analysis uncovered 518 splicing alterations in mRNA transcripts from 490 different genes with a change in percent spliced in (ΔPSI) value of at least 10% (Fig. 6A and Table S1). Several of the genes that are dependent on Esrp1 for proper splicing have known roles in inner ear development and in some cases cause hearing loss in humans when mutated (Fig. 6B,C).
Figure 6. Alternative splicing is impaired in the cochlear epithelium of Esrp1−/− embryos.

(A) Number and type of alternative splicing switches identified by MAJIQ between Esrp1−/− and control embryos in the cochlear epithelium at E16.5 (ΔPSI > 10%). (B) PSI (percent spliced in) values for selected genes with significant splicing differences between Esrp1−/− and control embryos determined by MAJIQ and subsequently validated by RT-PCR. The highlighted genes have known roles in inner ear development and/or auditory function. (C) Voila views (Vaquero-Garcia et al., 2016) of Esrp1 dependent alternative splicing events for four genes associated with hearing loss showing examples of mutually exclusive (Fgfr2, Eya4), complex (Col11a2) and single (Scrib) exon cassettes. Numbered exons for each gene are represented as rectangles. Numbers above splicing events indicate RNA-seq read counts. Violin plots represent PSI estimates. (D) Correlation plot of alternative splicing switches predicted by MAJIQ and validated by RT-PCR. The Pearson’s product-moment correlation and the line of best fit (least squares polynomial) were computed in R (P<0.0001). (E) Radioactive RT-PCR validation of differential splicing events in the cochlear epithelium of Esrp1−/− and control littermates at E16.5. (F) Comparison of PSI values for Fgfr1-3, exon III-b (epithelial) and exon III-c (mesenchymal) in cochlear epithelium from Esrp1−/− and control embryos as determined by RT-PCR (multiple t-test comparison, P<0.0001). See also Table S1.
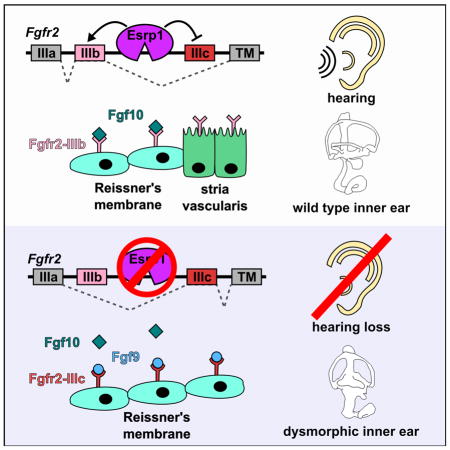
A switch in the epithelial to mesenchymal splicing pattern was confirmed by semi-quantitative RT-PCR analysis for 20 of these altered transcripts, consistent with the premise that Esrp1 regulates an epithelial specific splicing program (Fig. 6D,E). Fgfr2 exhibited the most dramatic change in PSI from 78% in control embryos to 7% in Esrp1−/− mutants (ΔPSI=71, Fig. 6B,C,F). Instead of expressing the Fgfr2-IIIb isoform typical of control cochlear epithelial cells, Esrp1 mutants inappropriately expressed the mesenchymal Fgfr2-IIIc isoform. This aberrant splicing switch in Fgfr2 is predicted to alter its binding specificity to Fgf ligands (Zhang et al., 2006).
The formation of Reissner’s membrane is dependent on Fgf10, which normally signals through Fgfr2-IIIb (Urness et al., 2015). This raises the question of how Reissner’s membrane might be expanded in Esrp1−/− mutants given the near complete replacement of Fgfr2-IIIb with Fgfr2-IIIc, an isoform that does not respond to Fgf10 (Zhang et al., 2006). It is unlikely that another Fgfr is compensating for the loss of Fgfr2-IIIb, since Fgfr1 and Fgfr3 predominantly express the IIIc isoform in the cochlear epithelium of wild type and Esrp1−/− embryos (Fig. 6F). Instead, we propose that ectopic signaling through a different Fgf ligand with affinity for Fgfr2-IIIc is mediating the expansion of Reissner’s membrane and subsequent loss of the stria vascularis in Esrp1−/− mutants. Fgf9 is an excellent candidate to be fulfilling this role given its expression in lateral cochlear wall progenitors and its ability to signal through Fgfr2-IIIc in the periotic mesenchyme (Fig. S6A,B,E,F; Pirvola et al., 2004; Zhang et al., 2006). In support of this model, ectopic expression of Fgf signaling effectors, Etv4 and Etv5, was observed along the lateral cochlear epithelium in Esrp1−/− embryos, concomitant with the expansion of Otx2 positive Reissner’s membrane (Fig. S6C–N).
To test the hypothesis that aberrant expression of Fgfr2-IIIc in Esrp1−/− embryos causes a gain in Fgf9 signaling within the lateral cochlear epithelium, we generated Esrp1−/−; Fgf9+/− compound mutants and observed a significant retraction of Reissner’s membrane (Otx2) and recovery of marginal (Nr3b2, Kcnq1, Barttin), intermediate (Cd44) and basal (Atp1a1) cell identities at E18.5 (Fig. 7A–V). Esrp1−/−; Fgf9+/− compound mutants also showed a reduction in the ectopic expression of Etv4 and Etv5 (Fig. S6O–T). These results support our model that ectopic Fgf9 signaling through an aberrantly spliced Fgfr2-IIIc isoform is responsible for the cell fate switch between Reissner’s membrane and marginal cells in Esrp1−/− mutants (Fig. 7W).
Figure 7. Ectopic signaling through Fgf9/Fgfr2-IIIc is responsible for the lateral cochlear wall defects in Esrp1 mutants.

(A–U) Transverse sections through the cochlear duct of control (n=15), Esrp1−/− (n=7) and Esrp1−/−;Fgf9+/− (n=6) embryos at E18.5 immunostained for E-cadherin (A,H,O), Otx2 (B,I,P) and cell type specific markers of the stria vascularis (C–G, J–N, Q–U). Scale bars = 50μm. (V) Quantification of cells expressing Otx2 and Nr3b2 represented as mean ± SD (***P<0.0001, ANOVA with Tukey’s test). (W) Schematic representation of the lateral cochlear wall phenotypes manifesting from altered Fgf signaling in Fgf10−/− and Esrp1−/− mutants compared to a control embryo. See also Figure S6.
Ectopic Fgf9/Fgfr2-IIIc signaling compensates for the loss of Fgfr2-IIIb to promote cochlear morphogenesis in Esrp1−/− mutants
Surprisingly, 46% (6/13) of Esrp1−/−; Fgf9+/− embryos also exhibited a cystic inner ear with no obvious vestibular or cochlear outgrowth (Fig. S7C). This phenotype is remarkably similar to that observed in 16% of Esrp1−/− mutants, as well as mouse embryos that specifically lack the Fgfr2-IIIb (epithelial) isoform (Fig. S7A–C; Pirvola et al., 2000). An important distinction between the Fgfr2-IIIb deletion line (De Moerlooze et al., 2000; Pirvola et al 2000) and Esrp1−/− mutants is that Fgfr2 expression is altogether absent in the otic epithelium of Fgfr2-IIIb mutants, but is present in the otic epithelium of Esrp1−/− embryos, albeit as the Fgfr2-IIIc (mesenchymal) isoform. This finding raises the intriguing possibility that Fgf9/Fgfr2-IIIc signaling is also compensating for other Fgfr2-IIIb dependent functions at earlier stages of inner ear morphogenesis. Consistent with this premise, all Esrp1−/−; Fgf9−/− double mutants examined (n=3) displayed cystic inner ears (Fig. S7D). Given that Fgf9 is normally dispensable for cochlear duct outgrowth (Pirvola et al., 2004), we attribute the fully penetrant cystic inner ear phenotype in Esrp1−/−; Fgf9−/− mutants to the loss of ectopic Fgf9/Fgfr2-IIIc signaling at early stages of inner ear development.
Discussion
We performed whole exome sequencing and identified mutations in ESRP1 that segregate with SNHL in a human pedigree. Our analysis of inner ear phenotypes in Esrp1−/− mouse embryos revealed possible pathogenic mechanisms for hearing loss, including defects in cochlear duct morphogenesis, auditory hair cell differentiation and marginal cell fate specification. However, the phenotypes observed in Esrp1−/− mouse embryos may not all occur with the same severity in affected children from the pedigree given that one of their mutant ESRP1 alleles (p.Leu259Val) is hypomorphic, as indicated by its reduced, but not absent, alternative splicing activity in patient derived iPSCs and other cell based assays. This residual ESRP1 function may have protected the affected children from cleft-lip and palate, which occurs in all Esrp1−/− newborn pups (Bebee et al., 2015). We therefore recommend that cases of congenital SNHL that are co-morbid with cleft-lip and palate be screened for mutations in ESRP1.
The most pronounced changes in gene expression within the cochlear duct of Esrp1−/− and control embryos were detected in nonsensory cells of the lateral cochlear epithelium. The expanded expression of Otx2 within Reissner’s membrane, and the absence of Nr3b2 expression in marginal cells of the stria vascularis suggested that the identity of these cells had been severely compromised in Esrp1−/− embryos. Nr3b2 is required for the expression of many ion channels and transporters that generate the high concentration of potassium ions in the endolymph, which is critical for hair cell mechanotransduction (Chen and Nathans, 2007). Several of these Nr3b2 dependent genes cause hearing loss when mutated and are downregulated in Esrp1−/− embryos. Thus, the reduced expression of Nr3b2, or any one of its essential transcriptional targets, is expected to cause severe hearing impairment in humans with mutations in ESRP1.
Alterations in marginal cell identity may also explain some of the non-cell autonomous phenotypes observed in Esrp1 mutants. Release of the Hepatocyte growth factor (Hgf) signal from marginal cells to its c-Met receptor on neural crest cell derived melanocytes is required for their recruitment to the intermediate cell layer of the stria vascularis (Shibata et al., 2016). Consequently, the loss of Hgf expression in Esrp1−/− embryos is likely responsible for the reduced number of intermediate cells.
Marginal cells are more severely affected in Esrp1−/− compared to Nr3b2−/− mutants, suggesting that other important determinants of their identity must also be misregulated in Esrp1−/− mutants. The expansion of Otx2 into the presumptive marginal cell territory in Esrp1−/− embryos represents the best explanation for why these cells failed to form. In wild type embryos, Otx2 is initially broadly expressed along the lateral wall of the cochlear duct at E13.5, including precursors of marginal cells and Reissner’s membrane. Otx2 expression is then downregulated from the marginal cell territory by E15.5, coincident with the onset of Nr3b2, but continues to be expressed in Reissner’s membrane. We propose that Otx2 regulates the timing of marginal cell development through a de-repression mechanism, whereby Otx2 expression must be extinguished from marginal cell progenitors in order for their development to proceed. In agreement with this model, a recent study demonstrated that mice lacking Otx2 in the inner ear showed ectopic expression of marginal cell markers along the entire lateral wall of the cochlear duct (Vendrell et al. 2015). Moreover, reduction of ectopic Otx2 expression in the marginal cell territory of Esrp1−/−;Fgf9+/− embryos coincided with the recovery of the stria vascularis. These data indicate that the dynamic expression of Otx2 along the cochlear lateral wall is important for regulating the developmental fates of marginal cells and Reissner’s membrane.
Reissner’s membrane does not form in Fgf10−/− embryos (Urness et al. 2015). A similar phenotype might have been expected in Esrp1−/− mutants given the aberrant splicing of Fgfr2, which reduces the abundance of Fgfr2-IIIb, the high affinity Fgf10 receptor (Zhang et al., 2006). Surprisingly, Reissner’s membrane still developed in Esrp1−/− embryos, albeit through an alternative signaling mechanism that benefitted from the ectopic expression of Fgfr2-IIIc in the cochlear epithelium and Fgf9, a ligand that normally signals to the otic mesenchyme and is mostly dispensable for cochlear but not vestibular development (Pirvola et al., 2004). However, a major consequence of utilizing Fgf9/Fgfr2-IIIc to compensate for Fgf10/Fgfr2-IIIb is the gain in signaling strength that results in expansion of Reissner’s membrane at the expense of marginal cells. Our study adds to the list of genetic disorders (e.g. Apert syndrome, Muenke syndrome) caused by a gain in Fgf signaling that results from alterations in Fgfr ligand-binding interactions (Yu et al., 2000; Mansour et al., 2013).
Fgfr2-IIIb is also required at early stages of inner ear morphogenesis as evidenced by the cystic inner ear phenotype displayed by embryos in which the Fgfr2-IIIb isoform was selectively deleted (Pirvola et al., 2000). Fgf3 and Fgf10 are the ligands that signal predominantly through Fgfr2-IIIb to promote early aspects of inner ear development (Alvarez et al., 2003; Wright and Mansour, 2003). One might expect cystic inner ears to be more prevalent in Esrp1−/− mutants given the depletion of Fgfr2-IIIb, however, only 16% of these embryos showed this phenotype. This observation raised the possibility that Fgf9/Fgfr2-IIIc signaling is also compensating for other Fgfr2-IIIb dependent functions at earlier stages of inner ear development in Esrp1 mutants. In support of this hypothesis, the removal of an allele of Fgf9 from the Esrp1−/− background (Esrp1−/−; Fgf9+/−) increased the penetrance of the cystic inner ear phenotype to almost 50%, whereas, 100% of Esrp1−/−; Fgf9−/− double mutants displayed cystic inner ears. These results indicate that dose dependent signaling through Fgf9/Fgfr2-IIIc is able to compensate for the loss of Fgfr2-IIIb to promote inner ear morphogenesis in Esrp1−/− mutants. This remarkable example of genetic compensation highlights the tremendous flexibility in Fgf signaling activity brought about by alternative splicing of receptor isoforms and the numerous genetically encoded ligands that bind to them.
Esrp1−/− embryos also displayed a marked decrease in the number of differentiated hair cells at the apex of the cochlear duct. This phenotype was attributed to a 48-hour delay in the onset of Atoh1 at the apical turn. Hair cell maturation was also delayed at the base and mid-base levels of the cochlear duct of Esrp1−/− embryos, but at a step downstream of Atoh1, as indicated by the 24-hour lag in Pou4f3 expression.
Sensory progenitors depend on the input of multiple signaling pathways to coordinate the expression of a set of transcriptional activators and repressors, the balance of which dictates the timing of Atoh1 transcription along the cochlear duct (Okano et al., 2011; Neves et al., 2012; Cai and Groves, 2015). The prevailing model stipulates that Sox2 functions through an incoherent feed forward loop to both directly activate Atoh1 transcription but also to promote the expression of Atoh1 repressors, including Hey1, Hey2 and Id1-3 (Neves et al., 2013; Benito-Gonzalez and Doetzlhofer, 2014). Once Atoh1 accumulates to a point where it can maintain its own expression, Sox2 is downregulated, so that its blockade on Atoh1 and other downstream components of the hair cell differentiation program can be released (Dabdoub et al., 2008; Ahmed et al., 2012; Neves et al., 2013; Puligilla and Kelley, 2016). Based on this model (Fig. 4Y), it is likely that the upregulation in Hey2 mRNA and Sox2 protein that we observed in sensory progenitors and hair cells, respectively, impedes Atoh1 expression and transcriptional activity, causing the delay in hair cell differentiation in Esrp1−/− embryos. Alternatively, the altered expression of Hey2 and Sox2 may be a secondary consequence of the delayed sensory development in Esrp1−/− embryos.
The aberrant splicing of one or more transcripts expressed within the sensory epithelium of the cochlear duct may contribute to the misregulation of Hey2 and Sox2 in Esrp1−/− embryos, although the identity of these transcripts remains to be determined. Esrp1 has also been shown to antagonize Sox2 translation by limiting the available pool of Sox2 transcripts for loading onto polysomes (Fagoonee et al., 2013). Future experiments will determine the precise mechanism responsible for the delay in hair cell differentiation in Esrp1−/− embryos and the consequence that this phenotype has on auditory function.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
For all reagent requests contact Douglas J. Epstein, Ph.D. (epsteind@mail.med.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Generation of Esrp1+/− mutant mice was described previously (Bebee et. al. 2015). Fgf9 mutant mice were procured from Jackson Laboratories (0224362; B6N(Cg)-Fgf9tm1b(KOMP)Wtsi/J; presented in text as Fgf9+/−). For all experiments control littermates have at least one wild-type allele of Esrp1. Pregnant dames were euthanized by carbon dioxide, and embryos were isolated and euthanized by decapitation in 4°C PBS. All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania.
Human Subjects
All human subjects involved in this study were enrolled under an IRB-approved protocol of informed consent (protocol number: IRB 00-002059) at the Children’s Hospital of Philadelphia.
METHOD DETAILS
Exome sequencing
Family members were enrolled under an IRB approved protocol of informed consent at the Children’s Hospital of Philadelphia (CHOP). Blood was obtained from all eight family members and genomic DNA was extracted from whole blood using standard procedures. Exomes were captured using Agilent SureSelect V4 and sequenced at 100X coverage using Illumina HiSeq 2000. Only coding regions and splice site variants at >4x coverage were analyzed, and the results were further filtered using a population frequency <3% in the 1000 Genomes and Exome Sequencing Project servers. Mutations that did not segregate in an autosomal recessive inheritance pattern were then filtered out. All putative causative mutations were validated with Sanger sequencing.
ESRP1 mutation screening
An additional cohort of 144 pediatric probands with bilateral SNHL of unknown molecular etiology was screened for mutations in ESRP1 and in the closely related gene ESRP2. These patients were all enrolled under an IRB approved protocol of informed consent at CHOP. PCR primers were designed for specific intronic sequences in ESRP1 and ESRP2 (Supplemental Table S2). DNA sequence analysis was performed using an ABI 3730XL automated DNA sequencer.
Generation of iPSCs
Peripheral blood mononuclear cells (PBMCs) were isolated from family members, transduced with EBV and transformed to lymphoblastoid cell lines (LCLs). The LCLs were maintained in RPMI media supplemented with 10% FBS, 1X Penn/Strep, 1X glutatmine in a 5% CO2 incubator. For iPSC generation, 2 million LCLs from the mother, father and 2 affected probands were transfected with nonintegrating, nontransmissible episomal plasmids expressing OCT4, SOX2, KLF4, MYCL, LIN28A, a p53 short hairpin (sh)RNA, and a green fluorescent protein (eGFP) via a single nucleofection (Hubbard et al., 2014). Transfected cells are then cultured in human embryonic stem cell (HES) media (DMEM/F12, 15% KOSR, 1X NEAA, 1X P/S, 1X glutamine, 2ME, 20ng/mL bFGF) in not adherent conditions for 6 days. Cells were then plated onto irradiated mouse embryonic fibroblasts (iMEFs) on day 6 in HES with the addition of 0.5mM sodium butyrate, and media was changed daily for a 14–17 days post-plating. iPSC-like colonies appeared between day 21–28 after nucleofection, colonies were isolated (~10/patient), and expanded on iMEFs using standard ES/IPSC culture conditions (Mills et al., 2014; Sullivan et al., 2013).
Repair of ESRP1 (c.665_683 del) allele in patient derived iPSCs
Patient derived iPSCs were maintained in feeder-free Geltrex (Life Technologies)-coated plastics and mTesr1 media (Stem Cell Technologies). For transfection, cells were dispersed with Accutase (Life Technologies) and re-suspended in P4 solution (Lonza) containing an ESRP1 sgRNA-expressing pX330-mCherry plasmid and the cognate repair ssODN (see Key Resources table), and electroporated using a 4D Nucleofector (Lonza). The repair template included a silent Cla I site which was used to test for integration (described below). Three days post-transfection, mCherry positive cells were recovered using a BD FACSJazz cell sorter and seeded onto a Geltrex-coated 10cm dish. One week post-seeding, colonies were manually picked for clonal expansion and genotyping. The site of potential repair was amplified and incubated with Cla I (see Key Resources table). Unmodified DNA was refractory to Cla I whilst repaired DNA was cleaved, yielding 235bp and 231bp fragments. Positive clones were expanded and Sanger sequenced to confirm repair of the paternal allele and absence of indel mutations on the maternal allele.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (dilution) | ||
| mouse anti-Atp1A1 (1:250) | Developmental Studies Hybridoma Bank | A5 |
| mouse anti-β-Actin M2 (1:10000) | Sigma | AC-74 |
| rabbit anti-Barttin (1:1000) | Thomas Jentsch (Leibniz-Institut für Molekulare Pharmakologie) | N/A |
| rat anti-CD44 (1:200) | BD Parmingen | 550538 |
| rabbit anti-Claudin-11 (1:100 +AR) | Novex | 364500 |
| mouse anti-Connexin-26/Gjb2 (1:200) | Thermo Scientific | 71-0500 |
| mouse anti-E-cadherin (1:200 +AR) | BD Transduction Laboratories | 610181 |
| mouse anti-Esrp1 (1:200) | Russ Carstens (UPenn) | N/A |
| rabbit anti-Esrrβ/Nr3b2 (1:500 +AR) | Jeremy Nathans (Johns Hopkins Univ.) | N/A |
| goat anti-Kcnq1 (1:300) | Santa Cruz | SC-10646 |
| rabbit anti-MyosinVIIa (1:500) | Proteus Biosciences | 25-6790 |
| mouse anti-Neurofilament (1:200) | Developmental Studies Hybridoma Bank | 2H3 |
| rabbit anti-P27kip1 (1:400) | Thermo Scientific | PA5-27188 |
| mouse anti-Pou4F3/Brn-3c (1:200 +AR) | Santa Cruz | sc-81980 |
| rabbit anti-Prox1 (1:500) | Chemicon Billerica | AB5475 |
| rabbit anti-Otx2 (1:1000) | Flora Vaccarino (Yale University) | N/A |
| Mouse anti-Sox2 (1:200 +AR) | R&D Systems | MAB2018 |
| donkey anti-mouse IGG Alexa488 (1:200) | Molecular Probes | A-21202 |
| donkey anti-rabbit IGG Alexa594 (1:200) | Molecular Probes | A-21207 |
| rabbit anti-goat IGG Alexa488 (1:200) | Molecular Probes | A-11078 |
| donkey anti-rat Alexa488 (1:200) | Molecular Probes | A-21208 |
| *AR, antigen retrieval – boil for 6 minutes in 10mM citric acid buffer (pH 6.0) | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phalloidin conjugated Alexa488 (1:50) | Molecular Probes | A-12379 |
| Critical Commercial Assays | ||
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | New England Biosystems | E7490S |
| Directional RNA Library Prep Kit for Illumina® | New England Biosystems | E7420S |
| Deposited Data | ||
| RNA-seq raw and analyzed data | This paper | GSE90821 |
| MAJIQ splicing analysis | This paper | http://majiq.biociphers.org/rohacek_2016/. |
| Experimental Models: Cell Lines | ||
| Human: patient derived IPSCs | This paper | N/A |
| Human: MDA-MB-231 cells | ATCC | HTB-26 |
| Mouse: Esrp1−/− embryonic stem cells | Cieply et al. 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Esrp1+/− | Bebee et al. 2015 | N/A |
| Mouse: Fgf9+/− - B6N(Cg)-Fgf9tm1b(KOMP)Wtsi/J | The Jackson Laboratories | 0224362 |
| Recombinant DNA | ||
| PMX-CFF-B | Modified from Toshio Kitamura | N/A |
| Sequence-Based Reagents | ||
| PCR Primers for human ESRP1 and ESRP2 exons see Table S3 | This paper | N/A |
| Primers used to detect gene specific alternative splicing switches by RT-PCR.see Table S4 | This paper | N/A |
| sgRNA oligos for ESRP1 (c.665_683 del)
CRISPR: F: caccGATGGAACTTATTGGGCACG R: aaacCGTGCCCAATAAGTTCCATC |
This paper | N/A |
| ESRP1 (c.665_683 del) repair template: TATTGAGTCCTTTGAAGAATCTTGCAATATCTTGATCTGAAGACTGCCATGGTAAACCTCGTGCCCTGACTACGGTGTTATCATCGATAAGTTCCATCTTGCTGCTGTGGATGAAGCAAAACTTCAGGTTGCATGATAAA | This paper | N/A |
| ESRP1 (c.665_683 del) repair PCR genotyping
primers: F: CACAGGCAGCCATGTTTCTA R: TAGGCAGTTGTCTTGCAGGG |
This paper | N/A |
| Software and Algorithms | ||
| RUM | Grant et al. 2011 | http://www.cbil.upenn.edu/RUM/ |
| edgeR | Robinson et al. 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| MAJIQ | Vaquero-Garcia et al., 2016 | http://majiq.biociphers.org/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Other | ||
Viral expression of ESRP1 mutation in human cell culture
Human ESRP1 full-length cDNA was cloned from the ORFeome Collection (RBP clone #290) using GATEWAY into a pIBX-cFFB-Emerald vector. The c.775C>G mutation was introduced into the ESRP1 cDNA by site directed mutagenesis. Wild type (WT) and mutant ESRP1 cDNAs were PCR amplified and cloned into pCRBlunt. The pMXs-cFF-B (IRESpuro) retroviral vector was cut with EcoRI, end-filled using Phusion, then cut with NotI. Wild type and mutant ESRP1 cDNAs were then subcloned from pCRBlunt using EcoRV and NotI. The resulting constructs include a Kozak, ESRP1 WT or c.775C>G mutant cDNA fused to a 2x FLAG tag at the carboxy terminus. Murine leukemia virus was generated using the viral packaging N16 cells were grown in DMEM, 10% FBS and transfected with the viral pMXs plasmids encoding WT or mutant ESRP1 in combination with VSV-G for pMXs EGFP, pMXs-cFF-B ESRP1 WT, and pMXs-cFF-B ESRP1 C>G (patient mutation). Virus was collected and applied in the indicated ratio with polybrene to triplicate wells of MDA-MB-231 cells grown in DMEM, 10% FBS, and Pennicillin/Streptomycin. Cells were collected into Trizol (Invitrogen) three days after infection. Total RNA was extracted for RT-PCR evaluation of endogenous ESRP splicing targets. Expression levels of WT and mutant ESRP1 C>G were confirmed by Western blot.
RT-PCR
Total RNA was extracted from purified cochlear epithelium and cultured cells (iPSCs, MDA-MB-231, mESCs) using Trizol-LS (Invitrogen), precipitated in the presence of glycogen and resuspended in 10mM Tris pH 8.0. For CRISPR corrected iPSCs and control lines, total RNA was extracted using an RNeasy mini kit (Qiagen). For synthesis of cDNA, 100ng of total RNA from cochlear epithelium or 1ug of total RNA from cell culture was used for random hexamer primed M-MLV reverse transcriptase (Promega) or Oligo(dT) primed Omniscript reverse transcriptase (Qiagen). Analysis of gene expression was performed using SYBR green master mix (AppliedBiosystems) and primer sequences listed in Table S4. Real-time RT-PCR analysis was performed using a 7500 Fast Realtime machine (AppliedBiosystems).
Semi-quantitative radioactive RT-PCR products were separated on 5% PAGE gels, dried and exposed on phosphor screens, scanned on a Typhoon FLA 9500, and quantified using ImageQuant TL, version 7.0. Splicing ratios are represented as Percent Spliced In (PSI) of the alternative exon for cassette exons and were normalized to RT-PCR product sizes. Quantification of exon IIIb and IIIc for Fgfr1, Fgfr2, and Fgfr3 required restriction enzyme digestion to discriminate the two isoforms. Fgfr2 PCR products were digested with AvaI (IIIb) or HincII (IIIc). Fgfr1 products were digested with BstXI (IIIb) and HincII (IIIc). Fgfr3 products were digested with StuI (IIIb) and PstI (IIIc) (all restriction digestions were performed according to NEB guidelines at 5U/digestion). Primer sequences are listed (Table S4).
Inner ear paint fill
Paint fills were performed essentially as described (Martin and Swanson, 1993) with the use of White-Out Plus (Bic, Milford, CT, USA) as the contrast medium. Cochlear length and width measurements were made using ImageJ.
In situ hybridization
Embryos were collected from timed pregnant females (vaginal plug = E0.5). The inner ears were dissected from the head and fixed for 2 hours in 4% paraformaldehyde at 4°C, then washed in PBS. Inner ears were cryoprotec ted overnight in 30% sucrose/PBS then snap frozen in OCT embedding compound (Sakura Finetek Torrence, CA). Inner ears were serially sectioned along the transverse plane from anterior to posterior. The position of a given section through the cochlear duct was calculated based on its distance (μm) from the first and last section (#sections x thickness = position within cochlear duct along anterior-posterior axis). Mid-modiolar sections containing all three turns of the cochlear duct (base, mid-base, apex) were confirmed using the above calculation to ensure comparison of equivalent sections through control and mutant embryos. Sections were hybridized with digoxigenin-UTP-labeled riboprobes as previously described (Nissim et al., 2007).
Whole mount cochlear preparations
Inner ears were dissected and fixed in 4% paraformaldehyde for 2 hours at 4°C, then washed in PBS. Cochleae were microdissected in 0.1% PBST to expose the sensory epithelium, and incubated with a rabbit polyclonal anti-Myosin VIIa antibody (1:500; Proteus Biosciences Inc, 25-6790) and a Phalloidin conjugated Alexa488 probe (1:50; Molecular Probes, A12379), followed by a donkey anti-rabbit IgG secondary antibody conjugated to Alexa594 (1:200). Hair cells were counted along the entire length of the cochlear duct, as well as per unit area (150μm) at base, mid and apical regions.
Immunohistochemistry
Inner ears were processed for immunohistochemistry in the same fashion as for in situ hybridization. Inner ear sections were stained with DAPI and antibodies found in Key Resources.
Western Blot
Total protein was extracted from patient derived and CRISPR corrected iPSCs in RIPA buffer. Lysate was separated on a Bis-Tris 4–12% gradient SDS-PAGE gel and transferred to nitrocellulose membrane. Membranes were blocked in 5% Non-fat Milk in 0.1% PBST then incubated overnight at 4°C in primary antibody and loading control (anti-Beta Actin M2; 1:10,000). Membranes were incubated in secondary Sheep anti-mIgG:HRP (1:2500; GE Healthcare) for one hour and protein detection was performed with ECL detection (Invitrogen) by chemiluminescence.
RNA-seq analysis
Cochlear epithelium from control and Esrp1−/− mouse embryos (n=3 pairs of biological replicates) were isolated at E16.5 exposed to collagenase P (0.1mg/ml) at 37°C for 15 min to remove surrounding mesenchyme, and snap frozen in Trizol-LS (Invitrogen). RNA was extracted and precipitated in the presence of glycogen and resuspended in 10mM Tris pH 8.0. Total RNA (200ng) was used for poly A selected RNA-seq library preparation using the NEBNext® Ultra™ Directional RNA Library Prep Kit from Illumina® (mRNA) (New England Biolabs) (products: NEBNext® Poly(A) mRNA Magnetic Isolation Module (E7490S) and NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (E7420S). Biological replicates were individually barcoded, pooled, and sequenced on two lanes of a HiSeq 2000 platform for 100x2 bp paired-end RNA-seq at the Penn Next Generation Sequencing Core (NGSC) Facility. Alignment and expression values were determined using RUM (Grant et al. 2011).
Characterization of alternative splicing switches
RNA-Seq reads were mapped to the reference mouse genome (mm10) using STAR with the option --alignSJoverhangMin 8 (Dobin et al., 2013). Alternative splicing events were analyzed using MAJIQ and VOILA with the default parameters (Vaquero-Garcia et al., 2016). Briefly, uniquely mapped, junction-spanning reads were used by MAJIQ to construct splice graphs for transcripts by using the Ensembl transcriptome annotation (release 82) supplemented with de-novo detected junctions. Here, de-novo refers to junctions that were not in the Ensembl transcriptome database, but had sufficient evidence in the RNA-Seq data (default: at least three reads mapping to at least two different start positions). The resulting gene splice graphs were analyzed for all identified local splice variations (LSVs), defined as splits in a splice graph to or from a given exon. Redundant LSVs were removed and each junction in the remaining LSVs was quantified for its expected percent spliced in (PSI) value in control and Esrp1−/− samples and expected change in PSI (ΔPSI) between WT and Esrp1−/− samples. PSI and ΔPSI correspond to the percent of isoforms and change in percent of isoforms, respectively, that use a specific junction compared to the other junctions in the given LSV. LSVs comprised of one or more junctions with an expected ΔPSI of at least 10% were then utilized by VOILA to produce gene and LSV splice graphs as well as violin plots representing PSI and ΔPSI quantifications (Table S1).
Quantification and Statistical Analysis
Statistical analysis of cell counts and splicing assays were performed using GraphPad Prism 7 software. Relevant information for each experiment including n-values, statistical tests and reported p-values are found in the legend corresponding to each figure. In all cases P<0.05 is considered statistically significant.
Cell counts were performed from equivalent sections through the cochlear duct using the cell counter feature in ImageJ. Cochlear length was measured from E14.5 paint fills and E18.5 and P0 cochlear epithelium preparations from the most basal to most apical turn of the cochlear duct using ImageJ.
RNA-seq results were processed using edgeR (Robinson et al., 2010) to detect differentially expressed transcripts with fold change >0.4 (P<0.05).
Data and Software Availability
RNA sequencing data is accessible at NCBI Gene Expression Omnibus under accession number: GSE90821
Interactive HTML output files for visualizing splicing variations between WT and Esrp1−/− samples are accessible at: http://majiq.biociphers.org/rohacek_2016/ and Table S1.
Supplementary Material
Table S1. Esrp1 dependent alternative splicing events in the cochlear epithelium. Related to Fig. 6. (see attached Excel file)
Highlights.
ESRP1 mutations segregate with sensorineural hearing loss
ESRP1 mutations disrupt alternative splicing in patient-derived iPSCs
FGFR2 alternative splicing is impaired in cochlear epithelium of Esrp1−/− embryos
Ectopic Fgf signaling via misspliced FGFR2 alters cell fate in Esrp1−/− cochleae
Acknowledgments
We thank Dr. Jason Mills, Director of the iPSC Core at the Center for Advanced Retinal and Ocular Therapeutics (Perelman School of Medicine, University of Pennsylvania), for deriving iPSC lines used in this study. This work was funded by grants from the National Institutes of Health, R01 DC006254 (DJE), R01 DE024749 (RPC), U01 HG006546 (IDK), R01 AG046544 (YB). AMR was supported by the Predoctoral Training Program in Genetics (T32 GM008216) and an NRSA fellowship F31DC014647.
Footnotes
Author Contributions
J.A.G, I.D.K, R.P.C. and D.J.E. conceived the study. J.A.G, R.K.T and I.D.K. characterized human clinical data. R.K.T, M.K., M.Z., and I.D.K. analyzed whole exome sequencing data and performed Sanger sequencing confirmation of ESRP1 mutations. T.W.B., N.P., B.C. and R.P.C. designed and performed RT-PCR and splicing studies. A.M.R and D.J.E designed and performed all mouse experiments. T.W.B. and A.M.R. generated and analyzed RNA-seq data. C.M.R. and Y.B. characterized alternative splicing data. C. M-R. and K.M. performed gene editing experiments in iPSCs. A.R.M, I.D.K, R.P.C and D.J.E, wrote the manuscript. All authors analyzed the data, discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed M, Wong EYM, Sun J, Xu J, Wang F, Xu PX. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev Cell. 2012;22:377–390. doi: 10.1016/j.devcel.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y, Alonso MT, Vendrell V, Zelarayan LC, Chamero P, Theil T, Bösl MR, Kato S, Maconochie M, Riethmacher D, et al. Requirements for FGF3 and FGF10 during inner ear formation. Development. 2003;130:6329–6338. doi: 10.1242/dev.00881. [DOI] [PubMed] [Google Scholar]
- Atik T, Bademci G, Diaz-Horta O, Blanton SH, Tekin M. Whole-exome sequencing and its impact in hereditary hearing loss. Genet Res. 2015;97:e4. doi: 10.1017/S001667231500004X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, Xing Y, Carstens RP. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife. 2015:4. doi: 10.7554/eLife.08954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee TW, Sims-Lucas S, Park JW, Bushnell D, Cieply B, Xing Y, Bates CM, Carstens RP. Ablation of the epithelial-specific splicing factor Esrp1 results in ureteric branching defects and reduced nephron number. Dev Dyn. 2016 Oct;245(10):991–1000. doi: 10.1002/dvdy.24431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito-Gonzalez A, Doetzlhofer A. Hey1 and Hey2 control the spatial and temporal pattern of mammalian auditory hair cell differentiation downstream of Hedgehog signaling. J Neurosci. 2014;34:12865–12876. doi: 10.1523/JNEUROSCI.1494-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Cai T, Groves AK. The Role of Atonal Factors in Mechanosensory Cell Specification and Function. Mol Neurobiol. 2015;52:1315–1329. doi: 10.1007/s12035-014-8925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Nathans J. Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell. 2007;13:325–337. doi: 10.1016/j.devcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B, Carstens RP. Functional roles of alternative splicing factors in human disease. RNA. 2015;6:311–326. doi: 10.1002/wrna.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B, Park JW, Nakauka-Ddamba A, Bebee TW, Guo Y, Shang X, Lengner CJ, Xing Y, Carstens RP. Multiphasic and Dynamic Changes in Alternative Splicing during Induction of Pluripotency Are Coordinated by Numerous RNA-Binding Proteins. Cell Rep. 2016 Apr 12;15(2):247–55. doi: 10.1016/j.celrep.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, Oostrik J, Lee K, Ahmed ZM, Caylan R, et al. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet. 2008;82:125–38. doi: 10.1016/j.ajhg.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabdoub A, Puligilla C, Jones JM, Fritzsch B, Cheah KSE, Pevny LH, Kelley MW. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc Natl Acad Sci U S A. 2008;105:18396–18401. doi: 10.1073/pnas.0808175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol. 2012;32:1468–1482. doi: 10.1128/MCB.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- Fagoonee S, Bearzi C, Di Cunto F, Clohessy JG, Rizzi R, Reschke M, Tolosano E, Provero P, Pandolfi PP, Silengo L, et al. The RNA binding protein ESRP1 fine-tunes the expression of pluripotency-related factors in mouse embryonic stem cells. PloS One. 2013;8:e72300. doi: 10.1371/journal.pone.0072300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, Stoeckert CJ, Hogenesch JB, Pierce EA. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. 2012;139:245–257. doi: 10.1242/dev.067074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hubbard JJ, Sullivan SK, Mills JA, Hayes BJ, Torok-Storb BJ, Ramakrishnan A. Efficient iPS cell generation from blood using episomes and HDAC inhibitors. J Vis Exp JoVE. 2014:e52009. doi: 10.3791/52009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Müller U. Sensing sound: molecules that orchestrate mechanotransduction by hair cells. Trends Neurosci. 2012;35:220–229. doi: 10.1016/j.tins.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, Stamm S. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, et al. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest. 2000;106:1447–55. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DR, Avraham KB. Hereditary hearing loss: from human mutation to mechanism. Hear Res. 2011;281:3–10. doi: 10.1016/j.heares.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Li M, Feng W, Zhang X, Yang Y, Wang K, Mort M, Cooper DN, Wang Y, Zhou Y, Liu Y. ExonImpact: Prioritizing Pathogenic Alternative Splicing Events. Hum Mutat. 2016 doi: 10.1002/humu.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher H, de Groot JCMJ, van Iperen L, Huisman MA, Frijns JHM, Chuva de Sousa Lopes SM. Development of the stria vascularis and potassium regulation in the human fetal cochlea: Insights into hereditary sensorineural hearing loss. Dev Neurobiol. 2015;75:1219–1240. doi: 10.1002/dneu.22279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SL, Li C, Urness LD. Genetic rescue of Muenke syndrome model hearing loss reveals prolonged FGF-dependent plasticity in cochlear supporting cell fates. Genes Dev. 2013;27:2320–2331. doi: 10.1101/gad.228957.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Swanson GJ. Descriptive and experimental analysis of the epithelial remodellings that control semicircular canal formation in the developing mouse inner ear. Dev Biol. 1993;159:549–558. doi: 10.1006/dbio.1993.1263. [DOI] [PubMed] [Google Scholar]
- Mehta D, Noon SE, Schwartz E, Wilkens A, Bedoukian EC, Scarano I, Crenshaw EB, Krantz ID. Outcomes of evaluation and testing of 660 individuals with hearing loss in a pediatric genetics of hearing loss clinic. Am J Med Genet A. 2016;170:2523–2530. doi: 10.1002/ajmg.a.37855. [DOI] [PubMed] [Google Scholar]
- Mills JA, Wang K, Paluru P, Ying L, Lu L, Galvão AM, Xu D, Yao Y, Sullivan SK, Sullivan LM, et al. Clonal genetic and hematopoietic heterogeneity among human-induced pluripotent stem cell lines. Blood. 2013;122:2047–2051. doi: 10.1182/blood-2013-02-484444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayedi Y, Basch ML, Pacheco NL, Gao SS, Wang R, Harrison W, Xiao N, Oghalai JS, Overbeek PA, Mardon G, et al. The candidate splicing factor Sfswap regulates growth and patterning of inner ear sensory organs. PLoS Genet. 2014;10:e1004055. doi: 10.1371/journal.pgen.1004055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Jahan I, Bonde G, Sun X, Hildebrand MS, Engelhardt JF, Smith RJH, Cornell RA, Fritzsch B, Bánfi B. A mutation in the Srrm4 gene causes alternative splicing defects and deafness in the Bronx waltzer mouse. PLoS Genet. 2012;8:e1002966. doi: 10.1371/journal.pgen.1002966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves J, Uchikawa M, Bigas A, Giraldez F. The prosensory function of Sox2 in the chicken inner ear relies on the direct regulation of Atoh1. PloS One. 2012;7:e30871. doi: 10.1371/journal.pone.0030871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves J, Vachkov I, Giraldez F. Sox2 regulation of hair cell development: incoherence makes sense. Hear Res. 2013;297:20–29. doi: 10.1016/j.heares.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Fauré S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–9. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Nissim S, Allard P, Bandyopadhyay A, Harfe BD, Tabin CJ. Characterization of a novel ectodermal signaling center regulating Tbx2 and Shh in the vertebrate limb. Dev Biol. 2007;304:9–21. doi: 10.1016/j.ydbio.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Jones SM, Johnson KR. Application of Mouse Models to Research in Hearing and Balance. J Assoc Res Otolaryngol JARO. 2016;17:493–523. doi: 10.1007/s10162-016-0589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano T, Xuan S, Kelley MW. Insulin-like growth factor signaling regulates the timing of sensory cell differentiation in the mouse cochlea. J Neurosci. 2011;31:18104–18118. doi: 10.1523/JNEUROSCI.3619-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Patuzzi R. Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hear Res. 2011;277:4–19. doi: 10.1016/j.heares.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, Fritzsch B, Dickson C, Ylikoski J. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. J Neurosci. 2000;20:6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Zhang X, Mantela J, Ornitz DM, Ylikoski J. Fgf9 signaling regulates inner ear morphogenesis through epithelial-mesenchymal interactions. Dev Biol. 2004;273:350–360. doi: 10.1016/j.ydbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Puligilla C, Kelley MW. Dual role for Sox2 in specification of sensory competence and regulation of Atoh1 function. Dev Neurobiol. 2016 doi: 10.1002/dneu.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Blencowe BJ. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron. 2015;87:14–27. doi: 10.1016/j.neuron.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Raviv D, Dror AA, Avraham KB. Hearing loss: a common disorder caused by many rare alleles. Ann N Y Acad Sci. 2010;1214:168–179. doi: 10.1111/j.1749-6632.2010.05868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickheit G, Maier H, Strenzke N, Andreescu CE, De Zeeuw CI, Muenscher A, Zdebik AA, Jentsch TJ. Endocochlear potential depends on Cl- channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008;27:2907–17. doi: 10.1038/emboj.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW, Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314–9. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- Schwander M, Kachar B, Müller U. Review series: The cell biology of hearing. J Cell Biol. 2010;190:9–20. doi: 10.1083/jcb.201001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Miwa T, Wu HH, Levitt P, Ohyama T. Hepatocyte Growth Factor-c-MET Signaling Mediates the Development of Nonsensory Structures of the Mammalian Cochlea and Hearing. J Neurosci. 2016;36:8200–8209. doi: 10.1523/JNEUROSCI.4410-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan-Heggen CM, Smith RJH. Navigating genetic diagnostics in patients with hearing loss. Curr Opin Pediatr. 2016;28:705–712. doi: 10.1097/MOP.0000000000000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SK, Mills JA, Koukouritaki SB, Vo KK, Lyde RB, Paluru P, Zhao G, Zhai L, Sullivan LM, Wang Y, et al. High-level transgene expression in induced pluripotent stem cell-derived megakaryocytes: correction of Glanzmann thrombasthenia. Blood. 2014;123:753–757. doi: 10.1182/blood-2013-10-530725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traunmüller L, Gomez AM, Nguyen TM, Scheiffele P. Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science. 2016;352:982–986. doi: 10.1126/science.aaf2397. [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, et al. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- Urness LD, Wang X, Shibata S, Ohyama T, Mansour SL. Fgf10 is required for specification of non-sensory regions of the cochlear epithelium. Dev Biol. 2015;400:59–71. doi: 10.1016/j.ydbio.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, Barash Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. eLife. 2016;5:e11752. doi: 10.7554/eLife.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrell V, López-Hernández I, Durán Alonso MB, Feijoo-Redondo A, Abello G, Gálvez H, Giráldez F, Lamonerie T, Schimmang T. Otx2 is a target of N-myc and acts as a suppressor of sensory development in the mammalian cochlea. Development. 2015;142:2792–2800. doi: 10.1242/dev.122465. [DOI] [PubMed] [Google Scholar]
- Vuong CK, Black DL, Zheng S. The neurogenetics of alternative splicing. Nat Rev Neurosci. 2016;17:265–281. doi: 10.1038/nrn.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Carstens RP. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT) Semin Cancer Biol. 2012;22:417–427. doi: 10.1016/j.semcancer.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130:3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Wu DK, Kelley MW. Molecular mechanisms of inner ear development. Cold Spring Harb Perspect Biol. 2012;4:a008409. doi: 10.1101/cshperspect.a008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Gan L, Li D, Chen ZY, Zhou L, O’Malley BW, Klein W, Nathans J. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci U S A. 1997;94:9445–9450. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease Science. 2015;347:1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hung LH, Licht T, Kostin S, Looso M, Khrameeva E, Bindereif A, Schneider A, Braun T. RBM24 is a major regulator of muscle-specific alternative splicing. Dev Cell. 2014;31:87–99. doi: 10.1016/j.devcel.2014.08.025. [DOI] [PubMed] [Google Scholar]
- Yang Y, Park JW, Bebee TW, Warzecha CC, Guo Y, Shang X, Xing Y, Carstens RP. Determination of a Comprehensive Alternative Splicing Regulatory Network and Combinatorial Regulation by Key Factors during the Epithelial-to-Mesenchymal Transition. Mol Cell Biol. 2016;36:1704–1719. doi: 10.1128/MCB.00019-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Goodrich LV. Morphological and physiological development of auditory synapses. Hear Res. 2014;311:3–16. doi: 10.1016/j.heares.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci U S A. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell. 2016;166:1147–1162. doi: 10.1016/j.cell.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Esrp1 dependent alternative splicing events in the cochlear epithelium. Related to Fig. 6. (see attached Excel file)