

Summary
Natural killer (NK) cells recognize and kill cancer cells and infected cells by engaging cell surface ligands that are induced preferentially or exclusively on these cells. These ligands are recognized by activating receptors on NK cells, such as NKG2D. In addition to activation by cell surface ligands, the acquisition of optimal effector activity by NK cells is driven in vivo by cytokines and other signals. This review addresses a developing theme in NK cell biology: that NK-activating ligands on cells, and the provision of cytokines and other signals that drive high effector function in NK cells, are driven by abnormalities that arise from transformation or the infected state. The pathways include genomic damage, which causes self DNA to be exposed in the cytosol of affected cells, where it activates the DNA sensor cGAS. The resulting signaling induces NKG2D ligands and also mobilizes NK cell activation. Other key pathways that regulate NKG2D ligands include PI-3 kinase activation, histone acetylation, and the integrated stress response. This review summarizes the roles of these pathways and their relevance in both viral infections and cancer.
Keywords: Natural killer cells, cancer, infectious disease, cell stress, Toll-like Receptors/Pattern Recognition Receptors, cGAS-STING
I. Introduction
A sea change in thinking about the immune response emerged in the late 1980s, with the proposal by Janeway that the immune response is not simply focused on foreign antigens, but also on the context in which a foreign entity is presented to the body (1). Like most immunologists at the time, Janeway was thinking about the adaptive immune response, and addressing the requirements to mount an antibody or T cell response. He pointed out that foreign antigens were not by themselves adequate for a response but instead required a context of infection. Experimentally, responses to purified protein antigens required an associated adjuvant, such as killed bacteria, to initiate the response. He posited that accessory cells for immune responses express “pattern recognition receptors” (PRRs), that recognize features of microbes and provide co-activating signals for T cells and B cells. His insights set the stage for the subsequent discovery of PRRs such as Toll Like receptors, NLRs, cytosolic RNA and DNA sensors and more (2), all of which can play a role in activating adaptive immune responses.
The discovery of PRRs had the additional impact of revealing the general importance of innate immune responses, which had been to a considerable extent ignored before this time. It was quickly appreciated that innate immune responses not only jump-start adaptive immune responses, but preceded the appearance of the adaptive immune response in evolution and function independently of it in many respects (3). PRRs underlie effective immune responses in animals that lack an adaptive immune system, and innate immune responses by themselves play protective roles against infections and cancer even in mammals.
A parallel set of developments in the understanding of innate immunity may ultimately be just as conceptually impactful. These findings emerged from the progression of studies of a previously discovered but then poorly understood component of the immune system, natural killer (NK) cells. Analysis of NK cell recognition of target cells revealed that the immune system responds to cellular abnormalities as well as to foreign pathogens (4). Among the abnormalities recognized by NK cells are molecules regulated by cellular stress pathways, which are often activated in unhealthy, infected or transformed cells.
NK cells were initially identified as cells that kill tumor cells without prior immunization, though it emerged later that they play an important role in controlling certain viral, bacterial and parasitic infections as well (4). Though recent studies suggest NK cells may in some cases exhibit adaptive properties, they are generally considered part of the innate immune system as they do not require the VDJ recombinase that creates highly diverse antigen receptors in T cells and B cells (4). As such, their mechanisms of target cell recognition would be expected to target predictable features. In some cases of recognition of virus-infected cells, NK cells directly engage virus-encoded proteins, an example being the recognition by the Ly49H NK receptor of the m157 protein encoded by mouse cytomegalovirus (MCMV) (5, 6). But direct recognition of microbes by NK cell receptors has only been documented in one or two cases, suggesting that other modes of recognition may be more important. Furthermore, NK cell killing of syngeneic tumors cells, without prior immunization, also suggested that strategies other than direct “antigen” binding often underlie NK cell recognition.
Critical early studies documented that NK cells preferentially kill MHC I-deficient cells, a mode of recognition called “missing self recognition” (7, 8). Even normal, untransformed MHC I deficient cells can be targeted (9, 10). To mediate missing self recognition, NK cells express receptors specific for MHC I molecules, which inhibit NK cell activation (11–14). Hence, loss of MHC I by a target cell relieves inhibition, and enhances NK cell activation. Tumor cells and virus-infected cells often downregulate MHC I, rendering them more susceptible to NK-mediated killing.
More central to the topics of this review, NK cells are also activated by target cells in which stress pathways have been activated or which have undergone malignant transformation. As will be discussed, recognition of stressed cells by NK cells was explicated by the analysis of the NKG2D receptor and its ligands (15–18). The appreciation has since grown that other components of the innate immune system can also target abnormalities resulting from infections or cancer rather than a specific foreign antigen (19). Therefore, events that accompany infection or transformation, rather than pathogens or antigens per se, can be targeted by the immune response. This review will focus on modes of action by NK cells, and in some cases T cells, that exemplify responses to abnormalities, as opposed to responses to pathogens per se.
The NKG2D activating receptor and its ligands
The NKG2D receptor plays an important role in tumor cell recognition. It is a type 2 transmembrane protein, expressed by essentially all NK cells, that pairs in the membrane with the signaling adapter molecule DAP10 (and in mice DAP12) (18). Receptor engagement by ligands expressed on other cells triggers target cell killing and release of cytokines such as interferon γ (IFN-γ) and tumor necrosis factor α (TNFα) by NK cells. NKG2D is also expressed by CD8 T cells and subsets of innate T cells such as NKT cells and gamma/delta T cells, where engagement of the receptor serves an accessory role in T cell function.
NKG2D binds to each of several MHC I-like ligands that are encoded by the host genome, including MICA, MICB, and ULBP1-6 in humans, and RAE-1α ε, H60a-c and MULT1 in mice (20). These NKG2D ligands are poorly expressed in most normal cells, but one or more of them are typically upregulated on the surface of most cancer cells and in cells infected with certain viruses, including herpesviruses such as cytomegaloviruses. As will be discussed below, NKG2D ligands are regulated in part by pathways induced by various forms of stress. Cell surface expression of NKG2D ligands by cells increases their sensitivity to killing by NK cells (15–17, 21–23). Consistent with a role of NKG2D in immunosurveillance of cancer, knockout mice lacking NKG2D exhibit a higher incidence or severity of cancer in several cancer models, including genetically engineered models of spontaneous cancer models such as the TRAMP model of prostate cancer and the Eμ-Myc model of lymphoma (24).
A common thread of NKG2D ligand regulation is that the ligands are induced by pathways indicative of cellular abnormalities (previously reviewed in (20), Table 1, Figure 1). In the present article, we will briefly review some previously described modes of such regulation and provide a somewhat more detailed discussion of stress pathways activated in tumor cells, with an emphasis on examples discovered since the topic was last extensively reviewed.
Table 1.
Regulation of cellular expression of NKG2D ligands by stress pathways
| NKG2D ligands | |||
|---|---|---|---|
| Stress Pathway | Mouse | Human | Citations |
| DNA damage response | RAE-1, H60, MULT1 | MICA/MICB, ULBP1 | (25, 32, 79) |
| Proliferative signals | RAE-1ε | MICA/MICB, ULBP2 | (26, 27) |
| Activated PI-3 kinase | RAE-1 | -- | (60, 77) |
| p53 | -- | ULBP1/ULBP2 | (31) |
| Integrated stress response (ER stress, amino acid starvation) | MULT1 | ULBP1 | (47, 49) |
| Heat shock stress | MULT1 | MICA/MICB | (27–30) |
| Reduced HDAC function | RAE-1 | MICA/MICB ULBP1-3 | (66, 74, 80) |
Figure 1.
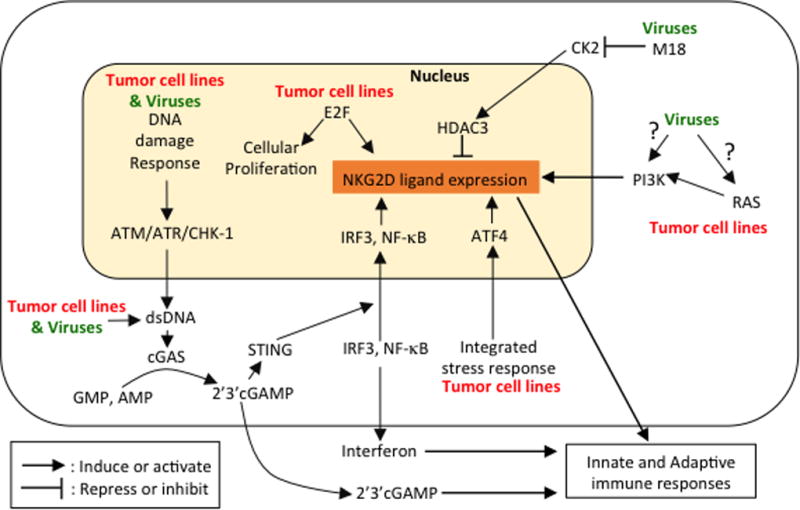
Summary of pathways and signals discussed in this review that induce NK cell activation and/or NKG2D ligand expression in tumor cells and in virus-infected cells. The pathways include the DNA damage response, the cGAS-STING pathway, proliferative signals, PI3-kinase activity, dysregulation of histone acetylation, and the integrated stress response. Most of these pathways to NK cell activation are deployed in both cancer cells and in viral infections, as indicated in the figure and in Table 2.
The six to eight NKG2D ligands expressed in each individual mouse or human show some shared patterns of regulation but most show distinctive mechanisms of regulation as well. An example of a stress pathway that regulates several or all ligands is DNA damage stress (25), discussed in more detail below. In other cases specific ligands are regulated by distinct stress pathways, indicating specialization of ligands with respect to the cues that regulate them. For example, powerful proliferative signals induce expression in fibroblasts of the murine RAE-1ε gene, but not other mouse NKG2D ligand genes, via direct transactivation of the Raet1e gene by E2F transcription factors (26). Proliferative signals also induce expression of human NKG2D ligands MICA/MICB (26, 27) and ULBP2 (26). Heat shock stress, in contrast, induced expression of MICA and MICB proteins on human cells (28) via the direct action of heat shock factor transcription factors (27). On mouse cells, heat shock induced the expression of the MULT1 protein, but the mechanism was distinct as it was mediated by a process that counters the ubiquitin-dependent degradation of MULT1 that occurs in unstressed cells (29, 30). In another example, p53 was shown to amplify expression of human ULBP1 and ULBP2 ligands (31), whereas p53 does not detectably regulate other human NKG2D ligands or mouse NKG2D ligands. These examples suggest that some stress events regulate one or a few specific NKG2D ligands and not others, showing a specialization of regulatory processes inherent to different ligands.
Genomic damage regulates NKG2D ligands
Numerous NKG2D ligands are upregulated in tumor cell lines exposed to DNA damage, or to agents that block DNA replication, which impart DNA replication stress (25, 32). Inhibitor and gene knockdown studies indicated that ligand upregulation in response to DNA damage and replication stress is mediated by the DNA damage response, which is activated by the ATM and ATR protein kinases and propagated by the checkpoint kinases CHK1 and CHK2 (25, 32). Blocking expression of ATR, ATM or CHK1 by shRNA knockdowns of gene expression or with inhibitors prevented ligand upregulation in cells subjected to DNA damage. Furthermore, constitutive expression of NKG2D ligands in several tumor cell lines was diminished by inhibitors of the DNA damage response (25), suggesting that DNA damage in tumor cell lines helps to sustain ligand expression in such cells. Interestingly, DNA damage by itself was inefficient at inducing NKG2D ligands in several normal cell types and some cell lines ((25), and data not shown), suggesting that additional signals work cooperatively with the DNA damage response to amplify expression of NKG2D ligands.
Interestingly, induction of NKG2D ligands imparted by the DNA damage response is at least partly dependent on the DNA sensing pathway, mediated by the cytoplasmic DNA sensor cGAS and downstream mediator STING. The cGAS-STING pathway has been shown to be critical for productive adaptive immune responses to many viruses and other pathogens (33), due to the appearance of cytosolic DNA in infected cells (33–36). In its well defined role, cGAS catalyzes the production of the cyclic dinucleotide 2’3’ cGAMP, which activates the STING protein, which in turn activates transcription factors IRF3 and NF-κB, leading to production of type I interferon and other cytokines (37–39). Knocking down expression of STING and IRF3 in mouse cell lines prevented DNA damage-induced upregulation of the RAE-1 ligand (40). Furthermore, knocking down STING and IRF3 in tumor cell lines that express RAE-1 constitutively resulted in decreased RAE-1 expression (40). These data suggested that induced DNA damage, via the cytosolic DNA sensing pathway, upregulates NKG2D ligands in cell lines that express low amounts of ligands. Furthermore, DNA damage associated with tumorigenesis appears to also activate the cytosolic DNA sensing pathway and sustains constitutive expression of NKG2D ligands in tumor cells (40–42). The mediators that link the cGAS-STING-IRF3 pathway to NKG2D ligand expression on the cell surface remain to be established.
DNA damage correlated with the appearance of DNA in the cytosol (40, 42–45). Inhibitors of the DNA damage kinases ATM and ATR prevented cytosolic DNA accumulation in tumor cells or in cells subjected to DNA damage (40). DNA mismanagement resulting from DNA damage and the DNA damage response may therefore result in the aberrant localization of DNA to the cytosol, activation of the cytosolic DNA sensing pathway and induction of NKG2D ligands. As a whole, these findings suggest that the DNA damage response and the cytosolic DNA sensing pathway work together to upregulate NKG2D ligands, and thus serve to alert NK cells and T cells to a cell’s aberrant status with respect to genomic integrity.
The integrated stress response regulates human ULBP1 expression in tumor cells
The pathways discussed above do not fully account for NKG2D ligand expression by all tumor cells. Blocking these pathways did not prevent ligand expression by certain tumor cell lines for example, suggesting the existence of additional pathways. Hence additional approaches were recently used to define the active pathways in such cells.
One approach examined HAP-1, a human haploid tumor cell line used for genetic screens after mutagenization with retroviral promoter trap vectors (46). HAP-1 cells constitutively express a few NKG2D ligands, including ULBP1. To identify the pathways that support ULBP1 expression, a library of HAP-1 cells that had been infected with the retroviral promoter trap vector to knock out genes at random was selected for cells that expressed ULBP1 poorly at the cell surface. High throughput sequencing of the selected cells, in comparison to unselected cells, revealed a number of genes with retroviral insertions that were enriched in the selected population, including the ATF4 gene (47).
ATF4 was of particular interest as it is a key transcription factor in the “integrated stress response” (ISR), which encompasses the unfolded protein response (also called endoplasmic reticulum (ER) stress) as well as responses of cells to amino acid starvation and heavy metals (48). When ATF4 was knocked out in each of three tumor cell lines, ULBP1 mRNA levels decreased by up to 10-fold, or as little as 1.5 fold, depending on the cell line. These findings suggested that ATF4 is constitutively activated to varying extents in different cell lines, and that it induces ULBP1 expression even when no purposeful stress was applied to the cells. When wildtype cells were further exposed to agents that impart ER stress or result in amino acid starvation, ULBP1 expression increased dramatically in all three cell lines, by 5–10 fold (47). The ATF4 mutant versions of each cell line failed to respond to these stressors, proving the role of ATF4. Finally, ChIP-seq experiments showed that ATF4 binds directly to the ULBP1 promoter, and luciferase reporter experiments showed that ATF4 transactivates the ULBP1 promoter directly (47). Taken together, these results suggested that tumor cells are subject to varying levels of stress that activates the integrated stress response and the ATF4 transcription factor, resulting in significant induction of ULBP1 expression.
Interestingly, ATF4 regulated ULBP1 but had little or no role in regulating other human NKG2D ligands, as tested by flow cytometry or RNA analysis (47). Furthermore, the ChIP-seq experiments showed that of all the functional NKG2D ligand genes, ATF4 binds only to the ULBP1 promoter. Thus, human ULBP1, but not the other human NKG2D ligand genes, is specifically regulated by the integrated stress response.
A recent report investigated the impact of the ER stress response in regulating NKG2D ligands and observed induction of human ULBP1, as well as mouse MULT1 (49). Mouse MULT1 is considered a homolog of human ULBP1. Other mouse NKG2D ligands were not induced. The authors implicate a distinct ER stress-induced transcription factor, CHOP, and not ATF4 in the induction of MULT1. Intestinal inflammation attributed to ER stress in a mouse model was dependent on NK1.1+ cells and not T or B cells, and was prevented by blockade of NKG2D in vivo.
Regulation of ULBP1 by RBM4, a regulator of RNA splicing
The screen in HAP-1 cells for cells with low expression of ULBP1 revealed a second regulator of interest: RBM4 (47). RBM4 regulates RNA processing (50) and (in a different context) translation of pre-formed mRNAs (51). Knockout cells lacking RBM4-expression showed a two-three fold reduction in properly spliced ULBP1 mRNAs and a similar reduction in cell surface expression of ULBP1 (47). The defect in expression was caused by dysfunction in splicing of a large fraction of ULBP1 transcripts, specifically in proper excision of the first intron. The aberrant splice event resulted in inclusion in the transcripts of a large segment of the intron, which contained in-frame stop codons. Interestingly, RBM4-deficiency altered the expression of ULBP1 but other human NKG2D ligands. RBM4 has been reported to support spicing of numerous other genes that suppress the transformed state, by impacting cellular migration, proliferation and survival (50).
Hence, RBM4 appears to suppress the transformed state. Based on our results, RBM4 is predicted to suppress cancer by yet another mechanism: upregulating ULBP1 expression and hence rendering the cells sensitive to cytolysis by NK cells (47). Consistent with a role in suppressing cancer, RBM4 downregulation in tumors is correlated with poor patient survival. Determining whether RBM4 expression or its splicing activity is regulated by stress pathways in cancer cells will be of interest for understanding its role in stress-induced immune responses.
Regulation of NKG2D ligands by PI3-Kinase in viral infections and cancer cells
NKG2D ligands are upregulated in cells undergoing certain infections. The most well studied examples are infections with herpesviruses such as the human and mouse cytomegaloviruses (52, 53). Infections of cells in culture results in a strong induction of several NKG2D ligand genes, as manifested by sharp increases in the corresponding mRNA levels. Interestingly, however, both the mouse and human cytomegaloviruses encode proteins that prevent expression of NKG2D ligands at the cell surface (52–56). Infection with viruses with induced mutations in those evasin genes can lead to virus-induced expression of NKG2D ligands at the cell surface, and greater NK-mediated suppression of the infection in vivo (52, 53, 56). These findings suggest that events associated with viral infection induce expression of NKG2D ligand genes as a protective mechanism, but the virus has evolved evasion mechanisms by encoding proteins that suppress the functional expression of the NKG2D ligand proteins.
The mechanisms whereby viral infections induce expression of NKG2D ligand genes are of interest. Some of the stress pathways already mentioned are known to be activated in virus-infected cells, an example being that rapid viral DNA replication may lead in some cases to the activation of the DNA damage response. Indeed, certain viral infections induce NKG2D ligands by activating the DNA damage response, including HIV (57), Abelson murine leukemia virus (58), and Kaposi’s sarcoma associated herpesvirus (59). It was therefore suspected that the DNA damage response would also account for induction of NKG2D ligands in cells infected with mouse cytomegalovirus (MCMV), as well. Interestingly, however, inhibitors of the DNA damage response had no detectable effect on ligand gene induction in cells infected with mouse cytomegalovirus (MCMV) (60). Indeed, it was found that induction of the mouse NKG2D ligand RAE-1 was associated with virus early gene expression, and did not require viral DNA replication or late gene expression (60).
The PI3 kinase (PI3K) pathway plays a central role in controlling cell growth, survival and cellular transformation, and is activated upon infection with numerous viruses (61). Several isoforms of PI3K with distinct biological roles vary based on the p110 subunit incorporated in the complex. A role for the PI3K pathway in the induction of RAE-1 after MCMV infection was shown using inhibitors. Specifically, inhibition of the p110α PI3 Kinase subunit prevented RAE-1 induction in MCMV infected cells, whereas inhibitors of p110β, δ or γ did not (60). Many viruses activate PI3K to enhance viral fitness (62, 63), suggesting a rationale for targeting this pathway for regulation of NKG2D ligands.
Notably, the p110α gene is commonly mutated to a constitutively active form in cancer cells (64, 65), suggesting the possibility that PI-3K also plays a role in NKG2D ligand expression by tumor cells. Indeed, it was found that steady state expression of RAE-1 in three tumor cell lines was sharply reduced by treating the cells with inhibitors of p110α, but not with inhibitors of p110β, δ or γ (60). The p110α inhibitors had a similar effect on expression of MULT1, another NKG2D ligand. Interestingly, RAE-1 NKG2D ligands are also induced in cells expressing the activated RAS oncogene, which is known to activate PI3-kinase. Inhibition of PI3K activity reduced RAE-1 expression in cells with activated RAS, cementing the conclusion that PI3K induces RAE-1 in cancer cells. Together, these data suggest that the p110α PI3K isoform plays a significant role in cell surface expression of NKG2D ligands induced by viral infection or constitutively expressed by cancer cell lines.
Regulation of NKG2D ligands by histone acetylation
Though activated PI3K plays a necessary role in RAE-1 expression in MCMV-infected cells, it is not sufficient, as shown by activating the pathway in uninfected cells. This finding set up a search for the additional required signals or pathways. Screens identified an MCMV deletion mutant that failed to induce RAE-1 after infection (66). Targeted gene disruptions ultimately demonstrated that a single viral gene, m18, was essential for induction of RAE-1 after infection. Transfection experiments showed that m18 was also sufficient for induction of RAE-1, and for increasing the sensitivity of the infected cells to being killed by NK cells. Somewhat surprisingly, therefore, RAE-1 induction was mediated primarily by a single viral gene that is not essential for viral replication in cultured cells. The gene likely plays a role in supporting virus infections in vivo.
Analysis showed that m18 stimulated transcription of a luciferase reporter driven by the RAE-1 gene promoter (66). Deletions of the promoter identified a short site necessary for m18-induced transcription, which contained a predicted Sp transcription factor binding site. The site was constitutively bound in vivo by one Sp family transcription factor, Sp3, as shown by chromatin precipitation assays. Expression in cells of a dominant negative variant of an Sp transcription factor lacking a transactivation domain reduced the expression of RAE-1 in cells also transfected with m18. Furthermore, mithramycin, an inhibitor of Sp factor binding to DNA, strongly blocked RAE-1 induction by m18. Together, these findings suggested Sp3 is required for m18-mediated expression of RAE-1..
Sp3 was associated with the RAE-1 promoter independent of m18 expression, suggesting its activity, rather than its association with the promoter, was regulated by m18. Sp factors may associate with histone deacetylases (HDACs), which regulate the activity of genes to which Sp3 is bound. Moreover, treatment of cell lines with HDAC inhibitors induced the expression of NKG2D ligand genes, including RAE-1 (25). Analysis indicated that m18 inhibits HDAC activity indirectly, by binding to casein kinase IIb and blocking phosphorylation of HDAC3, resulting in increased global histone H3 acetylation in cells and expression of RAE-1 (66). Interestingly, a number of herpesviruses inhibit HDACs, and such inhibition is often crucial to viral fitness (67, 68). In the case of m18 of MCMV, for example, viral mutants lacking functional m18 showed defects in viral titers in the salivary glands (66), which is predicted to impair viral transmission to other animals. Taken together, these collective results suggest that m18 is employed by the virus to inhibit HDACs and increase the expression of various host genes that favor viral fitness in vivo, and that NKG2D ligand genes are similarly induced to alert host immune cells to this danger. The expression of the NKG2D ligand genes would favor the elimination of the infected cells by NK cells, unless the virus also harbors evasin genes that prevent NKG2D ligand expression.
Induction of immune responses by transformed cells: role of the cytosolic DNA sensing pathway
Earlier in this review was a discussion of one mechanism of how tumorigenesis, via the DNA damage response and the cGAS-STING pathway, induces expression of NKG2D ligands. To reiterate the inferred events, DNA damage incurred in transformed cells results in access of DNA in the cytosol, which activates the DNA sensing enzyme cGAS. cGAS synthesizes the cyclic dinucleotide cGAMP, which activates STING, and consequently TBK1 and IRF3. Via an unknown mechanism, IRF3 induces expression of NKG2D ligands. This mechanism represents a cell intrinsic process in tumor cells to render them sensitive to immune control.
Interestingly, the DNA sensing pathway also acts to activate T cells and NK cells against tumors by a mechanism independent of its effects on NKG2D ligands. The initial finding was that mice with a homozygous STING mutation are highly impaired in generating CD8 T cell responses against transferred immunogenic tumors, such as B16 melanoma cells transduced to express epitopes recognized by CD8 T cells (69). Mice deficient for IRF3, which is downstream of STING, showed a similar defect. Both types of mice exhibited much weaker T cell priming, and failed to reject the tumors, whereas WT mice rejected these immunogenic tumors. In mice injected with tumor cells 1 day before, IFN-β mRNA was elevated in dendritic cells (DC) isolated from the tumors compared to DCs in the spleen or lymph nodes, and this was abrogated in STING-deficient mice. These data suggested that DCs in the tumor environment are induced via the STING pathway to produce IFN-β (and presumably other cytokines), which promotes T cell priming.
We have recently observed that the cGAS-STING pathway is similarly necessary for induction of spontaneous NK cell responses against cancer. NK cells have long been considered constitutively active. While it was known that innate immune responses via Toll-like receptors amplifies NK cell responses to viruses (70), it was not previously appreciated that innate responses are important for triggering anti-tumor activity of NK cells. We observed that spontaneous rejection by syngeneic mice of NK-sensitive tumors, including B16-BL6 melanoma cells and RMA-S lymphoma cells, is severely impaired in mice with a homozygous deficiency of STING (Marcus et al, submitted). The rejection of these tumors is not related to upregulation of NKG2D ligands on tumor cells, because these cell lines are unable to express NKG2D ligands. These findings indicate that STING in the cells of the recipient animal mediates activation of NK cells. Finally, it must be emphasized that in these studies, the defect in STING is in host cells as opposed to tumor cells, whereas STING acted in tumor cells to induce NKG2D ligands.
The findings that CD8 T cell and NK cell responses against tumors depend on host STING indicates that the transferred tumor cells somehow activate STING in host cells. It was proposed that DNA from tumor cells is transferred to the cytosol of host cells, where it activates cGAS, resulting in cGAMP production, STING activation and cytokine secretion (69). In contrast, our results suggest that cGAS acts in tumor cells, and cGAMP is transferred from tumor cells to host cells where it activates host cell STING (Marcus et al, submitted).
These findings suggest that genomic dysregulation in tumor cells is the danger signal that triggers the immune response, by activating cGAMP production in tumor cells, which is transferred to host cells to activate the immune response. A similar transfer mechanism may operate in cells that are exposed to DNA damage from environmental sources or from therapeutic cancer drugs, or in viral infections, where viral DNA activates cGAS activity in infected stromal cells.
A possible advantage for the host of this type of mechanism is that it is challenging for viruses or tumor cells to evolve products or mutations to evade it. In the process outlined here, STING functions in non-transformed or uninfected cells, where it cannot be easily inactivated by mutation and selection, or by a viral evasin. Inactivation or inhibition of cGAS in infected or transformed cells would, in contrast, abrogate the mechanism. However, the selective pressure for evolving such a mechanism, while potent, may be inefficient, since cGAMP produced in the infected or transformed cells ultimately acts in a cell extrinsic fashion. For example, in a tumor mass, a few cGAS-deficient variant tumor cells might not have a selective advantage, since neighboring cGAS-proficient cells could still produce cGAMP and activate an immune response that eliminates both types of tumor cells equally. On the other hand, several reports suggest that cGAS or STING function is suppressed in certain cancer cells, in some cases by epigenetic mechanisms (71, 72). It is possible that such inactivation arises primarily to counter the cell intrinsic actions of the cGAS STING pathway in infected or transformed cells, such as induction of NKG2D ligand expression, discussed earlier.
Commonalities of NK cell activation in viral infections and cancer
A fascinating aspect of the issues reviewed here is that the mechanisms of NKG2D ligand expression and NK cell activation uncovered in cancer cells also apply in some viral infections, and vice versa. Some of these similarities are listed in Table 2 and Figure 1, and will be summarized here.
Table 2.
Shared stress pathways and cellular aberrations that induce NKG2D ligands or NK responses in cancer and in infections
| Citations | ||
|---|---|---|
| Stress Pathway/aberration | Cancer | Infection |
| DNA damage response | (25, 79) | (57–59) |
| Proliferative signals | (26, 27) | ? |
| Activated PI-3 kinase | (60, 77) | (60) |
| p53 | (31) | ? |
| Integrated stress response (ER stress, amino acid starvation) | (47, 49) | ? |
| Heat shock stress | (27–30) | ? |
| Reduced HDAC function | (25, 74) | (66) |
| Cytosolic DNA sensor in immune cell activation | (69, 81), (Marcus et al, submitted) | (33–36) |
An obvious example of immune regulation that is shared by the immune responses to viruses and cancer is the DNA sensing pathway. As already mentioned, STING or cGAS-deficient mice show strong defects in T cell responses to viruses (33–36). Though less well studied, NK cell responses to viruses are also affected (A. Marcus, unpublished data). Spontaneous T cell responses and NK responses to cancer are similarly defective as a result of cGAS-STING deficiency ((69), Marcus, Vance and Raulet, submitted).
With respect to induction of NKG2D ligands, the role of the DNA damage response was first shown in cancer cells, but was subsequently extended to cells infected with various viruses, including human immunodeficiency virus (HIV) (57, 73), Abelson murine leukemia virus (A-MuLV) (58), and Kaposi sarcoma associated herpesvirus (KSHV) (59). These viruses inflict DNA damage or DNA replication stress, or activate the DNA damage response, by different mechanisms.
Histone acetylation was implicated in RAE-1 expression in viral infections (66), and it was separately shown that inhibitors of histone deacetylase enzymes (HDACs), which increase histone acetylation, induce NKG2D ligands in cultured cell lines (25, 74). Indeed, class I HDACs are elevated in cancers (75) leading to the proposal that they might suppress NKG2D ligands and aid in evasion of immunity by tumor cells (74). Importantly, HDAC inhibitors are being tested as cancer drugs (76), and while they suppress cancer by multiple mechanisms, their capacity to induce NKG2D ligands and enhance susceptibility of tumor cells to NK cells might be a contributory factor.
Another example concerns the role of PI3-Kinase in upregulation of NKG2D ligands, which was found in viral infections (60), but also applies for cancer cells (60, 77). The PI3K pathway is frequently mutated in cancer conferring constitutive activation that favors tumor cells survival and growth (64). Dysregulation of PI3K signaling may therefore be considered a shared pattern of pathogenesis of certain infected cells and cancer cells. Accordingly, PI3K inhibitors are being tested as possible cancer therapy drugs (78), but may have the unwelcome effect of reducing NKG2D ligands in tumor cells.
Conclusions
The research described herein emphasizes how cellular dysregulation, including the activation of various stress pathways, is tied to activation of NK cell responses against tumors and infected cells (Figure 1, Table 1, Table 2). The abnormalities targeted by NK cells are generally beneficial to a pathogen or to the transformed state, or are necessarily associated with it, examples being DNA damage in cancer, PI3K activation in cancer or infections or HDAC modulation in infections.
The findings emphasize that despite the fact that NK cells recognize predictable features of infected cells or cancer cells, they are not simply constitutively active surveyors of the body. Instead, dysregulated and unhealthy cells provide cues that activate NK cells to deploy their full activity. Furthermore, the expression of molecules on tumor cells and infected cells that can be targeted by activated NK cells, such as NKG2D ligands, are themselves regulated by cues emanating from the aforementioned stress pathways. While this review focuses primarily on NK cells, the same stress pathways impact the adaptive immune response. For example, NKG2D ligands induced by stress pathways engage NKG2D on T cells and provide costimulatory signals for these cells. Furthermore, genomic damage in tumor cells activates T cell responses as well as NK cell responses against tumors via the cGAS-STING pathway. These findings support the case that important components of the immune system respond not just to foreign entities such as antigens or pathogen-associated molecular patterns, but rather to cellular abnormalities, via the action of stress pathways.
Acknowledgments
We thank our colleagues and collaborators their contributions to the studies and ideas presented here. Our research was supported by NIH/NCI grants R01-AI113041 (DHR), R01-CA093678 (DHR), R01-AI039642 (DHR), and research grants from Aduro Biotech and Innate Pharma, SAS.
Footnotes
Conflict of Interest Statement
DHR is a co-founder of Dragonfly Therapeutics, and serves on the Scientific Advisory Boards of Innate Pharma, Aduro Biotech and Ignite Immmunotherapy; he has a financial interest in all four companies and received research support from a program supported by Aduro Biotech and Innate Pharma, and may benefit from commercialization of the results of this research. LC received research support from a program supported by Aduro Biotech. AM has no conflict of interest.
References
- 1.Janeway CA., Jr Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb Symp Quant Biol. 1989;LIV:1–13. [PubMed] [Google Scholar]
- 2.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 4.Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MG, Dokun AO, Heusel JW, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 6.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 7.Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 8.Ljunggren HG, Karre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 9.Bix M, Liao NS, Zijlstra M, Loring J, Jaenisch R, Raulet D. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature. 1991;349:329–331. doi: 10.1038/349329a0. [DOI] [PubMed] [Google Scholar]
- 10.Liao N, Bix M, Zijlstra M, Jaenisch R, Raulet D. MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science. 1991;253:199–202. doi: 10.1126/science.1853205. [DOI] [PubMed] [Google Scholar]
- 11.Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly-49+ IL-2 activated natural killer cells. Nature. 1992;358:66–70. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- 12.Colonna M, Samaridis J. Cloning of immunoglobulin-superfamily members associated with HLA-C and HLA-B recognition by human natural killer cells. Science. 1995;268:405–408. doi: 10.1126/science.7716543. [DOI] [PubMed] [Google Scholar]
- 13.Wagtmann N, Biassoni R, Cantoni C, et al. Molecular clones of the p58 natural killer cell receptor reveal Ig-related molecules with diversity in both the extra- and intra-cellular domains. Immunity. 1995;2:439–449. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 14.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 15.Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 16.Cerwenka A, Bakker ABH, McClanahan T, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 17.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 18.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 19.Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Song Y, Bakker AB, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 22.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerra N, Tan YX, Joncker NT, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–580. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med. 2012;209:2409–2422. doi: 10.1084/jem.20120565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol. 2007;178:961–969. doi: 10.4049/jimmunol.178.2.961. [DOI] [PubMed] [Google Scholar]
- 28.Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nice TJ, Coscoy L, Raulet DH. Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J Exp Med. 2009;206:287–298. doi: 10.1084/jem.20081335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nice TJ, Deng W, Coscoy L, Raulet DH. Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J Immunol. 2010;185:5369–5376. doi: 10.4049/jimmunol.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK Cells Are Alerted to Induction of p53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2. Cancer Res. 2011;71:5998–6009. doi: 10.1158/0008-5472.CAN-10-3211. [DOI] [PubMed] [Google Scholar]
- 32.Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood. 2007;110:606–615. doi: 10.1182/blood-2006-10-052720. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science. 2013 doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao D, Wu J, Wu YT, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konno H, Barber GN. The STING controlled cytosolic-DNA activated innate immune pathway and microbial disease. Microbes and infection / Institut Pasteur. 2014;16:998–1001. doi: 10.1016/j.micinf.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 39.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 40.Lam AR, Le Bert N, Ho SS, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74:2193–2203. doi: 10.1158/0008-5472.CAN-13-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Bert N, Lam AR, Ho SS, Shen YJ, Liu MM, Gasser S. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology. 2014;3:e29259. doi: 10.4161/onci.29259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho SS, Zhang WY, Tan NY, et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity. 2016;44:1177–1189. doi: 10.1016/j.immuni.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Shen YJ, Le Bert N, Chitre AA, et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 2015;11:460–473. doi: 10.1016/j.celrep.2015.03.041. [DOI] [PubMed] [Google Scholar]
- 46.Carette JE, Guimaraes CP, Varadarajan M, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 47.Gowen BG, Chim B, Marceau CD, et al. A forward genetic screen reveals novel independent regulators of ULBP1, an activating ligand for natural killer cells. Elife. 2015;4:e08474. doi: 10.7554/eLife.08474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 49.Hosomi S, Grootjans J, Tschurtschenthaler M, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J Exp Med. 2017 doi: 10.1084/jem.20162041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Chen D, Qian H, et al. The Splicing Factor RBM4 Controls Apoptosis, Proliferation, and Migration to Suppress Tumor Progression. Cancer Cell. 2014;26:374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin JC, Hsu M, Tarn WY. Cell stress modulates the function of splicing regulatory protein RBM4 in translation control. Proc Natl Acad Sci U S A. 2007;104:2235–2240. doi: 10.1073/pnas.0611015104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lodoen M, Ogasawara K, Hamerman JA, et al. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rolle A, Mousavi-Jazi M, Eriksson M, et al. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J Immunol. 2003;171:902–908. doi: 10.4049/jimmunol.171.2.902. [DOI] [PubMed] [Google Scholar]
- 54.Cosman D, Müllberg J, Sutherland CL, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 55.Wu J, Chalupny NJ, Manley TJ, Riddell SR, Cosman D, Spies T. Intracellular Retention of the MHC Class I-Related Chain B Ligand of NKG2D by the Human Cytomegalovirus UL16 Glycoprotein. J Immunol. 2003;170:4196–4200. doi: 10.4049/jimmunol.170.8.4196. [DOI] [PubMed] [Google Scholar]
- 56.Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med. 2004;200:1075–1081. doi: 10.1084/jem.20040583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward J, Davis Z, DeHart J, et al. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009;5:e1000613. doi: 10.1371/journal.ppat.1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gourzi P, Leonova T, Papavasiliou FN. A Role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24:779–786. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 59.Bekerman E, Jeon D, Ardolino M, Coscoy L. A role for host activation-induced cytidine deaminase in innate immune defense against KSHV. PLoS Pathog. 2013;9:e1003748. doi: 10.1371/journal.ppat.1003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tokuyama M, Lorin C, Delebecque F, Jung H, Raulet DH, Coscoy L. Expression of the RAE-1 Family of Stimulatory NK-Cell Ligands Requires Activation of the PI3K Pathway during Viral Infection and Transformation. PLoS Pathog. 2011;7:e1002265. doi: 10.1371/journal.ppat.1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yurochko AD. Human cytomegalovirus modulation of signal transduction. Curr Top Microbiol Immunol. 2008;325:205–220. doi: 10.1007/978-3-540-77349-8_12. [DOI] [PubMed] [Google Scholar]
- 62.Cooray S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. The Journal of general virology. 2004;85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 63.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nature reviews Microbiology. 2008;6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 65.Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. British journal of cancer. 2006;94:455–459. doi: 10.1038/sj.bjc.6602970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greene TT, Tokuyama M, Knudsen GM, et al. A Herpesviral induction of RAE-1 NKG2D ligand expression occurs through release of HDAC mediated repression. Elife. 2016;5:e14749. doi: 10.7554/eLife.14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nevels M, Paulus C, Shenk T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl Acad Sci U S A. 2004;101:17234–17239. doi: 10.1073/pnas.0407933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adib-Conquy M, Scott-Algara D, Cavaillon JM, Souza-Fonseca-Guimaraes F. TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol Cell Biol. 2014;92:256–262. doi: 10.1038/icb.2013.99. [DOI] [PubMed] [Google Scholar]
- 71.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016;14:282–297. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood. 2009;115:1354–1363. doi: 10.1182/blood-2009-08-237370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009;28:2370–2382. doi: 10.1038/onc.2009.117. [DOI] [PubMed] [Google Scholar]
- 75.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 76.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu XV, Ho SS, Tan JJ, Kamran N, Gasser S. Ras activation induces expression of Raet1 family NK receptor ligands. J Immunol. 2012;189:1826–1834. doi: 10.4049/jimmunol.1200965. [DOI] [PubMed] [Google Scholar]
- 78.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]