

Summary
Gain-of-function mutations in histone 3 (H3) variants are found in a substantial proportion of pediatric high-grade gliomas (pHGG), often in association with TP53 loss and PDGFRA amplification. Here, we describe a somatic mouse model wherein H3.3K27M and Trp53 loss alone are sufficient for neoplastic transformation if introduced in utero. H3.3K27M-driven lesions are clonal, H3K27me3-depleted, Olig2-positive, highly proliferative and diffusely spreading, thus recapitulating hallmark molecular and histopathological features of pHGG. Addition of wild-type PDGFRA decreases latency and increases tumor invasion, while ATRX knockdown associates with more circumscribed tumors. H3.3K27M-tumor cells serially engraft in recipient mice and preliminary drug screening reveals mutation-specific vulnerabilities. Overall, we provide a faithful H3.3K27M-pHGG model which enables insights into oncohistone pathogenesis and investigation of future therapies.
Keywords: Oncohistone, neurodevelopment, pediatric high-grade glioma, clonal, mosaic, invasion, PDGFRA
Graphical Abstract
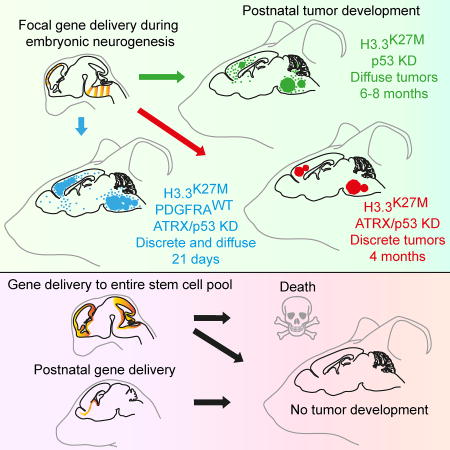
Pathania et al. create a model of pediatric high-grade glioma with H3.3K27M/TP53-mutation. Tumors occur only when alterations are introduced during a specific window of mouse development, and levels of PDGFRA and ATRX modulate tumor phenotype. They use the model to identify mutation-specific vulnerabilities.
Introduction
Pediatric High-Grade Glioma (pHGG) is a devastating disease representing the primary cause of cancer death in children (Ostrom et al., 2015; Ostrom et al., 2016). Despite striking molecular differences in their pathogenesis, current therapeutic approaches still follow recommendations for adult HGG and involve radiation therapy and other cytotoxic regimens (Faury et al., 2007; MacDonald et al., 2011; Paugh et al., 2010; Qu et al., 2010). Our group and others uncovered recurrent, somatic, gain-of-function mutations in H3 variants in pHGG (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012). Two mutations lead to specific amino acid changes in key residues of H3 variants and in specific brain regions. Indeed, more than 90% of pHGG in the brain midline, including the particularly deadly diffuse intrinsic pontine gliomas (DIPG), carry lysine 27 to methionine (K27M) mutations in an H3 variant (H3F3A/H3.3, HIST1H3B or HIST1H3C/H3.1 and rarely HIST2H3B/H3.2), while mutations of glycine 34 to arginine or valine (G34R/V) in H3.3 occur in 36% of pHGG in the cerebral hemispheres (Fontebasso et al., 2013; Fontebasso et al., 2014; Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Sturm et al., 2012). These “oncohistones” are found almost exclusively in pHGG and are mutually exclusive with the recurrent mutations in isocitrate dehydrogenase enzymes (IDH) identified in young adult gliomas (Yan et al., 2009), and the epigenetic heterogeneity seen in adult glioma (Gallo et al., 2015).
H3.3 is the predominant H3 variant mutated in pHGG and is a replication-independent histone variant loaded onto chromatin throughout the cell cycle predominantly by two distinct chaperone complexes, formed by either histone cell cycle regulator (HIRA)/Calcineurin binding protein 1 (CABIN1)/Ubinuclein (UBN1) or the death domain associated protein (DAXX)/α-thalassemia/mental retardation syndrome X-linked (ATRX) (Michod et al., 2012; Yuen and Knoepfler, 2013). H3.3 dynamically regulates gene transcription through both its post-translational modifications (PTMs) as well as its turnover within nucleosomes in a variety of cell types (Maze et al., 2014; Maze et al., 2015). While the function of H3.3G34R/V is still unclear, H3.3K27M has been shown to have gain-of-function activity inhibiting the Polycomb Repressive Complex 2 (PRC2) (Bender et al., 2013; Lewis et al., 2013), which in turn leads to a global reduction in H3K27me3 levels in primary tumors and tumor-derived primary cells. Rescue of H3K27me3 levels via pharmacological inhibition of H3K27me3 demethylases has anti-tumor effects against primary H3.3K27M pHGG cells, suggesting addiction to this mutation within established tumors (Hashizume et al., 2014). H3.3K27M pHGG cells are also sensitive to inhibition of histone deacetylases and menin, indicating the involvement of additional epigenetic changes in sustaining tumor growth (Funato et al., 2014; Grasso et al., 2015).
H3.3K27M-tumors and IDH-mutant gliomas possess co-occurring mutations in the same genes (Fontebasso et al., 2014; Khuong-Quang et al., 2012; Nikbakht et al., 2016; Wu et al., 2014). These include alterations affecting the p53 pathway in ~87% of H3.3K27M tumors (Fontebasso and Jabado, 2015; Fontebasso et al., 2014; Nikbakht et al., 2016) and loss-of-function mutations in ATRX, which occur with increasing frequency in older patients (Fontebasso et al., 2014; Khuong-Quang et al., 2012; Nikbakht et al., 2016). Moreover, amplifications and rare mutations of the platelet-derived growth factor receptor alpha gene (PDGFRA) often co-occur with H3.3K27M and TP53 mutations, indicating a potential role of this receptor in tumorigenesis (Flavahan et al., 2016; Khuong-Quang et al., 2012; Korshunov et al., 2015; Paugh et al., 2013).
H3.3 mutations are likely tied to the action of specific developmental programs. Attempts to model H3.3K27M-driven HGG formation in vivo via genetic recombination in neural progenitor/stem cells (NPCs) in the postnatal brain failed to demonstrate tumor-driving activity (Lewis et al., 2013), suggesting that the mutation event leading to HGG may actually occur during embryogenesis. A recent study reported that transduction of human embryonic stem cell-derived NPCs with H3.3K27M, p53 shRNA and constitutively active, mutant PDGFRAD842V led to neoplastic transformation, but only via in vitro passaging over several months (Funato et al., 2014). Furthermore, when transplanted into recipient immunocompromised mice, these genetically modified NPCs gave rise exclusively to low-grade glioma (LGG), not high-grade as seen in patients. Other attempts to model H3.3K27M-DIPG in vivo have required constitutive expression of the PDGF-B ligand, which is not present in pHGG and can induce tumors even with wild-type (WT) H3.3, suggesting that PDGFRA signalling drives rapid tumorigenesis and H3.3K27M is secondary in these models (Cordero et al., 2017; Hennika et al., 2017).
The lack of a truly representative in vivo model for mutant H3.3K27M-driven HGG limits our ability to dissect the underlying mechanisms instructing this archetypal epigenetic cancer and impedes our ability to develop therapies that have the best chance of success in the clinic. The main goal of the present study is to generate a mouse model recapitulating the histological and molecular characteristics of human H3.3-mutated pHGG.
Results
H3.3K27M and Trp53 loss induce diffuse tumorigenesis in both hindbrain and forebrain
To generate a mouse model of oncohistone pathogenesis, we employed several approaches. First, we knocked H3.3K27M into the H3f3a locus in mouse ES cells using Zinc Finger Nuclease technology (ZFN). However, this approach caused a severe embryonic phenotype wherein zygotes did not grow past the 4-cell stage (Figure S1A). Targeted expression of H3.3K27M downstream from either the Nes or Gfap promoter, which are active in both NPCs during development and the postnatal brain, also failed to induce tumors in mice up to 18 months old, even when the H3.3K27M transgene was expressed in combination with Trp53 loss (Figure S1B and data not shown). We also attempted a focal, somatic approach, based on in vivo electroporation of transposable vectors using the Sleeping Beauty (SB) system. Intracranial delivery and electroporation of SB-transposable H3.3K27M-encoding vectors together with ATRX/p53 knockdown constructs in neonatal mice generated small proliferative lesions around the injection site but failed to induce tumors (Figure S1C–E), in accordance with previous reports based on the Replication-Competent ASLV long terminal repeat (LTR) with a Splice acceptor (RCAS) system (Lewis et al., 2013).
Based on these findings, we reasoned that the cell of origin of pHGG is likely an embryonic precursor cell. To test this hypothesis, we performed in utero electroporation (IUE) of piggyBac transposon-based vectors (Chen et al., 2014; Chen and LoTurco, 2012) at E12.5-E13.5 to permanently overexpress GFP (empty vector), H3.3WT or H3.3K27M in NPCs and their progeny. As IUE carried out during neural development specifically targets transgenes to NPCs lining the ventricles (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001), only NPCs integrate the mutation-carrying transposons. After electroporation, the non-integrating transposase is rapidly diluted and the integrated mutation-carrying transgenes are stably inherited by successive progenies of the electroporated NPCs. Given that H3.3K27M tumors are predominantly found in the brain midline and pons, we used piggyBac IUE to target NPCs in the lower rhombic lip of the developing hindbrain, which contributes to the pontine nuclei as development proceeds (Figure 1A). We tested whether two drivers, transposable H3.3K27M and Trp53 loss using a non-transposable gRNA/Cas9 targeting the Trp53 locus (K27M-P, 2-hit), were sufficient to induce tumorigenesis in either hindbrain or cortex. Hindbrain IUE was performed at E12.5 and forebrain IUE was performed at E13.5, in order to introduce mutant H3.3K27M at the peak of cortical or pontine neurogenesis, respectively (Finlay and Darlington, 1995; Shinohara et al., 2013). Neoplastic transformation occurred with 100% penetrance in both hindbrain (Figure 1B and 1C) and cortex (Figure 1D), 6 and 8 months following surgery, respectively (Table S1). No tumors formed with empty vector GFP (EV), H3.3WT or H3.3G34R in combination with Trp53 loss (Table S1). Specifically, GFP+ K27M-P cells proliferated ectopically in both anatomical compartments, inducing both focal and diffuse tumors in the hindbrain and mainly diffuse tumors in the cortex (Ki67+; Figure 1B–D; Table S1). The hindbrain tumors also spread into the cerebellum and fourth ventricle (Figure 1C). Analysis of expressed variants by RNA-Seq showed no evidence of spontaneous mutations or CRISPR/Cas9 off-target effects that could underlie transformation (Table S2). Spontaneous non-recurrent events were detected in 11 genes that did not include oncogenes or tumor suppressors (Table S2). These data demonstrate that two genetic hits alone, H3.3K27M and Trp53 loss, can induce tumorigenesis in both supra- and infratentorial locations, suggesting that NPCs can be transformed by these genetic changes independent of anatomic location.
Figure 1. H3.3K27M expression and Trp53 loss are able to induce tumorigenesis in both cortex and hindbrain.

(A) Schematic describing the in utero electroporation strategy used to deliver piggyBac transposable-H3.3K27M and Trp53 CRISPR/Cas9 (K27M-P) into lower rhombic lip NPCs in vivo. (B) Coronal section prepared from a K27M-P hindbrain 6 months after electroporation showing immunofluorescent detection of GFP, Ki67 and DAPI. Orange arrowhead indicates GFP+Ki67+ cells in the pons, shown at higher magnification on the right. Diffusely spreading GFP+Ki67+ cells are also seen migrating dorsally within the hindbrain. Scale bars represent 100 µm (left) and 50 µm (right). (C) Histopathological analysis of a hindbrain K27M-P tumor harboring Ki67+, Olig2+ and GFAP+ cells. Scale bars represent 0.5 µm (left) and 75 µm (right). (D) Coronal view of cortical K27M-P driven tumorigenesis. 8 months following surgery GFP+Ki67+ cells are seen in cortical, ventral and contralateral locations (red arrows). Scale bars represent 1.5 mm. See also Figure S1 and Table S1.
Tumorigenesis induced by H3.3K27M in combination with Trp53 loss and ATRX knockdown
As H3.3-mutated pHGG is often associated with loss-of-function mutations in the chromatin-associated factor and H3.3 co-chaperone ATRX, we decided to model this additional genetic change using IUE. Based on similar transformation potential in both hindbrain and cortex and the important technical challenges of brainstem electroporations, we subsequently focused on forebrain electroporations at E13.5. We concomitantly introduced non-transposable gRNA/Cas9 targeting the Trp53 locus and a transposable shRNA against ATRX together with H3.3K27M (EV-AP/WT-AP/K27M-AP; 3-hit; Figure 2A). In GFP+ cells sorted and expanded from embryonic cortices 72 hr following IUE, efficient expression of HA-tagged H3.3WT, H3.3G34R or H3.3K27M and concomitant knockdown of both p53 and ATRX protein levels could be observed (Figure 2B).
Figure 2. H3.3K27M expression and knockdown of ATRX and p53 in embryonic NPCs leads to tumorigenesis in postnatal animals.

(A) Schematic describing the in utero electroporation strategy used to deliver piggyBac transposable-empty vector, H3.3WT or H3.3K27M along with ATRX shRNA and Trp53 CRISPR/Cas9 (EV/WT/K27M-AP) into cortical NPCs in vivo. (B) Validation of H3.3K27M–HA expression and knockdown of ATRX and p53 in ex vivo NPCs sorted and expanded 3 days following electroporation. (C) Coronal sections of EV-AP, WT-AP and two biological replicates of K27M-AP brains prepared 4 months following electroporation showing immunofluorescent detection of GFP, Ki67 and DAPI. GFP+Ki67+ cells in the cortex are indicated with an orange arrowhead, and contralaterally or ventrally migrating GFP+Ki67+ cells are indicated by the red arrows. Scale bars represent 1 mm. (D) Immunofluorescent overlap between GFP+, Ki67+ and Nestin+ cells in EV-AP, WT-AP and K27M-AP brains (two separate biological replicates shown for K27M-AP) 4 months following electroporation. Scale bars represent 50 µm. (E) Quantification of GFP+Ki67+ cells in EV-AP, WT-AP and K27M-AP electroporated brains at 4 months. Data are represented as mean ± SEM. (F) K27M-AP tumor appearance and immunofluorescence analysis, as depicted, at 9 months following electroporation. Scale bars represent 1.5 mm (left) and 50 µm (right). (G) Histology of K27M-AP tumors in comparison to a WT-AP electroporated brain. Scale bars represent 1 mm (top) and 100 µm (bottom). *p<0.05, **p<0.01, ****p<0.0001. See also Figure S2; Tables S1 and S2.
Following IUE, mice were allowed to deliver normally and were assessed at postnatal day 21 (P21), 4 months and 8–10 months of age. In H3.3K27M ATRX/p53-KD mice (K27M-AP), Ki67+ cells were marginally increased by P21. By 4 months, K27M-AP mice developed neoplastic lesions in the cortex and striatum (Figure 2C, orange arrowheads; Figure 2D) with 100% penetrance. These proliferative lesions were either focal (Figure 2C; left K27M-AP panel) or diffuse (Figure 2C; right K27M-AP panel). In both cases, small clusters of GFP+Ki67+ cells were also seen migrating along the corpus callosum into the contralateral cortex (Figure 2C, red arrows), mirroring the invasive nature of human pHGG. 20% of GFP+ cells were Ki67+ and Nestin+ in ectopic, non-neurogenic locations, thereby demonstrating stem cell-like characteristics (Figure 2D and 2E) and neoplastic transformation. In contrast, we did not observe GFP+Ki67+ cells outside the proliferative regions of the subventricular zone (SVZ) and dentate gyrus (DG) in EV-AP, WT-AP or G34R–AP (Figure S2A and S2B; Table S1). We also noted that WT-AP mice displayed a premature differentiation effect, with fewer GFP+ cells in the cortex and a larger neuron-to-astrocyte ratio at 4 months compared to EV-AP (Figure 2C and Figure S2A–C). In the olfactory bulb (OB) at 4 months, number of GFP+ neurons was decreased in WT-AP or K27M-AP compared to EV-AP (Figure S2D), correlating with altered neurogenic properties of NPCs in the SVZ upon H3.3 overexpression or mutation. By 9 months, the smaller proliferative lesions in K27M-AP cortex and striatum had progressed to fully developed HGG, with mice displaying neurological signs requiring euthanasia. These lesions were positive for Ki67, Olig2, Nestin and HA; and were depleted of K27me3 signal (Figure 2F). Tumors in these animals were also ATRX-depleted, as expected, and showed increased proliferation and contralateral infiltration, thus resembling H3.3K27M pHGG, (Figure 2G). Taken together, these data indicate that in utero delivery of H3.3K27M, ATRX knockdown and Trp53 loss can induce glioma by 4 months.
PDGFRAWT overexpression shortens latency of tumorigenesis
PDGFRA plays a major role in early brain development and whilst activating mutations are rare, amplifications of the PDGFRA locus are often seen in pediatric glioma in the context of H3.3 mutations (Fontebasso et al., 2014; Khuong-Quang et al., 2012; Paugh et al., 2013; Wu et al., 2014). To determine whether overexpression of PDGFRA further modifies the tumorigenic properties of the K27M-AP (3 hit) model, we overexpressed a WT PDGFRA cDNA using piggyBac IUE (EV/WT/K27M-APP; 4-hit; Figure 3A and 3B). Overexpression of PDGFRA at E13.5 in all conditions greatly increased the numbers of neurons, astrocytes and oligodendrocytes in the electroporated region at both P21 and 4 months, when compared to the K27M-AP/3-hit model (Figure 3C vs Figure 2C). It also increased the thickness of the electroporated neocortex as well as the size of the ipsilateral hemisphere in relation to the contralateral hemisphere (Figure 3C). In several cases, this effect was so extensive that the lateral ventricle became occluded with supernumerary striatal cells on the electroporated side (Figure 3C). At P21, ectopic proliferation was only seen in K27M-APP-electroporated animals (Figure 3C), showing a significantly decreased latency compared to the K27M-AP model where ectopic proliferation was only seen at 4 months (Figure 3C vs Figure 2C). By 4 months, 100% of K27M-APP animals developed extensive proliferative lesions migrating into the contralateral hemisphere (Figure 3C; Table S1). 15% of cells in these lesions were Ki67+GFP+ (Figure 3D and 3E), and as expected, had increased levels of PDGFRA (Figure 3D). At 4 months, EV-APP and WT-APP brains also showed low-penetrance tumors (10–25%; Table S1), but penetrance did not increase up to 8–10 months. This is in contrast to K27M-APP which showed 100% tumor penetrance at 4 and 8–10 months (Table S1). By 9 months, K27M-APP tumors were significantly progressed, with very high numbers of Ki67+Nestin+ cells and multiple necrotic foci (Figure 3F and 3G). These lesions were intermittently positive for GFAP, but were universally depleted of MBP and K27me3 signal (Figure 3G). Histology revealed K27M+ and Ki67+ cells colocalizing with GFP (Figure 3H). Thus, addition of WT PDGFRA shortens the latency of tumorigenesis and acts synergistically with H3.3K27M to transform NPCs in vivo.
Figure 3. Addition of PDGFRAWT overexpression to H3.3K27M and ATRX/p53 KD results in shorter latency of tumorigenesis.

(A) Schematic describing the in utero electroporation strategy used to deliver piggyBac transposable-empty vector, H3.3WT or H3.3K27M along with PDGFRAWT, ATRX shRNA and Trp53 CRISPR/Cas9 (EV/WT/K27M-APP) into cortical NPCs in vivo. (B) Validation of PDGFRAWT overexpression in ex vivo NPCs sorted and expanded 3 days following electroporation. (C) Coronal sections of EV-APP, WT-APP and K27M-APP brains prepared 21 days and 4 months following electroporation showing immunofluorescent detection of GFP, TdTomato, Ki67 and DAPI. Scale bars represent 1.5 mm (multi-color panels) and 200 µm (black-and-white panels). (D) Immunofluorescent overlap between GFP+, Ki67+ and PDGFRA in WT-APP and K27M-APP brains. Scale bars represent 50 µm. (E) Quantification of GFP+Ki67+ cells in EV-APP, WT-APP and K27M-APP electroporated brains at 4 months. Data are represented as mean ± SEM. (F) Comparison of Ki67 and Nestin immunofluorescence levels in WT-APP and K27M-APP at 9 months following surgery. Scale bars represent 50 µm. (G) Left: low magnification view of K27M-APP tumors at 9 months. Right: immunofluorescence analysis of K27M-APP tumors, as depicted, 4 months following surgery. Scale bars represent 1.5 mm (left) and 50 µm (right). (H) Histology of K27M-APP tumors. Scale bars represent 100 µm. *p<0.05, **p<0.01, ****p<0.0001. See also Figure S3 and Table S1.
We then investigated PDGFRAD842V activating mutations previously shown to cooperate with H3.3K27M and p53 knockdown to induce transformation in human NPCs in vitro (Funato et al., 2014). In contrast to WT PDGFRA, overexpression of PDGFRAD842V led to 100% penetrant, high-grade lesions also in WT-APP and EV-APP mice (Figure S3A and S3B, Table S1), suggesting that, unlike WT PDGFRA, PDGFRAD842V does not require H3.3K27M to drive tumorigenesis in vivo (Cordero et al., 2017; Hennika et al., 2017; Paugh et al., 2013).
H3.3K27M tumor cells can serially engraft in recipient mice and can be used for targeted drug screening
Next, we set out to determine the engrafting capacity of H3.3K27M-driven tumor cells upon orthotopic transplantation as further proof of their neoplastic nature. K27M-P, K27M-AP and K27M-APP tumor cells were injected into the striatum of NOD-SCID mice, and brains were collected 45–70 days after transplantation. Histopathological analysis revealed that K27M-P, K27M-AP and K27M-APP cells formed 100% diffuse tumors with extensive spreading through striatal, dorsolateral and dorsomedial cortical regions, similar to what is observed in pHGG (Figure 4A and 4B) (Buczkowicz et al., 2014; Louis et al., 2016; Nikbakht et al., 2016).
Figure 4. Allografted K27M-P, K27M-AP and K27M-APP tumor cells generate diffusely spreading Ki67+ lesions in NOD/SCID mice, demonstrating their transformed status.

(A) Low magnification view of K27M-AP tumor cell detection using GFP immunohistochemistry in recipient cortex. Scale bars represent 1 mm. (B) Higher magnification views of GFP immunohistochemistry showing regions of dorsolateral and dorsomedial frontal cortex in K27M-P, K27M-AP and K27M-APP tumor cell-injected mice. Scale bars represent 100 µm. (C) A small molecule library of 430 kinase inhibitors was applied to WT-AP, WT-APP, K27M-AP tumor and K27M-APP tumor cells, as well as EV control cells. Changes in viability are shown as a screen-wide image of z-scores (left) and a normal Q-Q plot (right). The normalized percent inhibition method was used to compare z-scores of WT-AP and K27M-AP responses, as well as the z-scores of WT-APP and K27M-APP responses. (D) Quantification of the effect of vacquinol-1 (left) and Akti-1/2 (right) on viability of K27M-AP and K27M-APP tumor cells, respectively, at a range of concentrations. Data are represented as mean ± SEM. *p<0.05, **p<0.01, ****p<0.0001. See also Figure S4 and Table S3.
To demonstrate the utility of our model for high-throughput screening approaches, we performed a proof-of-principle comparison of the sensitivity of K27M-AP and K27M-APP tumor cells to a library of 430 small molecule kinase inhibitors (Figure 4C and 4D; Figure S4; Table S3). This library was chosen as kinases are actionable and several have been implicated in pHGG. Results revealed a specific vulnerability of K27M-AP tumor cells to Vacquinol-1, compared to WT-AP cells (Figure 4D and 4E; Table S3; z-score 0.98). In addition, K27M-APP were specifically vulnerable to Akt inhibition using Akti-1/2, compared to WT-APP cells (Figure 4D and 4E; Table S3; z-score 0.96). These preliminary results demonstrate that our model can be used for screening approaches.
H3.3K27M-mediated tumorigenesis in mice is clonal, recapitulates features of invasive human pHGG
To address the clonal evolution and/or the homogeneity of the mutational landscape of H3.3K27M tumors, we performed whole exome sequencing (WES) of 10 biopsies from a K27M-AP tumor, spatially distributed along the rostral-caudal axis (Figure S5A). Single nucleotide variants (SNVs) exhibited very high concordance across all samples (≥ 98% concordance between any pair of biopsies, Table S4). Clonality analysis revealed 7 distinct clusters of non-synonymous, exonic SNVs, and 6 of 7 clusters exhibit a consistent cellular prevalence across all biopsies (Figure S5B). The cellular prevalence of the remaining minor cluster varied between 25% and 40%, with no variant belonging to this cluster overrepresented in any of the samples (Figure S5A and S5B). Altogether, these results indicate that H3.3K27M-driven tumors are clonal and derive from the originating mutations introduced during IUE.
Given that H3K27me3 levels are drastically decreased in human H3.3K27M gliomas due to the inhibitory effect of this mutant histone on EZH2 (Lewis et al., 2013), we analyzed this mark in 2-, 3- and 4-hit H3.3K27M cells. Ex vivo, H3K27me3 levels were reduced in preneoplastic NPCs and further reduced in cells isolated from K27M-P, K27M-AP and K27M-APP tumors (Figure 5A). In vivo, H3K27me3 levels were also depleted in K27M-AP and K27M-APP cells compared to WT-AP and WT-APP cells at 4 months and 21 days, respectively (Figure 5B and 5C). Furthermore, this depletion was progressive, as in 10-month K27M-AP tumors there was further depletion of this repressive PTM compared to tumors at 4 months (K27M-AP vs K27M-AP tumor 10 mo.; Figure 5C). We also observed that K27M-AP and K27M-APP cells preferentially migrated along CD31+ blood vessels (Figure 5D and 5E).
Figure 5. K27M-AP and K27M-APP tumor cells have reduced K27me3 levels and migrate along blood vessels.

(A) Acid-extracted histone preparations made from EV-AP, WT-AP and K27M-AP preneoplastic cells and K27M-P, K27M-AP and K27M-APP tumor cells were probed for K27me3, HA, K27ac, K36me3 and total H3 levels. (B) Left: representative images of K27me3 immunofluorescence in WT-AP and K27M-AP cells at 4 months following electroporation. Right: representative images of K27me3 immunofluorescence in WT-APP and K27M-APP cells at 21 days following electroporation. Cell bodies are highlighted within circles. Scale bars represent 50 µm. (C) Quantification of K27me3 immunofluorescence signal intensity in WT-AP vs K27M-AP and WT-APP vs K27M-APP. Data are represented as mean ± SEM. (D) Immunofluorescent detection of K27M-AP tumor cells, K27me3 and CD31+ blood vessels (indicated by white arrows). Scale bars represent 50 µm. (E) Top panel: immunofluorescence of K27M-APP tumor cells expressing GFP and CD31+ blood vessels (indicated by white arrows). Bottom panel: immunofluorescence of K27me3 and HA signal (indicated by white arrows) in GFP+ K27M-APP tumor cells. Scale bars represent 50 µm. *p<0.05, **p<0.01, ****p<0.0001. See also Figure S5 and Table S4.
K27M-AP and K27M-APP tumors were also significantly enriched in cells expressing Olig2, similar to human DIPG (Figure 6A and 6B). The increase in Olig2 expression was associated with extensive migration away from the electroporation site in K27M-APP cells, as more Olig2+GFP+ cells were seen in the contralateral cortex compared to the ipsilateral one (Figure 6B). In contrast, this contralateral migration or invasion never occurred in EV-APP or WT-APP at 4 months. We confirmed the increased invasiveness of K27M-APP vs K27M-AP tumor cells in vitro (Figure 6C). The extent of GFP+ cell migration was also increased in vivo in K27M-APP brains compared to EV-APP and WT-APP (Figure S6A and S6B). Migration into the OB was also increased in K27M-APP (Figure S6C). Furthermore, ATRX knockdown conditions (K27M-AP and K27M-APP) generated increased numbers of focal or localized cortical tumors compared to K27M-P (0% K27M-P vs 65% K27M-AP; Table S1 and Figure 6D, p<0.0001). These data demonstrate that K27M-AP and K27M-APP tumors progressively lose H3K27me3 genome-wide as transformation proceeds, that K27M-APP tumor cells are intrinsically more invasive than K27M-AP cells, and that addition of ATRX knockdown to H3.3K27M and Trp53 loss can reduce the diffuse nature of the resulting lesions.
Figure 6. Olig2 levels are increased in K27M-AP and K27M-APP tumor cells.

(A) Representative images and quantification of Olig2+GFP+ cells in EV-AP, WT-AP and K27M-AP brains at 4 months following electroporation. Scale bars represent 50 µm. Data are represented as mean ± SEM. (B) Representative images and quantification of Olig2+GFP+ cells in ipsilateral and contralateral cortical regions of K27M-APP brains at 4 months following electroporation. PDGFRA immunofluorescence is also shown. Scale bars represent 50 µm. Data are represented as mean ± SEM. (C) Relative invasion index of K27M-APP vs K27M-AP tumor cells. Data are represented as mean ± SEM. (D) Quantification of the incidence of focal tumors correlating with ATRX loss. Chi-squared test. *p<0.05, **p<0.01, ****p<0.0001. See also Figure S6 and Table S1.
Molecular cross-species analysis shows that H3.3K27M mouse tumors recapitulate human H3K27M pHGG and uncovers disease-associated pathways
To identify specific genes and pathways deregulated in preneoplastic and tumor models, we performed genome-wide transcriptional profiling (RNA-seq) of GFP+ cells sorted and expanded ex vivo from embryonic cortices of electroporated mice at 72 hr (preneoplastic) and 5–9 months (tumor) post-IUE. Principal component analysis (PCA) and t-Distributed Neighbor Embedding (t-SNE) revealed that H3.3K27M induced global changes in gene expression, with K27M cells segregating from controls (Figure 7A and 7B). We observed a progression from the untransformed to the transformed state, with large transcriptional changes evident already at the preneoplastic stage. Cells harboring three hits (K27M-AP) showed a variable degree of transformation (Figure S7A), while cells harboring all four hits (K27M-APP) consistently clustered close to H3.3K27M-transformed tumor cells (Figure S7A and Figure 7A and 7B), in line with the shorter latency observed for PDGFRA expressing tumors. Elevated levels of PDGFRA were confirmed in RNA-seq data (Figure S7B).
Figure 7. H3.3K27M-dependent transformation induces transcriptomic changes that are recapitulated in human tumors.

(A) Principal component analysis (PCA) of murine samples based on transcriptome-wide expression level data is shown. Curved arrow indicates a progression in global transcriptomic patterns from the untransformed to the transformed state. Control: EV-AP/EV-APP. WT: WT-AP. K27M: K27M-AP/K27M-APP. K27M–T: K27M-AP T / K27M-APP T. (B) Two-dimensional visualization of the expression dataset was performed by t-SNE on the PCA space, using the first 50 PCs. 5,000 iterations were performed with perplexity=6 and theta=0.5. (C) t-SNE visualization of human normal brain, H3.3WT pediatric High Grade Glioma (pHGG) and H3.3K27M pHGG samples based on the expression of human orthologs of the murine K27M–T. 10,000 iterations were performed on the PCA space (first 50 PCs) with perplexity=2, theta=0. (D) Top: bubble plot summarizing the number of overlapping genes between the signatures derived from murine and human tumors. Bottom: Random expectations for the number of overlapping genes (N) with pGBM signatures were derived by iteratively sampling N genes from the human genome and computing the overlap with human tumor signatures (blue histograms, 10,000 iterations). Red vertical lines correspond to the observed overlaps (K27M–T signature). P-values indicate the probability of observing these overlaps by chance based on random expectations. (E) Gene expression changes (log2 FC) of the human orthologs of pathway-specific gene subsets extracted from the murine K27M–T signature using functional annotation in H3.3K27M pHGG with respect to normal brain are shown. Red: K27M–T upregulated genes. Blue: K27M–T downregulated genes. Filed circles: statistically significant genes. Open circles: non-significant genes. Solid line: median log2 FC of significant genes. See also Figure S7; Tables S5, S6 and S7.
To directly assess the disease relevance of our mouse models, we derived gene signatures based on the human orthologs of differentially expressed genes in the mouse models (Table S5). Our results show that the global H3.3K27M-dependent transcriptional changes were recapitulated in human tumors (Figure 7). PCA and t-SNE of human samples based on the expression of human orthologs of the murine K27M tumor signature (K27M–T) clearly segregated normal brain, H3.3WT and H3.3K27M pHGG (Figure 7C, Figure S7C–E). We then focused on individual genes to identify those that were consistently deregulated in mouse and human samples (preserved direction of change; statistical significance with adjusted p<0.05). Figure 7D shows that human tumors bearing the K27M mutation had the largest overlap with murine K27M tumor signatures (significant overlap p<0.0001; 2.6 times larger than with WT pHGG; Table S6), indicating that the genes identified in our mouse models specifically reflect K27M human tumor biology and not merely a general oncogenic transformation.
To validate this result and identify specific pathways recapitulated in pHGG, we performed functional annotation of the murine K27M–T signature (Table S7), and assessed the concordance of specific sets of genes in pHGG through single-sample gene set enrichment analysis (ssGSEA) (Barbie et al., 2009) and through direct assessment of expression changes in H3.3K27M pHGG relative to normal brain. Among the deregulated genes consistent with human pHGG, we identified groups of genes related to chromatin developmental pathways including Wnt and Hox genes, DNA binding, extracellular matrix proteins and signal peptides (Figure 7E, Figure S7F, Table S7).
In human H3.3K27M pHGG, H3K27me3 levels are significantly reduced. We used ChIP-seq to assess K27me3 levels in H3.3K27M-expressing tumor and preneoplastic cells, H3.3WT and EV cells. We observed marked overall downregulation of H3K27me3 in H3.3K27M samples (Figure 8A), confirming the reduction seen by immunofluorescence (Figure 5C) and mirroring findings in human H3.3K27M pHGG .
Figure 8. K27M–induced alteration of genome-wide H3K27me3 deposition is captured by the mouse model.

(A) ChIP-Rx Ratio: ratio of sequencing reads mapping to mouse genome and drosophila genome, normalized by input. (B) Heatmaps illustrating H3K27me3 levels at transcription start sites (TSSs). H3K27me3 levels in regions of 10Kb surrounding TSSs (in rows) were normalized locally on a per-sample basis (in columns), and genes were subsequently clustered by k-means (k=12) based on average H3K27me3 levels in each sample. The five gene clusters most recapitulative of human K27M tumor H3K27me3 loss are shown, alongside the magnitude of changes in expression of their constituent genes (right). Only significant gene expression changes are reported, in the form of log2 fold-change in K27M–T relative to baseline controls. H3.3 WT: EV-ctl, EV-PDGFRA. K27M: K27M-AP T /K27M-APP T. (C) Human genes were classified as in (B) into four tiers (k=4). K27me3 ChIP-Seq data was obtained from (Bender et al., 2013). A lower k was used since the lower coverage of this data set prevented a high resolution clustering of genes. Note that the number of annotated genes for mouse and human differ, and thus the heatmaps are not scaled to proportion. (D) Pairwise similarity matrix between murine (rows) and human (columns) gene clusters. Similarity was measured as the proportion of mouse genes with human orthologs present in each human cluster. (E) H3K27me3 levels found at the loci of selected genes showing increased (Pdgfra, Satb2) or decreased (Wnt6, Wnt10a, Tacr3) mark deposition. Coverage is normalized across all samples. See also Figure S8 and Table S8.
This decrease affected the promoter region of a large proportion of the genes, with a clear depletion of the mark surrounding their transcription start sites (TSS). Clustering of genes based on K27me3 patterns revealed that loss of the mark at the TSS is associated with upregulation of expression (clusters M1-M3; Figure 8B and Figure S8A and S8B), consistent with the repressive role of this mark. On the other hand, genes with lower levels of K27me3 in WT samples (clusters M5-M7, M10-M12; Figure S8A and S8B) or situated at K27me3 boundaries (cluster M4) showed variable changes in expression, consistent with the idea of K27me3-independent regulation. Importantly, we observed high concordance in K27me3 deposition patterns between our mouse model and human tumors, particularly for genes under the regulatory control of PRC2, i.e. with high H3K27me3 in H3.3WT samples. To assess this, we classified human genes into four tiers based on H3K27me3 ChIP-Seq signal from a previously published dataset (Bender et al., 2013) (Figure 8C) and computed the proportion of overlap of each murine cluster with each of these four tiers (Figure 8D). Cluster M1, which has the highest level of K27me3, is strongly conserved, with 78% of genes falling into human cluster H1. This cluster contains the top homeobox genes deregulated in our dataset (i.e. ALX4, IRX2, SIX4, POU4F1, ISL2), as well as some of the Wnt pathway genes (i.e. WNT6 and WNT10A). M5, on the other hand, is depleted in K27me3 and shows 69% overlap with H3-H4. A general agreement is observed for all clusters with clear K27me3 patterns (Figure 8D). As expected, clusters with less defined patterns show lower level of conservation (Figure S8C).
Finally, we found rare instances of increased deposition of K27me3 despite the known inhibitory effects of the K27M mutation on the SET domain of EZH2. We detected a significant increase in K27me3 levels at the PDGFRA promoter in samples overexpressing PDGFRA (Figure 8E). This indicates that a dynamic inhibitory feedback loop can be activated in response to exogenous overexpression of PDGFRA so as to repress its endogenous expression despite EZH2-inhibiting H3.3K27M. Active deposition of the H3K27me3 mark at selected sites remains possible in H3.3K27M cells (Figure 8E; Figure S8D), possibly by residual EZH2 activity, as recently suggested (Mohammad et al., 2017), or by continued activity of EZH1-containing complexes (Shen et al., 2008).
Discussion
Despite landmark discoveries in the genetics of rare cancers over the past 35 years, subsequent development of effective, targeted therapeutics has been rare. This is in part due to a lack of preclinical in vivo models that fully recapitulate the genetic and histopathological features of human tumors, thereby preventing the identification and validation of high-confidence treatment strategies. For pHGG in particular, there are currently no in vivo models simulating H3.3K27M-driven brain tumorigenesis. In this study, we generate a preclinical model of H3.3K27M oncohistone-driven neoplastic transformation, from which cells can be engrafted in recipient animals and that recapitulates histologic features of human pHGG.
Tumorigenesis occurs only when H3.3K27M is introduced during development, as very early expression of this oncogene is lethal to mouse postzygotic cells whereas postnatal NPCs seem refractory to its oncogenic effects. Therefore, our data support an embryonic origin of H3.3K27M–driven tumors in mice, suggesting that the H3.3K27M mutation arises early in actively dividing neural precursors during CNS development where it produces a tumorigenic event by disrupting normal neurogenesis. This is supported by anecdotal findings of “pre”-DIPG tumors in very young children prior to clinical manifestations (Wright et al., 2015) and explains why initial attempts to model K27M–driven tumorigenesis postnatally using both Sleeping Beauty (our work) and the RCAS systems (Lewis et al., 2013) were unsuccessful. Furthermore, our data implicate a specific window during NPC lineage specification for tumorigenesis to occur, as classical transgenic approaches using both the Nes and Gfap promoters in combination with Trp53 loss failed to induce tumors. Since we used a ubiquitous promoter to drive expression of H3.3K27M embryonically, the precise cell within the NPC lineage that can initiate tumorigenesis remains to be discovered. Interestingly, we show that NPCs in the forebrain and hindbrain are equally susceptible to neoplastic transformation upon concomitant introduction of H3.3K27M and its associated genetic changes. The tumors generated are clonal and derive from those stem cells expressing mutant H3.3 that were targeted during development using IUE. Hence, they elucidate a possible mechanism by which H3.3K27M tumors initiate in human pHGG as well, i.e. during gestation.
Our data suggest that the H3.3K27M requires only one additional genetic hit in the form of Trp53 loss to initiate tumorigenesis. While the process of transformation begins during development, it progresses slowly, taking 4 months in mice to advance to high-grade lesions. By 9 months, these lesions are spread throughout the rostrocaudal axis of the neocortex. It should be noted that although this model is the most representative of pHGG it still takes 4 months in mice. However, this incubation period is in accordance with most, if not all, other autochthonous models of medulloblastoma or adult glioma and may represent a fundamental aspect of brain cancer modeling in mice (Alcantara Llaguno et al., 2009; Hill et al., 2015).
Addition of ATRX knockdown to H3.3K27M expression and Trp53 loss promotes focal or localized tumor formation, suggesting that ATRX loss diminishes the invasive potential of tumors. We have reported elsewhere that a large proportion of thalamic H3.3K27M tumors, and the majority of cortical H3.3G34R/V tumors, are more focal at initial presentation and display loss of ATRX (Fontebasso et al., 2014). This is in contrast to H3.3K27M DIPG where ATRX loss is rare (less than 10%) and subclonal (Nikbakht et al., 2016), potentially in accordance with our model. Furthermore, in adult HGG, ATRX loss associates with IDH mutations and TP53 loss and is also associated with improved prognosis (Pekmezci et al., 2017; Wiestler et al., 2013), potentially due to better resection of focal tumors during surgery. On the other hand, the addition of WT PDGFRA overexpression significantly decreases tumor latency while drastically promoting invasion when co-expressed with H3.3K27M.
H3.3K27M-bearing mouse tumors induced using piggyBac IUE recapitulate many aspects of human H3.3K27M pHGG. This includes 1) the characteristic invasive nature of these tumors, 2) migration along blood vessels, 3) colonization of the contralateral hemisphere, 4) upregulation of Olig2, an established diagnostic marker for H3.3K27M pHGG (Sturm et al., 2012), which positively correlates with the extent of invasion of transformed cells in vivo, and 5) scattered GFAP expression is in tumor samples.
Molecular analysis of mouse datasets also showed that our model closely recapitulates human H3.3K27M mutant pHGG. Cross-species analysis of transcriptomic signatures and H3K27me3 deposition in mice and in human pHGG shows that H3.3K27M-dependent transformation induces epigenetic and transcriptomic changes that are recapitulated in human tumors. Notably, our data indicate that de novo deposition of this mark is still possible despite the strong inhibitory effects of the H3.3K27M mutation on EZH2 methyltransferase activity. Indeed, H3K27me3 is deposited at the endogenous PDGFRA promoter to decrease PDGFRA expression in response to overexpression of ectopic PDGFRA. This negative feedback loop demonstrates a previously underappreciated dynamic deposition of H3K27me3 even in H3.3K27M mutant cells. The mechanisms behind this effect are unknown and are the subject of active investigations.
In conclusion, we describe a faithful preclinical model of H3.3K27M-driven pHGG and provide a tool for studying molecular mechanisms underlying the transforming function of H3.3K27M. Our findings will enable the elucidation of developmental and tissue homeostasis mechanisms altered by H3.3K27M in pHGG, and promote discoveries targeting this devastating pediatric brain cancer.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Paolo Salomoni (p.salomoni@ucl.ac.uk).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were housed, bred and subjected to listed procedures according to institutional, Animal Welfare and Ethical Review Body (AWERB) and UK Home Office guidelines (Project licence 70/8240, 70/7428 and 80/2325). Female and male C57BL/6J mice were used for in utero electroporations. Female and male NOD SCID mice were used for allotransplantation experiments. Mice were monitored daily for neurological symptoms of brain tumors: weight loss, epilepsy, altered gait, lethargy; and euthanized immediately when recommended by veterinary and biological services staff members.
METHOD DETAILS
Vector Construction
The piggyBac donor and helper vector system was used to transduce NPCs in utero as described previously (Chen and LoTurco, 2012). CAG-PBase and PBCAG-GFP were a kind gift from F. Chen and J. LoTurco. PBCAG-H3.3WTHA-2A–GFP and PBCAG-H3.3K27MHA-2A–GFP were generated by linearising PBCAG-GFP using EcoRI/NotI and using HIFI Gibson assembly (NEB) to insert cDNA encoding C-terminal HA-tagged Drosophila His3.3A (a highly conserved ortholog of mammalian H3f3a) followed by a P2A self-cleaving linker and the EGFP sequence. K27M mutants were generated using Quikchange Lightning site-directed mutagenesis (Agilent). The stop codon was removed following H3.3HA to allow for polycistronic mRNA translation. Two ATRX shRNA vectors with equivalent knockdown efficiencies were obtained by cloning the following sequences into the short hairpin RNA construct PBmU6pro (kind gift from J. Loturco), cut with BbsI/XbaI: 5’-TTCATTTACATTCTCATCCGTG-3’; 5’-TCATTTACATTCTCATCCG-3’. A control (ctl) shRNA vector was constructed using a scrambled shRNA sequence (5’-CCAACTCTTGTTTCAGCCG-3’) lacking homology to any known mouse and human genes as described previously (Singh et al., 2010). CRISPR/Cas9 pX330 vectors containing negative control (5’-GCGACCAATACGCGAACGTC-3’) or Trp53-targeting gRNA (5’-ACAGCCATCACCTCACTGCA-3’) sequences were a kind gift from J. Gronych (Zuckermann et al., 2015). PBCAG-PDGFRA was constructed by removing EGFP from PBCAG-GFP using AgeI/NotI to remove problematic restriction sites, blunt-ligating and then inserting PCR-amplified murine mPDGFRA from pcDNA5FRT-EF-Pdgfralpha-EGFPN (L. Pederson, Addgene #66787) into the KPnI/XmaI-digested vector. The PBCAG-PDGFRA-PQR-tdTomato vector was constructed by inserting a self-cleaving PQR sequence into AgeI/NotI-digested PBCAG-GFP, then inserting the tdTomato-encoding sequence (M. Davidson and R. Tsien, Addgene #54642) downstream from the PQR using NheI/NotI and finally inserting the mPDGFRA sequence upstream from the PQR using KPnI/XmaI. The stop codon was removed following mPDGFRA to allow for polycistronic mRNA translation.
In Utero Electroporation
In utero electroporation was performed using sterile technique on isoflurane/oxygen-anesthetized pregnant females at E12.5 (hindbrain) or E13.5 (cortex) as described previously (Nitarska et al., 2016). Analgesic support was also provided pre-emptively (subcutaneous delivery of Vetergesic and Carpofen at 0.1 mg/kg and 5 mg/kg, respectively). Uterine horns were exposed through a 1 cm incision and individual embryos were digitally manipulated into the correct orientation for intraventricular injection. Pulled borosilicate capillaries were loaded with endotoxin-free DNA and Fast Green dye (0.05%, Sigma) for visualisation, and a microinjector (Eppendorf) was used to inject either the lateral or fourth ventricles with the DNA-dye mixture. 3–5 plasmids were injected simultaneously, each at a final concentration of 2 µg/µl and 1–2 µl of total solution was injected per embryo. DNA was electroporated into cortical neural progenitors using 5 mm tweezertrodes (BTX), or into lower rhombic lip progenitors using 3 mm tweezertrodes, applying 5 square pulses at 35 V, 50 ms each with 950 ms intervals. The embryos were returned into the abdominal cavity, the muscle and skin were sutured and the animal was monitored until fully recovered from the procedure.
Ex vivo NPC Isolation and Culture
Animals were euthanized by CO2 exposure and/or cervical dislocation and cortices were rapidly dissected in ice-cold dissociation medium containing: 20 mM glucose, 81.8 mM Na2SO4, 30 mM K2SO4, 5.8 mM MgCl2, 250 µM CaCl2, 1 mM HEPES, 160 µM NaOH, 0.8 mM kynurenic acid, 50 µM D-APV, 100 U/mL penicillin, 100 µg/mL streptomycin, 5 µg/ml plasmocin and 100 µg/ml primocin. GFP+ regions were microdissected under an epifluorescence stereomicroscope in the same medium and enzymatically digested into a single-cell suspension using the Papain Dissociation System (Worthington Biochemicals) followed by mechanical trituration through a series of fire-polished Pasteur pipettes. The dissociated cell solution was then separated on an OptiPrep density gradient to remove debris and GFP+TdTomato+ cells were sorted using a FACSAria III or LSRFortessa X-20 (BD Biosciences) into Hibernate-E media (Life Technologies) containing 2% B-27, 2 mM Glutamax, 0.8 mM kynurenic acid, 50 µM D-APV, 100 U/mL penicillin, 100 µg/mL streptomycin, 5 µg/ml plasmocin and 100 µg/ml primocin. Sorted cells were plated into NeuroCult NSC proliferation media (STEMCELL Technologies) containing 10 ng/ml of both epidermal growth factor (EGF, Peprotech) and basic fibroblast growth factor (bFGF, Peprotech) in laminin-coated culture vessels (Sigma).
Western Blot Analyses
Cells were scraped on ice in lysis buffer containing 50 mM Tris (pH 7.5), 0.5% Triton-X, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF and 1X Halt protease and phosphatase inhibitor (Thermo Fisher Scientific). Lysates were rotated for 30 min, sonicated briefly, cleared by centrifugation at 13000 rpm for 10 min, quantified using the BCA assay and then boiled in Laemmli buffer for 3 min. SDS-PAGE was carried out with 20 µg of protein per sample, transferred to nitrocellulose and then blocked in 5% skimmed milk or BSA dissolved in PBS-Tween or TBS-Tween (0.1% Tween), respectively, for 1 hr at RT. Membranes were incubated with primary antibodies at 4 °C overnight, washed in PBS-Tween/TBS-Tween and then incubated with IRDye-conjugated anti-rabbit and anti-mouse secondary antibodies (LI-COR) for 1 hr at RT. Signal detection and quantification was performed using an Odyssey infrared detection instrument (LI-COR) and immunoblots were performed at least thrice to confirm reproducibility.
Nuclear extraction and co-immunoprecipitation was performed using the Universal Magnetic Co-IP kit (Active Motif). Briefly, cells were lysed in a hypotonic solution and nuclear extracts were prepared using an enzymatic shearing cocktail designed to preserve complexes of DNA-binding proteins. Nuclear extracts were incubated with primary antibody at 4 °C overnight after which magnetized Protein G beads were added to the mixture for a further 4 hr. The beads were then washed several times before boiling in reducing buffer and SDS-PAGE analysis.
Histone acid extraction was performed as previously described (Shechter et al., 2007). Briefly, cell pellets were lysed in hypotonic buffer containing 10 mM Tris-Cl (pH 8.0), 1 mM KCl, 1.5 mM MgCl2, 1 mM DTT and 1X Halt protein and phosphatase inhibitor, the nuclei were resuspended in 0.4 N H2SO4 and incubated overnight to solubilise histones and then precipitated using trichloroacetic acid. Precipitated histones were washed in acetone and resuspended in ddH2O, quantified and then boiled in Laemmli buffer prior to SDS-PAGE analysis.
RNA Extraction and qPCR
Total RNA was extracted using the RNeasy Plus Mini kit (Qiagen). RNA was quantified using an ND-1000 spectrophotomoter (NanoDrop) or on a 2100 Bioanalyzer (Agilent). cDNA was prepared using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-Time PCR was performed using Fast SYBR Green Master Mix (Applied Biosystems). Relative expression levels were normalised to B2m within samples and calculated using 2-ΔΔCt method as previously described (Livak and Schmittgen 2001).
Allograft of Tumor Cells
15 µl of 1×106 tumor cells were injected into the left striatum of adult NOD SCID mice (bregma, 1.5 mm lateral, 2 mm deep), using a 22 G needle and a 25 µl Hamilton syringe. Mice were examined twice weekly for the development of neurological signs of intracranial pressure before euthanasia.
Immunostaining
P21 - 12 month old mice under terminal anaesthesia were transcardially perfused with ice-cold PBS followed by ice-cold 4% paraformaldehyde. Brains were dissected and postfixed in 4% paraformaldehyde overnight before sectioning on a VF-700 compresstome (Precisionary Instruments) into 50–100 µm thick free-floating sections. For embryonic mice, brains were rapidly dissected in ice-cold PBS and drop-fixed in 4% paraformaldehyde overnight before transferring to a 30% sucrose-PBS solution. After complete permeation with sucrose solution, brains were frozen in O.C.T. (Sakura) on liquid nitrogen-cooled isopentane and sectioned on a Leica 3050S cryostat (Leica Biosystems) into 20 µm thick slide-mounted sections.
For immunofluorescence, free-floating or slide-mounted sections were incubated in a blocking solution (10% goat or donkey serum, 3% BSA, 0.3% Triton-X in PBS) for 1 hr at RT and then incubated with primary antibodies at 4 °C o vernight. Sections were washed in PBS-Tween (0.05%) before addition of Alexa Fluor- or DyLight-conjugated secondary antibodies in blocking solution for 1 hr at RT. Following washing, sections were mounted in ProLong Gold mounting reagent containing DAPI (Thermo Fisher Scientific) and imaged on a confocal microscope (Zeiss LSM 700/880).
For histological analysis of tumors, mice were perfused with ice-cold 10% formalin, brains were dissected and embedded in paraffin, cut into 3 µm sections and processed for haematoxylin-eosin (HE) staining. Immunohistological detection of various antigens was performed on the Ventana Discovery automated staining platform (Ventana Medical Systems) using biotin-free secondary antibodies coupled to streptavidin-horseradish peroxidase and diaminobenzidine as a chromogen. Slides were digitized on a Leica SCN400 scanner and viewed/exported using Slidepath software (Leica Biosystems).
Cell Counts and Image Quantification
GFP+Ki67+Nestin+ and GFP+Olig2+ cells in cortex and striatum were manually counted using the multi-point tool in ImageJ in a blinded manner. GFP+ neurons, astrocytes and oligodendrocytes were identified using both morphology and cell type-specific markers (neurons: NeuN; astrocytes: GFAP/glutamine synthetase; oligodrendrocytes: Olig2/PDGFRA) and manually counted in a similar manner. Intensity of H3K27me3 fluorescence signal in HA+ cells was quantified using both ImageJ and Metamorph software (Molecular Devices). 2–4 single-plane confocal images with a z-step of 2 µm were combined for fluorescence intensity analysis (total depth less than 10 µm). All cell counting and fluorescent intensity analyses were carried out on images from a minimum of 3 mice per condition.
RNA-sequencing
Total RNA was extracted from cell pellets and mouse tumors using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) according to instructions from the manufacturer. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicentre) to achieve greater coverage of mRNA and other long non-coding transcripts. Paired-end sequencing was performed on the Illumina HiSeq 2500 platform.
Exome-sequencing
Genomic DNA was extracted from cell pellets and frozen tumor tissue and the Agilent SureSelect Reagent Exome kit (Agilent) was used to prepare DNA libraries according to the manufacturer’s instructions. Sequencing was performed on Illumina HiSeq 2000 using rapid-run mode with 100 bp paired-end reads.
Kinase inhibitor screening assay
The compounds from the Life Kinase Inhibitor Library-Z198511 (Selleckchem) were dispensed into 96-well plates at a final concentration of 10µM, using an acoustic liquid handler Echo 550 (Labcyte Inc. Sunnyvale, CA). DMSO was used at the final concentration of 0.001% and celecoxib (positive control) at the final concentration of 20µM. Plates were prepared in triplicate. Cells were seeded into the plates at density of 5000 to 7500 cells/well in volume of 90µl/well using a multiflo dispenser (Biotek) at low speed. Cells were then incubated at 37C, 5% CO2 for a period of 72 hr. After the exposure period, 10% final PrestoBlue (Life Technologies) was added to each well and the plates were returned to the incubator for 6 hr. The fluorescence intensity was measured using a multiwell plate reader (Envision, Perkin Elmer) at excitation 544nm and emission 620nm. The celHTS2 software package was used for drug screen analysis and generation of z-scores (Boutros et al., 2006).
ChIP-sequencing
Cells were fixed with 1% formaldehyde (Sigma). Fixed cell preparations were washed, pelleted and stored at −80°C. Sonication of lysed n uclei (lysed in a buffer containing 1% SDS) was performed on a BioRuptor UCD-300 for 60 cycles, 10s on 20s off, centrifuged every 15 cycles, chilled by 4 °C water cooler. Sampl es were checked for sonication efficiency using the criteria of 150–500bp by gel electrophoresis. After the sonication, the chromatin was diluted to reduce SDS level to 0.1% and before the ChIP reaction 2% of sonicated drosophila S2 cell chromatin was spiked into the samples for quantification of total levels of histone mark after sequencing (see below).
The ChIP reaction was performed on a Diagenode SX-8G IP-Star Compact using Diagenode automated Ideal ChIP-seq Kit. 25µl Protein A beads were washed and then incubated with 5µg of H3K27me3 antibody (Cell Signaling Technologies, #9733) and 2 million cells of sonicated cell lysate combined with protease inhibitors for 10 hr, followed by 20 min wash cycle with provided wash buffers. Reverse cross linking took place on a heat block at 65°C for 4 hr. ChIP samples were then treated with 2µl RNase Cocktail at 65°C for 30 min followed by 2µl Proteinase K at 65°C for 30 min. Samples were the n purified with QIAGEN MiniElute PCR purification kit as per manufacturers’ protocol. In parallel, input samples (chromatin from about 50,000 cells) were reverse crosslinked and DNA was isolated following the same protocol.
Library preparation was carried out using Kapa HTP Illumina library preparation reagents. Briefly, 25µl of ChIP sample was incubated with 45µl end repair mix at 20°C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50µl buffer enzyme mix for 30°C 30 min, followed by PE G/NaCl purification. Adaptor ligation: bead bound sample was incubated with 45µl buffer enzyme mix and 5µl of different TruSeq DNA adapters (Illumina) for each sample, for 20°C 1 5 min, followed by PEG/NaCl purification (twice). Library enrichment: 12 cycles of PCR amplification. Size selection was performed after PCR using a 0.6x/0.8x ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments.
ChIP libraries were sequenced using Illumina HiSeq 2000 at 50bp SE reads.
Bioinformatics –RNA-Seq Data Processing
RNA sequencing reads were trimmed using Trimmomatic (v0.32) (Bolger et al., 2014) , removing adaptor and other Illumina-specific sequences as well as the first four bases from the start of each read, and low-quality bases at the end of each read, using a 4bp sliding window to trim where average window quality fell below 30 (phred33 < 30). In addition, an additional 3bp was clipped from the start and end of a read if found of low quality. Trimmed reads < 30 bp were discarded. The resulting clean set of reads were then aligned to the reference genomes using STAR (v2.3.0e) (Dobin et al., 2013) with default parameters. Reads mapping to more than 10 locations in the genome (MAPQ < 1) were discarded. The reference genome build used was hg19 (GRCh37) for human samples and mm10 (GRCm38) for mouse samples.
Gene expression levels were estimated by quantifying uniquely mapped reads to exonic regions (the maximal genomic locus of each gene and its known isoforms) using featureCounts (v1.4.4) (Liao et al., 2014) and the Ensembl gene annotation set. Normalization (mean of ratios) and variance-stabilized transformation of the data were performed using DESeq2 (Love et al., 2014). Multiple control metrics were obtained using FASTQC (v0.11.2), samtools (v0.1.19) (Li et al., 2009), BEDtools (v2.17.0) (Quinlan and Hall, 2010) and custom scripts. For visualization, normalized Bigwig tracks were generated using BEDtools and UCSC tools. Integrative Genomic Viewer (Thorvaldsdóttir et al., 2013) was used for data visualization.
Bioinformatics –RNA-Seq Clustering
Global changes in expression levels were evaluated using the t-Stochastic Neighbor Embedding (t-SNE; perplexity=6; N=5000 iterations; algorithm applied to the PCA space by taking the first 50 PCs) (Garcia-Alonso et al., 2014) and principal component analysis (PCA) dimensionality-reduction techniques on normalized expression data coupled with variance-stabilized transformation (DESeq2) (Love et al., 2014)).
Bioinformatics –Gene Signatures
Differential expression analysis was performed using DESeq2 (Love et al., 2014). Statistically significant (adjusted p value ≤ 0.05) genes with large expression changes (absolute fold change ≥ 2) that are expressed above a threshold (average normalized expression across samples ≥ 50) were identified for all mouse models and human samples. Gene signatures for each of the murine models (i.e. K27M-AP T and K27M-APP T) were derived by differential expression of each relative to baseline controls (EV ctl) and subsequent removal of genes present in the WT-AP signature (i.e. differentially expressed genes between WT-AP and baseline controls, representing genes deregulated simply by overexpression of H3.3 and knockdown of ATRX/p53). Extended gene signatures for K27M tumors (K27M–T, pooling together K27M-AP T and K27M-APP T) were derived by differential expression relative to extended controls (i.e. EV ctl and EV-PDGFRA). Similarly, gene signatures corresponding to human H3.3WT pHGG and H3.3K27M pHGG were derived by differential expression relative to tumor-matched normal brain controls (NB).
Each of the gene signatures was functionally annotated using DAVID (Huang et al., 2007), computing their enrichments against a background set of genes. Background genes for mouse signatures were defined as genes expressed across all mouse samples (i.e. in the upper 3 quartiles of expression for at least one sample in the entire dataset). Background genes for human signatures were similarly obtained. Specific signatures corresponding to defined pathways of interest (e.g. K27M–T DNA-binding) were then obtained by subsetting the corresponding genes from each signature based on functional annotations.
Bioinformatics –Comparison of Human and Mouse Expression Data
To assess the relevance of the genes identified in our mouse cell and tumor models in human patient data, we employed three distinct approaches.
Unsupervised clustering of human samples based on expression levels of human orthologs of genes belonging to each murine signature using the t-SNE (perplexity=2; N=10000 iterations; using the first 50 PCs of expression data) and PCA dimensionality-reduction techniques; exact, case insensitive match of their corresponding HGNC symbols (i.e. murine Sox10 to human SOX10) was required (Figure 7C).
Computation of overlaps of genes (exact, case insensitive match of HGNC symbols as well as agreeing directions of change in expression; Figure 7D and Figure S8D). In order to assess the significance of the reported overlaps in each human-murine signature comparison, we computed an empirical p value for the reported overlap in each comparison. For each comparison, we obtained a null distribution of overlaps by repeatedly resampling (N=10,000 iterations) murine signatures of the same length as the original murine signature, computing for each the number of overlapping gene orthologs in the human signature. The murine genes available for resampling were limited to those that are expressed in corresponding murine comparison by filtering out those with a null fold-change.
Projection of murine signatures across human samples using single sample gene set enrichment analysis (ssGSEA) as described before (Barbie et al., 2009). Briefly, a score is defined to represent the degree of enrichment of a given gene set in a sample: gene expression values are rank-normalized for each sample, and an enrichment score is produced using the Empirical Cumulative Distribution Function (ECDF) of genes, with the final score obtained by integrating the difference between a weighted ECDF of genes in the signature and the ECDF of the remaining genes. This calculation is computed for each signature and each sample, using the ssGSEAProjection (v6) module of the GenePattern suite (Kuehn et al., 2008). Genes in the murine signatures were converted to their human orthologs using biomaRt (v2.26.1) (Smedley et al., 2015) prior to ssGSEA enrichment analysis (Figure S8E).
Bioinformatics –H3K27me3 ChIP-Seq Data Processing
ChIP-seq reads were aligned to the mouse reference genome build mm10 (GRCm38) using bwa (v0.7.10-r789) (Jo and Koh, 2015) with default settings. BigWig tracks were generated for each sample, with coverage scaled according to ChIP-Rx ratio (Orlando et al., 2014). The tracks were then visualized in the Integrative Genomics Viewer (Thorvaldsdóttir et al., 2013).
Bioinformatics –Calculation of ChIP-Rx Ratio
The spiked-in drosophila chromatin was used for quantification of histone mark, as described in Orlando et al., 2014 (Orlando et al., 2014). After sequencing, the reads were aligned to mouse and drosophila genomes, then the number of reads mapping to each genome (from H3K27me3 and input samples) were used to compute the relative levels of histone mark using the following formula for each sample:
An in-depth calculation of the ChIP-Rx ratio is provided in Table S8.
Bioinformatics –H3K27me3 ChIP-Seq Clustering
H3K27me3-level based clustering of genes was performed using ngs.plot (v2.61) (Shen et al., 2014). Gene intervals were obtained from the Ensembl gene annotation set release 75 for the mouse genome build mm10 (GRCm38). Intervals extending from 10kb upstream to 10kb downstream of the transcription start sites (TSS) of all genes were considered in the summarization of H3K27me3 levels for each gene. Intervals were binned into 100bp windows, and ChIP-Seq reads normalized locally on a per-sample basis into Z-scores, considering both IP and input reads. Genes were then clustered by k-means (k=12) based on average perigenic H3K27me3 levels in each sample.
Bioinformatics –Comparison of Human and Mouse H3K27me3 Data
In order to assess the agreement of H3K27me3 distribution in genic regions between our mouse model and human patient data, we obtained H3K27me3 ChIP-seq data for H3.3WT and H3.3K27M pHGG through European Genome-phenome Archive accession number EGAS00001000578 (Bender et al., 2013). We clustered these human tumor-derived H3K27me3 data into k=4 classes based on average perigenic H3K27me3 levels using the same method as mentioned above, but with gene intervals obtained from the Ensembl gene annotation set release 75 for the human genome build hg19 (GRCh37).
Next, we estimated agreement between clusters found in human and mouse data by computing the proportion of genes in each mouse cluster that have orthologs in each of the 4 human clusters. The mouse-to-human ortholog association map were again obtained using the R Bioconductor package biomaRt (v2.26.1) (Smedley et al., 2015). Finally, we considered only one-to-one associations in order to obtain a conservative measure of agreement between cross-species H3K27me3 cluster gene assignment.
Bioinformatics –De Novo Deposition of H3K27me3
Peaks were called on each murine ChIP-seq sample using the callpeak function of MACS2 (v2.1.1.20160309) (Zhang et al., 2008) with the “broad” and “nomodel” flags as well as a default FDR cutoff of 0.05. Both IP and input reads were considered for peak calling and the mouse genome build mm10 (GRCm38) was used as a reference genome. Differential binding analysis was then performed in different groupings of H3.3K27M samples (all K27M samples, K27M tumors-only, and individual tumors) relative to H3.3WT (EV-ctl, EV-PDGFRA, WT-AP, WT-APP) samples based on the peak calls using the R Bioconductor package DiffBind (v2.5.4), which was modified to allow libraries to be normalized according to their respective ChIP-Rx scaling factors. Peaks significantly increased (FDR < 0.05) in tumor samples with respect to controls were inspected manually using IGV and reported in Figure S9D.
Bioinformatics –Whole Exome-Seq Data Processing
Exome sequencing reads were processed using the same approach as for RNA-seq with the exception that the first 4 bases at the start of each read are retained. The resulting reads were then aligned to the mm10 (GRCm38) reference genome using BWA-MEM v0.7.13 (Li and Durbin, 2010) using default parameters.
Bioinformatics –WES and RNA-Seq Variant Calling
The split’N’Trim function of the Genome Analysis Toolkit (GATK) (v3.2-2) (Van der Auwera et al., 2013) was used to split reads that have introns and reassign the mapping quality from 255 to 60. The IndelRealigner function of GATK was then used to realign indels. Duplicate reads were marked using the Picard toolset (v1.118). Variants are called using the mpileup function of samtools (v0.1.19) (Li et al., 2009) and using Annovar (February 2, 2016 version) (Wang et al., 2010) over the mm10 refGene annotation.
For WES samples, positions where coverage is greater than 1000 were considered to be sequencing artefacts and thus ignored. Finally, we applied multiple levels of filters to remove likely false positive variant calls on WES or RNA-seq samples, retaining only variants with coverage ≥ 10 reads, alternative nucleotide count ≥ 3, SNV ratio ≥ 5%, indel ratio ≥ 15%, variant quality ≥ and mapping quality ≥ 15.
Bioinformatics –CRISPR Off-Target and Spontaneous SNV Analysis
In order to evaluate the presence of potential off-target effects of the CRISPR construct targeting Trp53 (Zuckermann et al., 2015), we obtained a list of 40 possible off-targets using the Cas-OFFinder web tool (Bae et al., 2014) based on the gDNA sequence, allowing up to 3 mismatches of the gDNA sequence. We found that none of the potential off-target genomic locations harbored variants in K27M-P T RNA-seq sample that were not also found in the Trp53+/+ control samples (EV-ctl and EV-PDGFRA). We performed a similar analysis in order to detect the occurrence of spontaneous SNVs, where we report variants occurring in exonic regions that are found in the K27M-P T RNA-seq sample but not in the Trp53+/+ control samples (prior to variant filtration; EV-ctl and EV-PDGFRA). Additionally, we provide the number tumor Trp53−/− K27M that also habor these variants (after filtration).
Bioinformatics –Clonality Analysis
From the filtered list of putative SNVs obtained for each of the 10 WES samples derived from spatial biopsies of one K27M-AP tumour, we discarded all the but the most reliable SNVs, that is, non-synonymous variants occurring in exonic regions. Using PyClone (v0.13.0) (DOI:10.1038/nmeth.2883) to infer the clonal structure of the tumor, we obtained a final set of 336 variants that were clustered into 10 clusters. The number of clusters was reduced to 7 after discarding the clusters consisting of a single SNV (Figure S5).
To assess the similarity of the complete mutational landscape of the tumor beyond that of only non-synonymous exonic variants, we determined a complete set of genomic positions harboring variants in at least one of the 10 biopsies (i.e. union of the good quality variants) and then calculated the variant allele frequency (VAF) of each site for each biopsy, discarding the indels in the process. Next, to obtain a measure of similarity between samples, we calculated for each pair i, j (respectively rows and columns in Table S4) of biopsies the proportion of good quality variants in the i-th biopsy that were also present in the j-th biopsy with VAF > 0%.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values and graphs are expressed as mean ± SEM (standard error of the mean) and statistical analyses were performed using unpaired two-tailed t tests, ordinary one-way ANOVA followed by Tukey post hoc analysis or the Chi-squared test (GraphPad Prism). A statistically significant difference was indicated by a p value less than 0.05.
DATA AND SOFTWARE AVAILABILITY
All sequencing data have been deposited on GEO (accession number GSE95169) and can be accessed via the following link.
Supplementary Material
Table S2. Related to Figure 2. CRISPR off-target and spontaneous SNV analysis.
Table S3. Related to Figure 4. List of drugs identified in the kinase inhibitor library screen and their z-scores.
Table S4. Related to Figure 5. Concordance matrix of SNVs.
Table S5. Related to Figure 7. Gene signatures.
Table S6. Related to Figure 7. Signature overlaps.
Table S7. Related to Figure 7. Functional annotations.
Table S8. Related to Figure 8. ChIP-seq normalization factors.
Significance.
Mutations in histone 3 variants are believed to drive development of high-grade glioma in children. Despite extensive efforts, no truly representative model demonstrating the oncogenic activity of mutant histone 3 variants has been described. Here we report a somatic mouse model of oncohistone-driven tumorigenesis that recapitulates hallmark features of human pHGG. Our findings support the concept that these mutations act specifically in utero to drive changes in neurodevelopmental programs that lead to full blown transformation postnatally. Ultimately, this model provides insights into mechanisms of transformation in the brain and may enable identification of targets for more effective therapeutic intervention.
Highlights.
Mosaic H3.3K27M and Trp53 loss drives transformation during neurodevelopment in utero
Expression of H3.3K27M postnatally or in the entire stem cell pool fails to transform
Adding PDGFRA overexpression accelerates disease onset and increases tumor invasion
H3.3K27M-driven tumors are clonal and recapitulate key features of pediatric HGG
Acknowledgments
We thank Juan P Martinez-Barbera, Teresa Marafioti (University College London, UK), Fuyi Chen, Joseph LoTurco (University of Connecticut), Jan Gronych (DKFZ), Melania Capasso and Daniele Bano (DZNE) for reagents and/or critical discussion. A special thanks to all members of PS, NJ, CK, AR and SB labs and the CRUK UCL Centre Core Services and the UCL Scientific Services. This work was in part funded by: the European Research Council Consolidator Award (H3.3Cancer to PS); funding from: US National Institutes of Health (NIH; P01CA196539 to N.J), the Canadian Institutes for Health Research (CIHR; MOP 286756, N.J.), I-CHANGE consortium, the Fonds de Recherche du Québec- Santé (salary awards, C.L.K, N.D.J), the UK Medical Research Council to the MRC-UCL University Unit (Ref. MC_U12266B). Computational infrastructure was provided by Compute Canada. PS is head of the Samantha Dickson Brain Cancer Unit and supported by Brain Tumour Charity (formerly known as Samantha Dickson Brain Tumour Trust) and the Biomedical Research Council. N.J. is a member of the Penny Cole Laboratory and the recipient of a Chercheur Boursier, Chaire de Recherche Award from the Fond de la Recherche du Québec en Santé. All sequencing data have been deposited on GEO (accession number GSE95169) and can be accessed via the following reviewer link
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
MP contributed to design of experiments, supervision of NM, performed experiments, analyzed and contributed to data interpretation, writing first draft, and review and editing further drafts
NDJ substantially contributed to data analysis, to writing of the first draft and review and editing of further drafts
NM performed experiments and contributed to data analysis, contributed to review and editing of drafts of the manuscript
ASH performed experiments
JN provided training and resources
PP contributed to design of methodology and data analysis
SH contributed to data analysis
LGM contributed to project administration
ARL provided resources and contributed to data collection
YZ performed experiments
JRC performed experiments and contributed to data analysis
SH contributed to data analysis
SK performed experiments
NS performed experiments
JH contributed to supervision
AR provided resources, training and contributed to supervision
SA contributed to data analysis and interpretation
RK contributed to supervision, data interpretation
SB provided resources and contributed to data analysis and interpretation
CLK provided supervision, contributed to data analysis and interpretation, writing initial and subsequent drafts, project management and funding acquisition
NJ co-led the project with PS, provided supervision, formulation and evolution of overarching research goals, contributed to experimental design, data analysis and interpretation, writing initial and subsequent drafts, project management and funding acquisition
PS is the Lead Contact, co-led the project with NJ, provided supervision, formulation and evolution of overarching research goals, contributed to experimental design, data analysis and interpretation, writing initial and subsequent drafts, project management and funding acquisition
References
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usade IB. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Bras LP, Huber W. Analysis of cell-based RNAi screens. Genome Biol. 2006;7:R66. doi: 10.1186/gb-2006-7-7-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta neuropathologica. 2014;128:573–581. doi: 10.1007/s00401-014-1319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Becker AJ, Loturco JJ. Contribution of Tumor Heterogeneity in a New Animal Model of CNS Tumors. Mol Cancer Res. 2014;12:742–753. doi: 10.1158/1541-7786.MCR-13-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, LoTurco J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J Neurosci Methods. 2012;207:172–180. doi: 10.1016/j.jneumeth.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, Murphy SK, Hashizume R, Becher OJ. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-16-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faury D, Nantel A, Dunn SE, Guiot MC, Haque T, Hauser P, Garami M, Bognar L, Hanzely Z, Liberski PP, et al. Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:1196–1208. doi: 10.1200/JCO.2006.07.8626. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB. Linked regularities in the development and evolution of mammalian brains. Science. 1995;268:1578–1584. doi: 10.1126/science.7777856. [DOI] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Jabado N. Pediatric Brain Tumors: Genomics and Epigenomics Pave the Way. Critical reviews in oncogenesis. 2015;20:271–299. doi: 10.1615/critrevoncog.2015013565. [DOI] [PubMed] [Google Scholar]
- Fontebasso AM, Liu XY, Sturm D, Jabado N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain pathology. 2013;23:210–216. doi: 10.1111/bpa.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature genetics. 2014;46:462–466. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, Remke M, Li R, Takayama N, Desai K, et al. MLL5 Orchestrates a Cancer Self-Renewal State by Repressing the Histone Variant H3.3 and Globally Reorganizing Chromatin. Cancer cell. 2015;28:715–729. doi: 10.1016/j.ccell.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Garcia-Alonso CR, Perez-Naranjo LM, Fernandez-Caballero JC. Multiobjective evolutionary algorithms to identify highly autocorrelated areas: the case of spatial distribution in financially compromised farms. Ann Oper Res. 2014;219:187–202. [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–559. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. nat Med. 2014;20:1394–1396. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennika T, Hu G, Olaciregui NG, Barton KL, Ehteda A, Chitranjan A, Chang C, Gifford AJ, Tsoli M, Ziegler DS, et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS One. 2017;12:e0169485. doi: 10.1371/journal.pone.0169485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer cell. 2015;27:72–84. doi: 10.1016/j.ccell.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo H, Koh G. Faster single-end alignment generation utilizing multi-thread for BWA. Biomed Mater Eng. 2015;26:S1791–1796. doi: 10.3233/BME-151480. [DOI] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, Sturm D, Meyer J, Jones C, Zheludkova O. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta neuropathologica. 2015;131:137–146. doi: 10.1007/s00401-015-1493-1. [DOI] [PubMed] [Google Scholar]
- Kuehn H, Liberzon A, Reich M, Mesirov JP. Using GenePattern for gene expression analysis. Curr Protoc Bioinformatics. 2008 doi: 10.1002/0471250953.bi0712s22. Chapter 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski L, Garcia B, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;40:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro Oncol. 2011;13:1049–1058. doi: 10.1093/neuonc/nor092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Noh K-M, Soshnev A, Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nature genetics. 2014;15:259–271. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh K-M, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL, et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron. 2015;87:77–94. doi: 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michod D, Bartesaghi S, Khelifi A, Bellodi C, Berliocchi L, Nicotera P, Salomoni P. Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron. 2012;74:122–135. doi: 10.1016/j.neuron.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. EZH2 is a potential therapeutic target for H3K27M–mutant pediatric gliomas. Nat Med. 2017 doi: 10.1038/nm.4293. [DOI] [PubMed] [Google Scholar]
- Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho CY, Kambhampati M, Hwang EI, Faury D, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nature communications. 2016;7:11185. doi: 10.1038/ncomms11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitarska J, Smith JG, Sherlock WT, Hillege MM, Nott A, Barshop WD, Vashisht AA, Wohlschlegel JA, Mitter R, Riccio A. A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development. Cell Rep. 2016;17:1683–1698. doi: 10.1016/j.celrep.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, Guenther MG. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014;9:1163–1170. doi: 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, De Blank PM, Finlay JL, Gurney JG, McKean-Cowdin R, Stearns DS, Wolff JE, Liu M, Wolinsky Y, et al. American Brain Tumor Association Adolescent and Young Adult Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;18:i1–i50. doi: 10.1093/neuonc/nov297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016;18:v1–v75. doi: 10.1093/neuonc/now207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73:6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, Sicotte H, Kollmeyer TM, McCoy LS, Sarkar G, et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta neuropathologica. 2017;133:1001–1016. doi: 10.1007/s00401-017-1690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu HQ, Jacob K, Fatet S, Ge B, Barnett D, Delattre O, Faury D, Montpetit A, Solomon L, Hauser P, et al. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 2010;12:153–163. doi: 10.1093/neuonc/nop001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014;15 doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Zhu Y, Murakami F. Four-dimensional analysis of nucleogenesis of the pontine nucleus in the hindbrain. J Comp Neurol. 2013;521:3340–3357. doi: 10.1002/cne.23353. [DOI] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh M, Baldock R, Barbiera G, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–598. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43 doi: 10.1002/0471250953.bi1110s43. 11 10 11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, Weller M, Wick W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta neuropathologica. 2013;126:443–451. doi: 10.1007/s00401-013-1156-z. [DOI] [PubMed] [Google Scholar]
- Wright KD, Sabin ND, Cheuk D, McNall-Knapp R, Shurtleff SA, Gajjar A, Broniscer A. Incidental Diagnosis of Diffuse Intrinsic Pontine Glioma in Children. Pediatr Blood Cancer. 2015;62:1081–1083. doi: 10.1002/pbc.25408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature genetics. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BTK, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based Analysis of ChIP-Seq (MACS) Genome Biology. 2008;9 doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DT, Tschida B, Moriarity B, et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nature communications. 2015;6:7391. doi: 10.1038/ncomms8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Related to Figure 2. CRISPR off-target and spontaneous SNV analysis.
Table S3. Related to Figure 4. List of drugs identified in the kinase inhibitor library screen and their z-scores.
Table S4. Related to Figure 5. Concordance matrix of SNVs.
Table S5. Related to Figure 7. Gene signatures.
Table S6. Related to Figure 7. Signature overlaps.
Table S7. Related to Figure 7. Functional annotations.
Table S8. Related to Figure 8. ChIP-seq normalization factors.