

Abstract
Understanding the skin mycobiome (fungal communities) is important because both commensal and pathogenic fungi can drive cutaneous disease depending on host status and body sites, including the scalp, feet, and groin. Interestingly, age may also affect skin fungal infections as certain dermatophytoses (i.e. tinea capitis) are more frequent in children than adults. We previously described the skin mycobiomes in healthy adults, showing lipophilic fungi Malassezia predominate in most skin sites. Since children have less sebaceous skin before puberty, we compared the fungal communities of primary clinical samples from healthy children and adults, based on sequencing of a fungal phylogenetic marker. While Malassezia predominated on trunk, head and arm skin of adults (age 20s–30s), children (age <14) had more diverse fungal communities, for example, Eurotiomycetes which includes common dermatophytes. Species-level classification showed M. globosa predominated in children. Collectively, our findings indicate that prepubertal skin is colonized by diverse fungi, whereas adult skin is predominantly obligatory lipophilic Malassezia, suggesting that fungal communities on skin profoundly shift during puberty. Mycobiome shifts during puberty are likely due to alterations in sebaceous gland activation and sebum composition. This study provides a foundational framework for studies investigating interactions between fungi, skin, and pediatric dermatophytosis.
Introduction
Human skin is a dynamic habitat hosting a wide variety of microbes, including bacteria, fungi, viruses, and parasites (Belkaid and Segre, 2014). Although bacteria often predominate in the ‘skin microbiome’ and have been the major focus of most studies (Grice and Segre, 2011; Oh et al., 2014), understanding the fungal communities - mycobiome - on skin is also important given the close association of commensal and pathogenic fungi, e.g. Malassezia, Candida, and dermatophytes, with various skin disorders, such as seborrheic dermatitis, atopic dermatitis, and dermatophytosis (Gaitanis et al., 2012; Gupta et al., 2004; Havlickova et al., 2008; Seebacher et al., 2008; White et al., 2014). However, in contrast to bacterial communities, less is known about the structure and dynamics of the skin mycobiome in health and disease.
Sequence-based skin mycobiome studies have revealed that Malassezia is commonly found on healthy adult skin at most body sites, along with Aspergillus and Penicillium as less abundant commensals (Findley et al., 2013; Paulino et al., 2006; Underhill and Iliev, 2014; Zhang et al., 2011). Interestingly, previous culture-based and targeted PCR-based studies demonstrated that the prevalence of major commensal Malassezia fungi on the skin of prepubertal children was lower than on adults (Faergemann and Fredriksson, 1980; Gupta and Kohli, 2004; Jang et al., 2009; Sugita et al., 2010), and the incidence of fungus-associated diseases, such as tinea capitis, tinea corporis, and tinea versicolor, varies among different age groups (Hawkins and Smidt, 2014; Kyriakis et al., 2006; Shy, 2007), suggesting differential fungistatic properties of the skin in children (prepubertal) and adults (postpubertal). In addition, we previously showed the striking differences in the nares and skin bacterial communities in adults versus children (Oh et al., 2012). Collectively, these observations suggest that the skin mycobiome may differ between young children and adults, but the potential differences remain unknown.
The fungal internal transcribed spacer-1 (ITS1) sequence is a taxonomic signature enabling identification at the genus, and sometimes species level. Using ITS1 sequencing, we compared taxonomic relative abundance, diversity and similarity of healthy children (HC) mycobiomes with those of healthy adults (HA). We found the mycobiomes of children were more diverse than those of adults, who were colonized almost exclusively with Malassezia. Within-group comparisons revealed that children had higher interpersonal variation than adults. Furthermore, species-level classification showed that M. globosa was the predominant species of Malassezia on children across all skin sites tested, whereas M. restricta constituted a significant proportion of several skin sites in adults. Together, our results demonstrate that children possess distinct and diverse skin mycobiomes as compared to adults. To our knowledge, this study represents the initial sequence-based approach to analyzing skin fungal communities based on age and sexual maturation.
Results
Overview of dataset and taxonomic analysis
To investigate whether skin fungal communities of healthy children (HC) differed from healthy adults (HA), samples from nine skin sites [four from the head (external auditory canal (Ea), forehead (Fh), occiput (Oc), and retroauricular crease (Ra); sebaceous), two from the trunk (back (Ba) and manubrium (Mb); sebaceous), two from folds in extremities (antecubital fossa (Ac) and inguinal crease (Ic); moist), and one from flat surfaces of extremities (volar forearm (Vf); dry)] and nares of 33 individuals (19 HAs and 14 HCs) were collected and sequenced using the internal transcribed spacer-1 (ITS1) region to investigate fungal diversity (Table 1 and Supplementary Table S1). Sequencing data were collected using both the Roche 454 and Illumina MiSeq instruments, due to changes in sequencing technologies capabilities (see Materials and Methods for platform comparability test). A total of 16,561,411 reads passed initial quality control (quality trimming for 454, and quality trimming and end-joining for MiSeq). To explore fungal communities on skin in different age groups, all sequence data were taxonomically classified to the genus level using a previously established database (Findley et al., 2013). Rarefaction curves indicated that taxonomic diversity plateaued near 3,000 reads for all samples (Supplementary Figure S1). Therefore, all samples were randomly subsampled to 3,000 reads/sample for further analysis, seven samples were excluded because of low read numbers (Supplementary Table S1).
Table 1.
Characteristics of the participants.
| Characteristic | Healthy adults (HA) | Healthy children (HC) |
|---|---|---|
| Total subjects analyzed | 19 | 14 |
| Male:Female | 12: 7 | 8: 6 |
| Tanner stage, (Age, median (range)) |
5 (28yr (19–33)) |
1 – 3 (Stg 1: 12.5yr (12–13) Stg 2: 11yr (10–12) Stg 3: 10yr (7–11)) |
Fungal communities differ in children and adults
To determine whether fungal communities differed between children and adults, genus-level taxonomic abundance and Shannon diversity, an ecological measure of community richness and evenness, were evaluated (Figure 1a and b, and Supplementary Figure S2a and b). As previously reported, Malassezia fungi predominated on the skin of adults (Ac: mean 80.40% (95% CI 76.10–84.71), Ba: mean 99.41% (95% CI 99.22–99.60), Ea: mean 99.84 (95% CI 99.80–99.88), and Vf: mean 82.79 (95% CI 78.14–87.43)) (Findley et al., 2013). In contrast, Malassezia had a lower relative abundance on children (Ac: mean 36.50 (95% CI 29.8–43.72), Ba: mean 76.65 (95% CI 71.55–81.76), Ea: mean 64.58 (95% CI 54.86–74.30), and Vf: mean 35.02 (95% CI 7.40–42.63)). Furthermore, diverse Ascomycota fungi including Aspergillus, Epicoccum, and Phoma constituted commensal flora on children, with levels >5% in 40.2% of samples from children as compared to only 9.5% of adult samples (Figure 1a and Supplementary Figure S2a). Similarly, fungal diversity was significantly increased in children (Figure 1b and Supplementary Figure S2b). For clinical samples from children, decreased diversity was correlated with increased relative abundance of Malassezia, especially on sebaceous sites (Figure 1b). Given the predominance of Malassezia on sebaceous skin, it is possible that reduction in diversity was attributed to relative overexpansion of Malassezia. Normalized diversity (recalculated after excluding Malassezia sequences) was not correlated with Malassezia abundance (Supplementary Figure S2c), suggesting independence of fungal community diversity from Malassezia predominance.
Figure 1.
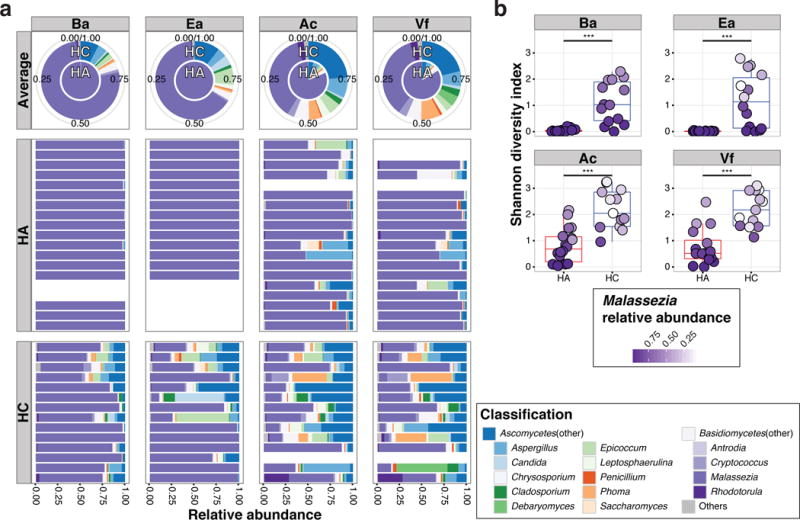
Relative abundances and Shannon diversity of fungal genera at different skin sites of healthy adults (HA) and children (HC). (a) Relative abundances of the 14 major genera from 4 representative body sites, back/Ba (trunk, sebaceous), external ear canal/Ea (head, sebaceous), antecubital fossa/Ac (extremity, moist), and volar forearm/Vf (extremity, dry) for HA and HC: average (upper pie charts) and individual relative abundances (lower bar charts). (b) Box-plot of Shannon diversity in each group. The box represents the lower quartile, median, and upper quartile. Color intensity of each dot represents relative abundance of Malassezia in each sample. Statistical significance tested with Wilcoxon rank-sum test; ***P≤0.001.
Similarities between samples were determined by principal coordinate analysis using taxonomic abundance-based Yue-Clayton Theta (θ) similarity coefficient (Figure 2a and Supplementary Figure S2d) (Yue and Clayton, 2005). Fungal communities of adults clustered tightly together, suggesting high similarity among subjects. On the other hand, fungal communities in children were scattered throughout both axes without noticeable clustering, suggesting high within-group variation. Concordantly, pairwise comparison of Theta similarity showed that adults were similar to each other while within-group variation were higher in children (Figure 2b). Taken together, these results suggest that healthy children have more diverse and divergent fungal communities on skin than healthy adults.
Figure 2.
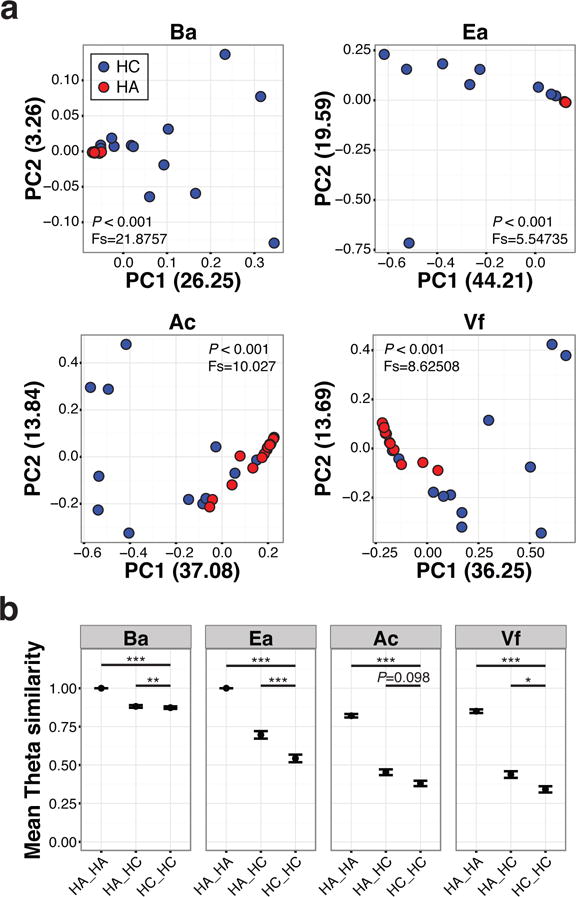
Dissimilarity comparison between HA and HC. (a) Samples clustered using principle coordinate analysis (PCoA) of the Theta similarity coefficient. Difference of population tested with Analysis of Molecular Variance (AMOVA) method. (b) Mean Theta similarity index of pairwise comparisons of within or between individuals of each group. A value of 1 indicates identical fungal community structure. Error bars represent the SEM. Statistical significance tested with Wilcoxon rank-sum test; *P≤0.05, **P≤0.01, ***P≤0.001.
Nares fungal communities differ in children and adults
The anterior nares have been regarded as a physiologically important microbial habitat, because the nares can serve as a reservoir for colonization of potential bacterial pathogens such as Staphylococcus aureus and Streptococcus pneumoniae (Archer and Climo, 2001; von Eiff et al., 2001). With stratified, non-cornified epithelium, the nares also represented a different physiological niche as compared to other sites studied in this cohort. Furthermore, a previous report determined that prominent bacterial shifts occurred in the nares during puberty (Oh et al., 2012). Therefore, we investigated whether fungal profiles in the nares differed between children and adults (Figure 3a–c). Fungal communities of nares were less Malassezia predominant (56.1% (95% CI 48.87–63.34) in children, versus 92.3% (95% CI 89.12–95.50) in adults) and colonized by diverse fungi, similar to other body sites.
Figure 3.
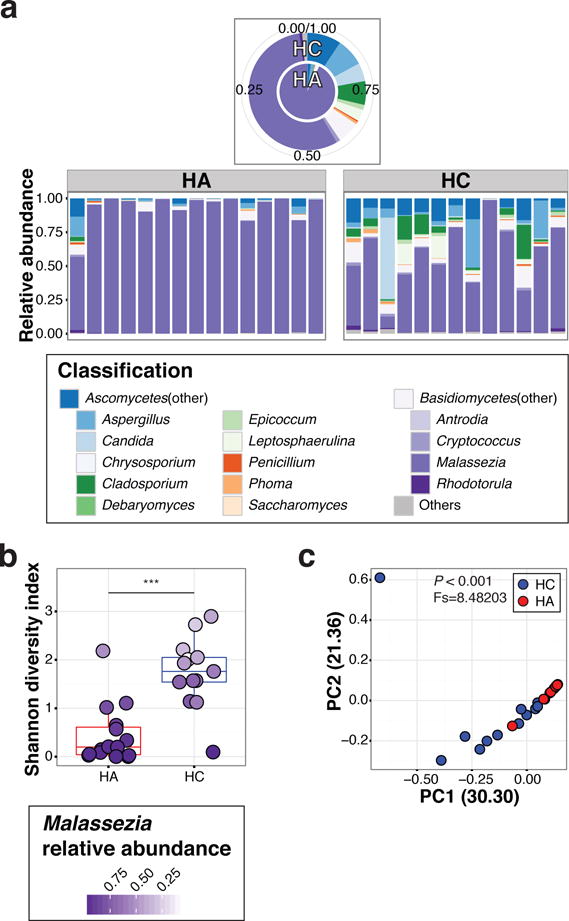
Fungal communities in nares. (a) Relative abundance of major genera in nares; average (upper pie chart), and individual abundance (lower bar charts). (b) Box-plot of Shannon diversity in HA and HC. Color intensity of each dot represents abundance of Malassezia in each sample. Statistical significance tested with Wilcoxon rank-sum test; ***P≤0.001. (c) PCoA plot of nares using Theta coefficient. Difference of population tested with AMOVA.
M. globosa predominates on the skin of healthy children
Previously, using a curated reference database of species-level Malassezia ITS1 sequences, we observed that M. restricta predominated at Ea, Ra, and Fh (head-sebaceous sites), and M. globosa predominated at Ba and Oc in healthy adults (Findley et al., 2013). Various reports have shown that distinct Malassezia species exhibit different lipid-dependency and are potentially implicated in various cutaneous diseases including seborrheic dermatitis (Gupta et al., 2004). Since our current results showed that fungal communities in children were considerably different from those in adults, we next interrogated species-level abundances of Malassezia fungi. Consistent with previous reports (Findley et al., 2013; Wu et al., 2015), among sebaceous sites in adults, Ea, Ra, and Fh exhibited M. restricta predominance (mean: 90.0%, 74.4%, and 85.2% of M. restricta, respectively) whereas M. globosa predominated on Ba, Oc, and Mb (mean: 61.1%, 78.4%, and 53.5% of M. globosa, respectively) (Figure 4a and b, and Supplementary Figure S3). While M. restricta was identified on children, M. globosa was the only predominant Malassezia species independent of body site (mean, Ba: 59.1%, Ra: 66.3%, Fh: 72.5%, Ba: 84.0%, Oc: 92.4%, and Mb: 79.9% of M. globosa) (Figure 4a and b, and Supplementary Figure S3).
Figure 4.
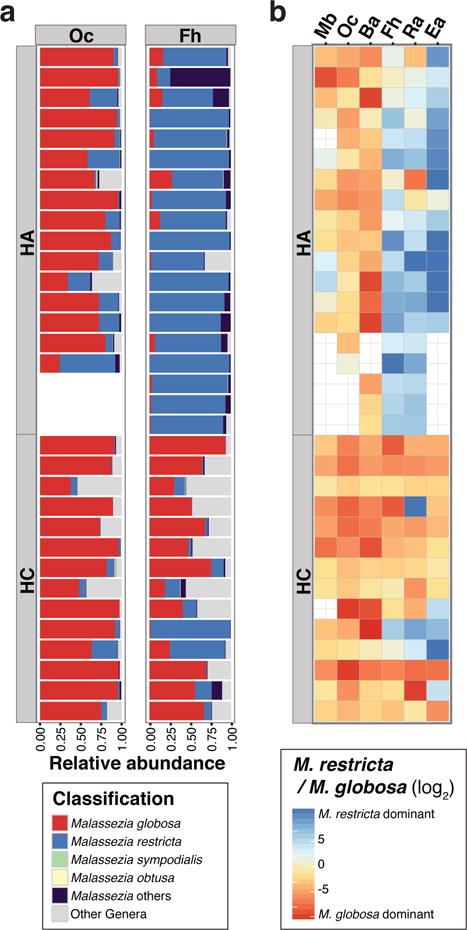
Comparison of relative abundance of Malassezia species between HA and HC. (a) Relative abundance plot of the major Malassezia species on representative sebaceous skin sites. (b) Heatmap of relative predominance of M. globosa versus M. restricta on 6 sebaceous sites. Red color indicates M. globosa predominance.
Effect of gender on the fungal community
Since children have higher fungal diversity than adults and are less similar to each other, clinical metadata were examined as potential factors contributing to the diversity and structure of fungal skin communities in children (Supplementary Table S1). Interestingly, linear discriminant analysis (LDA) effect size (LEfSe, LDA score (log 10) > 3) test demonstrated that there were discriminatory taxa between boys and girls (Segata et al., 2011). Epicoccum and Cryptococcus genera were enriched (11.6 and 9.6 fold, respectively) in sebaceous sites of boys, whereas Malassezia was enriched (1.4 fold) in girls (Supplementary Figure S4). These results suggested that gender may affect mycobiome structures during sexual maturation.
Discussion
Fungi are clinically important constituents of the skin microbiome. In this report, using ITS1 sequencing, we demonstrated differences in skin fungal communities of healthy children and adults. In accordance with our previous report, Malassezia predominated on all body and arm sites of all individuals (Findley et al., 2013). However, unlike adult subjects, children exhibited more diverse fungal communities on skin, as shown by both relative abundance and community diversity (Figure 1, and 2 and Supplementary Figure S2). Within-group analysis showed that the interpersonal variation of children was significantly higher than adults (Figure 2), and species-level analysis indicated that children were predominantly colonized by M. globosa (Figure 4). Together, these observations suggested age-dependent differences in skin physiology and mycobiome. This initial investigation of the skin mycobiomes of healthy children and adults strongly suggests an association between sexual maturity, skin mycobiome and physiology.
Prior culture-based studies have reported the presence versus absence of Malassezia in different age groups, such that the percentage of individuals with Malassezia increased during puberty (36.3% (age < 14) and 90.7% (age 15–25) in one study, 55.7% (age<10), 68% (age 11–20) and 75% (age 21–30) in another study), and decreased after the third decade of life (Faergemann and Fredriksson, 1980; Gupta and Kohli, 2004; Jang et al., 2009). In contrast, our sequence-based, culture-independent mycobiome analyses revealed that all subjects are essentially positive for Malassezia. Because Malassezia spp. are fastidious microbes in vitro and various fungi optimally grow in different conditions (Gupta et al., 2004), sequence-based studies provide a culture-independent and global view of the mycobiome. In addition, since Malassezia is an obligatory lipophilic fungus, differential Malassezia abundance might be due to the full activation of sebaceous glands during puberty (Pochi et al., 1979). Therefore, it would be intriguing to identify the sebaceous gland activity and sebum signatures during childhood in conjunction with sequence-based mycobiome analysis.
In this report, we demonstrated that diverse and divergent fungi can colonize the skin of children. The major taxa on each child were highly variable (Figure 1a and Supplementary Figure S2a). Malassezia was less frequent, and diverse fungi, such as Aspergillus, Epicoccum, Cladosporium, Phoma, and Cryptococcus, contributed to the fungal flora of healthy children. Pairwise Theta index comparison also indicated that within-group similarity of children was even lower than similarity between children and adults (Figure 2b). We propose a possible relationship of these observations to dermatoses. Several fungal skin infections (dermatophytoses), such as tinea capitis and tinea corporis, are more frequently seen in children (Havlickova et al., 2008; Seebacher et al., 2008). This epidemiological dichotomy in fungal infections may relate to the physiologic characteristics of younger skin, which appears more permissive to colonization by diverse fungi. Conversely, tinea versicolor (pityriasis versicolor), which is primarily caused by mycelial growth of Malassezia spp., is more prevalent in adults (20s–40s) (Kyriakis et al., 2006). More detailed studies are necessary to elucidate the relationship between fungistatic property of skin and its link to pathogenic fungal colonization.
Along with finding distinct skin mycobiomes of children, we also identified features of mycobiome that relate to gender (Supplementary Figure S4). In adults, the skin of both genders was predominantly colonized by Malassezia (except foot sites). In children, however, taxonomic comparison showed Cryptococcus relative abundances were significantly higher in boys (Supplementary Figure S4), consistent with epidemiologic findings that Cryptococcus neoformans, a human pathogen underlying both meningitis and cryptococcosis, preferentially colonizes males (Lortholary et al., 2002; McClelland et al., 2013). Although our sample size was not powered to extensively compare fungal communities on girls versus boys, additional investigations of the fungal communities of different genders with a larger cohort size and more in-depth evaluation of sexual maturation status, i.e. endocrine studies, would be of great interest.
In this study, two different sequencing platforms were used: 454 pyrosequencing and Illumina MiSeq. With increasing technical advances and rapid platform turnover, more studies may utilize more than one sequencing platform. We extensively tested platform comparability, and confirmed statistically significant high correlation between the platforms (r=0.75–0.88, P < 2.2e-16; See Materials and Methods, and Supplementary Figure S5 and S6). Sequencing standardized control and mock community samples on each platform ensured optimal comparative analyses. Of note, the Illumina read-joining methods can alter which sequences pass quality control and discernably alter downstream results describing the fungal community composition (Supplementary Figure S6a and b).
It was especially striking to identify preferential colonization of prepubertal skin with M. globosa. This result suggested a selective colonization of Malassezia on the skin of children. M. globosa was previously reported to have stronger lipase activity compared to M. restricta, based on p-nitrophenyl olate metabolism in vitro (DeAngelis et al., 2007). More recently, Wu et al. showed different lipid assimilation specificity of Malassezia species, confirming lipid preferences of each Malassezia species (Wu et al., 2015). In the same report, the authors sequenced and compared the genomes of 14 known Malassezia species, and identified variable lipase gene family numbers in different species. Therefore, expansion and colonization of Malassezia may be affected by the surrounding niche, especially composition of lipid source. Investigating Malassezia colonization along with lipid composition of the skin in different age groups may provide insights into understanding molecular determinants of Malassezia colonization and pathogenesis of fungus-associated diseases, such as dandruff, atopic dermatitis and dermatitis (Gaitanis et al., 2012).
Materials and Methods
Subjects
Healthy adults (age 20s–30s, 19 individuals) and children (age <14, 14 individuals) volunteers were recruited from the Washington, DC metropolitan region, United States, from January 2011 to August 2015. Sample collection was approved by the Institutional Review Board of the National Human Genome Research Institute (http://www.clinicaltrials.gov/ct2/show/NCT00605878. Written informed consent was obtained from subjects (for adults) or parents or guardians of the subjects (for children). When age appropriate, children also provided assent. Subjects provided medical and medication history and underwent a dermatologic examination. Exclusion criteria included use of antibiotics within one year prior to skin sampling. Subjects bathed and/or showered with only non-antibacterial soap or cleansers for 7 days prior to sample collection. No bathing, shampooing or moisturizing was permitted for 24 hours before sample collection.
Sample collection, DNA extraction, PCR amplification and sequencing
Samples (including negative controls) were collected and processed similarly to those described previously (Findley et al., 2013; Grice et al., 2009; Oh et al., 2014). Specifically, Catch-All Sample Collection Swabs (Epicentre) were pre-moistened with Yeast Cell Lysis Solution (Epicentre) prior to sample collection. Sampling methods were comparable (data not shown; (Grice et al., 2008)). At each sampling event, controls were obtained by pre-moistening swabs and exposing them to air without skin contact to monitor for potential contamination. Swabs were stored in lysis solution at −80°C following collection. For extractions, skin swabs were incubated in Yeast Cell Lysis buffer and ReadyLyse Lysozyme Solution (Epicentre) for 1 h with shaking at 37°C. 5-mm steel beads were added to mechanically disrupt fungal cell walls using a Tissuelyser (Qiagen) for 2 minutes at 30 Hz, followed by 30 minutes incubation at 65°C for complete lysis. MPC Reagent was added to samples and resulting supernatant was processed using the PureLink Genomic DNA Kit (Invitrogen). DNA was eluted in DNA-Free PCR Water (MoBio). Control swabs also underwent DNA extraction processes and sequencing along with experimental samples, and no contamination from either reagents or experimental procedures were observed.
The ITS1 region was amplified using primers modified with 454 adapters 18S-F (5′-GTAAAAGTCGTAACAAGGTTTC) and 5.8S-1R (5′-GTTCAAAGAYTCGATGATTCAC) as previously described (Findley et al., 2013; Khot et al., 2009; Lennon et al., 2010). The following PCR conditions were used: 2.5ul 10X AccuPrime Buffer II, 0.2ul Accuprime Taq (Invitrogen), 0.1ul 18SF (100 mM), 2ul barcoded 5.8S-1R (5uM), 16.2ul PCR Water (MoBio), and 4ul of DNA. Reactions were performed in duplicate for 32 cycles, combined, purified using Agencourt AmpureXP (Beckman Coulter), and quantified using the Quant-IT dsDNA Kit (Invitrogen). Equivalent amounts of amplicons were pooled together, purified (MinElute PCR Purification Ki; Qiagen), and sequenced on a Roche 454 GS20/FLX platform.
For Illumina MiSeq sequencing, the ITS1 region was amplified with the primers 18S-F and 5.8S-1R using the strategy described (Fadrosh et al., 2014). The following PCR conditions were used: 2.5ul 10X PCR Buffer, 4ul dNTP Mix, 0.25ul Takara LA Taq Polymerase (Clontech), 1ul 18S-F (10uM), 1ul 5.8S-1R (10uM), 13.75ul PCR Water (MoBio), and 2.5ul DNA. Reactions were performed in duplicate for 30 cycles, combined, purified using Agencourt AmpureXP (Beckman Coulter), and quantified using the Quant-IT dsDNA Kit (Invitrogen). Equivalent amounts of amplicons were pooled together, purified with MinElute PCR purification kit (Qiagen), and sequenced on an Illumina MiSeq.
Sequence analysis pipeline
For ITS1 sequence analysis, a mothur-based pipeline was used, as previously described (Findley et al., 2013; Schloss et al., 2009). Briefly, sequences were pre-processed to remove primers and barcodes and subsampled to 3000 sequences per sample. Chimeras from PCR artifacts were identified and removed using UCHIME in mothur (Edgar et al., 2011). Next, remaining sequences (median 2995 reads, range 2877–3000) were classified down to the genus level with reference database from Findley, et al (Findley et al., 2013).
Malassezia ITS1 sequences were classified to species level as previously described (Findley et al., 2013). First, only Malassezia sequences were acquired via get.lineage command in mothur, and each sequence was classified phylogenetically using manually curated Malassezia database and pplacer software package (likelihood score >0.65) (Matsen et al., 2010). Later, species information was merged into mothur-compatible taxonomy file for further community analysis.
Platform comparability test
Due to evolution of sequencing technologies, two different sequencing platforms were used in this study: 454 pyrosequencing and Illumina MiSeq. To assess whether ITS1 amplicon sequencing from 454 and MiSeq were comparable, we analyzed results from two different microbial sources: i) samples from human skin, ii) Fungal Mock Community (FMC). For MiSeq, several paired-end joining methods (single read only, FLASh, mothur, PANDAseq, and PEAR) were tested for comparability with 454 sequencing results. Results from 40 human skin samples (5 sites each from 8 subjects) indicated that correlations of relative abundance of 25 major taxa (15 genera and 10 phyla) with 454 sequencing were significantly correlated to MiSeq (Supplementary Figure S5, r=0.75–0.88, P < 2.2e-16). Among tested joining methods, the mothur, PEAR and PANDAseq showed similar level of correlation with 454 data. Also, relative abundances of major taxa of FMC showed that 454 data is highly similar when MiSeq sequences were joined by mothur (Supplementary Figure S6a–c). Therefore, we used mothur-based paired-end joining method for MiSeq data in current analysis. We also tested the robustness of our reference database, by comparing results with the recent version of other ITS1 databases: UNITE (version 7, released at 08/01/2015) (Koljalg et al., 2005), and ITSoneDB (ITS1 GenBank annotation, released at 06/2014) (Santamaria et al., 2012) were similar with our database (Supplementary Figure S6d).
In addition, we tested possible effects of confounders, including sequencing platforms, collection dates and sample processing dates, on the dataset; however, no bias was detected.
Data access
The sequence data from this study have been submitted to NCBI BioProject under accession number 46333.
Community analysis
Community analysis was done with genus-level taxonomic classifications. R software was implemented to generate all plots including relative abundance of fungal genera and species. To estimate sampling saturation and subsampling size, rarefaction curves were generated for each sample, using rarefaction.single command in mothur. For alpha diversity (community evenness and richness), Shannon index was used, and for beta diversity (shared community structure and membership), Theta index was used (Yue and Clayton, 2005).
Statistics
All statistical analyses in figures are represented as mean ± standard error of the mean unless otherwise indicated, values for relative abundance in the text are represented as mean and a 95% confidence interval (CI) calculated using the normal approximation formula:
where n = number of samples, and σ = standard deviation. For statistical testing of differences in abundance and Shannon index between groups, non-parametric Wilcoxon rank sum test was used (wilcox.test in the statistical software R). Analysis of molecular variance (AMOVA) was used with theta index to test for statistically significant differences between groups (Excoffier et al., 1992).
Supplementary Material
Acknowledgments
We thank Mark C. Udey, Keisuke Nagao, and Carmen Contreras-Sesvold for their helpful discussions; and Sheila Phang and Amynah Pradhan for their underlying efforts. This work was supported by NIH NCI/CCR and NHGRI Intramural Research Program and a grant of the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI15C1095; J-HJ). Sequencing was funded by grants from the National Institutes of Health (1UH2AR057504-01 and 4UH3AR057504-02). This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Abbreviations
- Ac
Antecubital fossa
- Ba
Back
- Ea
External auditory canal
- Fh
Forehead
- Ic
Inguinal crease
- Mb
Manubrium
- Oc
Occiput
- Ra
Retroauricular crease
- Vf
Volar forearm
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Archer GL, Climo MW. Staphylococcus aureus bacteremia–consider the source. The New England journal of medicine. 2001;344(1):55–6. doi: 10.1056/NEJM200101043440110. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;346(6212):954–9. doi: 10.1126/science.1260144. [DOI] [PubMed] [Google Scholar]
- DeAngelis YM, Saunders CW, Johnstone KR, Reeder NL, Coleman CG, Kaczvinsky JR, Jr, et al. Isolation and expression of a Malassezia globosa lipase gene, LIP1. The Journal of investigative dermatology. 2007;127(9):2138–46. doi: 10.1038/sj.jid.5700844. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131(2):479–91. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faergemann J, Fredriksson T. Age incidence of Pityrosporum orbiculare on human skin. Acta dermato-venereologica. 1980;60(6):531–3. [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–70. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. The Malassezia genus in skin and systemic diseases. Clinical microbiology reviews. 2012;25(1):106–41. doi: 10.1128/CMR.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–2. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Renaud G, Young AC, Program NCS, Bouffard GG, et al. A diversity profile of the human skin microbiota. Genome research. 2008;18(7):1043–50. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA. The skin microbiome. Nature reviews Microbiology. 2011;9(4):244–53. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta AK, Batra R, Bluhm R, Boekhout T, Dawson TL., Jr Skin diseases associated with Malassezia species. Journal of the American Academy of Dermatology. 2004;51(5):785–98. doi: 10.1016/j.jaad.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Kohli Y. Prevalence of Malassezia species on various body sites in clinically healthy subjects representing different age groups. Medical mycology. 2004;42(1):35–42. doi: 10.1080/13693780310001610056. [DOI] [PubMed] [Google Scholar]
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(Suppl 4):2–15. doi: 10.1111/j.1439-0507.2008.01606.x. [DOI] [PubMed] [Google Scholar]
- Hawkins DM, Smidt AC. Superficial fungal infections in children. Pediatric clinics of North America. 2014;61(2):443–55. doi: 10.1016/j.pcl.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Jang SJ, Lim SH, Ko JH, Oh BH, Kim SM, Song YC, et al. The Investigation on the Distribution of Malassezia Yeasts on the Normal Korean Skin by 26S rDNA PCR-RFLP. Annals of dermatology. 2009;21(1):18–26. doi: 10.5021/ad.2009.21.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khot PD, Ko DL, Fredricks DN. Sequencing and analysis of fungal rRNA operons for development of broad-range fungal PCR assays. Applied and environmental microbiology. 2009;75(6):1559–65. doi: 10.1128/AEM.02383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, et al. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. The New phytologist. 2005;166(3):1063–8. doi: 10.1111/j.1469-8137.2005.01376.x. [DOI] [PubMed] [Google Scholar]
- Kyriakis KP, Terzoudi S, Palamaras I, Pagana G, Michailides C, Emmanuelides S. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49(6):517–8. doi: 10.1111/j.1439-0507.2006.01285.x. [DOI] [PubMed] [Google Scholar]
- Lennon NJ, Lintner RE, Anderson S, Alvarez P, Barry A, Brockman W, et al. A scalable, fully automated process for construction of sequence-ready barcoded libraries for 454. Genome biology. 2010;11(2):R15. doi: 10.1186/gb-2010-11-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortholary O, Improvisi L, Fitting C, Cavaillon JM, Dromer F. Influence of gender and age on course of infection and cytokine responses in mice with disseminated Cryptococcus neoformans infection. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2002;8(1):31–7. doi: 10.1046/j.1469-0691.2002.00375.x. [DOI] [PubMed] [Google Scholar]
- Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland EE, Hobbs LM, Rivera J, Casadevall A, Potts WK, Smith JM, et al. The role of host gender in the pathogenesis of Cryptococcus neoformans infections. PloS one. 2013;8(5):e63632. doi: 10.1371/journal.pone.0063632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Deming C, Conlan S, Program NCS, Kong HH, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514(7520):59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome medicine. 2012;4(10):77. doi: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino LC, Tseng CH, Strober BE, Blaser MJ. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. Journal of clinical microbiology. 2006;44(8):2933–41. doi: 10.1128/JCM.00785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochi PE, Strauss JS, Downing DT. Age-related changes in sebaceous gland activity. The Journal of investigative dermatology. 1979;73(1):108–11. doi: 10.1111/1523-1747.ep12532792. [DOI] [PubMed] [Google Scholar]
- Santamaria M, Fosso B, Consiglio A, De Caro G, Grillo G, Licciulli F, et al. Reference databases for taxonomic assignment in metagenomics. Briefings in bioinformatics. 2012;13(6):682–95. doi: 10.1093/bib/bbs036. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75(23):7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008;166(5–6):335–52. doi: 10.1007/s11046-008-9100-9. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy R. Tinea corporis and tinea capitis. Pediatrics in review/American Academy of Pediatrics. 2007;28(5):164–74. doi: 10.1542/pir.28-5-164. [DOI] [PubMed] [Google Scholar]
- Sugita T, Suzuki M, Goto S, Nishikawa A, Hiruma M, Yamazaki T, et al. Quantitative analysis of the cutaneous Malassezia microbiota in 770 healthy Japanese by age and gender using a real-time PCR assay. Medical mycology. 2010;48(2):229–33. doi: 10.1080/13693780902977976. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nature reviews Immunology. 2014;14(6):405–16. doi: 10.1038/nri3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. The New England journal of medicine. 2001;344(1):11–6. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- White TC, Findley K, Dawson TL, Jr, Scheynius A, Boekhout T, Cuomo CA, et al. Fungi on the skin: dermatophytes and Malassezia. Cold Spring Harbor perspectives in medicine. 2014;4(8) doi: 10.1101/cshperspect.a019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Zhao H, Li C, Rajapakse MP, Wong WC, Xu J, et al. Genus-Wide Comparative Genomics of Malassezia Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin. PLoS genetics. 2015;11(11):e1005614. doi: 10.1371/journal.pgen.1005614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue JC, Clayton MK. A similarity measure based on species proportions. Commun Stat-Theor M. 2005;34(11):2123–31. [Google Scholar]
- Zhang E, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiology and immunology. 2011;55(9):625–32. doi: 10.1111/j.1348-0421.2011.00364.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.