


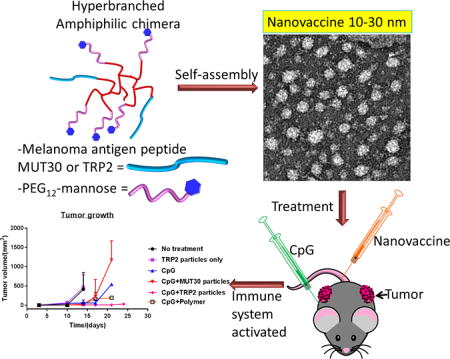
Abstract
Personalized cancer vaccines (PCVs) are receiving attention as an avenue for cancer immunotherapy. PCVs employ immunogenic peptide epitopes capable of stimulating the immune system to destroy cancer cells with great specificity. Challenges associated with effective delivery of these peptides include poor solubility of hydrophobic sequences, rapid clearance, and poor immunogenicity, among others. The incorporation of peptides into nanoparticles has the potential to overcome these challenges, but the broad range of functionalities found in amino acids presents a challenge to conjugation due to possible interferences and lack of reaction specificity. Herein, a facile and versatile approach to generating nanosized PCVs under mild nonstringent conditions is reported. Following a simple two-step semibatch synthetic approach, amphiphilic hyperbranched polymer–peptide conjugates were prepared by the conjugation of melanoma antigen peptides, either TRP2 (hydrophobic) or MUT30 (hydrophilic), to an alkyne functionalized core via strain-promoted azide–alkyne click chemistry. Self-assembly of the amphiphiles gave spherical nanovaccines (by transmission electron microscopy) with sizes in the range of 10–30 nm (by dynamic light scattering). Fluorescently labeled nanovaccines were prepared to investigate the cellular uptake by antigen presenting cells (dendritic cells), and uptake was confirmed by flow cytometry and microscopy. The TRP2 nanovaccine was taken up the most followed by MUT30 nanoparticles and, finally, nanoparticles without peptide. The nanovaccines showed good biocompatibility against B16–F10 cells, yet the TRP2 peptide showed signs of toxicity, possibly due to its hydrophobicity. A test for immunogenicity revealed that the nanovaccines were poorly immunogenic, implying the need for an adjuvant when administered in vivo. Treatment of mice with melanoma tumors showed that in combination with adjuvant, CpG, groups with the peptide nanovaccines slowed tumor growth and improved survival (up to 24 days, TRP2) compared to the untreated group (14 days).
Graphical Abstract
INTRODUCTION
Immunotherapy has attracted significant interest as an avenue for cancer treatment and vaccination.1 The appeal emanates from the exploitation of the body’s own immune system to overcome cancer by stimulating the production of cytotoxic T-cells capable of destroying tumor cells with impeccable specificity.2 Strategies employed to manipulate the immune system in cancer immunotherapy include the use of subunit antigens such as peptide epitopes of tumor-associated antigens or mutated neoantigens, dendritic-cell-based vaccines, wholetumor adoptive T-cell therapy, and immune checkpoint blockades, among others.3–6 Peptide-based antigens are probably the most popular and widely studied due to the simplicity of their synthesis and general biocompatibility.7 Eliciting an immune response with peptide antigens requires the peptide to be delivered to antigen-presenting cells (APCs), primarily dendritic cells (DCs), which are professional APCs and a key initiator of the adaptive immune response.8 Once endocytosed by DCs, the peptides are processed for presentation intracellularly and loaded onto the major histocompatibility complex (MHC) leading to activation of T-cells, which can destroy tumor cells.9 However, the efficacy of antigen peptides tends to be negatively affected by rapid clearance, extracellular and enzymatic degradation, poor solubility (hydrophobic peptides), poor immunogenicity, and poor uptake of the peptide by DCs.10,11 With current advances in cancer biology pointing toward the need for personalized vaccines based on tumor-derived mutated peptide sequences,12–17 biocompatible and nontoxic delivery systems that can efficiently deliver peptides and neoantigens to DCs could help overcome the current shortcomings of antigenic therapies.
The use of nanoparticles as peptide-antigen delivery vehicles and carriers is currently receiving attention to overcome the aforementioned issues.10,18,19 Nanoparticles can improve circulation time, protect peptides against enzymatic degradation, and improve the bioavailability and solubility of hydrophobic antigen peptides.20 To this end, peptide vaccine nanoparticle systems based on lipids, polymers, inorganic nanoparticles, self-assembled polypeptides, or carbon nanotubes have been reported, with lipids and polymers being the most widely used.21,22 Successful fabrication and implementation of such systems requires consideration of variables such as material toxicity, particle size, particle shape, surface charge, and stiffness or rigidity.23,24 For efficient delivery, size is a major parameter. Small particles in the 10–50 nm size range selectively accumulate in the lymphatic system and lymph nodes, where DCs are present in large numbers, whereas larger particles are mostly taken up by macrophages.25,26 Targeting to DCs is also possible by decorating the nanoparticle surface with ligands such as mannose, DEC-205, and CD40.27–29 Tethering or encapsulating multiple peptide antigens or adjuvants can be easily accomplished during the formulation and fabrication of nanoparticulate vaccines. Furthermore, the multivalent display of antigens on the surface of nanoparticles has the potential for enhancing peptide vaccine immunogenicity.30,31
Among the materials employed, combining polymers with immunogenic peptides presents a versatile route for the production of a wide range of peptide-based vaccine nanoparticles.32 The incorporation of the peptides with polymers affords polymer–peptide conjugates that can be fabricated into nanoparticles via self-assembly, thus ensuring that the majority of the immunostimulating peptide sequences identified during sequencing can be utilized for personalized cancer treatment.16,33 Advances in synthetic polymer chemistry have afforded polymers with an extensive array of architectures (e.g., linear, star, block, hyperbranched, etc.),34,35 (bio)-degradability, biocompatibility, and functionalities that allow the simple conjugation of peptides through well-established chemistries.36,37 Thus, major nanovaccine design variables such as particle size, toxicity, and shape can be easily controlled.38 Hyperbranched polymers represent an interesting class of polymers that can be easily adapted to peptide nanovaccines in a similar manner to dendrimers.39–42 However, labor-intensive synthesis, limited commercial availability, and steep prices negatively impacts the widespread use of dendrimers as nanovaccine polymer constructs. However, hyperbranched polymers are easier to prepare and not as time-consuming as dendrimers. Hyperbranched amphiphiles can exist as unimolecular micelles or aggregates of several amphiphilic (micelles) nanoparticles; thus, multiantigen presentation (multivalent display) akin to the cell surface is possible, which can lead to enhanced immunogenicity.43 A notable feature of these materials is that the disaggregation of the aggregates (particles) yields unimolecular micelles, which also exhibit multivalent antigen display. Furthermore, the intrinsic structure of hyperbranched amphiphiles prevents antigen entanglement, thus ensuring recognition upon uptake by DCs.44
Various approaches to coupled hyperbranched polymers with immunostimulating peptide epitopes had been reported. Pioneering reports relied on coupling the N-terminal −NH2 groups of the peptides to carboxylic acid groups (or succinimide activated esters) on the surface of the branched core.45–48 Although the resulting conjugates were shown to be immunostimulating, a limitation in terms of functionalities existed. For instance, the coupling of peptide sequences with multiple glutamic acid and lysine units would require the protection and deprotection chemistry to work as they bear functional groups that also undergo amide formation. Considering the myriad of peptide sequences generated upon screening for personalized vaccines, the amide formation approach would thus require modifications based on the sequence(s) of the peptide(s) to be attached to ensure orthogonality. Conjugation via the disulfide formation of immunostimulating cysteine containing peptides to a protected thiol functionalized hyperbranched core has also been reported.48,49 This approach, however, suffers from potential self-coupling (disulfide formation) of the immunostimulating peptide, which can reduce the yield. Glaffig et al. reported the preparation of antitumor vaccines by conjugating hyper-branched polymers with an antigen via a copper-catalyzed “click” reaction.44,50–53 In their work, a hyperbranched polyglycerol terminated with alkyne groups was first prepared and subsequently conjugated to an antigen, which is a water-soluble glycosylated tumor-associated mucin 1 peptide (MUC1) fused with a T-cell immunostimulating epitope derived from tetanus toxoid. The click-chemistry approach is advantageous in terms of reaction yield, its tolerance to a wide variety of functionalities, and mild reaction conditions. Thus, this approach accommodates a greater variety of peptide sequences. However, the use of copper in biological systems poses toxicity concerns, and moreover, the presence of copper binding functionalities in the peptide sequences can limit the application of this approach.54 It is noteworthy that in the work available in the literature, both the hyperbranched cores and the immunostimulating peptides used were water-soluble. The conjugates, hence, would not be amphiphilic, which is a requisite for self-assembly to form nanoparticles, and no information on the obtained sizes is available.
As research on cancer immunotherapy gathers momentum toward personalized cancer vaccines,15–17 peptide epitopes of varying solubility, degradability and toxicity are continually emerging, necessitating the need for versatile, efficient approaches for the fabrication of nontoxic, low-cost and biocompatible nanoparticulate systems with effective delivery capabilities. Thus, a synthetic approach, which is facile, can be carried out under mild conditions, circumvents toxicity concerns, and is tolerant to the wide range of functionalities encountered in amino acids without the need for modification, would be ideal. Furthermore, the system should be able to accommodate the incorporation and delivery of both hydrophilic as well as the more-challenging hydrophobic peptides. For instance, the administration of hydrophobic peptides, such as the tumor-associated antigen peptide derived from tyrosinase-related protein 2 (TRP2, with a sequence of SVYDFFVWL) and pan DR T-helper epitope (PADRE), presents a challenge due to poor solubility in aqueous solutions.55,56 Herein, we demonstrate a facile yet versatile approach to preparing antigen peptide delivering nanovaccines based on amphiphilic hyperbranched polymer–peptide conjugates (chimeras) synthesized via copper-free click chemistry. In this approach, we prepared antigen peptide containing hyperbranched chimeras and showed that the material could form nanoparticles capable of triggering an immune response in vivo against melanoma cancer. To construct the amphiphiles, we used the well-established but powerful biorthogonal strain promoted azide–alkyne click chemistry (SPAAC), which negates the use of potentially toxic copper that is widely employed in metal-catalyzed click reactions.52,57–59 Moreover, SPAAC is tolerant to all functionalities encountered in amino acids, can be conducted under very mild conditions, and does not require a complex reaction setup. Starting from a commercially available nontoxic and biodegradable hyper-branched polymer, Boltorn H40, we prepared amphiphilic conjugates that form nanoparticles with sizes of 10–30 nm, which is within the range (10–50 nm) advocated for efficient antigen transportation to DCs. The multiple reaction sites also allow for multiple peptide epitope attachment. Furthermore, our approach is adaptable to both hydrophobic and hydrophilic antigen peptides, yielding particles within 10–50 nm, which we demonstrate using the hydrophobic TRP2 peptide and a mutated polar neoantigen sequence, MUT30, PSKPSFQEFVD-WENVSPELNSTDQPFL in murine B16-F10.60 We were able to obtain water-soluble TRP2 nanoparticles and show that they elicit an immune response in vivo against melanoma. We also synthesized neoantigen (MUT30) nanoparticles, although these did not elicit a notable immune response, possibly due to poor or low expression of the mutated sequence in the cell line.
RESULTS AND DISCUSSION
Preparation and Characterization of Amphiphilic Hyperbranched Macromolecular Chimeras
To obtain the amphiphilic polymer–peptide conjugates, we leveraged strain promoted azide–alkyne click chemistry (SPAAC) following the strategy outlined in Figure 1. SPAAC offers high yields under mild conditions with great selectivity that eliminates interference from amino-acid functionalities.
Figure 1.

Simplified concept for the design of antigen-decorated nanoparticles and their intended application as cancer immunotherapy agents.
Synthesis and Characterization of the Azide-Reactive Hyperbranched Polymer Core
An azide-reactive lipophilic core was constructed starting from the commercially available hyperbranched biodegradable polyester, Boltorn H40. Boltorn H40 has 64 terminal hydroxyl groups available for functionalization that were converted into carboxylic groups via an esterification reaction with succinic anhydride (Scheme 1A). Conversion of the hydroxyl groups into carboxyl groups was ascertained by 1H nuclear magnetic resonance (NMR) (Figures 2A,B) and Fourier transform infrared (FTIR) (Figure 2D). The −CH2− protons from the ring-opened succinic anhydride were observed at ca. 2.4 ppm by 1H NMR, while a carboxylic acid stretch (3500–2500 cm−1) and a carbonyl stretch due to the C═O of the carboxylic acid at ca. 1600 cm−1, which are absent in the spectrum of the starting materials, were observed in the FTIR spectrum of the resulting product. The cyclooctyne, dibenzocyclooctyne–amine (DBCOA), was coupled to the terminal carboxyl groups on the polymer via the carbodiimide-mediated coupling, and formation of the desired product was confirmed by 1H NMR (Figure 2C) and FTIR (Figure 2D). Peaks in the aromatic region of the NMR, which were absent in the spectra of the carboxyl functionalized polymer, were observed due to the presence of the phenyl rings in the cyclooctyne. In addition, a peak due to amide bond formation was observed at ca. 7.8 ppm and another peak at 5.3 ppm due to the CH2 adjacent to the nitrogen in the cyclooctyne. The carboxylic acid stretch was absent in the spectrum of the alkyne product, while the appearance of a new peak at 3250 cm−1 due to amide bond formation was observed, indicating consumption of the carboxyl groups. Furthermore, a slight shift in number-average molecular weight, Mn, from 7670 to 14200 g/mol was observed via size-exclusion chromatography (SEC) (Figure 2E), indicating an increase in the hydrodynamic volume of the polymer. The number of available alkynes on the hyper-branched core was quantified by measuring the amount of phenyl 6-azidohexanoate (Figure S1) consumed in a reaction with a known amount of polymer. By calculating the initial and final azide molecule concentrations based on high-performance liquid chromatography (HPLC), the amount of azide consumed could be established and was related to the number of alkynes per given mass. The amount of alkynes per milligram was calculated to be 1.42 µmol/mg, equating to 37 alkyne groups per hyperbranched core (based on MALDI-TOF, Mn = 2000 Da for starting Boltorn H40). Analysis of different batches gave results in the range of 1.2–1.5 µmol/mg.
Scheme 1.

Synthetic Strategy Employed to Obtain the Polymer–Peptide Conjugatesa
a(A): (i) Esterification of Boltorn H40 (1) with succinic anhydride to generate −COOH terminal groups, (ii) coupling of DBCOA to the carboxylic acid functionalized Boltorn core (2), (iii) SPAAC of alkyne functionalized hyperbranched core (3) with azido antigen peptide targeting 10% of available alkynes to obtain peptide-functionalized hyperbranched peptide functionalized polymer (4) and (iv) SPAAC of remaining alkyne groups on hyperbranched core with PEG mannose to yield the amphiphilic hyperbranched chimera (5). In panel B, the synthesis of the non-azide functionalized peptides (MUT30 and TRP2) and azido peptides (MUT30-azide and TRP2-azide) via SPPS on a Wang resin (TRP2 and azido TRP2) and on a Rink amide resin (MUT30 and azido MUT30).
Figure 2.

1H NMR spectra of pure Boltorn H40 before modification (A), after esterification (B), and after coupling DBCOA (C); FTIR spectra (D) showing changes functional groups starting from pure Boltorn H40 before modification after esterification and functionalization with DBCOA; and gel permeation chromatography (GPC) chromatograms (E) of Boltorn H40 (Mn = 7671 g/mol, PDI = 1.97) before and after alkyne functionalization, B64DBAC (Mn = 14183 g/mol, PDI = 2.70).
Synthesis and Characterization of Azide-Function-alized Peptides
We proceeded to prepare the azide-functionalized segments for conjugation to the alkyne-functionalized polymer starting with the peptides (Scheme 1B). The TRP2 peptide and its azide-functionalized analog were prepared via the Fmoc-strategy solid-phase peptide synthesis (SPPS), starting from a Wang resin via microwave-assisted peptide synthesis (Scheme 1B). For the azido peptides, we incorporated a PEG spacer and azidovaleric acid to avoid burying peptide deeply in the hydrophilic layer of the conjugate upon particle formation (vide infra). Thus, Fmoc–N-amido–dPEG4–acid was coupled at the N-terminus of the TRP2 peptide prior to adding the terminal azide functionality by coupling azidovaleric acid as the last “amino acid”. The peptides were afforded by cleavage from the resin in acidic conditions followed by purification using preparative HPLC. Analysis of the obtained product via matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF), liquid chromatography–mass spectrometry (LC–MS), and amino acid analysis (AAA) revealed the desired masses and high purity (see Figures S2–S5 and Tables S1–S4). Similarly, the MUT30 and azido MUT30 peptides were prepared and characterized as mentioned for TRP2 peptides but on a Rink amide resin. In addition, the azido peptides were also analyzed via FTIR, which revealed the presence of the azide stretch at 2109 cm−1.
Preparation of Azide-Functionalized PEG Mannose
Following the preparation of the azido peptides, we prepared the other component of the amphiphile, bifunctional azido PEG12 capped with mannose (Scheme 1A). Mannose was included to cap the PEG as a neutral but polar molecule because a strongly negative-charged moiety could negatively affect the particle uptake. Moreover, the presence of mannose also augments uptake by DCs due to the presence of mannose receptors on the DCs. The product of the coupling reaction was judged to have been formed based on mass spectrometry with the peak of the desired product appearing at m/z = 827.41 Da [M + Na]+, which is higher than that of the starting polymer and mannose amine hydrochloride (Figure S6). We observed that for the subsequent reaction of this product with the alkyne, it is not necessary to purify the product, thanks to the selectivity of the SPAAC reaction, which also contributes toward the simplification of the material-preparation process.
Synthesis of Amphiphilic Hyperbranched Macromolecular Chimeras via SPAAC
With the requisite building blocks constructed, conjugation via SPAAC ensued. Conjugation of the azido peptides and azido PEG mannose to the alkyne polymer was done in two steps via a semibatch process starting with the peptide (Scheme 1). First, the peptide was conjugated to the polymer on the basis of a target functionalization of 10% mol of the alkyne functional groups (i.e., 0.1 equiv). The reaction was found to be quantitative based on the disappearance of the azide peak in the FTIR spectrum (Figure 3) as well as the disappearance of the peptide mass signal upon analysis by MALDI. However, we could not ascertain the mass of the conjugate based on MALDI due to poor ionization of the conjugates. The low peptide loading ensured quantitative consumption of the peptide by the polymer and at the same time reduced surface coverage by the peptide, which would help minimize chances of interaction and entanglement between the peptide epitopes. Second, the azido PEG mannose was conjugated to the polymer by adding 3 equiv (compared to the starting alkyne functional groups) of crude azide PEG mannose to saturate the remaining alkyne groups (90% mol). A polymer-only conjugate was also prepared following a similar approach, wherein the peptide step was skipped and the alkyne core was reacted with 3 equiv of azido PEG mannose. After 3 days of reaction, analysis by FTIR (Figure 3A,C,E) revealed the presence of an azide peak due to the presence of excess azido PEG mannose in solution, which was then removed by dialysis to afford the pure amphiphiles. Analysis of the amphiphiles via FTIR (Figure 3) confirmed a complete removal of the free PEG mannose (disappearance of the azide peak at 2109 cm−1), and a successful conjugation of PEG mannose onto polymer (a strong signal of the hydroxyl groups of mannose at 3500 cm−1). From 1H NMR (Figure 3B,D), the peaks belonging to the protons of the starting alkyne hyperbranched polymer disappeared, while a strong ethylene −CH2− signal due to PEG (from PEG mannose, at 3.7 ppm) as well as the peaks of the peptide were observed. SEC also showed a clear shift in Mn, from the low Mn of the alkyne hyperbranched polymer (∼14200 g/mol) to a higher Mn for the conjugates, which is evidence of successful conjugation (Figure 3F). The bimodal SEC peaks can be explained by taking into account the imperfect nature of hyperbranched polymers (compared to dendrimers) and, hence, the difference in number of terminal functionalities per hyperbranched molecule. Molecules that bear more terminal groups lead to a greater Mn shift upon their functionalization than ones with less terminal functionalities resulting a in shoulder being observed after coupling with DBCOA (retention time ca. 14 min). Similarly, further functionalization with larger molecules (the azido peptide and the PEG), which are conjugated randomly, result in an even-greater Mn shift and further broadening of the peaks.
Figure 3.

FTIR spectra obtained at different stages of preparation of the polymer amphiphile– and polymer–peptide conjugates (A, C, E) (DBACB64–MUT30 crude and DBACB64–TRP2 crude refer to crude reaction mixtures following the SPAAC reaction between the alkyne core and the respective azido peptides. The reaction with the azido-PEG mannose (DBACB64–PEG–MUT30 crude and DBACB64–PEG–TRP2 crude) then follows, and analysis is done on the reaction mixture before purification (crude) and after purification (pure). 1H NMR spectra obtained for the azido peptides, azido-PEG12–mannose, DBACB64, and of the polymer–peptide conjugates (B and D) and GPC chromatograms (F) of the polymer amphiphile (DBACB64–PEG–mannose, Mn = 39565 g/mol, PDI = 2.62) and polymer–peptide conjugates (DBACB64–PEG–MUT30, Mn = 39041 g/mol, PDI = 2.01 and DBACB64–PEG–TRP2, Mn = 31489 g/mol, PDI = 1.91) compared to the starting polymer B64DBAC (Mn = 14183 g/mol, PDI = 2.70).
Nanovaccine Formation via Self-Assembly
The amphiphilic conjugates were self-assembled in aqueous solution, and the size and morphology of the formed nanoparticles were assessed by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Figures 4A,B). Small nanoparticle sizes (10–50 nm) are considered to be ideal as antigen nanocarriers aimed at delivering antigens to the DCs. This was taken into consideration in the design of the conjugates leading to the selection of short-chain PEG arms (dPEG4) for the hydrophilic segment of the conjugate. The spacers incorporated in the peptides were chosen to be of sufficient length to ensure that the peptide would be on the periphery when the nanoparticles were formed but not too long to significantly increase the size. In addition to minimizing entanglement, the incorporation of a low amount of peptide also ensured that amphiphilicity would be maintained even when the conjugated peptide is hydrophobic. Upon self-assembly, the TRP2 particles had a size of 11 nm, measured by DLS, while MUT30 particles were 19 nm (Figure 4B). In both cases, a negative ζ potential (−7.5 mV for TRP2 particles and −17.5 mV for MUT30 particles) (Figure 4B) was observed. This negative charge is due to the negative peptide charge (calculated at pH 7 as −1 for TRP2 and −4 for MUT30); in addition, a small amount of the azido–dPEG12–NHS ester could have undergone hydrolysis of the NHS ester, resulting in the formation of carboxylic acid groups. The polymer-only conjugates also formed nanoparticles with a DLS size of 29 nm and a negative ζ potential due to potential PEG NHS ester hydrolysis as discussed above. For all conjugates, nearly spherical particles were observed via TEM with sizes ranging from 13 to 18 nm (Figure 4A). It is noteworthy that among different batches, the sizes obtained were within the 10–30 nm range, ideal for the delivery of antigens. The stability of the particles in aqueous solution (PBS, pH = 7.4) was determined by comparing the sizes and ζ potentials soon after preparation and 7 days later (Figure 4B). No precipitation was observed, and only small variations in size (<6 nm) were observed indicating good stability over the 1 week period. However, the ζ potential showed some significant changes possibly due to slow hydrolytic degradation of the peptide over time. After self-assembly and purification by dialysis, we confirmed the integrity of the structure of the peptide by amino acid analysis, which revealed the presence of expected amino acids suggesting that the peptide does not undergo any appreciable degradation during the processing and purification steps (Tables S2 and S4). Of interest to note is that the approach yields reproducible results with very little variations in size and morphology between batches. Moreover, we were also able to prepare a nanovaccine using another azide-modified neoantigen, MUT44 (EFKHIKAFDRTFANNPGPMVVFATPGM),60 which gave nanoparticles in the 10–30 nm range (results not shown). Substituting mannose with serinol, which carries two hydroxyl groups for the modification of the azido PEG, also resulted in nanoparticles in the same size range for the MUT30, TRP2, and MUT44 peptides (results not shown). The quantities of peptide obtained by amino acid analysis for different batches were typically 8–12% (w/w) of the polymer-peptide conjugates.
Figure 4.

TEM images of polymer particles and polymer–peptide conjugates nanoparticles (A). Size and ζ potential of nanoparticles obtained via DLS soon after preparation and 7 days later (B). UV–vis spectra of pHrodo red labeled nanoparticles (strong signal) and nonfluorescently labeled nanoparticles (flat lines) (C). GPC spectra of pHrodo red labeled polymer only and polymer–peptide conjugates (strong UV and RI signals) and nonfluorescently labeled polymer only and polymer–peptide conjugates (strong RI signal and negligible UV signal). (D, E) Particle size, ζ potential (top table), and TEM images (i–iv) of pHrodo red labeled nanoparticles ((i) polymer–pHrodo nanoparticles, (ii) MUT30–pHrodo nanoparticles, and (iii–iv) TRP2 pHrodo nanoparticles from different portions of the grid representative of the bimodal distribution with most parts of the grid represented by panel iii).
Preparation of Fluorescently Labeled Chimeras for Cell-Uptake Assay
To have an effect on the immune system, the particles need to be taken up by APCs, especially DCs. The ability of the prepared nanoparticles to be taken up by mouse DCs ex vivo was thus assessed. Nanoparticles were labeled with a fluorescent pH-sensitive dye (pHrodo red) to be tracked by microscopy and flow cytometry. The pHrodo red fluorescent dye has the advantage of being poorly fluorescent at neutral or basic pH (≥ 7), and it becomes strongly fluorescent once it enters an acidic environment. The resulting change in fluorescence can thus be attributed to the intracellular uptake of the particle into the endocytic lysosomal system.61 Thus, azido pHrodo red was prepared by coupling the commercially available pHrodo red activated ester with amino–PEG3–azide. However, the pHrodo red structure is proprietary, so it was not possible to obtain standard characterization information. The crude product was purified by preparative HPLC to determine which fraction had the desired product. Subsequently, the collected fractions were freeze-dried and reacted with the alkyne core on a small scale followed by analysis of the products by SEC while monitoring the RI signal and UV detector signal at 570 nm. Considering that the alkyne-functionalized polymer does not absorb at 570 nm but pHrodo red has a strong absorption signal at that wavelength, we were able to identify fractions with the desired product by comparing the UV detector signal against the refractive index detector signal. When the dye is conjugated to the alkyne polymer, signals are observed in both the RI and UV detector. Without dye conjugation, only the RI signal is observed (Figure 4C,D).
Formation of Fluorescently Labeled Nanoparticles
pHrodo red-labeled nanoparticles were therefore prepared using a similar approach to that employed for conjugating peptides and PEG mannose. Azido pHrodo red was conjugated to the alkyne core targeted at 20% mol of the alkyne moieties, and after completion of the reaction, based on the disappearance of the azide peak by FTIR, the resultant molecule was assembled as previously discussed. We were able to obtain fluorescently labeled nanoparticles with sizes in the 19–37 nm range by DLS. However, by DLS, the TRP2–pHrodo particles exhibited a bimodal distribution with a 19 nm population as well as a second population at 76 nm probably existing as clusters of smaller micelles. We turned to TEM for an indication of the distribution and morphology. The individual particles were observed to be nearly spherical by TEM, with most of the nanoparticles having diameters ranging between 12 and 26 nm (Figure 4E). Interestingly, the DLS result for TRP2–pHrodo particles was corroborated by TEM, which showed the presence of some larger particles (48 nm) in some parts of the TEM grid in addition to the majority of the particles, which were small (23 nm). Sizes observed by TEM were generally smaller than those obtained via DLS due to TEM sizes being measured in the dehydrated state, whereas the DLS measures size in the hydrated state. The ζ potential of polymer–pHrodo particles was observed to be significantly decreased compared to its nonlabeled counterpart, while peptide-containing nanoparticles showed similar ζ potentials regardless of dye labeling. This could be attributed to the azido pHrodo red conjugate and the azido mannose being of nearly the same length. Taking into account the fact that the molar mass of pHrodo red succinimide is given by the manufacturer as ca. 650 g/mol, while that of the 11-azido-3,6,9-trioxaundecan-1-amine used for azide modification is 218 g/mol, the conjugate of the two gives a compound with a molar mass of ca.750 g/mol, which is close to that of the azido-PEG-mannose, ca. 800 g/mol. Therefore, in the case of the polymer-only nanoparticles, unlike where there are peptides that are longer, the particles possibly form with both the pHrodo red and the polymer extended out to a similar length, and thus, the pHrodo red also significantly impacts the surface charge. Without the knowledge of the structure of the pHrodo red (protected by the manufacturer), it is, however, difficult to say what its influence on the ζ potential is. In samples with peptides, the peptides are extended out and influence the surface charge to a larger extent; hence, the ζ potential differences are minimal between the labeled and unlabeled samples. To confirm that the particles maintained the expected characteristics of pHrodo red, we incubated the particles in aqueous solution at pH 7.4 and pH 5 at the same concentration then measured their fluorescence on a Tecan plate reader. Fluorescence intensities at pH 5 were 20 times or greater than those observed at pH 7.4, indicating that the characteristics of the dye had not changed (Figure S7).
Cellular Uptake of Fluorescently Labeled Nano-particles
The cellular uptake of the pHrodo-labeled nanoparticles was assessed by incubation with bone marrow derived murine DCs at 4 °C and at 37 °C. Normal cellular uptake of particles is expected at 37 °C, while at 4 °C, cellular internalization is diminished albeit adherence of particles to the surface remains possible.62 Thus, if particles are not internalized by cells, a similar dye signal is expected at both temperatures at any specific time point. However, if cellular uptake occurs, the dye signal will differ significantly, with a stronger fluorescence intensity expected at 37 °C. Indeed, by flow cytometry, the cellular pHrodo fluorescence signal intensity was greater at 37 °C than at 4 °C, after both 4 and 24 h of incubation, for all nanoparticles tested (Figure 5A). At 37 °C, particle uptake was observed to increase with time as evidenced by the increase in fluorescence intensity with the time of incubation. TRP2–pHrodo particles showed the strongest signal, indicating more intracellular uptake than MUT30–pHrodo particles, which were in turn taken up more than the polymer–pHrodo particles based on fluorescence mean intensities at 4 and 24 h (see Figure S8). Because all of the nanoparticles were negatively charged, the superior uptake of the TRP2–pHrodo particles can be attributed to the combination of the small nanoparticle size and the presence of the hydrophobic peptide. Smaller nanoparticles are generally taken up more than larger particles, while hydrophobic molecules are known to cause membrane destabilization, which could have enhanced uptake of TRP2–pHrodo particles compared to the other conjugates with primarily polar surfaces.63,64 However, although MUT30–phrodo and polymer–pHrodo particles are both negatively charged, the greater negative charge on the polymer–pHrodo particles (−41 mV) compared to that on the MUT30–pHrodo particles (−20 mV) could have reduced the uptake of the polymer-pHrodo particles. Cellular uptake was also monitored by fluorescence microscopy in samples incubated at 37 °C. At 4 h, the strong red fluorescence signal of endocytosed pHrodo red particles is observed, while the control shows no fluorescence (Figure 5B). Results at 0 and 2 h are available in Figure S9 and Figure S10. In agreement with the flow cytometry results, TRP2–pHrodo particles displayed the strongest cellular uptake. Overall, the antigen containing nanoparticles were effectively taken up by DCs, indicating their potential to deliver antigens to DCs.
Figure 5.

Dendritic cell uptake of pHrodo red labeled nanoparticles obtained at 4 °C and at 37 °C via flow cytometry at different time points (A) and obtained via microscopy (B) at 4 h (37 °C), with endocytosed nanoparticles visible as the red signal in the fluorescence and merged channels.
Cytotoxicity of Peptides and Nanoparticles
Materials to be used in biological systems are required to have low or no toxicity; thus, the conjugates and peptides were evaluated for toxicity against B16F10 melanoma cells in vitro via an MTT cell viability assay (Figure 6A,B). The cell viabilities observed for the particles and the MUT30 peptide all remained above 60% over a 48 h period, even at high concentrations (1 mg/mL particles or 0.5 mg/mL peptide), indicating their suitability for application in biological systems. However, after incubation with free TRP2 peptide, cell viability was lower than all other materials at all concentrations. This is most likely attributable to precipitation or flocculation of the peptide due to its poor solubility in aqueous media and possibly cell distress due to membrane disruption as a result of the peptide’s hydrophobicity. The observed result indicates that integrating the TRP2 peptide into the nanoparticle lowered its toxicity probably as a result of aqueous solubilization. The dosing utilized in the viability studies was comparable to the concentrations in the blood pool used in later in vivo studies (400–600 µg of material or 0.27–0.4 mg/mL for a 20 g mouse, ca. 1.5 mL blood pool volume). At these concentrations, cell viability remained ca. 70% or more for 48 h. If the complete body mass is taken into account, the in vivo concentration would, thus, be much lower, which would, in turn, reduce toxicity.
Figure 6.

B16-F10 melanoma cell viability obtained at 24 h (A) and 48 h (B) after incubation with peptides or nanoparticles. The concentration of the peptide in the conjugate is ca. 10% (w/w) of the conjugate concentration based on AAA (hence, the maximum peptide concentration for the conjugates is ca. 0.1 mg/mL, which is equivalent to the second point in the curves for the peptides, MUT30 and TRP2). The results of an in vitro DC maturation study (C) measured various dendritic cell maturation markers as an indicator of a nanoparticle’s ability to generate an immune response without an adjuvant. The composite maturation index reported in this figure represents an unweighted average of the expression of CD80, CD86, and MHCII normalized to the iDC (untreated) population. iDC: immature DCs; LPS: lipopolysaccharide (* represents p < 0.05 compared to the iDC population, as analyzed using ANOVA with a Tukey post-test); see Figure S10.
In Vitro Immunogenicity Assay
We proceeded to investigate the immunostimulatory potential of the prepared materials by assessing the immunophenotype of bone marrow derived mouse DCs after co-culturing with the nanoparticles (Figures 6C and S11). When the particles and peptides are taken up by DCs in vivo, it is expected that immature DCs (iDC) undergo maturation and migrate to lymph nodes. Upon maturation, DCs acquire an enhanced capacity to form and accumulate peptides, major histocompatibility complex (MHC) class II molecules, and co-stimulatory molecules such as CD40, CD80, and CD86.65 Thus, by looking at the up-regulation of the co-stimulatory molecules (CD80 and CD86) and MHCII, the extent of maturation can be determined as a composite maturation index.66 iDCs were thus treated with PBS (a negative control) and lipopolysaccharide (LPS, 1.5 µg/mL, a TLR-4 agonist, a positive control) or prepared polymer-only and polymer-peptide nanoparticles. At 6 and 24 h, the LPS-treated DCs show an expected high maturation index, while the polymer only and polymer–peptide particle-treated DCs show a response similar to that of the negative control. Only MUT30 particles show a slightly greater maturation index than the iDCs in PBS albeit not statistically significant. These results suggest the particles are poorly immunogenic, which was also observed when we analyzed the peptides alone (data not shown) and indicate the need for an adjuvant to realize a response in vivo with the MUT30 and TRP2 peptide epitopes.
Assessment of in Vivo Efficacy of Nanoparticles against Melanoma (B16-F10) Model
The route of vaccine administration tends to have a significant effect on its efficacy. Thus, we initially compared two common routes of vaccine administration, subcutaneous (s.c.) and intravenous (i.v.), and we observed that injecting mixed CpG plus TRP2 particles subcutaneously did not perform as well as the combination of CpG (s.c.) plus TRP2 particles (i.v.) against B16-F10 melanoma (unilateral tumor) (Figure S14). Thus, for in vivo studies, mice with bilateral tumors were teated via i.v. injection with polymer nanoparticles or nanoparticles of the polymer–peptide conjugates and s.c. injection of CpG as the adjuvant, as shown in Figure 7A. Following the injection of the vaccine, the mouse weight remained constant or increased (Figure S13). The bilateral approach was used because it facilitates the assessment of local as well as systemic responses. Most peptide vaccines tend to suffer from poor immunogenicity, and as was observed during the DC maturation studies (Figure 6C), the nanoparticles with or without the peptides were not immunogenic, indicating the need for an adjuvant to observe an effect. Indeed, when TRP2 nanoparticles were employed without an adjuvant, the survival and tumor growth results obtained were similar to those observed in the nontreated mice group (Figure 7B–D). CpG, a TLR9 agonist that can stimulate the immune system, was thus employed as an adjuvant and injected on the left side (treated side) subcutaneously, and the tumor growth on both sides was followed by ultrasound imaging. Among the groups treated in combination with the adjuvant, TRP2 particles had the longest median survival of 23 days with 50% of the animals surviving up to day 24, compared to the no-treatment (NTC) group, which had a median survival of only 14 days (Figure 7B), and the CpG-only group that survived to 19 days. The difference in survival curves between the TRP2 particles group and the NTC group was statistically significant (p = 0.0058) based on the log-rank (Mantel-Cox) test. Treatment with MUT30 nanoparticles and CpG also resulted in an improved median survival, 21 days, compared to the NTC, although only 25% of the animals survived up to day 24, which is less than those that remained for the TRP2 group. The result for the MUT30 nanoparticles plus CpG was, however, not statistically significant albeit there was a trend toward an effect (p = 0.0171). This observation could be due to poor or low expression of that particular mutated sequence or possibly its absence in cells used for these experiments because we did not perform sequencing prior to experiments. We instead used previously reported sequencing data. The performances of the rest of the groups were not statistically significant in comparison to the NTC group. The median survival for polymer particles with CpG was 17 days, and for both CpG-treated and polymer plus CpG treated mice, no animals survived beyond 21 days. A comparison of the best-performing treatment, TRP2 nanoparticles plus CpG, with adjuvant alone showed pCpG = 0.0598, which was strongly trending toward an effect indicating the effectiveness of the CpG plus TRP2 particle combination compared to the adjuvant alone.
Figure 7.

Treatment protocol for in vivo studies with four animals per group (A). Kaplan–Meier survival plots (p ≤ 0.01 (Bonferroni corrected for five comparisons) was considered statistically significant with comparison to the no-treatment (NTC) group. TRP2 particles (p = 0.180), CpG (p = 0.031), CpG plus polymer particles (p = 0.042), CpG plus MUT30 particles (p = 0.0171) and CpG plus TRP2 particles (p = 0.0058) (asterisks indicate p < 0.01). For comparison against CpG, pCpG ≤ 0.05 considered to be statistically significant, CpG plus TRP2 particles versus CpG (pCpG = 0.0598) ) (B). Tumor sizes of the left side (CpG-injected tumor) (C) and right side (distant) tumor (D). Histology results showing hematoxylin and eosin (H&E)-stained tumor tissue from a mouse in the no-treatment group (E), CpG plus MUT30 nanoparticle-treated group (F), and H&E- (G) and F4/80- (macrophage marker) stained fat pads from a mouse in the CpG plus TRP2-nanoparticle-treated group (H); the black arrows show a rim of dead and dying cells surrounding a small central mass of viable tumor cells after treatment (ellipse-like shape, with the white arrow showing the major axis). Raw tumor sizes and body weights are available in Figures S12 and S13.
It is noteworthy that the survival results herein are based on the systemic response because the distant (right side) tumors grew faster and reached burden before the adjuvant-injected (left side) tumors (Figure 7C,D). The tumor growth curves indicate a slower growth of the treated tumor than on the distant tumor, suggesting the systemic response was generally weaker than the local response. Overall, the results indicate that, when administered with CpG, TRP2 particles suppressed tumor growth and significantly improved survival. At the end of the study, tumors were collected for histological analysis. The H&E stained tumors from the left side of mice in the no treatment, MUT30 particles plus CpG and TRP2 particles plus CpG groups are shown in Figure 7E–G. The histology results indicated an extensive viable tumor in the no-treatment and MUT30 plus CpG nanoparticles groups, whereas viable tumor was greatly reduced in the TRP2 particles plus CpG treated tumor (shown by black arrows). Tumor tissues were also stained with the F4/80 macrophage marker to detect macrophage presence in the tumor, which is expected to be pronounced upon activation of the immune system. Indeed, for the TRP2 particles plus CpG group, the presence of macrophages was observed in most parts of the tumor and surrounding fat pad, as evidenced by the positive F4/80 staining (brown-colored spots) (Figure 7H). CD8+ T cells demonstrate a similar distribution to the macrophages in the region of remaining tumor, with few cells infiltrating the center and a larger number surrounding the remaining tumor (Figure S15). The presence of viable tumor after treatment is possibly due to the tumor developing an immunosuppressive environment or developing tolerance, suggesting the need for combining peptide vaccine treatment with other therapies, such as checkpoint blockade or gene silencing, to mitigate tolerance or immune suppression.67–69
CONCLUSIONS
In summary, we have shown a simple, yet versatile, approach for the realization of self-assembling hyperbranched polymer-peptide amphiphiles that can be applied as nanovaccines for immunotherapy. Using the SPAAC reaction, azido melanoma antigen peptides and azido PEG mannose were conjugated to a hyperbranched cyclooctyne core, in a simple two-step semi-batch fashion, under extremely mild and nonstringent conditions to obtain amphiphilic hyperbranched polymer–peptide constructs. The macromolecular conjugates self-assembled in aqueous solution into nanoparticles within the 10–30 nm range by DLS and were assessed as spherical via TEM. By incorporating either a well-known hydrophobic melanoma tumor-associated antigen peptide TRP2 or a polar 27 amino acid melanoma neoantigen peptide MUT30, potential melanoma cancer nanovaccines were fabricated. Based on in vitro studies, the nanovaccines were successfully taken up by DCs but did not display immunogenicity. The nanoparticles showed minimal toxicity, although the TRP2 peptide alone proved to be toxic possibly due to its hydrophobicity. When we incorporated the peptide into the nanoparticle construct, we saw a reduction in hydrophobicity and toxicity. CpG adjuvant was required to enhance efficacy against B16-F10 melanoma in vivo. The combination of CpG with TRP2 particles suppressed tumor growth, leading to improved survival compared with both the nontreated and CpG-only treated groups. However, based on histology, most of the tumor had been eliminated, but some viable tumor remained after treatment with TRP2 nanoparticles. This could possibly be due to immune tolerance or immune suppression, suggesting that the nanoparticles could potentially be used as one component of a melanoma vaccine, in combination with other therapies that counter tolerance and immune suppression. Overall, our synthetic approach is facile, uses mild conditions, is tolerant to a wide range of functionalities and polarities, and can allow the incorporation of more than one peptide epitope, making it attractive as a simple and versatile approach for fabricating personalized cancer nanovaccines.
EXPERIMENTAL SECTION
Reagents
Boltorn H40 (Sigma-Aldrich) was purified by precipitation from acetone and diethyl ether and dried under a vacuum at room temperature before use. Amino acids, coupling agents and the Wang resin (0.87 mmol/g) were obtained from EMD Millipore USA and used as received. The Rink amide resin (Rink amide chemMatrix, 0.49 mmol/g) was purchased from Biotage. Fmoc–N–amido–dPEG4–acid (Quantabiode-sign), mannosamine hydrochloride (Aldrich), triethylamine (TEA, Aldrich), dimethylamino pyridine (Aldrich), azido-dPEG12–NHS ester (Quanta Biodesign), pHRodo red succinimidyl ester (Thermofisher), O-(2-azidoethyl)-O′-methyl-triethylene glycol (Aldrich), 1,2-ethanedithiol (EDT, Al-drich), phenol (Acros Organics), diisopropylethylamine (DIPEA, Aldrich), trifluoroacetic acid (TFA, Alfa Aesar), thioanisole (Aldrich), triisopropylsilane (TIPS, Aldrich), succinic anhydride (Acros Organics), N-(3-(dimethylamino)-propyl)-N′-ethylcarbodiimide hydrochloride, EDC·HCl (Al-drich), N-hydroxybenzotriazole (HOBT, Aldrich) and dibenzylcylooctyne (Click Chemistry Tools) were used as received. Methanol (Fisher) and dimethylformamide (Fisher) were dried over molecular sieves (Aldrich) of 3 and 4 Å, respectively. Azidovaleric acid was synthesized in accordance with a previously published protocol.70 All other solvents were purchased from Fisher at the highest purity available and used as received.
Cells for in Vitro Studies
B16-F10 murine melanoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, no. CRL-6475) and cultured in high-glucose Dulbecco’s modified Eagle media (Invitrogen, Carlsbad, CA) supplemented with 1% penicillin–streptomycin (10 000 U/mL, Invitrogen, Carlsbad, CA) and 10% fetal bovine serum (Omega Scientific Inc., Tarzana, CA). Dendritic cells (DCs) were obtained from 8–12 weeks old, female, C57BL/6J mice in accordance with guidelines approved by UC Davis Teaching and Research Animal Care Services using a modified 10 day protocol (see the Supporting Information).66,71
Animal Experiments
All animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee of the University of California, Davis. To generate the subcutaneous B16–F10 model of melanoma, 7–8 week old female C57BL/6J mice were used (The Jackson Laboratory, Bar Harbor, ME). At the time of injection, animals were anesthetized with 3% isoflurane (in oxygen; flow rate: 2 L/min) and maintained at 1.5–2.0% and 1 × 105 cells in 50 µL 1:1 PBS/Matrigel (BD Biosciences, San Jose, CA) were injected subcutaneously into the left and right flank regions. CpG–ODN 1826 (5′-tccatgacgttcctgacgtt-3′; total backbone phosphorothioated) was purchased from InvivoGen (San Diego, CA).
Esterification of Boltorn H40 with Succinic Anhydride (B64–COOH)
To a solution of Boltorn H40 (0.20 g, 0.1 mmol) in dry dimethylformamide (DMF) (20 mL) under nitrogen was added succinic anhydride (0.915 g, 9.15 mmol), triethylamine (1.28 mL), and 4-N,N-dimethylaminopyridine (0.1116 g). The reaction was allowed to proceed at room temperature for 48 h before water (2 mL) was added, and the reaction was left to stir for 15 min. The reaction mixture was precipitated in water twice with the product being recovered by centrifugation. The recovered product was dissolved in dimethyl sulfoxide (DMSO) and subjected to dialysis against water for 24 h using Spectra Por dialysis tubing with MWCO 2 kDa, after which the contents of the dialysis bag were centrifuged at 4000 rpm and the sedimented product was collected. The product was again dissolved in DMSO and dialyzed for 24 h. The contents of the dialysis bag were carefully transferred into a Falcon tube and lyophilized to afford the esterified Boltorn H40 (B64–COOH) as a pale brown-white powder with a yield of 0.3 g. The conversion of −OH to −COOH was considered to be quantitative based on the appearance of the ethylenic protons of the succinic anhydride and the disappearance of the peaks due to −OH in the 1H NMR of the product (Figure 1).
Dibenzylcyclooctyne-Modified Boltorn H40 (B64DBAC)
B64–COOH (0.025 g, 3 µmol) was dissolved in dry DMF (15 mL) under nitrogen before dibenzylcyclooctyne amine (DBCOA) (0.050 g, 181 µmol) was added. The temperature of the solution was lowered to 0 °C using an ice bath and EDC·HCl (0.0552 g, 288 µmol), DIPEA (0.0744 g, 576 µmol, 101 µL) and HOBT (0.0390 g, 288 µmol) were added. The reaction was allowed to reach room temperature and left to stir for 48 h. From the reaction solution, 5 µL was obtained and spotted on a thin-layer chromatography (TLC) plate against a solution of DBCOA in DMF of the same concentration as that at the start of the reaction. The TLC was carried out using a solvent composed of DCM/MeOH (4:1), and the plate was treated with ninhydrin staining solution to determine the presence of free amines (RfDBCOA = 0.6). A very faint orange-greenish spot was observed on the TLC plate for the reaction solution compared to the DBCOA solution. The removal of the DMF was done on a rotary evaporator, and the resulting residue was suspended in dichloromethane (50 mL) followed by washing with 0.001 N HCl (3 × 50 mL), water (3 × 50 mL), and brine (1 × 50 mL). The organic layer was dried over sodium sulfate, and the solvent volume was lowered in vacuo followed by precipitation in diethyl ether. The precipitate obtained was dissolved in dichloromethane, and the product was again precipitated in diethyl ether. Precipitation was conducted one more time, and the solvent was removed in vacuo to obtain the desired product as a pale brown amorphous solid with a yield of 87 mg.
Synthesis of Phenyl 6-Azidohexanoate
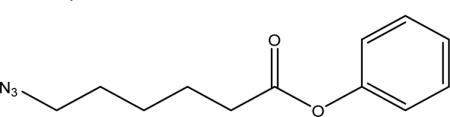
To a solution of azidopentanoic acid (4.0 g, 26 mmol) in anhydrous THF (10 mL) was added an excess of phenol (7.3 g, 130 mmol) under nitrogen, and the solution was cooled to 0 °C in an ice bath. The reaction was stirred for 10 min at 0 °C, and then DMAP (1.54 g, 8.4 mmol) and EDC·HCl (2.4 g, 25.2 mmol) were added. The reaction was allowed to gradually reach room temperature and left to stir for 16 h, after which the solvent was removed in vacuo. The residue was dissolved in 50 mL of DCM and extracted with 0.001 M NaOH (3 × 50 mL), 0.1 M HCl (1 × 50 mL), water (10 × 50 mL), and brine (1 × 50 mL). The organic layer was dried over anhydrous Na2SO4, which was filtered off followed by the removal of solvent in vacuo to afford the product as a colorless oil (3.3 g, 82%). 1H NMR (800 MHz, CDCl3, Figure S1, δ ppm from TMS): 1.73 (m, 2H, −CH2), 1.85 (m, 2H, −CH2), 2.61 (t, J = 7.457 Hz, 2H, −CH2), 3.35 (t, J = 6.779 Hz, 2H, −CH2), 7.07 (m, 2H, −CH), 7.23 (tt, J1 = 1.140, J2 = 7.530, J3= 14.968 Hz, 1H, −CH), 7.38 (m, 2H, −CH). See Figure S5 for 1H NMR spectrum.
Determination of B64DBAC Alkyne Loading
To B64DBAC (0.784 mg, 0.03 µmol) in DMF (150 µL, 5.2267 mg/mL stock solution) was added phenyl 6-azidohexanoate (0.42 mg, 1.92 µmol) dissolved in DMF (50 µL, 0.0084 mg/mL stock solution). The resulting solution was left on a shaker for 3 days at room temperature with an aliquot (25 µL) withdrawn every 24 h and analyzed by HPLC (eluents, A (0.1% TFA in water) and B (0.1% TFA in acetonitrile); gradient of 50 to 100% B over 45 min, λ = 254 nm). Maximum conversion was achieved after 24 h based on changes in the peak area of phenyl 6-azidohexanoate (tR = 20 min). The peak area at 3 days was used to determine the concentration of free phenyl 6-azidohexanoate remaining in solution based on a calibration curve by analyzing solutions of known concentrations of phenyl 6-azidohexanoate.
Synthesis of Peptides
Solid-phase peptide synthesis was performed on a Biotage Initiator Alstra automated microwave peptide synthesizer using the Fmoc strategy. For coupling, 2-(1H–benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluoro-phosphate–DIPEA was employed, and for deprotection, 20% piperidine in DMF was used. The Wang resin (TRP2 and azido TRP2) was preloaded with the first amino acid prior to automated synthesis (details available in the Supporting Information).
Synthesis of Azido–PEG12–Mannose
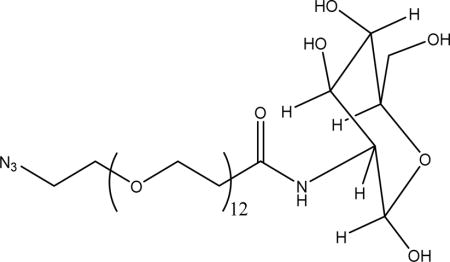
Under an inert atmosphere, mannosamine hydrochloride (2.5 equiv) was dissolved in dry methanol and added to a vial containing azido–dPEG12–NHS ester (1 equiv) dissolved in dry methanol via a cannula. The resulting solution was mixed thoroughly on a vortex mixer and cooled to 0 °C in an ice bath. Triethylamine (2.5 equiv) was then added to the cold solution, and the reaction was allowed to gradually warm up to room temperature and left to stir over 24 h. At the end of the reaction, the solvent was removed. A total of 4 mL of a cold solution (0 °C) composed of acetonitrile/water/TFA (1:1:0.05) v/v/v was added, and the resulting solution was lyophilized to give the crude product as a viscous pale yellow oil that was used for click reactions without purification. The presence of the product was verified by mass spectrometry (Figure S6, [M + Na]+ = 827.41 Da, expected mass = 804.42 Da).
Conjugation of N3–PEG–Mannose to BH64DBAC via the SPAAC Reaction (Polymer Amphiphile)
To B64DBAC (1 equiv of alkyne units) in DMF was added the azido–PEG polymer (3 equiv) dissolved in DMF. The resulting solution was left on a shaker at room temperature, and an aliquot (50 µL) was withdrawn after 4 days and analyzed by FTIR (azide peak observed at 2100 cm−1). The reaction was assumed to have reached the maximum conversion and the crude product was purified by dialysis against water for 3 days (MWCO 2 kDa). The solution obtained was lyophilized to obtain a very viscous oil that was analyzed by FTIR and 1H NMR.
Preparation of Polymer–Peptide Conjugates via the Conjugation of Peptides and N3–PEG12–Mannose to B64DBAC via the SPAAC Reaction
Peptide Conjugation
To B64DBAC (1 equiv of alkyne units) in DMF was added the azido–peptide (0.1 equiv) dissolved in DMF. The resulting solution was left on a shaker for 4 days at room temperature. An aliquot (50 µL) was withdrawn and analyzed by HPLC (eluents, A (0.1% TFA in water) and B (0.1% TFA in acetonitrile); gradient of 50 to 100% B over 45 min; λ = 254 nm), MALDI, and FTIR to determine the extent of the reaction. Conversion was quantitative based on the disappearance of the azide peak at 2100 cm−1 in the IR spectrum, the MALDI spectrum, and the absence of the peak due to the pure peptide in the HPLC chromatogram.
Polymer Conjugation
To the polymer–peptide conjugation reaction solution (above) was added azido–PEG polymer (3 equiv based on starting alkyne units, an excess) dissolved in DMF. The resulting solution was left on a shaker for 4 days at room temperature. An aliquot (50 µL) was withdrawn and analyzed by FTIR (azide peak observed at 2100 cm−1). The reaction was considered to have reached the maximum conversion, and the crude product was purified by dialysis against water for 3 days (MWCO 3.5 kDa). The solution obtained was lyophilized to obtain a white solid that was analyzed by FTIR, 1H NMR, SEC (main text, Figure 3), and AAA (Tables S2 and S4).
Self-Assembly of Macromolecular Chimeras and Polymer Amphiphiles
The macromolecular chimera and polymer amphiphile (5 mg) was dissolved in previously degassed DMF (500 mL) and left to stir for 4 h. Filtered (0.2 µm sterile filter), deoxygenated water (4.5 mL) was then added to the solution using a syringe pump (0.03 mL/min) while stirring at 500 rpm. The solution was placed in a dialysis bag and dialyzed against 4 L of filtered deoxygenated deionized distilled water (Millipore Steritop, 0.45 µm, sterile filter) overnight (MWCO 3.5 kDa). The water was then replaced with filtered deoxygenated deionized distilled water, and dialysis ensued for a further 6 h, after which the contents of the dialysis bag were collected and centrifuged at 3000 rpm for 2 min. The volume of solution was ca. 5 mL (ca. 1 mg/mL macromolecular chimera). An aliquot of the solution was obtained for analysis via TEM and another, mixed with 100 µL of PBS buffer (pH 7.4, 25 mmol NaCl), was analyzed via DLS (size and ζ potential).
Synthesis of azido-PEG3–pHrodo Red
pHrodo red NHS ester (1.00 mg, 1.54 µmol) was dissolved in dry DMF (500 µL), from which 10 µL was withdrawn for analytical purposes. To the remainder of the dye solution was added O-(2-azidoethyl)-O′-methyl-triethylene glycol (0.43 mg, 1.2 equiv) dissolved in dry DMF (500 µL) followed by triethylamine (50 µL). The resulting solution was left on a shaker at room temperature for 24 h and an aliquot (50 µL) was withdrawn to determine the extent of reaction via HPLC. No peak was observed at the retention time expected for the pure succinimidyl–pHrodo red (or its hydrolyzed form) after 24 h of reaction. The reaction was stopped, and the product was purified via preparative HPLC to afford the product as a red solid after freeze-drying of the collected fractions (azide peak observed at 2100 cm−1) with a yield of 1.1 mg. NB: The structure of pHrodo red succinimidyl ester is protected by the manufacturer, so no further chemical characterization was conducted.
Preparation of Fluorescently Labeled Polymer–Peptide Conjugates via the Conjugation of azido-pHrodo Red, Peptides and N3–PEG–Mannose to B64DBAC via the SPAAC Reaction
Fluorescent Dye Conjugation
To B64DBAC (1 equiv of alkyne units) in DMF was added the azido–pHrodo red (0.2 equiv) dissolved in DMF. The resulting solution was left on a shaker for 2 days at room temperature. An aliquot (50 µL) was withdrawn and analyzed by HPLC (eluents A (0.1% TFA in water) and B (0.1% TFA in acetonitrile); gradient of 50% to 100% B over 45 min, λ = 570 nm) and gel permeation chromatography (GPC) to determine the extent of the reaction. Maximum conversion was achieved based on the absence of the peak due to the pure dye in the HPLC and GPC chromatograms.
Peptide Conjugation
To dye-labeled B64DBAC (1 equiv of alkyne units) in DMF was added the azido–peptide (0.1 eq of starting alkyne) dissolved in DMF. The resulting solution was left on a shaker for 4 days at room temperature. An aliquot (50 µL) was withdrawn and analyzed by HPLC (a gradient of 50 to 100% B over 45 min, λ = 254 nm), MALDI, and FTIR to determine the extent of the reaction. Maximum conversion was achieved based on the disappearance of the azide peak at 2100 cm−1 in the IR spectrum, the MALDI spectrum, and the absence of the peak due to the pure peptide in the HPLC chromatogram.
Polymer Conjugation
To the polymer–peptide conjugation reaction solution (above) was added azido–PEG polymer (3 equiv based on starting alkyne units, an excess) dissolved in DMF. The resulting solution was left on a shaker for 4 days at room temperature. An aliquot (50 µL) was withdrawn and analyzed by FTIR (azide peak observed at 2100 cm−1). The reaction was considered to have reached the maximum conversion and the crude product was purified by dialysis against water for 3 days (MWCO 3.5 kDa). The solution obtained was lyophilized to obtain a red solid that was analyzed by GPC and FTIR.
Cellular Uptake and Internalization of Nanoparticles
Nanoparticle Formation
For in vitro experiments, nanoparticles (with and without fluorescent label) were prepared following the self-assembly procedure described above. To obtain the nanoparticle solutions in PBS at the desired concentrations, 1 mg/mL, solvent exchange (water to PBS) was completed using Amicon centrifugal filters (MWCO 3 kDa) centrifuged at 4000 rpm. Nitrogen-degassed filtered PBS buffer was added to replace the water after each concentration cycle.
Cellular Uptake Studies via Flow Cytometry
Mouse bone marrow derived DCs were plated at 1.4 × 105 cells per well in 24-well tissue culture plates 24 h prior to experiments. Particles were incubated with cells continuously at 37 °C in complete media (100 µg/mL in 300 µL for) for up to 24 h. Cells were rinsed once with PBS and collected at 0, 4, and 24 h in 170 µL TrypLE Express dissociation buffer (ThermoFisher Scientific, Waltham MA). Samples were then analyzed on a FACScan flow cytometer.
Cellular Internalization Studies via Microscopy
The internalization of nanoparticles was confirmed using fluorescent microscopy. Mouse bone marrow derived DCs were plated at 2 × 105 cells per dish in 35 mm dishes 24 h prior to experiments. DCs were continuously incubated with pHrodo red labeled nanoparticles at (100 µg/mL in 1 mL) for 24 h in the BZ-X710 Live Imaging Microscope (Keyence, Elmwood Park, NJ) with 5% CO2 and at 37 °C. Images were acquired in the bright-field and fluorescent channel at 0, 2, and 4 h. The merged images were analyzed using ImageJ by dividing the total fluorescence by the cell number per image.
Cell Viability Studies
B16-F10 murine melanoma cells (ATCC, Manassas, VA, no. CRL-6475) were plated at 2000 cells in 100 µL media per well in 96-well tissue culture plates 24 h prior to peptide and particle addition. Free peptide or particles were added to each well in 100 µL of complete media. Cells were incubated continuously with either free peptide (0.5, 0.25, 0.1, 0.05 mg/mL) or particles (1, 0.5, 0.25, and 0.1 mg/mL) for either 24 or 48 h at 37 °C in a 5% CO2 incubator. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Invitrogen, Carlsbad, CA) was added to media at a concentration of 0.5 mg/mL, and cells were incubated 2 h at 37 °C in a 5% CO2 incubator. Media was removed, and formazan crystals dissolved in DMSO (100 µL/well, Sigma-Aldrich, St. Louis, MO). Absorbance was measured using a Tecan (San Jose, CA) Infinite M1000 microplate reader.
Dendritic Cell Phenotype and Maturation Markers (In Vitro Test for Immunogenicity)
For these studies, nanoparticles were added to mouse DC cultures at 37 °C for a period of 24, 72, or 120 h prior to analysis. Nanoparticles were co-cultured at either 50 or 100 µg/mL. In these experiments, half of the media was discarded and replaced with fresh media every 2 days, and the particles were sedimented at 16g and then resuspended in fresh media. Dendritic cell immunophenotype was quantified by measuring cell-surface marker expression by flow cytometry. Following nanoparticle incubation, DCs were lifted by incubating with a 5 mM Na2EDTA (Fisher Scientific) in PBS solution at 37 °C for 10 min. DCs were then washed with 1% fetal bovine serum in PBS and incubated with antibodies against CD16/CD32 (Fcγ III/II Receptor) (clone 2.4G2, IgG2b, k); (BD Pharmingen, CA) for 15 min at 4 °C to block Fcγ receptors on DCs. Cells were washed and then stained with antibodies against CD80 (clone 16-10A1, IgG2, k), CD86 (clone GL1, IgG2a, k), I-A/I-E (clone M5/114.15.2, IgG2b, k), and CD11c (clone HL3, IgG1, l2) for 30 min at 4 °C. Data acquisition was performed using flow cytometry (Attune NxT). Statistical analyses were performed using general linear model ANOVA followed by post-hoc pair-wise comparisons using the Tukey test. Differences were considered significant when p ≤ 0.05 using Prism (Version 7, GraphPad, La Jolla, CA).
In Vivo Efficacy
Therapeutic Treatment Protocol
Therapeutic treatment with nanoparticles (equivalent to 50 µg peptide for peptide containing nanoparticles in 50–150 µL of solution for polymer-only particles), 400 µg of material were injected in 50–150 µL of solution (PBS) or control (PBS only). Injections occurred on days 3 and 10 after B16-F10 cells were injected. For all relevant groups, CpG was injected subcutaneously into the left flank at a dose of 100 µg/50 µL water on days 3 and 10 after B16-F10 cell implantation. Once tumors were palpable, ultrasound imaging was used to obtain the volume size of tumors twice per week using an Acuson Sequoia 512 system (Siemens Medical Solution USA, Inc., Issaquah, WA). Mice were euthanized and tumors harvested before total tumor burden reached 2.0 cm3.
Nanoparticle Preparation
Nanoparticles for in vivo experiments were prepared following the self-assembly procedure described above. To obtain the nanoparticle solutions in PBS at the desired concentrations, solvent exchange (water to PBS) was effected by using Amicon centrifugal filters (MWCO 3 kDa) that were centrifuged at 4000 rpm. Nitrogen-degassed filtered PBS buffer was added to replace the water after each concentration cycle. Peptide presence and content were ascertained by AAA (Tables S2 and S4).
Data Analysis
Kaplan–Meier survival plots were obtained using GraphPad Prism software and compared via the log rank (Mantel–Cox) test.
Histological Analysis
Tumor samples were fixed in neutral buffered formalin. A Tissue-Tek VIP autoprocessor (Sakura, Torrance, CA) was used to process tumors, which were then embedded in Paraplast paraffin (melting temperature 56–60 °C), sectioned to 4 µm, and mounted on glass slides. Tumor sections were then stained using Mayer’s hematoxylin and eosin to facilitate histology and morphology evaluation.72
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health (NIHR01CA112356, NIHR01CA199658, NIHR01CA211602, NIHR01CA210553, and NIHR01CA134659).
Footnotes
ASSOCIATED CONTENT
- Details of experimental equipment, experimental synthesis and characterization, microscopy and flow cytometry cellular uptake results, in vitro DC maturation and nanovaccine efficacy studies, and raw tumor growth curves and animal weights. (PDF)
The authors declare no competing financial interest.
References
- 1.Couzin-Frankel J. Cancer Immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Fan Y, Moon J. Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy. Vaccines. 2015;3:662–685. doi: 10.3390/vaccines3030662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucka K, Banchereau J. Dendritic cell-based cancer therapeutic vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2016;16:489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YT, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 9.Yoshizaki Y, Yuba E, Sakaguchi N, Koiwai K, Harada A, Kono K. Potentiation of pH-sensitive polymer-modified liposomes with cationic lipid inclusion as antigen delivery carriers for cancer immunotherapy. Biomaterials. 2014;35:8186–8196. doi: 10.1016/j.biomaterials.2014.05.077. [DOI] [PubMed] [Google Scholar]
- 10.Kapadia CH, Perry JL, Tian S, Luft JC, DeSimone JM. Nanoparticulate immunotherapy for cancer. J. Controlled Release. 2015;219:167–180. doi: 10.1016/j.jconrel.2015.09.062. [DOI] [PubMed] [Google Scholar]
- 11.Slingluff CL. The Present and Future of Peptide Vaccines for Cancer: Single or Multiple, Long or Short, Alone or in Combination? Cancer J. 2011;17:343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura T, Egawa S, Uemura H. Personalized peptide vaccines and their relation to other therapies in urological cancer. Nat. Rev. Urol. 2017;14:501. doi: 10.1038/nrurol.2017.77. [DOI] [PubMed] [Google Scholar]
- 13.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 14.Griewank KG, Schilling B. Next-Generation Sequencing to Guide Treatment of Advanced Melanoma. Am. J. Clin. Dermatol. 2017;18:303–310. doi: 10.1007/s40257-017-0260-6. [DOI] [PubMed] [Google Scholar]
- 15.Sahin U, Derhovanessian E, Mller M, Kloke B-P, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 16.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015;115:11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-Based Immunotherapy for Cancer. ACS Nano. 2015;9:16–30. doi: 10.1021/nn5062029. [DOI] [PubMed] [Google Scholar]
- 20.Zhu G, Zhang F, Ni Q, Niu G, Chen X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano. 2017;11:2387–2392. doi: 10.1021/acsnano.7b00978. [DOI] [PubMed] [Google Scholar]
- 21.Zhao L, Seth A, Wibowo N, Zhao C-X, Mitter N, Yu C, Middelberg APJ. Nanoparticle vaccines. Vaccine. 2014;32:327–337. doi: 10.1016/j.vaccine.2013.11.069. [DOI] [PubMed] [Google Scholar]
- 22.Parry AL, Clemson NA, Ellis J, Bernhard SSR, Davis BG, Cameron NR. ‘Multicopy Multivalent’ Glycopol-ymer-Stabilized Gold Nanoparticles as Potential Synthetic Cancer Vaccines. J. Am. Chem. Soc. 2013;135:9362–9365. doi: 10.1021/ja4046857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris AS, Wongrakpanich A, Geary SM, Salem AK. Microparticles and Nanoparticles for Cancer-Targeting Vaccines. In: Toth I, editor. Micro and Nanotechnology in Vaccine Development. William Andrew Publishing; Norwich, NY: 2017. pp. 171–183. [Google Scholar]
- 24.Saleh T, Shojaosadati SA. Multifunctional nanoparticles for cancer immunotherapy. Hum. Vaccines Immunother. 2016;12:1863–1875. doi: 10.1080/21645515.2016.1147635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caucheteux SM, Mitchell JP, Ivory MO, Hirosue S, Hakobyan S, Dolton G, Ladell K, Miners K, Price DA, Kan-Mitchell J, et al. Polypropylene Sulfide Nanoparticle p24 Vaccine Promotes Dendritic Cell-Mediated Specific Immune Responses against HIV-1. J. Invest. Dermatol. 2016;136:1172–1181. doi: 10.1016/j.jid.2016.01.033. [DOI] [PubMed] [Google Scholar]
- 26.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 27.Dagvadorj N, Deuretzbacher A, Weisenberger D, Baumeister E, Trebing J, Lang I, Kochel C, Kapp M, Kapp K, Beilhack A, et al. Targeting of the WT191-138 fragment to human dendritic cells improves leukemia-specific T-cell responses providing an alternative approach to WT1-based vaccination. Cancer Immunol. Immunother. 2017;66:319–332. doi: 10.1007/s00262-016-1938-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diebold SS, Kursa P, Wagner E, Cotten M, Zenke M. Mannose polyethylenimine conjugates for targeted DNA delivery into dendritic cells. J. Biol. Chem. 1999;274:19087–19094. doi: 10.1074/jbc.274.27.19087. [DOI] [PubMed] [Google Scholar]
- 29.Tillman BW, Gruijl TDd, Bakker SAL-d, Scheper RJ, Pinedo HM, Curiel TJ, Gerritsen WR, Curiel DT. Maturation of Dendritic Cells Accompanies High-Efficiency Gene Transfer by a CD40-Targeted Adenoviral Vector. J. Immunol. 1999;162:6378–6383. [PubMed] [Google Scholar]
- 30.Huang Z-H, Sun Z-Y, Gao Y, Chen P-G, Liu Y-F, Chen Y-X, Li Y-M. Strategy for Designing a Synthetic Tumor Vaccine: Multi-Component, Multivalency and Antigen Modification. Vaccines. 2014;2:549–562. doi: 10.3390/vaccines2030549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rad-Malekshahi M, Fransen MF, Krawczyk M, Mansourian M, Bourajjaj M, Chen J, Ossendorp F, Hennink WE, Mastrobattista E, Amidi M. Self-Assembling Peptide Epitopes as Novel Platform for Anticancer Vaccination. Mol. Pharmaceutics. 2017;14:1482–1493. doi: 10.1021/acs.molpharmaceut.6b01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao G, Chandrudu S, Skwarczynski M, Toth I. The application of self-assembled nanostructures in peptide-based subunit vaccine development. Eur. Polym. J. 2017;93:670–681. doi: 10.1016/j.eurpolymj.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye Z, Xu L, Dong Z, Xiang P. Designing polyethylenes of complex chain architectures via Pd-diimine-catalyzed ″living″ ethylene polymerization. Chem. Commun. 2013;49:6235–6255. doi: 10.1039/c3cc42517g. [DOI] [PubMed] [Google Scholar]
- 35.Matyjaszewski K, Müller AHE, editors. Controlled and Living Polymerizations: From Mechanisms to Applications. Wiley VCH; Weinheim, Germany: 2009. [Google Scholar]
- 36.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 37.Braunecker WA, Matyjaszewski K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007;32:93–146. [Google Scholar]
- 38.Liu H, Irvine DJ. Guiding Principles in the Design of Molecular Bioconjugates for Vaccine Applications. Bioconjugate Chem. 2015;26:791–801. doi: 10.1021/acs.bioconjchem.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heegaard PMH, Boas U, Sorensen NS. Dendrimers for Vaccine and Immunostimulatory Uses. A Review. Bioconjugate Chem. 2010;21:405–418. doi: 10.1021/bc900290d. [DOI] [PubMed] [Google Scholar]
- 40.Skwarczynski M, Zaman M, Urbani CN, Lin IC, Jia Z, Batzloff MR, Good MF, Monteiro MJ, Toth I. Polyacrylate Dendrimer Nanoparticles: A Self-Adjuvanting Vaccine Delivery System. Angew. Chem., Int. Ed. 2010;49:5742–5745. doi: 10.1002/anie.201002221. [DOI] [PubMed] [Google Scholar]
- 41.Liu T-Y, Hussein WM, Jia Z, Ziora ZM, McMillan NAJ, Monteiro MJ, Toth I, Skwarczynski M. Self-Adjuvanting Polymer–Peptide Conjugates As Therapeutic Vaccine Candidates against Cervical Cancer. Biomacromolecules. 2013;14:2798–2806. doi: 10.1021/bm400626w. [DOI] [PubMed] [Google Scholar]
- 42.Liu T-Y, Hussein WM, Giddam AK, Jia Z, Reiman JM, Zaman M, McMillan NAJ, Good MF, Monteiro MJ, Toth I, et al. Polyacrylate-Based Delivery System for Self-adjuvanting Anticancer Peptide Vaccine. J. Med. Chem. 2015;58:888–896. doi: 10.1021/jm501514h. [DOI] [PubMed] [Google Scholar]
- 43.Kurniasih IN, Keilitz J, Haag R. Dendritic nanocarriers based on hyperbranched polymers. Chem. Soc. Rev. 2015;44:4145–4164. doi: 10.1039/c4cs00333k. [DOI] [PubMed] [Google Scholar]
- 44.Glaffig M, Palitzsch B, Stergiou N, Schull C, Straβburger D, Schmitt E, Frey H, Kunz H. Enhanced immunogenicity of multivalent MUC1 glycopeptide antitumour vaccines based on hyperbranched polymers. Org. Biomol. Chem. 2015;13:10150–10154. doi: 10.1039/c5ob01255d. [DOI] [PubMed] [Google Scholar]
- 45.Wilkinson KA, Hudecz F, Vordermeier HM, Ivanyi J, Wilkinson RJ. Enhancement of the T cell response to a mycobacterial peptide by conjugation to synthetic branched polypeptide. Eur. J. Immunol. 1999;29:2788–2796. doi: 10.1002/(SICI)1521-4141(199909)29:09<2788::AID-IMMU2788>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 46.Hilbert Á, Hudecz F, MezÔ G, Mucsi I, KajtÁR J, Kurucz I, Gergely J, RajnavÖLgyi É. The Influence of Branched Polypeptide Carriers on the Immunogenicity of Predicted Epitopes of HSV-1 Glycoprotein D. Scand. J. Immunol. 1994;40:609–617. doi: 10.1111/j.1365-3083.1994.tb03512.x. [DOI] [PubMed] [Google Scholar]
- 47.Mező G, Dalmadi B, Mucsi I, Bősze S, Rajnavölgyi É, Hudecz F. Peptide based vaccine design: Synthesis and immunological characterization of branched polypeptide conjugates comprising the 276–284 immunodominant epitope of HSV-1 glycoprotein D. J. Pept. Sci. 2002;8:107–117. [PubMed] [Google Scholar]
- 48.Wilkinson KA, Vordermeier H, Ivanyi J, Hudecz F. Synthesis and in Vitro T-Cell Immunogenicity of Conjugates with Dual Specificity: Attachment of Epitope Peptides of 16 and 38 kDa Proteins from Mycobacterium tuberculosis to Branched Polypeptide. Bioconjugate Chem. 1998;9:539–547. doi: 10.1021/bc970159+. [DOI] [PubMed] [Google Scholar]
- 49.Mezö G, Mihala N, Andreu D, Hudecz F. Conjugation of Epitope Peptides with SH Group to Branched Chain Polymeric Polypeptides via Cys(Npys) Bioconjugate Chem. 2000;11:484–491. doi: 10.1021/bc990116o. [DOI] [PubMed] [Google Scholar]
- 50.Glaffig M, Palitzsch B, Hartmann S, Schull C, Nuhn L, Gerlitzki B, Schmitt E, Frey H, Kunz H. A Fully Synthetic Glycopeptide Antitumor Vaccine Based on Multiple Antigen Presentation on a Hyperbranched Polymer. Chem. - Eur. J. 2014;20:4232–4236. doi: 10.1002/chem.201400256. [DOI] [PubMed] [Google Scholar]
- 51.Jewett JC, Sletten EM, Bertozzi CR. Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones. J. Am. Chem. Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 53.Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 54.Rubino JT, Chenkin MP, Keller M, Riggs-Gelasco P, Franz KJ. A comparison of methionine, histidine and cysteine in copper(i)-binding peptides reveals differences relevant to copper uptake by organisms in diverse environments. Metallomics. 2011;3:61–73. [PubMed] [Google Scholar]
- 55.Vasievich EA, Ramishetti S, Zhang Y, Huang L. Trp2 Peptide Vaccine Adjuvanted with (R)-DOTAP Inhibits Tumor Growth in an Advanced Melanoma Model. Mol. Pharmaceutics. 2012;9:261–268. doi: 10.1021/mp200350n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander J, del Guercio M-F, Maewal A, Qiao L, Fikes J, Chesnut RW, Paulson J, Bundle DR, DeFrees S, Sette A. Linear PADRE T Helper Epitope and Carbohydrate B Cell Epitope Conjugates Induce Specific High Titer IgG Antibody Responses. J. Immunol. 2000;164:1625–1633. doi: 10.4049/jimmunol.164.3.1625. [DOI] [PubMed] [Google Scholar]
- 57.Agard NJ, Prescher JA, Bertozzi CR. A Strain-Promoted [3 + 2] Azide–Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 58.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 59.Ning X, Guo J, Wolfert MA, Boons G-J. Visualizing Metabolically Labeled Glycoconjugates of Living Cells by Copper-Free and Fast Huisgen Cycloadditions. Angew. Chem., Int. Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012;72:1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 61.Miksa M, Komura H, Wu R, Shah KG, Wang P. A novel method to determine the engulfment of apoptotic cells by macrophages using pHrodo succinimidyl ester. J. Immunol. Methods. 2009;342:71–77. doi: 10.1016/j.jim.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karin M, Mintz B. Receptor-mediated endocytosis of transferrin in developmentally totipotent mouse teratocarcinoma stem cells. J. Biol. Chem. 1981;256:3245–3252. [PubMed] [Google Scholar]
- 63.Lin Y-L, Jiang G, Birrell LK, El-Sayed MEH. Degradable, pH-sensitive, membrane-destabilizing, comb-like polymers for intracellular delivery of nucleic acids. Biomaterials. 2010;31:7150–7166. doi: 10.1016/j.biomaterials.2010.05.048. [DOI] [PubMed] [Google Scholar]
- 64.Bhattacharjee S, Ershov D, Fytianos K, van der Gucht J, Alink GM, Rietjens IMCM, Marcelis ATM, Zuilhof H. Cytotoxicity and cellular uptake of tri-block copolymer nanoparticles with different size and surface characteristics. Part. Fibre Toxicol. 2012;9:11. doi: 10.1186/1743-8977-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stockwin LH, McGonagle D, Martin IG, Blair GE. Dendritic cells: Immunological sentinels with a central role in health and disease. Immunol. Cell Biol. 2000;78:91–102. doi: 10.1046/j.1440-1711.2000.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. J. Mater. Chem. B. 2014;2:2562–2574. doi: 10.1039/C3TB21460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016;22:1402–1410. doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, Wang L, Kowolik C, Deng J, Figlin R, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010;70:7455–7464. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Z, Wang Y, Zhang L, Huang L. Nanoparticle-Delivered Transforming Growth Factor-β siRNA Enhances Vaccination against Advanced Melanoma by Modifying Tumor Micro-environment. ACS Nano. 2014;8:3636–3645. doi: 10.1021/nn500216y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kakwere H, Payne RJ, Jolliffe KA, Perrier S. Self-assembling macromolecular chimeras: controlling fibrillization of a β-sheet forming peptide by polymer conjugation. Soft Matter. 2011;7:3754–3757. [Google Scholar]
- 71.Lewis JS, Dolgova NV, Chancellor TJ, Acharya AP, Karpiak JV, Lele TP, Keselowsky BG. The effect of cyclic mechanical strain on activation of dendritic cells cultured on adhesive substrates. Biomaterials. 2013;34:9063–9070. doi: 10.1016/j.biomaterials.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fraser CC, Altreuter DH, Ilyinskii P, Pittet L, LaMothe RA, Keegan M, Johnston L, Kishimoto TK. Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non-human primates. Vaccine. 2014;32:2896–2903. doi: 10.1016/j.vaccine.2014.02.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.