

Abstract
Objective
Animal models recapitulating post-traumatic osteoarthritis (OA) suggest that subchondral bone (SCB) properties and remodeling may play major roles in disease initiation and progression. Thus, we investigated the role of SCB properties and its effects on load-induced OA progression by applying a tibial loading model on two distinct mouse strains treated with alendronate (ALN).
Design
Cyclic compression was applied to the left tibia of 26-week-old male C57Bl/6 (B6, low bone mass) and FVB (high bone mass) mice. Mice were treated with ALN (26μg/kg/day) or vehicle (VEH) for loading durations of 1, 2, or 6 weeks. Changes in articular cartilage and subchondral and epiphyseal cancellous bone were analyzed using histology and microcomputed tomography.
Results
FVB mice exhibited thicker cartilage, a thicker SCB plate, and higher epiphyseal cancellous bone mass and tissue mineral density than B6 mice. Loading induced cartilage pathology, osteophyte formation, and SCB changes; however, lower initial SCB mass and stiffness in B6 mice did not attenuate load-induced OA severity compared to FVB mice. In contrast, FVB mice exhibited less cartilage damage, and slower-growing and less mature osteophytes. In B6 mice, inhibiting bone remodeling via ALN treatment exacerbated cartilage pathology after 6 weeks of loading, while in FVB mice, inhibiting bone remodeling protected limbs from load-induced cartilage loss.
Conclusions
Intrinsically lower SCB properties were not associated with attenuated load-induced cartilage loss. However, inhibiting bone remodeling produced differential patterns of OA pathology in animals with low compared to high SCB properties, indicating that these factors do influence load-induced OA progression.
Keywords: Osteoarthritis, mouse, subchondral bone, bone remodeling, bone properties, mechanical loading
Introduction
Osteoarthritis (OA) is a degenerative joint disease that clinically presents as radiographic narrowing of the joint space with accompanying subchondral bone (SCB) thickening and osteophyte formation1,2. Its exact etiology has been long debated, despite preclinical and clinical studies intended to elucidate the factors responsible for OA disease initiation and progression.3–5. Risk factors include traumatic injuries5, occupational activities6, and obesity7, suggesting that mechanical loading plays a major role in OA initiation. An abnormal joint mechanical environment could initiate cell-mediated processes leading to both cartilage damage and SCB adaptation; however, the tissue in which the disease initiates is still controversial.
Given the clinical evidence of SCB thickening in OA patients, historically, the hypothesis has been that disease initiation is associated with increased mass and apparent stiffening of the SCB plate, diminishing its ability to act as an effective shock absorber for the cartilage8–10. However, recent studies suggest that SCB stiffening may not influence the stresses engendered on the cartilage surface3,11 leading to the conclusion that OA joint pathology initiates in the articular cartilage rather than the SCB. In more advanced stages of OA, abnormal mechanical forces can contribute to articular cartilage loss via the initiation of microcracks in the SCB plate that activate a bone remodeling response, leading to tidemark advancement and subsequent thinning of the cartilage3,12–14. These findings implicate a contributory role for bone remodeling in the pathogenesis of OA. Further evidence implicating bone remodeling in OA development is provided by the observation that in several animal models of OA, SCB mass is reduced at disease initiation, followed by thickening as the disease progresses15,16. Furthermore, in animal models, the inhibition of bone remodeling with pharmacological agents that impair osteoclast-mediated bone resorption attenuates the progression of OA17–21.
The role of bisphosphonates, which inhibit osteoclast-mediated bone resorption, has been studied in multiple animal models of OA17,18,20,21. Bisphosphonates bind to the surface of mineralized bone and are metabolized by osteoclasts during bone remodeling, leading to impaired osteoclast activity and/or apoptosis22,23. Although bisphosphonates were effective in attenuating OA progression in preclinical post-traumatic OA models, clinical studies in human subjects failed to show attenuation of cartilage loss assessed radiographically, despite the evidence that the treatment inhibited bone remodeling24,25. The discrepancies in the efficacy of bisphosphonates between preclinical and clinical models could be due to multiple factors including the use of invasive injury to induce OA in the animal models and the diverse stages of OA progression in the patient cohorts at the time of treatment intervention26. Previous studies examining the effect of inhibiting bone remodeling with bisphosphonates on attenuating OA disease progression have not used a controlled, non-invasive, preclinical OA model.
We and others have recently developed a non-invasive load-induced model of OA in the mouse27,28, based on controlled cyclic compression of the tibia and initially intended for bone adaptation studies29,30. Using a peak load of 9N for 1200 cycles, this model induced controlled instabilities in the knee joint31, and recapitulated OA pathology in the cartilage and SCB after 1, 2, and 6 weeks of daily loading in adult C57BL/6 mice27,28. A single bout of loading also induced disease initiation and progression, demonstrating that OA pathology in this model can be initiated by cell-mediated processes that are activated by mechanical loading32.
In the present study, we sought to elucidate the role of SCB properties and remodeling on OA initiation and progression using our controlled, non-invasive, preclinical OA model. We utilized two mouse strains with different bone properties and used alendronate (ALN) treatment to inhibit bone remodeling to examine the respective roles of SCB properties and SCB remodeling on temporal changes in SCB plate and cartilage pathology. We hypothesized that mice with initially stiffer SCB would exhibit more severe disease and that the inhibition of bone remodeling using ALN would attenuate load-induced OA progression.
Methods
Mechanical Loading and Treatment Conditions
To examine the role of SCB properties on OA progression, we subjected two strains of mice with different bone mass and stiffness to compressive joint loading. We used 26-week-old male C57Bl/6 (B6) and FVB/NJ (FVB) mice, with FVB having higher bone mass and stiffness compared to B6 mice33 (Fig. 1A). To examine the role of SCB remodeling on OA, mice from both strains were randomly divided into 2 treatment groups: alendronate (ALN) to inhibit bone remodeling or vehicle saline control (VEH).
Figure 1.

A) 26 week-old male C57Bl/6 (red) and FVB mice (blue) were administered alendronate (26μg/kg/day) or vehicle saline treatment for 1, 2, and 6 weeks (5 days/week). B) Concurrently, all mice were subjected to compressive tibial loading of the left limb at a peak load of 9N (B6) or 10.3N (FVB). The right limb served as the contralateral control. C) Mice were euthanized after 1, 2, and 6 weeks of loading and treatment (n = 5=7/group)
All mice were housed by strain in groups of four to five per cage with ad libitum access to food and water. At the start of the experiment, B6 and FVB mice weighed 30.7 ± 2.4g and 32.9 ± 2.6g, respectively. Body mass was measured 5 days/week to monitor the general health of each mouse over the duration of the experiment. A sample size of n = 6–7 was used per group based on a power analysis from previous data27. All experimental procedures occurred in the morning in a veterinary research facility. Mice were subjected to loading and treatment in random order within each cage. All procedures were approved by the Institutional Animal Care and Use Committee.
At 26 weeks, the left hindlimb of each mouse was subjected to in vivo cyclic compressive loading across the knee joint for 1, 2, or 6 weeks, 5 days/week (Fig. 1B, C, n = 6–7/treatment/duration). Under general anesthesia (2% isoflurane, 1.0L/min, Webster, Devens, MA), B6 mice were loaded at 9.0N peak load, and FVB mice were loaded at 10.3N. These peak forces correspond to the loads required to generate 1200με of tension at the medial midshaft of the tibia based on previous in vivo strain measurements with B6 mice27,34 and a pilot strain gauge study with FVB mice. The loading protocol was applied for 1200 cycles (5 minutes) at a frequency of 4Hz based on previous studies27. The right limb served as a contralateral control. Concurrent with loading, each mouse was treated 5 days/week with ALN (26μg/kg/day ip, Sigma-Aldrich, St. Louis, MO) or VEH based on previous studies35. After 1, 2, or 6 weeks, the mice were euthanized. Both knee joints were dissected and fixed in 10% formalin overnight. One B6 mouse and three FVB mice were excluded, due to anesthesia-related death or excessive weight loss during loading.
Cartilage and Subchondral Bone Assessment
To assess bone morphological changes, intact joints were transferred to 70% ethanol after tissue fixation overnight, and scanned by microcomputed tomography (microCT) with a 10μm isotropic voxel resolution (μCT35, Scanco: 55kVp, 145μA, 600ms integration time). A 0.5mm aluminum filter was used to reduce the effects of beam hardening. In addition, with a scan resolution of 10μm, the voxel size is appropriately small relative to the cortical thickness, minimizing any error due to partial volume36. Knee joints were then decalcified in formic acid/sodium citrate for one week, dehydrated in an ethanol gradient, and embedded in paraffin. Serial coronal 6μm-thick sections were obtained (Leica RM2255, Germany). Safranin O/Fast green staining was performed on sections at 90μm intervals to assess cartilage morphology in the tibial plateau. Cartilage degradation was examined by a blinded observer using a modified murine cartilage histological scoring system37 on the most posterior 180μm of the tibial plateau. Scores from the posterior medial and lateral plateaus were summed for our analyses, as these regions exhibited the most cartilage damage in previous studies27,32.
Tibial SCB morphology was assessed using microCT in two volumes of interest (VOI): 1) the SCB plate, extending from the joint space to the epiphyseal cancellous bone, and 2) the epiphyseal cancellous bone. Mineralized tissue from the SCB plate and cancellous epiphysis were segmented using global thresholds. For the SCB plate VOI, cortical bone was manually contoured to assess average cortical plate thickness and tissue mineral density (TMD, mg HA/cm3) in the medial and lateral tibial plateau. For the epiphyseal cancellous bone VOI, cancellous bone within the epiphysis was manually identified to measure cancellous bone volume fraction (BV/TV, mm3/mm3), trabecular thickness (Tb.Th, μm), trabecular separation (Tb.Sp, μm), and TMD.
We assessed localized cartilage and SCB plate thicknesses and osteophyte formation using sections stained with Safranin O/Fast green. The tibial plateau was divided into medial and lateral halves, and then further divided into anterior, middle, and posterior regions, resulting in six tibial plateau regions for evaluation. A single representative slide from each region was used to measure cartilage and local SCB plate thicknesses using previously established protocols27. In addition, osteophyte formation was examined at the margin of the posterior medial tibial plateau. Osteophyte maturity was measured based on previously established protocols38 with scores of 0 (no osteophyte), 1 (mainly cartilaginous), 2 (cartilaginous/mineralized mixed tissue), and 3 (predominately mineralized osteophyte). Osteophyte size was measured as the maximum medial-lateral width of the tissue.
Statistical Analysis
All statistical analysis was performed using linear regression models (JMP Pro 10.0, SAS Institute Inc.). First, the effects of mouse strain and/or treatment were examined in the control (right) limbs using a multiple linear regression model with fixed effects of mouse strain, treatment, duration, and their interactions. Then, to determine the effect of loading, differences between loaded and control limbs were calculated for each metric ([Loaded – Control] limb) and used in a multiple linear regression model with fixed effects as outlined above. In addition, a mixed multiple linear regression model was examined with fixed effects of limb, strain, treatment, duration, and interactions; a random mouse effect accounted for the repeated left-right limb measurement. Each model was optimized using backward elimination of interaction effects. For each linear regression model, we performed a residual analysis to ensure that the residuals were normally distributed and that the data exhibited homogenous variance. In the case of the bone morphology metrics (cancellous and cortical bone), one sample in the 1-week ALN-treated B6 group was an outlier, based on the residual analysis, and was excluded from all analyses of bone morphology. Tukey post-hoc comparisons were performed when interaction effects were significant. p <0.05 indicated significance. All results presented are statistically significant unless otherwise stated.
Results
Intrinsic Differences in Bone and Cartilage due to Mouse Strain and Treatment
Control limbs of B6 and FVB mice had intrinsic differences in bone and cartilage morphology. FVB mice had significantly thicker SCB plates and higher epiphyseal cancellous bone mass than B6 mice due to thicker trabeculae and reduced Tb.Sp (Fig. 2A–C, E). TMD was also higher in SCB and epiphyseal cancellous bone from FVB mice (Fig. 2D, F). In both mouse strains, epiphyseal cancellous bone mass in control limbs decreased over time in VEH-treated mice. ALN treatment prevented age-related reductions in bone mass by increasing Tb.Th and decreasing Tb.Sp (Fig. 2A–C). ALN also significantly increased SCB plate and cancellous TMD over time (Fig. 2D, F). SCB plate thickness increased with ALN only in control limbs of FVB mice, but not B6 control limbs (Fig. 2E). Intrinsic cartilage properties differed in the mouse strains. Cartilage was thicker on the posterior, middle, and anterior aspects of the joint in FVB mice compared to B6 (Fig. 3, middle & anterior data not shown). Based on average thickness values, cartilage thickness was not different in either mouse strain with ALN treatment in this study. Generally, FVB mice had higher bone mass and thicker cartilage compared to B6 mice, and ALN prevented age-related cancellous bone loss in both mouse strains.
Figure 2.

In control (right) limbs, FVB mice exhibited higher cancellous and cortical bone mass than B6 mice, and ALN treatment inhibited bone remodeling. A) ALN prevented a decrease in BV/TV after 6 weeks, and FVB mice exhibited higher cancellous bone mass due to B) higher trabecular thickness and C) lower trabecular separation. FVB mice also had higher D) cancellous and F) cortical tissue mineral density and E) a thicker SCB plate, which was further increased with ALN treatment in FVB mice only. p < 0.05 for ^strain, +duration, §treatment, %strain*duration, #strain*treatment, ¶duration*treatment. Means sharing the same letter are not significantly different from each other by Tukey’s HSD: A>B>C, p<0.05).
Figure 3.
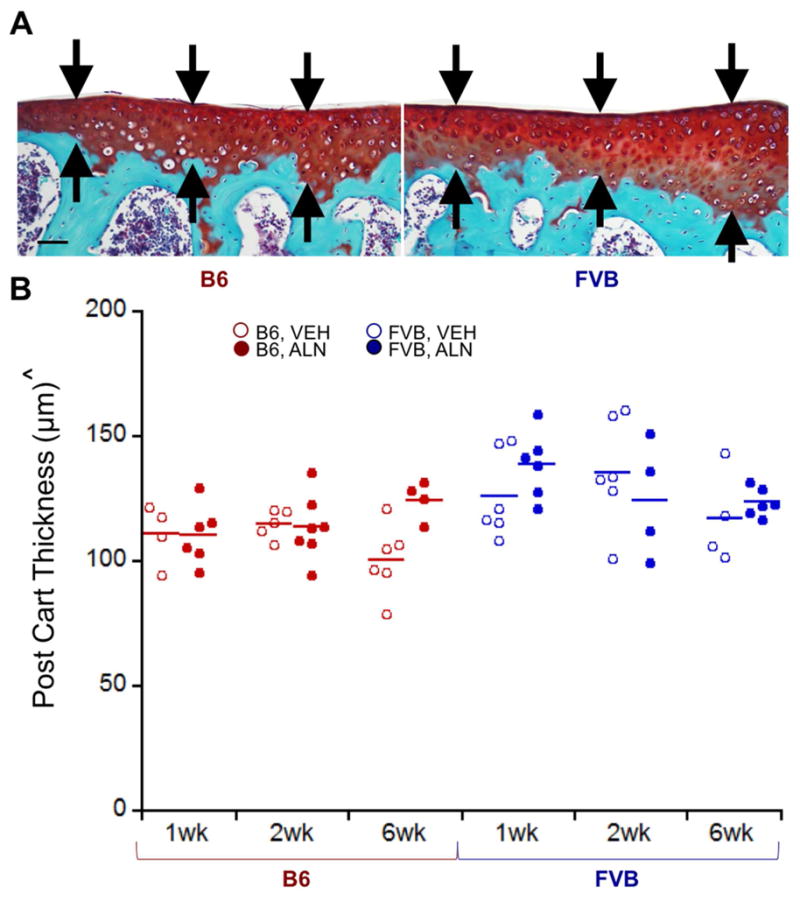
In control (right) limbs, cartilage was thicker in the posterior medial quadrant of FVB limbs than in the same region in B6 limbs. A) Representative posterior medial cartilage histology for B6 and FVB control limbs treated with VEH at 1 week, and B) quantitative cartilage thickness in B6 and FVB control limbs treated with VEH and ALN after 1, 2, and 6 weeks. Scale bar = 50μm. p < 0.05 for ^strain.
Load-Induced Subchondral Bone Adaptation was Mouse Strain-Specific
Loading and ALN treatment induced differential effects on SCB changes in the two mouse strains (Fig. 4). Loading significantly thinned the SCB plate only in B6 mice after 6 weeks (Fig. 4A), resulting in a 13% decrease in mean thickness regardless of treatment based on microCT measurements (Supplemental Table 1). Analysis of the local SCB plate thickness in B6 mice using histology showed a decrease in all aspects of the tibial plateau ranging from 2 – 50%, with the most thinning occurring in medial-middle and medial-anterior aspects. In contrast, SCB plate thickness was not altered with loading in FVB mice. Unlike SCB plate thickness, epiphyseal cancellous bone mass was not affected by loading in B6 mice; however, loading decreased cancellous bone mass in FVB mice regardless of treatment (Fig. 5A). Cancellous and SCB plate TMD were also generally reduced in loaded limbs (Fig. 5D, F). ALN treatment did not attenuate the load-induced reduction in SCB plate TMD in either strain. Loading decreased SCB plate TMD more over time in B6 compared to FVB limbs (Fig. 5F). Loading affected only the SCB plate in B6 and only the cancellous bone in FVB mice. These load-induced responses were not attenuated by ALN treatment.
Figure 4.

Control (right limb, black) and loaded (left limb) data shown. A) Loading thinned SCB plate thickness at 6 weeks in B6 mice only. B) Loading damaged cartilage in most groups except the FVB, ALN group. p < 0.05 for ^strain, +duration, §treatment, %strain*duration, #strain*treatment, ¶duration*treatment, *load. Means sharing the same letter are not significantly different from each other by Tukey’s HSD: A>B>C, p<0.05).
Figure 5.

Loading affected cancellous and cortical SCB morphology in B6 and FVB mice treated with VEH and ALN after 1, 2, and 6 weeks. Δ = [Loaded – Control] (left – right limb) data shown. A) Loading decreased cancellous bone volume fraction in FVB mice only, due to combined effects in B) trabecular thickness and C) trabecular separation. D) Loading decreased cancellous TMD and E) SCB plate thickness more so in B6 mice than FVB. F) Cortical TMD was also generally decreased with loading. p < 0.05 for ^strain, +duration, §treatment, %strain*duration, #strain*treatment, ¶duration*treatment. L indicates load effect (p<0.05).
Articular Cartilage Pathology with Loading
Loading generally increased cartilage matrix loss and thinning over time. As in our previous study27, cartilage damage was localized to the posterior aspect of the tibial plateau, with more damage occurring on the medial posterior aspect. The degree of cartilage pathology depended on mouse strain and treatment (Fig. 6). Specifically, on the posterior aspect of the tibia, loading increased cartilage matrix loss compared to contralateral limbs in all groups except FVB mice treated with ALN (pooled across all treatment durations) (Fig. 4B). FVB mice exhibited less cartilage pathology with loading compared to B6 mice (32% lower histological damage score). ALN-treated B6 mice had the most extensive cartilage matrix changes compared to all other groups after 6 weeks of loading. Local cartilage thinning also occurred with loading and increased with loading duration particularly on both the lateral and medial posterior joint aspects, regardless of mouse strain or treatment (Supplemental Table 1). Cartilage thickness changes with loading ranged from a 21% decrease in the posterior medial aspect to a 23% increase in the anterior lateral aspect. While loading induced cartilage damage in both mouse strains, FVB mice exhibited less pathology compared to B6 mice.
Figure 6.

Loading damaged posterior cartilage matrix in B6 and FVB mice treated with VEH and ALN after 1, 2, and 6 weeks. Δ = [Loaded – Control] (left – right limb) data shown. (A) In most groups, loading created cartilage damage that increased over time as was reflected in the histological scores. (B) FVB mice treated with ALN did not exhibit cartilage damage with loading (pooled group means summarized in box plot). (C) Loading also decreased posterior cartilage thickness over time. Scale bar = 50μm. p<0.05 for ^strain, +duration, §treatment, %strain*duration, #strain*treatment, ¶duration*treatment. L indicates load effect (p<0.05). Yellow arrowheads indicate areas of cartilage damage.
Osteophyte Formation with In Vivo Loading
Loading induced pre-osteophyte or osteophyte formation in all but one mouse (FVB mouse, 1 week, VEH-treated). Osteophytes matured from primarily cartilaginous to mineralized tissue over longer load durations (Fig. 7). Osteophytes in FVB mice were less mature, smaller, and slower growing compared to those in B6 mice (Fig. 7B, C). ALN inhibited osteophyte maturation compared to VEH treatment, but did not affect osteophyte size. Osteophytes were absent in control limbs. Osteophytes occurred with loading, indicative of OA pathology; however, osteophytes in loaded FVB limbs were less mature and smaller than those in B6 limbs.
Figure 7.

Loading induced osteophytes, which were smaller in FVB mice. A) Loading induced visible osteophytes that matured and grew over time. B) ALN treatment slowed maturation of osteophytes, which were also C) smaller in FVB mice. Scale bar = 250μm. p<0.05 for ^strain, +duration, §treatment, %strain*duration, #strain*treatment, ¶duration*treatment.
Discussion
In this study, we examined the role of SCB properties and changes in OA initiation and progression. We used two mouse strains with different bone properties and ALN treatment to inhibit bone changes, with the objective of examining OA pathology in both cartilage and SCB morphology. Our results confirmed the presence of significant intrinsic differences between FVB and B6 mouse strains in bone mass and stiffness and in responses to the inhibition of bone remodeling with ALN treatment. In control limbs, FVB mice had a thicker SCB plate, higher epiphyseal cancellous bone mass, and higher bone mineral density than B6 mice. Furthermore, FVB mice had stiffer diaphyseal cortical bone as reflected by the higher load (10.3N) needed to engender +1200με at the mid-diaphysis of the tibia. While we did not directly measure SCB plate stiffness in this study, differences in cortical bone material properties between FVB and B6 mice were assessed by our strain calibration and have been assessed previously by mechanical loading33. Based on these diaphyseal data, we assume that FVB mice had stiffer SCB compared to B6 mice. Similar to previous preclinical studies39, ALN prevented age-related reductions in cancellous bone mass. ALN treatment also increased the cancellous and SCB plate TMD over time in control limbs, indicating that the treatment was indeed effective in inhibiting bone remodeling in both cancellous and cortical bone35,40. These findings support the validity of our experimental approach to examine the role of intrinsic differences in bone properties and bone remodeling on the progression of load-induced OA joint pathology.
Non-invasive cyclic compression induced OA cartilage pathology and osteophyte formation in both mouse strains. We did not observe any ligament tears in this study. Similar to previous studies27,28, loading generally led to reduced proteoglycan content, cartilage surface fibrillation, cartilage matrix thinning, osteophyte formation, and subchondral and epiphyseal cancellous bone adaptation, recapitulating OA progression. Lower initial SCB mass and stiffness in B6 mice did not attenuate load-induced OA severity compared to FVB mice. In fact, FVB mice exhibited less cartilage pathology and slower-growing and less mature osteophytes, consistent with diminished OA severity.
Cyclic loading induced differential effects on bone adaptation in the tibiae of B6 and FVB mice. In B6 mice, loading thinned the SCB plate, particularly after 6 weeks. In contrast, loading decreased only the epiphyseal cancellous bone mass in FVB mice and did not affect SCB plate thickness. No increase in bone mass was detected in either strain over time. Ko et al.27 reported a reduction in epiphyseal cancellous bone with daily loading in B6 mice, accompanied by localized thickening of the SCB plate. These contradictory outcomes could reflect the difficulty of distinguishing between calcified cartilage and SCB using microCT; however, localized SCB plate thickness measured by histology also did not increase with loading (Supplemental Table 1). In several preclinical studies15,16 SCB plate thickness decreased initially, followed by thickening as OA progressed. Thus, either our time points were too distant to detect subtle temporal changes in SCB plate thickness, or at 6 weeks post-loading the mice were still in the early stages of OA development.
Inhibiting bone remodeling had differential effects on cartilage and bone adaptation to loading in the two mouse strains. ALN treatment exacerbated cartilage pathology in B6 mice after 6 weeks of loading, but protected FVB limbs from load-induced cartilage changes. Unlike other preclinical studies17–21, ALN treatment in our study did not consistently protect against cartilage pathology during OA progression. However, the lack of chondro-protection with ALN treatment is similar to the results found in a comprehensive clinical study25. Changes in cancellous and SCB plate bone mass and mineralization with loading depended on the mouse strain. Loading initially decreased cancellous TMD in both ALN- and VEH-treated groups; however, cancellous TMD was maintained without further loss thereafter with ALN treatment. Differences in these data compared to results obtained in post-traumatic injury models of OA may reflect the non-invasive nature of our model compared to the surgical intervention required in other models32. In this study, ALN generally did not inhibit load-induced changes in bone and had differential effects on cartilage changes depending on mouse strain. The limited effect of ALN treatment on OA pathology could be due to the timing of treatment. Pre-treatment of bisphosphonates prior to OA induction may be more effective at attenuating bone changes and OA pathology41. Future studies could examine the use of higher doses or longer term ALN treatment to effectively inhibit load-induced changes in bone.
The results of our study do not support our initial hypothesis that intrinsically lower SCB mass and stiffness attenuate OA progression. Radin and Rose9 first hypothesized that increased SCB mass and stiffness would diminish shock absorption by bone and increase stresses in the cartilage surface. However, Burr and others,3,11 employing a model involving the insertion of a metal plug in the subchondral cancellous bone, demonstrated that increasing apparent SCB stiffness did not exacerbate cartilage damage. Our results using mice with intrinsically different SCB stiffness led to a similar conclusion.
The use of two mouse strains to test the contribution of intrinsic bone and cartilage physical properties to the development of OA joint pathology did not account for differences in intrinsic cartilage thickness and potential differences in bone and cartilage metabolism between the two strains. ALN treatment was chondro-protective in FVB mice as a group (all ALN treatment durations pooled), but exacerbated cartilage pathology in B6 mice. This seemingly contradictory result suggests that alternate factors determined the severity of OA progression in the different mouse strains, possibly related to differences in intrinsic strain, differences in cartilage thickness, or genetic variations in bone and cartilage homeostasis. Specifically, B6 mice with intrinsically thinner cartilage exhibited significant thinning of the SCB plate with loading, accompanied by severe cartilage pathology and osteophyte formation. In contrast, FVB mice with intrinsically thicker cartilage, when treated with ALN, did not display significant changes in SCB plate thickness or mineralization over time and exhibited diminished OA severity. Similar findings were reported in another load-induced OA model in which intrinsically thicker cartilage in Str/ort mice correlated with diminished cartilage loss42. Based on FEA simulations in that prior study, increased cartilage thickness reduced the contact stresses, which accounted for the attenuated cartilage damage. Furthermore, genetic differences in bone remodeling between the two mouse strains were not examined and could play a significant role in our results. Future studies using mouse strains with established differences in cartilage thickness and/or differential patterns of bone and cartilage metabolism would permit assessment of these factors. Alternately, these factors could be minimized to eliminate their potential confounding contribution to load-induced OA.
While ALN treatment effectively reduced bone remodeling with age in control limbs, changes in bone with loading were still present. In addition, although we used ALN in our studies to target SCB remodeling, ALN treatment may not exclusively affect bone and may also directly affect cartilage metabolism43. We did not specifically examine the effect of ALN treatment on chondrocytes and macrophages, but we saw no effect of treatment on cartilage structure by histology. While ALN could affect chondrocytes and macrophages, these cells might not be involved in load-based adaptation, as is the case with the studies of Sugiyama et al.44 in which they speculate that the increase in bone with loading was mediated by down-regulation of sclerostin in osteocytes, an effect that was not blocked by bisphosphonate treatment. Regardless of the ALN treatment effect on these cells, their role in load-induced tissue changes is unknown. Future studies should investigate the role of chondrocytes and macrophages on load-induced tissue adaptation. Although ALN effectively reduced bone remodeling by inhibiting bone resorption, alternate approaches for modulating SCB bone properties, for example by inhibiting sclerostin activity represent additional experimental approaches to test our hypothesis.
We used two different peak loads based on in vivo strain gauge data for B6 and FVB mice. Whereas the use of different peak loads for each mouse strain may appear to be a limitation, we intentionally controlled the strain induced on the bone. Bone surface strains during peak activity are remarkably well conserved across mammals45. Therefore, we used the loads to induce +1200 μstrain at the mid-diaphysis as a metric to equilibrate the applied loads across animals of different strains and ages. The body mass and skeleton of the FVB mouse are larger than those of the B6 mouse, suggesting that the loads engendered during normal activities would be higher and consistent with the higher loads needed to produce similar mechanical strains in the two mouse strains. Furthermore, we were interested in the effect of bone on OA progression in the cartilage, thus we controlled the stimulation (strain) engendered on the bone. Because we equalized the stimulation on the bone regardless of mouse strain, we can distinguish the role of bone mass/stiffness on cartilage degradation.
In conclusion, contrary to our prediction, we found that intrinsically lower SCB properties were not associated with attenuated load-induced cartilage pathology. This result may be related, in part, to intrinsic differences in cartilage thickness, although this hypothesis needs to be tested. Our findings that inhibition of bone remodeling produced differential patterns of OA pathology in animals with low or high SCB properties indicate that SCB properties and remodeling do affect the progression of load-induced OA cartilage pathology. These data support the utility of the compressive loading model for defining the roles of SCB plate properties and remodeling on the pathogenesis of OA.
Supplementary Material
Posterior, medial, and lateral cartilage score, thickness, and SCB plate measurements of control and loaded limbs in B6 and FVB mice treated with VEH and ALN after 1, 2, and 6 weeks of loading. p<0.05 for astrain, blimb, cstrain*limb, dstrain*treatment, eduration*treatment, flimb*duration, gstrain*duration*limb hstrain*treatment*limb, istrain*duration*treatment.
Acknowledgments
We thank Lyudmila Lukashova of the HSS microCT core facility, Amanda Rooney, Naa Shidaa Armar, Allison Hsia, Dan Loeffler, the Cornell CARE staff and the Cornell Statistical Consulting Unit for their experimental assistance.
Role of the funding source
This work was supported by NIH R21-AR064034 (MCHM), the Robert & Helen Appel Fellowship, NSF GRFP (OOA, DGE-1144153), and the Sloan Graduate Fellowship (OOA).
Footnotes
Contributions
Conception and design: OOA, FCK, SRG, MBG, TMW, MCHM
Acquisition, analysis, and interpretation of the data: OOA, FCK, PTW, SRG, MBG, TMW, MCHM
Drafting and critical revision of the article for important intellectual content: OOA, FCK, PTW, SRG, MBG, TMW, MCHM
Final approval of the article: OOA, FCK, PTW, SRG, MBG, TMW, MCHM
Conflict of interests
The authors have no conflict of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, Jordan JM, Katz JN, Kremers HM, Wolfe F National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA. Osteoarthritis - an untreatable disease? Nat Rev Drug Discov. 2005;4(4):331–344. doi: 10.1038/nrd1693. [DOI] [PubMed] [Google Scholar]
- 3.Burr DB, Schaffler MB. The Involvement of Subchondral Mineralized Tissues in Osteoarthrosis3: Quantitative Microscopic Evidence. Microsc Res Tech. 1997;37:343–357. doi: 10.1002/(SICI)1097-0029(19970515)37:4<343::AID-JEMT9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Culley KL, Dragomir CL, Chang J, Wondimu EB, Coico J, Plumb DA, Otero M, Goldring MB. Mouse models of osteoarthritis: surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol Biol. 2015;1226:143–173. doi: 10.1007/978-1-4939-1619-1_12. [DOI] [PubMed] [Google Scholar]
- 5.Felson DT. Osteoarthritis: New Insights. Part 1: The Disease and Its Risk Factors. Ann Intern Med. 2000;133(8):635. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 6.Cameron KL, Hsiao MS, Owens BD, Burks R, Svoboda SJ. Incidence of physician-diagnosed osteoarthritis among active duty United States military service members. Arthritis Rheum. 2011;63(10):2974–2982. doi: 10.1002/art.30498. [DOI] [PubMed] [Google Scholar]
- 7.Lohmander LS, Gerhardsson de Verdier M, Rollof J, Nilsson PM, Engström G. Incidence of severe knee and hip osteoarthritis in relation to different measures of body mass: a population-based prospective cohort study. Ann Rheum Dis. 2009;68(4):490–496. doi: 10.1136/ard.2008.089748. [DOI] [PubMed] [Google Scholar]
- 8.Radin E, Paul I, Rose R. Role of mechanical factors in pathogenesis of primary osteoarthritis. Lancet. 1972;299(7749):519–522. doi: 10.1016/S0140-6736(72)90179-1. [DOI] [PubMed] [Google Scholar]
- 9.Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986;(213):34–40. [PubMed] [Google Scholar]
- 10.Radin EL, Martin RB, Burr DB, Caterson B, Boyd RD, Goodwin C. Effects of mechanical loading on the tissues of the rabbit knee. J Orthop Res. 1984;2(3):221–234. doi: 10.1002/jor.1100020303. [DOI] [PubMed] [Google Scholar]
- 11.Brown TD, Radin EL, Martin RB, Burr DB. Finite element studies of some juxtarticular stress changes due to localized subchondral stiffening. J Biomech. 1984;17(1):11–24. doi: 10.1016/0021-9290(84)90075-7. [DOI] [PubMed] [Google Scholar]
- 12.Burr DB. The importance of subchondral bone in the progression of osteoarthritis. J Rheumatol Suppl. 2004;70:77–80. [PubMed] [Google Scholar]
- 13.Li G, Yin J, Gao J, Cheng TS, Pavlos NJ, Zhang C, Zheng MH, Grynpas M, Alpert B, Katz I, Lieberman I, Pritzker K, Suri S, Walsh D, Madry H, van Dijk C, Mueller-Gerbl M, Burr D, et al. Subchondral bone in osteoarthritis: insight into risk factors and microstructural changes. Arthritis Res Ther. 2013;15(6):223. doi: 10.1186/ar4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawcak CE, McIlwraith CW, Norrdin RW, Park RD, James SP. The role of subchondral bone in joint disease: a review. Equine Vet J. 2010;33(2):120–126. doi: 10.1111/j.2042-3306.2001.tb00589.x. [DOI] [PubMed] [Google Scholar]
- 15.Hayami T, Pickarski M, Zhuo Y, Wesolowski GA, Rodan GA, Duong LT. Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone. 2006;38(2):234–243. doi: 10.1016/j.bone.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Botter SM, Van Osch GJVM, Clockaerts S, Waarsing JH, Weinans H, Van Leeuwen JPTM. Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: An in vivo microfocal computed tomography study. Arthritis Rheum. 2011;63(9):2690–2699. doi: 10.1002/art.30307. [DOI] [PubMed] [Google Scholar]
- 17.Muehleman C, Green J, Williams JM, Kuettner KE, Thonar EJ-MA, Sumner DR. The effect of bone remodeling inhibition by zoledronic acid in an animal model of cartilage matrix damage. Osteoarthr Cartil. 2002;10(3):226–233. doi: 10.1053/joca.2001.0506. [DOI] [PubMed] [Google Scholar]
- 18.Hayami T, Pickarski M, Wesolowski GA, Mclane J, Bone A, Destefano J, Rodan GA, Duong LT. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 2004;50(4):1193–1206. doi: 10.1002/art.20124. [DOI] [PubMed] [Google Scholar]
- 19.Hayami T, Zhuo Y, Wesolowski GA, Pickarski M, Duong LT. Inhibition of cathepsin K reduces cartilage degeneration in the anterior cruciate ligament transection rabbit and murine models of osteoarthritis. Bone. 2012;50(6):1250–1259. doi: 10.1016/j.bone.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 20.Myers SL, Brandt KD, Burr DB, O’Connor BL, Albrecht M. Effects of a bisphosphonate on bone histomorphometry and dynamics in the canine cruciate deficiency model of osteoarthritis. J Rheumatol. 1999;26(12):2645–2653. [PubMed] [Google Scholar]
- 21.Siebelt M, Waarsing JH, Groen HC, Müller C, Koelewijn SJ, de Blois E, Verhaar JAN, de Jong M, Weinans H. Inhibited osteoclastic bone resorption through alendronate treatment in rats reduces severe osteoarthritis progression. Bone. 2014;66:163–170. doi: 10.1016/j.bone.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Fleisch H. Bisphosphonates: Mechanisms of Action. Endocr Rev. 1998;19(1):80–100. doi: 10.1210/edrv.19.1.0325. [DOI] [PubMed] [Google Scholar]
- 23.Russell RGG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int. 2008;19(6):733–759. doi: 10.1007/s00198-007-0540-8. [DOI] [PubMed] [Google Scholar]
- 24.Spector TD, Conaghan PG, Buckland-Wright JC, Garnero P, Cline GA, Beary JF, Valent DJ, Meyer JM, Dixon T, Shaw M, Ebrahim S, Dieppe P, Millar W, Melzer D, Guralnik J, Brock D, Quam J, Michet C, et al. Effect of risedronate on joint structure and symptoms of knee osteoarthritis: results of the BRISK randomized, controlled trial [ISRCTN01928173] Arthritis Res Ther. 2005;7(3):R625. doi: 10.1186/ar1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bingham CO, Buckland-Wright JC, Garnero P, Cohen SB, Dougados M, Adami S, Clauw DJ, Spector TD, Pelletier JP, Raynauld JP, Strand V, Simon LS, Meyer JM, Cline GA, Beary JF. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: Results of the two-year multinational knee osteoarthritis st. Arthritis Rheum. 2006;54(11):3494–3507. doi: 10.1002/art.22160. [DOI] [PubMed] [Google Scholar]
- 26.Holyoak DT, Tian YF, van der Meulen MCH, Singh A. Osteoarthritis: Pathology, Mouse Models, and Nanoparticle Injectable Systems for Targeted Treatment. Ann Biomed Eng. 2016;44(6):2062–2075. doi: 10.1007/s10439-016-1600-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko FC, Dragomir C, Plumb DA, Goldring SR, Wright TM, Goldring MB, van der Meulen MCH. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheum. 2013;65(6):1569–1578. doi: 10.1002/art.37906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulet B, Hamilton RW, Shefelbine S, Pitsillides AA. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 2011;63(1):137–147. doi: 10.1002/art.27765. [DOI] [PubMed] [Google Scholar]
- 29.Turner CH, Akhter MP, Raab DM, Kimmel DB, Recker RR. A noninvasive, in vivo model for studying strain adaptive bone modeling. Bone. 1991;12(2):73–79. doi: 10.1016/8756-3282(91)90003-2. [DOI] [PubMed] [Google Scholar]
- 30.Fritton J, Myers E, Wright T, van der Meulen M. Loading induces site-specific increases in mineral content assessed by microcomputed tomography of the mouse tibia. Bone. 2005;36(6):1030–1038. doi: 10.1016/j.bone.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Adebayo OO, Ko FC, Goldring SR, Goldring MB, Wright TM, van der Meulen MCH. Kinematics of meniscal- and ACL-transected mouse knees during controlled tibial compressive loading captured using roentgen stereophotogrammetry. J Orthop Res. 2016;(April 2016):1–23. doi: 10.1002/jor.23285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko FC, Dragomir CL, Plumb DA, Hsia AW, Adebayo OO, Goldring SR, Wright TM, Goldring MB, van der Meulen MCH. Progressive cell-mediated changes in articular cartilage and bone in mice are initiated by a single session of controlled cyclic compressive loading. J Orthop Res. 2016;(October 2015) doi: 10.1002/jor.23204. n/a–n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wergedal JE, Sheng MH-C, Ackert-Bicknell CL, Beamer WG, Baylink DJ. Genetic variation in femur extrinsic strength in 29 different inbred strains of mice is dependent on variations in femur cross-sectional geometry and bone density. Bone. 2005;36(1):111–122. doi: 10.1016/j.bone.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Lynch ME, Main RP, Xu Q, Schmicker TL, Schaffler MB, Wright TM, van der Meulen MCH. Tibial compression is anabolic in the adult mouse skeleton despite reduced responsiveness with aging. Bone. 2011;49(3):439–446. doi: 10.1016/j.bone.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misof BM, Roschger P, Baldini T, Raggio CL, Zraick V, Root L, Boskey AL, Klaushofer K, Fratzl P, Camacho NP. Differential effects of alendronate treatment on bone from growing osteogenesis imperfecta and wild-type mouse. Bone. 2005;36(1):150–158. doi: 10.1016/j.bone.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 37.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative – recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil. 2010;18:S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 38.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, Shah M, Thompson EW. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodan GA, Seedor JG, Balena R. Preclinical pharmacology of alendronate. Osteoporos Int. 1993;3(S3):7–12. doi: 10.1007/BF01623001. [DOI] [PubMed] [Google Scholar]
- 40.Camacho NP, Raggio CL, Doty SB, Root L, Zraick V, Ilg WA, Toledano TR, Boskey AL. A controlled study of the effects of alendronate in a growing mouse model of osteogenesis imperfecta. Calcif Tissue Int. 2001;69(2):94–101. doi: 10.1007/s002230010045. [DOI] [PubMed] [Google Scholar]
- 41.Strassle BW, Mark L, Leventhal L, Piesla MJ, Jian Li X, Kennedy JD, Glasson SS, Whiteside GT. Inhibition of osteoclasts prevents cartilage loss and pain in a rat model of degenerative joint disease. Osteoarthr Cartil. 2010;18(10):1319–1328. doi: 10.1016/j.joca.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Poulet B, Westerhof TaT, Hamilton RW, Shefelbine SJ, Pitsillides aa. Spontaneous osteoarthritis in Str/ort mice is unlikely due to greater vulnerability to mechanical trauma. Osteoarthr Cartil. 2013;21(5):756–763. doi: 10.1016/j.joca.2013.02.652. [DOI] [PubMed] [Google Scholar]
- 43.Dombrecht EJ, De Tollenaere CB, Aerts K, Cos P, Schuerwegh AJ, Bridts CH, Van Offel JF, Ebo DG, Stevens WJ, De Clerck LS. Antioxidant effect of bisphosphonates and simvastatin on chondrocyte lipid peroxidation. Biochem Biophys Res Commun. 2006;348(2):459–464. doi: 10.1016/j.bbrc.2006.07.075. [DOI] [PubMed] [Google Scholar]
- 44.Sugiyama T, Meakin LB, Galea GL, Jackson BF, Lanyon LE, Ebetino FH, Russell RGG, Price JS. Risedronate does not reduce mechanical loading-related increases in cortical and trabecular bone mass in mice. Bone. 2011;49(1):133–139. doi: 10.1016/j.bone.2011.03.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubin CT. Skeletal strain and the functional significance of bone architecture. Calcif Tissue Int. 1984;36(S1):S11–S18. doi: 10.1007/BF02406128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Posterior, medial, and lateral cartilage score, thickness, and SCB plate measurements of control and loaded limbs in B6 and FVB mice treated with VEH and ALN after 1, 2, and 6 weeks of loading. p<0.05 for astrain, blimb, cstrain*limb, dstrain*treatment, eduration*treatment, flimb*duration, gstrain*duration*limb hstrain*treatment*limb, istrain*duration*treatment.