

Abstract
We describe a facile, scalable route to access functional group-rich gem-difluoroalkenes. Using visible-light activated catalysts in conjunction with an arsenal of carbon-radical precursors, an array of trifluoromethyl-substituted alkenes undergoes radical defluorinative alkylation. Non-stabilized primary, secondary, and tertiary radicals can be used to install functional groups in a convergent manner that would otherwise be challenging via two-electron pathways. The process readily extends to other perfluoroalkyl-substituted alkenes. In addition, we report the development of an organotrifluoroborate reagent to expedite the synthesis of the requisite trifluoromethyl-substituted alkene starting materials.
Keywords: Photoredox, Organofluorine, Alkenes, Alkylation, Radicals
Graphical Abstract
CF3 today, CF2 tomorrow: We describe a facile, scalable route to access functional group-rich gem-difluoroalkenes. Using visible-light activated catalysts in conjunction with an arsenal of C-radical precursors, an array of trifluoromethyl-substituted alkenes undergo radical defluorinative alkylation. Non-stabilized primary, secondary, and tertiary radicals can be used install functional groups in a convergent manner that would otherwise be challenging to access via two-electron pathways. The process readily extends to other perfluoroalkyl-substituted alkenes. In addition, we report the development of an organotrifluoroborate reagent to expedite synthesis of the requisite trifluoromethyl-substituted alkene starting materials.
Optimizing oral bioavailability is an important facet of developing molecules for use as therapeutic agents.[1] Although ketone functional groups can be vital for binding and substrate recognition,[2] they are uncommon in pharmaceutical compounds because of their reduction to the corresponding alcohol during phase I metabolism by mammalian NAD(P)H-dependent reductases.[3] This process facilitates inactivation and excretion, which leads to diminished bioavailability.[3]
Consequently, efforts have been made to identify carbonyl mimics that preserve the electronics and geometry of the C=O unit but are less susceptible to in vivo metabolism.[4] The gem-difluoroethylene moiety, an isopolar analog that may still engage in hydrogen bonding,[5] is one example. Replacement of a carbonyl with the corresponding gem-difluoroalkene has been demonstrated to provide bioactive substrates that are still recognized by their target; however, only a few examples have been reported in the literature.[6] In the case of exo-difluoroethylene artemisinin, the prolonged in vivo antimalarial activity may arise from resistance to metabolic processes (Figure 1).[6a] Additionally, this motif has proved beneficial in several niche applications.[7]
Figure 1.

Representative applications of gem-difluoroalkenes.
One reason that this potentially valuable carbonyl mimic has not been more extensively evaluated may be that conventional routes to construct the gem-difluoroethylene motif typically involve a functional group interconversion that relies upon highly reactive intermediates, organometallic reagents, or harsh conditions, thus hampering their applicability (Scheme 1a).[8] Although several convergent methods exist, they do not lend themselves to late-stage functionalization or to the incorporation of sensitive functional groups (Scheme 1b). [8]
Scheme 1.

Typical routes to gem-difluoroalkenes.
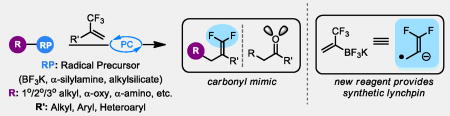
Our laboratory has demonstrated that the regulated generation of carbon-centered radicals from bench-stable progenitors via photoredox catalysis proceeds under very mild conditions.[9] The reactions of these radicals occur readily at room temperature and typically have a broader substrate scope than analogous two-electron pathways because radicals are not prone to acid/base processes. Thus, we anticipated that the radical alkylation of trifluoromethylalkenes would facilitate a convergent synthesis of functional group-rich gem-difluoroalkenes.
The requisite radical for such an alkylation event can be readily generated via the single electron oxidation of carbon-radical precursors such as those depicted in Scheme 2. Following radical addition, the resulting α-CF3 radical would likely undergo SET reduction by the reduced state of the photocatalyst,[10] generating a carbanion. Although protonation is viable, we anticipated the known propensity for such anions to undergo E1cB-type fluoride elimination[11] to predominate and provide the desired gem-difluoroalkene via a radical/polar crossover process. Indeed, such a radical process is viable for the addition of acyl and stabilized α-amino radicals to trifluoromethyl styrenes, but a general method for the utilization of 1°, 2°, and 3° radicals does not exist.[12] Moreover, we reasoned that using an array of radical precursors would impart several benefits such as additive-free reactions at near neutral pH, the ability to use inexpensive photocatalysts, and the opportunity to install a wide range of functional groups.
Scheme 2.

A convergent route to gem-difluoroalkenes via radical alkylation.
To evaluate the tenability of this approach, a trifluoromethyl styrene bearing two readily manipulated functional handles (1a), in conjunction with three classes of radical precursors that produce a byproduct that can efficiently sequester fluoride upon homolytic fragmentation, were subjected to a range of photoredox conditions. Pleasingly, both the inexpensive organometallic photocatalyst, Ru(bpy)3(PF6)2, and the organic photocatalyst 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN)[13] facilitated the desired transformation without the need for additives (Scheme 2). Control studies verified that each component of the reaction mixture was necessary.
Next, the scope of this process was evaluated. Various aminomethyl moieties were introduced via the corresponding α-trimethylsilylamines (Table 1). These amines readily undergo desilylative fragmentation following single electron oxidation of the nitrogen’s lone pair.[14] Difluoroalkenes bearing cyclic and acyclic amines were isolated in good yields.
Table 1.

|
All values indicate isolated yields.
General reaction conditions: radical precursor (1.4 equiv), perfluoroalkyl-substituted alkene (1 equiv, 0.25–0.5 mmol scale), Ru(bpy)3(PF6)2 (2.5 mol %) or 4CzIPN (5 mol %) DMF or DMSO (0.05–0.1 M), rt, 18–36 h at rt, irradiating with blue LEDs (6 to 40 W), see Supporting Information for details.
Ratio of gem-difluoroalkene to trifluoromethyl alkane by 19F NMR.
1.2 equiv of LiBF4 added
Identiy of major and minor isomers determined by1H–19F HOESY, see Supporting Information for details.
Potassium organotrifluoroborates also served as competent precursors for a variety of 1°, 2°, and 3° carbon-centered radicals using the organic photocatalyst 4CzIPN. Cyclic secondary radicals provided the desired gem-difluoroalkenes in good yields. An acyclic, secondary radical bearing an unprotected alcohol at the radical center was also well-tolerated (3k). Products resulting from the addition of primary α-thio- and α-oxy-radicals were obtained in reasonable yields (3l–3n). The addition of a 3° radicals also proceeded smoothly (3g,3p, and 3q). These difluoroalkenes would be challenging to construct using conventional approaches, which is a testament to the functional group tolerance of this approach.
Given their unique ability to furnish non-stabilized 1° radicals, alkylbis(catecholato)silicates were next examined.[15] A variety of functional groups were installed with ease. Protected secondary amines (3w, 3x), a lactam (3y), a carbazole (3u), and a free aniline (3t) were all tolerated under the standard reaction conditions. The distinct advantage of this approach is well illustrated here: many of the alkylbis(catecholato)silicates examined have protic functional groups and, further, all have Brønsted acidic ammonium counter-ions. The absence of appreciable protonation (i.e., generation of an alkyl-CF3) during this process indicates that SET reduction/fluoride elimination is a fast process. In addition, a pyridine (3v), an acetate (3s) and an ether (3r) were also successfully incorporated. Lastly, a secondary norbornane ring (3z) was introduced in good yield.
The generality of the radical scope prompted us to evaluate the trifluoromethylalkene scope. Styryl systems bearing a 1,3-benzodioxole (4a), an acetamide (4b), a phenol (4c), and a cyclic amide (4d) were all effectively transformed to their corresponding gem-difluoroalkenes. An exocyclic, disubstituted gem-difluoroalkene (4k) was obtained from a trifluoromethylalkene derived from α-tetralone. Heteroaromatics such as quinoline (4g), indole (4f), indazole (4i), and an aminopyridine (4h) were well-tolerated. In a select case, appreciable formation of the undesired alkyl-CF3 by-product was observed (4j, 82:18 alkene/alkane). Although we are unsure why this particular substrate deviated substantially from all other cases, it may result from a resonance stabilized, longer-lived anion.
Moving beyond styryl systems, a variety of 2-trifluoromethyl-1,3-enynes also underwent successful alkylation without concomitant addition to the alkynyl moiety. Substrates bearing groups such as a protected piperidine (4l), a tertiary propargylic alcohol (4m), or a protected furanose (4n) were readily transformed into the corresponding 1,1-difluoro-1,3-enyne. Also, 3° and α-amino radicals added to non-styryl alkyltrifluoromethylalkenes in good yields (4o, 4p). Finally, the reaction was not limited to trifluoromethyl alkenes. Both CF2H- and CF2CF3-subsituted alkenes provided monofluoro- and (fluoro)trifluoromethylalkenes, respectively, (4q, 4r).
To assess the utility of the described defluorinative alkylation further, the process was scaled to 3 mmol using 4CzIPN for practical reasons [~$5/mmol vs $125/mmol for Ru(bpy)3(PF6)2] (Figure 2). This modification did not compromise the yield, and the product obtained from this scale-up was then further elaborated via Ni/photoredox dual catalysis to forge a Csp2–Csp3 bond.[16] The combination of these two modes enables rapid diversification and illustrates the relative stability of gem-difluoroalkenes under conditions where additional radicals are generated.
Figure 2.

Scale-up of defluorinative alkylation with subsequent cross-coupling.
Finally, during the course of our studies it became clear that most methods to synthesize trifluoromethyl styrenes in a convergent manner relied on the Pd-catalyzed coupling of low-boiling 2-bromo-3,3,3-trifluoro-1-propene with various organoboron compounds.[17] The umpolung approach, wherein the organometallic partner bears the 3,3,3-trifluoro-1-propenyl moiety, is virtually unknown.[18] This relates to the known instability of the corresponding organometallic (i.e., organolithium, -magnesium, etc.).[19] Thus, development of the corresponding potassium 1-trifluoromethylethenyltrifluoroborate would not only enable this umpolung approach to be executed with ease on commercially ubiquitous (hetero)aryl bromides, but also provide an easily handled, eminently stable source of the trifluoromethylvinyl fragment. A reliable protocol for the synthesis of this crystalline, bench-stable species was established, enabling multi-gram quantities to be prepared over three steps (see Supporting Information for details). This organotrifluoroborate was subjected to standard Suzuki cross-coupling conditions with an aryl bromide whose boron-substituted counterpart is prohibitively expensive. The desired trifluoromethyl alkene 1s was obtained in a good yield (76%) and was next subjected to radical alkylation to furnish the corresponding gem-difluoroalkene 4t in 77% yield (Figure 3). These results indicate that potassium trifluoromethylvinyltrifluoroborate serves as a unique synthon with the potential for enabling the construction of a diverse array of trifluoromethyl-substituted styrenes and subsequently for the rapid assembly of gem-difluoroalkenes.
Figure 3.

Development and implementation of an organotrifluoroborate for trifluoromethyl-substituted styrene synthesis.
The defluorinative alkylation protocol developed herein has resulted in the generation of a vast library of functional group-rich gem-difluoroalkenes. Installation of numerous complex fragments was achieved from a variety of carbon-radical progenitors, each from its own unique chemical feedstock. This process is readily scaled and can be paired with existing photoredox technology to enhance structural diversity. Finally, an organotrifluoroborate reagent was developed to expedite trifluoromethyl alkene synthesis and provide a new approach to (trifluoromethyl)vinylation.
Supplementary Material
Acknowledgments
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878) and NIH S10 OD011980. C.B.K. is grateful for an NIH NRSA postdoctoral fellowship (F32 GM117634−01). R. J. W. is grateful for an NSF GRFP fellowship. We sincerely thank Dr. David Primer, Dr. James Phelan, and Dr. Álvaro Gutierrez-Bonet of the University of Pennsylvania (UPenn) for useful discussions. We thank Mr. Mark Campbell (UPenn) for assistance in preparing 3y. Frontier Scientific is acknowledged for their generous donation of potassium organotrifluoroborates. We thank Dr. George Furst for help in the NMR elucidations of 4q and 4r. We thank Dr. Charles W. Ross, III (UPenn) for assistance in obtaining HRMS data.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.a) Veber DF, Johnson SR, Cheng H-Y, Smith BR, Wards KW, Kopple KD. J. Med. Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]; b) Leeson PD, Springthorpe B. Nat. Rev. Drug Discov. 2007;11:881. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]; c) Agoram B, Woltosz WS, Bolger MB. Adv. Drug Deliv. Rev. 2001;50:S41. doi: 10.1016/s0169-409x(01)00179-x. [DOI] [PubMed] [Google Scholar]; d) Aungst BJ. J. Pharm. Sci. 2017;106:921. doi: 10.1016/j.xphs.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 2.a) Veale CA, Damewood JR, Jr, Steelman GB, Bryant C, Gomes B, Williams J. J. Med. Chem. 1996;38:86. doi: 10.1021/jm00001a014. [DOI] [PubMed] [Google Scholar]; b) Madsen AS, Olsen CA. Med. Chem. Commun. 2016;7:464. [Google Scholar]; c) Ottosen ER, Sørensen MD, Björkling F, Skak-Nielsen T, Fjording MS, Aaes H, Binderup L. J. Med. Chem. 2003;46:5651. doi: 10.1021/jm030851s. [DOI] [PubMed] [Google Scholar]; d) Goldstein DM, Alfredson T, Bertrand J, Browner MF, Clifford K, Dalrymple SA, Dunn J, Freire-Moar J, Harris S, Labadie SS, La Fargue J, Lapierre JM, Larrabee S, Li F, Papp E, McWeeney D, Ramesha C, Roberts R, Rotstein D, Pablo BS, Sjogren EB, So O-Y, Talamas FX, Tao W, Trejo A, Villasenor A, Welch M, Welch T, Weller P, Whiteley PE, Young K, Zipfel S. J. Med. Chem. 2006;49:1562. doi: 10.1021/jm050736c. [DOI] [PubMed] [Google Scholar]
- 3.a) Malátková P, Wsól V. Drug Metab. Rev. 2014;46:96. doi: 10.3109/03602532.2013.853078. [DOI] [PubMed] [Google Scholar]; b) Matsunaga T, Shintani S, Hara A. Drug Metab. Pharmacokinet. 2006;21:1. doi: 10.2133/dmpk.21.1. [DOI] [PubMed] [Google Scholar]; c) Barski OA, Tipparaju SM, Bhatnagar A. Drug Metab. Rev. 2008;40:553. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meanwell NA. J. Med. Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 5.Uneyama K. Organofluorine Chemistry. Blackwell Publishing; Oxford, UK: 2006. Hydrogen Bonding in Organofluorine Compounds. [Google Scholar]
- 6.a) Magueur G, Crousse B, Ourévitch M, Bonnet-Delpon D, Bégué J-P. J. Fluorine Chem. 2006;127:637. [Google Scholar]; b) Leriche C, He X, Chang C-w, Liu H-w. J. Am. Chem. Soc. 2003;125:6348. doi: 10.1021/ja021487+. [DOI] [PubMed] [Google Scholar]
- 7.(a) Altenburger J-M, Lassalle GY, Matrougui M, Galtier D, Jetha J-C, Bocskei Z, Berry CN, Lunven C, Lorrain J, Herault J-P, Schaeffer P, O’Connor SE, Herbert J-M. Bioorg. Med. Chem. 2004;12:1713. doi: 10.1016/j.bmc.2004.01.016. [DOI] [PubMed] [Google Scholar]; (b) Pan Y, Qiu J, Silverman RB. J. Med. Chem. 2003;46:5292. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 8.a) Chelucci G. Chem. Rev. 2012;112:1344. doi: 10.1021/cr200165q. [DOI] [PubMed] [Google Scholar]; b) Zhang X, Cao S. Tetrahedron Lett. 2017;58:375. doi: 10.1016/j.tetlet.2017.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hu M, Ni C, Li L, Han Y, Hu J. J. Am, Chem. Soc. 2015;137:14496. doi: 10.1021/jacs.5b09888. [DOI] [PubMed] [Google Scholar]
- 9.For an overview see: Matsui JK, Lang SB, Heitz DR, Molander GA. ACS Catal. 2017;7:2563. doi: 10.1021/acscatal.7b00094.
- 10.Although the reduction potential of α-trifluoromethyl radicals are unknown, they certainly lie below the known potential for their corresponding alkyl congeners and thus would be within the window of the reduced ground state of many photocatalysts see: Wayner DDM, McPhee DJ, Griller D. J. Am. Chem. Soc. 1988;110:132. and Zhang X-M. J. Org. Chem. 1998;63:3590. doi: 10.1021/jo981129i.; Similarly, we have observed that α-CF3 radicals are much more difficult to form oxidatively from the corresponding organotrifluoroborates, indicating the destabilizing nature of the α-CF3 group see: Ryu D, Primer DN, Tellis JC, Molander GA. Chem.–Eur. J. 2016;22:120. doi: 10.1002/chem.201504079.
- 11.This type of reactivity has been observed with organolithium species previously, see: Begue J-P, Bonnet-Delpon D, Rock MH. Tetrahedron Lett. 1995;36:5003.Begue J-P, Bonnet-Delpon D, Rock MH. J. Chem. Soc., Perkin Trans. 1996;1:1409.; For a theoretical study indicating that β-fluoride elimination proceeds preferentially by an E1cB type mechanism see: Alunni S, De Angelis F, Ottavi L, Papavasileiou M, Tarantelli F. J. Am. Chem. Soc. 2005;127:15151. doi: 10.1021/ja0539138.
- 12.a) Xiao T, Li L, Zhou L. J. Org. Chem. 2016;81:7908. doi: 10.1021/acs.joc.6b01620. [DOI] [PubMed] [Google Scholar]; b) Li L, Xiao T, Chen H, Zhou L. Chem.–Eur. J. 2017;23:2249. doi: 10.1002/chem.201605919. [DOI] [PubMed] [Google Scholar]
- 13.Luo J, Zhang J. ACS Catal. 2016;6:873. [Google Scholar]
- 14.a) Brumfield MA, Quillen SL, Yoon UC, Mariano PS. J. Am. Chem. Soc. 1984;106:6856. [Google Scholar]; b) Pandey G, Kumaraswamny G, Bhalerao UT. Tetrahedron Lett. 1989;30:6059. [Google Scholar]; c) Su Z, Mariano PS, Falvey DE, Yoon UC, Oh SW. J. Am. Chem. Soc. 1998;120:10676. [Google Scholar]; d) Espelt LR, McPherson IS, Wiensch EM, Yoon TP. J. Am. Chem. Soc. 2015;137:2452. doi: 10.1021/ja512746q. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hsieh S-Y, Bode JW. Org. Lett. 2016;18:2098. doi: 10.1021/acs.orglett.6b00722. [DOI] [PubMed] [Google Scholar]; f) Remeur C, Kelly CB, Patel NR, Molander GA. ACS Catal. 2017;7:6065. doi: 10.1021/acscatal.7b01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka D, Nishigaichi Y. Chem. Lett. 2014;43:559.Matsuoka D, Nishigaichi Y. Chem. Lett. 2015;44:163.Corcé V, Chamoreau L-M, Derat E, Goddard J-P, Ollivier C, Fensterbank L. Angew. Chem., Int. Ed. 2015;54:11414. doi: 10.1002/anie.201504963.Jouffroy M, Primer DN, Molander GA. J. Am. Chem. Soc. 2016;138:475. doi: 10.1021/jacs.5b10963. These species have also been used for radical addition into imines, see Patel NR, Kelly CB, Siegenfeld AP, Molander GA. ACS Catal. 2017;7:1766. doi: 10.1021/acscatal.6b03665.
- 16.Reviews on Ni/photoredox cross-coupling: Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Acc. Chem. Res. 2016;49:1429. doi: 10.1021/acs.accounts.6b00214.Gui Y-Y, Sun L, Lu Z-P, Yu D-G. Org. Chem. Front. 2016;3:522.Skubi KL, Blum TR, Yoon TP. Chem. Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018.Kelly CB, Patel NR, Primer DN, Jouffroy M, Tellis JC, Molander GA. Nat. Protoc. 2017;12:472. doi: 10.1038/nprot.2016.176.Levin MD, Kim S, Toste FD. ACS Cent. Sci. 2016;2:293. doi: 10.1021/acscentsci.6b00090.Shaw MH, Twilton J, MacMillan DWC. J. Org. Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449.
- 17.Pan R-q, Liu X-x, Deng M-z. J. Fluorine Chem. 1999;95:167. [Google Scholar]
- 18.Several examples of couplings of α-(trifluoromethyl)ethenyl boronic acid exist. The material is never isolated because of its inherent instability. It is always used crude directly after its synthesis and often in large excess, see: Jiang B, Wang Q-F, Yang C-G, Xu M. Tetrahedron Lett. 2001;42:4038.Ichitsuka T, Fujita T, Arita T, Ichikawa J. Angew. Chem., Int. Ed. 2014;53:7564. doi: 10.1002/anie.201402695.
- 19.3,3,3-Tritluoroisopropenyllithium is notoriously unstable and decomposes to generate allenes even at temperatures below −90 °C, see: Drakesmith FG, Stewart OJ, Tarrant P. J. Org. Chem. 1968;33:280.; Although the corresponding organozinc is known, its preparation is quite arduous and can only be used in cross-coupling with a very limited number of systems. Moreover, this reagent must be stored in solution under argon, see: Jiang B, Xu Y. J. Org. Chem. 1991;56:7336.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.