

Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) commonly results in end-stage renal disease (ESRD), yet a long-term treatment that is well tolerated is still lacking. In a small randomized trial in children and adolescents pravastatin administration for 3 years was associated with reduced renal cyst growth, but no large trial has tested the effect of statins in adults.
Methods
We performed a post-hoc analysis of the HALT PKD trials to compare outcomes of participants who never used statins with those who used statin for at least 3 years. Because statins were not randomly allocated we used propensity score models with inverse probability of treatment weighting to account for imbalances between the groups. For subjects in Study A (preserved renal function, n=438) relevant outcomes were percent change in total kidney and liver volume and the rate of decline in estimated glomerular filtration rate (eGFR); for those in Study B (reduced renal function, n=352) we compared time to the composite endpoint of death, ESRD or 50% decline in eGFR. Follow-up was 5–8 years.
Results
There was no difference in any outcome between the 2 groups. However, limitations of this analysis are the small number of statin users in Study A, different statin drugs and doses used, non-randomized allocation and advanced disease stage in Study B.
Conclusions
Although this post-hoc analysis of the HALT PKD trials does not demonstrate a benefit of statin therapy, conclusions remain preliminary. A larger randomized trial in young people with ADPKD is necessary to answer the question whether statins can slow renal cyst growth and preserve kidney function.
Keywords: Autosomal dominant polycystic kidney disease, end-stage renal disease, glomerular filtration rate, HALT PKD trials, hydroxymethylglutaryl-CoA reductase inhibitors, total kidney volume
Graphical abstract
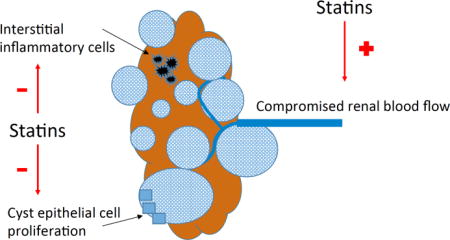
Statins have pleiotropic effects which include inhibition of cell proliferation and inflammation. Statins have been shown to improve endothelial dysfunction and increase renal blood flow. We tested the hypothesis that statins prevent progression of ADPKD, by performing a secondary analysis of the HALT PKD Trials.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic and potentially fatal disease, with an estimated prevalence of 1 in 400 to 1 in 2,500 in populations worldwide1–3. Progressive development and enlargement of renal cysts eventually destroys the normal parenchyma, leading to end-stage renal disease (ESRD) in the majority of afflicted patients. Despite recent progress in understanding the genetic basis and pathophysiological mechanisms of this disease, the incidence of ESRD and age at onset of ESRD may not have changed significantly4–6.
Several new medications have been tested in interventional trials. Some may slow but not halt the progression of ADPKD and all have substantial side effects7–10. Although tolvaptan has been approved in Japan, Canada and Europe for rapidly progressive ADPKD, it is not approved in the United States for this indication, and the mainstay of therapy remains control of hypertension to prevent left ventricular hypertrophy and cardiovascular complications11–13. Additional well tolerated therapies to slow the progression of ADPKD are urgently needed.
Studies in animal models of ADPKD have shown that statin treatment decreases cyst formation, preserves renal blood flow and mitigates interstitial inflammation14–16. In a randomized placebo-controlled trial in 110 young (age 8–22 years) patients with ADPKD, treatment with pravastatin for 3 years reduced the increase in height-adjusted total kidney volume (TKV) measured by magnetic resonance imaging (MRI)17. No statin treatment trial with sufficient statistical power has been performed in adults with preserved renal function. Although the SHARP (Study of Heart and Renal Protection) trial included 675 patients with polycystic kidney disease, the mean estimated glomerular filtration rate (eGFR) at randomization was 27 ml/min/1.73 m2, reflecting disease too far advanced for any intervention to alter the course18.
Therefore we undertook a secondary analysis of the HALT PKD trials which involved 1044 adult subjects participating for 5–8 years (ClinicalTrials.gov numbers NCT00283686 for Study A and NCT01885559 for Study B, see below)19,20. The primary objective of these trials was to compare aggressive blood pressure (BP) control and intensive blockade of the renin-angiotensin-aldosterone system (RAAS) with less rigorous therapy21. We examined whether subjects who took a statin drug for at least 3 years had slower progression of ADPKD compared to those who did not, the cutoff being based on the pravastatin trial in children and young adults.
SUBJECTS AND METHODS
The HALT PKD trials were randomized, double-blind, multicenter trials to test the hypothesis that intensive BP control using single or double RAAS blockade can retard the progression of kidney disease in hypertensive patients with ADPKD19–21. The 2 trials involved individuals at different disease stages: Study A randomized 558 young (15–49 years, mean age 36 years) subjects with preserved renal function (eGFR > 60 mL/min/1.73 m2) in a 2×2 factorial design to either a low BP goal (95/60–110/75 mm Hg) or standard BP goal (120/70–130/80 mm Hg) using either lisinopril (an angiotensin converting enzyme inhibitor) and placebo or the combination of lisinopril and telmisartan (an angiotensin-2 receptor blocker), with other medications added as needed to achieve the BP goal. Study B randomized 486 older (18–64 years, mean age 48 years) patients with reduced renal function (eGFR 25–60 mL/min/1.73 m2) to either lisinopril and placebo or lisinopril and telmisartan to achieve a single BP goal of 120–130/70–80 mm Hg. Known coronary artery disease and diabetes were exclusion criteria for HALT, therefore subjects with a strong indication for statin use were excluded from both Study A and B.
All participants gave informed consent and the trials were conducted according to the principles of the Helsinki Declaration.
Cardiac and renal MRI was obtained at baseline and after 2, 4 and 5 years in Study A participants using methods established by the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP)22. Estimated GFR (eGFR) was calculated for both Study A and B using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, based on centralized serum creatinine determinations by isotope dilution mass spectrometry (IDMS)23.
The primary outcome for Study A was percent change in TKV, for Study B the composite of time to ESRD, death, or 50% reduction in eGFR. Secondary outcomes were rate of change in eGFR, and for Study A change in height-adjusted total liver volume (htTLV) and in left ventricular mass index (LVMI). Follow-up time was 5–8 years.
Participants were evaluated at 7 study centers at baseline, at 4, 7 and 12 months, and then every 6 months until the end of the trial in 2014 (Study A) or until a subject met an endpoint (Study B). At each study visit all concomitant medications were documented and entered into the database. Statin use was defined by the presence of any hydroxymethylglutaryl-CoA-reductase inhibitor on the medication list at any time point of follow up. The duration of statin use was calculated by adding up the number of 6-month periods (regardless whether consecutive or not) where participants reported their use. The HALT study protocol did not make any recommendation regarding statin therapy, and serum cholesterol measurements were not obtained. Prescription of a statin drug was at the discretion of the patient’s private physician.
Statistical analysis
For this post-hoc analysis of the HALT PKD trials we categorized participants into 2 groups based on statin use: 1) No Use (never used) and 2) Statin Use (at least 3 years of use, based on the pravastatin trial in children and young adults17), which led to the exclusion of 113 participants (37 for Study A; 76 for Study B) who used statins for less than 3 years (mean years of use for Study A and B: 0.9 ± 0.8 and 1.1 ± 0.8, respectively). We compared demographic and clinical baseline characteristics between the 2 groups using analysis of variance and Chi-square tests of significance, or their nonparametric counterparts when necessary. Several variables such as TKV, htTLV and urine albumin were log-transformed in order to normalize. Because of imbalances in baseline characteristics between the 2 groups we utilized a propensity score model with inverse probability of treatment weighting (IPTW) to create a cohort of participants who were well-balanced on all baseline covariates24. For both Study A and B, the propensity of taking statins for at least 3 years was calculated for each participant using logistic regression as a function of the following baseline predictors: age, gender, body mass index (BMI), eGFR, urine albumin, home systolic and diastolic BP, and study drug arm. For Study A only, LVMI, TKV, and BP arm were also included. Because not all participants provided their home BP measurements at baseline, we used the first non-missing value as long as it occurred prior to their first statin use. For each participant, the estimated propensity score was weighted by the inverse probability of being in the No Use or Statin Use group. In order to assess for balance between groups, we calculated weighted standardized mean differences for each of the baseline covariates included in the model and compared the magnitude of imbalance to the unweighted differences.
For Study A, linear mixed models were used to assess whether changes in outcomes (TKV and eGFR) were different between the 2 groups after accounting for the inverse probability of treatment weights. Predictors included month, statin use group, and their interaction. Of interest was whether the interaction was significant, which would indicate differences in annual percent increase of TKV or rate of decline in eGFR. In addition, we examined whether statin use had any effect on htTLV using similar linear mixed models.
For Study B a Cox proportional hazards model was used to investigate whether statin use was a predictor of time to composite endpoint after accounting for the inverse probability of treatment weights. The only predictor was statin use group.
RESULTS
The most commonly prescribed statins were simvastatin (36%) and atorvastatin (35.6%), followed by pravastatin (11%), rosuvastatin (10.4%), and lovastatin (6%). Fluvastatin and pitavastatin were used by one individual each. Unfortunately the doses of each drug were not adequately documented.
In Study A only 59 of 558 (10.5%) of participants used a statin for at least 3 years (mean 5.3 ± 1.5 years), whereas 462 (83%) were in the No Use group. Statin users were more often male and significantly older than non-users, had higher baseline (or first non-missing) systolic BP, and lower baseline eGFR (Table 1), but after accounting for the propensity weights the standardized mean differences between statin use groups were attenuated, with all below 0.20 (Figure 1 a). Average home systolic and diastolic BPs during the trial were similar in the 2 groups (data not shown). Due to missing data on any of the covariates used in the propensity score model, 83 Study A participants were not included in the IPTW analyses. These participants were similar to those 438 included, with the exception that the latter group had a higher baseline BMI (25.8 ± 4.7 kg/m2 vs. 27.3 ± 5.2 kg/m2; p=0.02; Table 2).
Table 1.
Baseline demographic and clinical characteristics of Study A participants by use of statin drugs, before and after IPTW
| a. Categorical measures: | |||||||
|---|---|---|---|---|---|---|---|
| BEFORE IPTW | AFTER IPTW | ||||||
| Never used (n=462) |
Used 3 or more years (n=59) |
Never used (n=382) |
Used 3 or more years (n=56) |
||||
| Characteristic | Category | n (%) | n (%) | p value | % | % | p value |
| Sex | Male | 218 (47.2%) | 40 (67.8%) | 0.0029 | 49.6% | 42.4% | 0.5059 |
| Female | 244 (52.8%) | 19 (32.2%) | 50.4% | 57.6% | |||
| PKD genotype | NMD | 35 (8.4%) | 6 (10.2%) | 0.8975 | 7.9% | 6.9% | 0.4952 |
| PKD1 | 312 (74.8%) | 43 (72.9%) | 74.8% | 81.8% | |||
| PKD2 | 70 (16.8%) | 10 (16.9%) | 17.2% | 11.3% | |||
| Treatment group | Lisinopril/Telmisartan | 223 (48.3%) | 31 (52.5%) | 0.5363 | 49.7% | 44.9% | 0.6747 |
| Lisinopril/Placebo | 239 (51.7%) | 28 (47.5%) | 50.3% | 55.1% | |||
| Blood pressure group | Standard BP | 237 (51.3%) | 30 (50.8%) | 0.9479 | 50.3% | 41.9% | 0.4388 |
| Low BP | 225 (48.7%) | 29 (49.2%) | 49.7% | 58.1% | |||
| CAD or Angina | Yes | 2 (0.4%) | 0 (0.0%) | 0.6122 | 0.7% | 0% | – |
| No | 459 (99.6%) | 59 (100.0%) | 99.3% | 100.0% | |||
| b. Continuous measures: | ||||||||
|---|---|---|---|---|---|---|---|---|
| Before IPTW | After IPTW | |||||||
| Never used (n=462) |
Used 3 or more years (n=59) |
Never used (n=382) |
Used 3 or more years (n=56) |
|||||
| Measure | n | Mean ± SD | n | Mean ± SD | p value | Mean (SE) | Mean (SE) | p value |
| Age at baseline (years) | 462 | 35.8 ± 8.5 | 59 | 40.7 ± 6.2 | <.0001 | 36.6 (0.4) | 35.3 (3.3) | 0.6935 |
| BMI (kg/m2) | 453 | 27.0 ± 5.2 | 58 | 28.2 ± 5.2 | 0.0907 | 27.3 (0.3) | 27.2 (1.2) | 0.9323 |
| Home average systolic BP (mmHg)* | 430 | 120.8 ± 10.4 | 59 | 123.5 ± 9.0 | 0.0649 | 121.2 (0.6) | 122.1 (1.1) | 0.4882 |
| Home average diastolic BP (mmHg)* | 430 | 80.6 ± 8.2 | 59 | 82.2 ± 7.8 | 0.1595 | 80.7 (0.4) | 79.7 (1.7) | 0.5698 |
| CKD EPI eGFR (mL/min/1.73m2) | 461 | 93.0 ± 17.7 | 59 | 84.3 ± 15.3 | 0.0004 | 91.4 (0.9) | 94.9 (7.2) | 0.6345 |
| Urine albumin (mg/24 hrs) Median (p25, p75) |
448 | 18.5 (12.1, 32.8) | 58 | 16.4 (12.6, 34.0) | 0.6158** | 34.8 (2.6) | 35.9 (8.2) | 0.9839** |
| Total kidney volume (mL) | 456 | 1187.4 ± 722.2 | 58 | 1393.2 ± 788.5 | 0.0437 | 1228 (38.8) | 1224 (122.2) | 0.9746 |
| Logarithm total kidney volume (mL) | 456 | 6.9 ± 0.6 | 58 | 7.1 ± 0.6 | 0.0352 | 7.0 (0.0) | 7.0 (0.1) | 0.9324 |
| Height-adjusted TKV (mL/m) | 448 | 679.9 ± 400.7 | 57 | 786.4 ± 434.5 | 0.0617 | 701.5 (21.2) | 703.3 (65.1) | 0.9788 |
| Renal blood flow (mL/min/1.73m2) | 309 | 607.4 ± 208.8 | 44 | 608.5 ± 212.0 | 0.9752 | 599.3 (12.5) | 722.2 (93.1) | 0.1919 |
| Left ventricular mass index (g/m2) | 446 | 63.7 ± 12.8 | 58 | 65.5 ± 15.4 | 0.3264 | 63.8 (0.7) | 63.2 (2.5) | 0.8222 |
| CKD EPI eGFR at F5 (mL/min/1.73m2) | 422 | 90.4 ± 18.6 | 59 | 81.9 ± 16.6 | 0.0009 | 89.5 (1.0) | 93.4 (7.6) | 0.6207 |
| Statin Use (# years of any use) | 462 | N/A | 59 | 5.3 ± 1.5 | — | — | 4.9 (0.4) | — |
BP: blood pressure; CAD: coronary artery disease; IPTW: inverse probability of treatment weighting; NMD: no mutation detected.
At the visit a participant first reported a home systolic and diastolic BP reading (for 96% of participants, this was at the baseline (71%) or 4 month (25%) visit)
p value from log transformed variable
BMI: body mass index; BP: blood pressure; CKD EPI eGFR: estimated glomerular filtration rate using the Chronic Kidney Disease Epidemiology Collaboration equation; F5: Follow-up visit 5 (after 4 months in the study); IPTW: inverse probability of treatment weighting; p25: 25th percentile.
Figure 1.
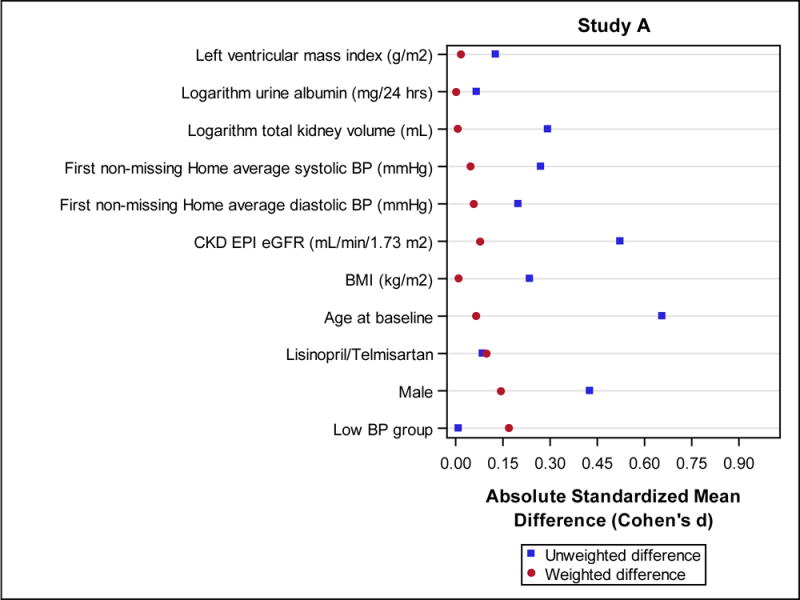
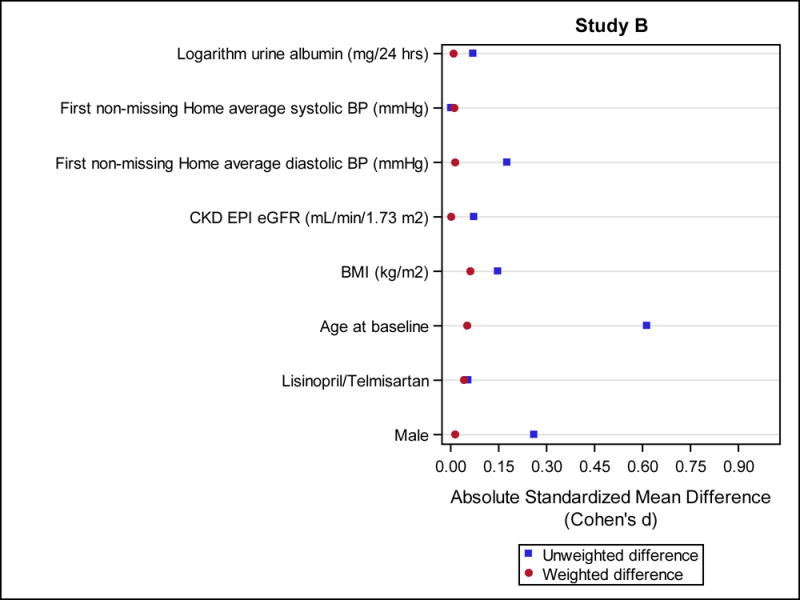
Standardized mean differences with (weighted differences) and without (unweighted differences) accounting for the inverse probability of treatment weight (IPTW). Baseline characteristics which were used in the propensity score models are shown for Study A (Fig. 1 a) and Study B (Fig. 1 b).
Table 2.
Baseline demographic and clinical characteristics of Study A participants by included vs not included in IPTW analysis
| a. Categorical measures: | ||||
|---|---|---|---|---|
| Not included (n=85) |
Included (n=438) |
|||
| Characteristic | Category | n (%) | n (%) | p value |
| Sex | Male | 41 (48.2%) | 217 (49.5%) | 0.8253 |
| Female | 44 (51.8%) | 221 (50.5%) | ||
| PKD genotype | NMD | 6 (15.4%) | 35 (8.0%) | 0.2528 |
| PKD1 | 28 (71.8%) | 327 (74.7%) | ||
| PKD2 | 5 (12.8%) | 76 (17.4%) | ||
| Treatment group | Lisinopril/Telmisartan | 38 (44.7%) | 217 (49.5%) | 0.4142 |
| Lisinopril/Placebo | 47 (55.3%) | 221 (50.5%) | ||
| Blood pressure group | Standard BP | 48 (56.5%) | 220 (50.2%) | 0.2920 |
| Low BP | 37 (43.5%) | 218 (49.8%) | ||
| b. Continuous measures: | |||||
|---|---|---|---|---|---|
| Not included (n=85) |
Included (n=438) |
||||
| Measure | n | Mean ± SD | n | Mean ± SD | p value |
| Age at baseline (years) | 85 | 35.4 ± 8.7 | 438 | 36.6 ± 8.3 | 0.2158 |
| BMI (kg/m2) | 75 | 25.8 ± 4.7 | 438 | 27.3 ± 5.2 | 0.0202 |
| Home average systolic BP (mmHg)* | 51 | 120.6 ± 9.4 | 438 | 121.2 ± 10.4 | 0.7095 |
| Home average diastolic BP (mmHg)* | 51 | 81.1 ± 7.1 | 438 | 80.7 ± 8.2 | 0.7641 |
| CKD EPI eGFR (mL/min/1.73 m2) | 84 | 94.5 ± 17.5 | 438 | 91.5 ± 17.6 | 0.1578 |
| Logarithm urine albumin (mg/24 hrs) | 69 | 3.3 ± 1.0 | 438 | 3.0 ± 0.9 | 0.0833 |
| Logarithm total kidney volume (mL) | 78 | 6.9 ± 0.6 | 438 | 7.0 ± 0.6 | 0.1569 |
| Left ventricular mass index (g/m2) | 68 | 64.9 ± 12.8 | 438 | 63.7 ± 13.2 | 0.5072 |
At the visit a participant first reported a home systolic and diastolic BP reading (for 96% of participants, this was at the baseline (71%) or 4 month (25%) visit)
BMI: body mass index; BP: blood pressure; CKD EPI eGFR: estimated glomerular filtration rate using the Chronic Kidney Disease Epidemiology Collaboration equation; IPTW: inverse probability of treatment weighting; NMD: no mutation detected.
The rate of increase in TKV (Figure 2) and htTLV (Figure 3) was not significantly different between the 2 groups after accounting for the propensity weights. TKV growth was 6.5% vs. 6.2% per year in the Statin Use and No Use groups, respectively (p=0.51). Increase in htTLV was 0.8% and 1.0% per year in the Statin Use and No Use groups (p=0.54). eGFR declined slightly faster (3.06 ml/min/year) in the Statin Use group than the No Use group (2.87 ml/min/year), but this was not significant (p=0.57) (Figure 4).
Figure 2.
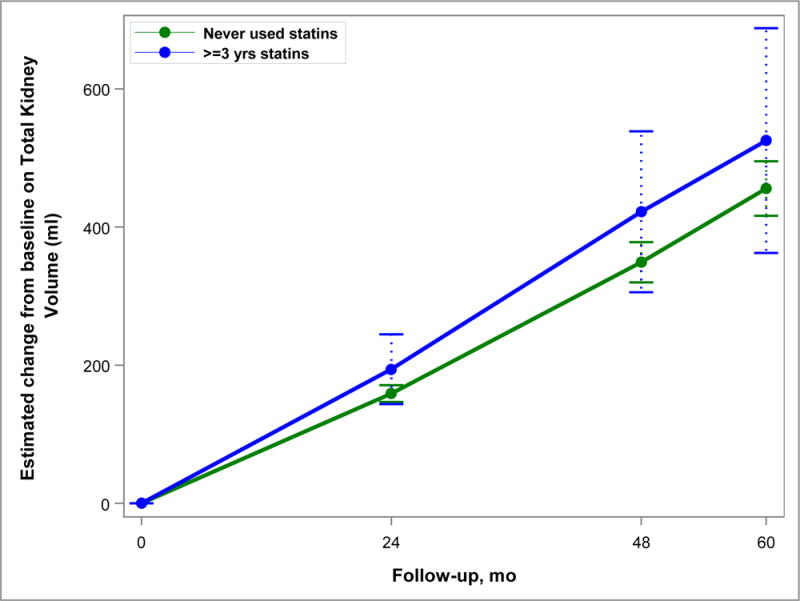
Model-based estimates of change in total kidney volume (TKV) from baseline over 60 months in Study A. Point estimates and 95% confidence intervals derived from linear mixed models accounting for the inverse probability of treatment weights and including predictors for month, statin use group, and their interaction. The difference between statin users (for at least 3 years) and never users was not significant (p=0.51).
Figure 3.
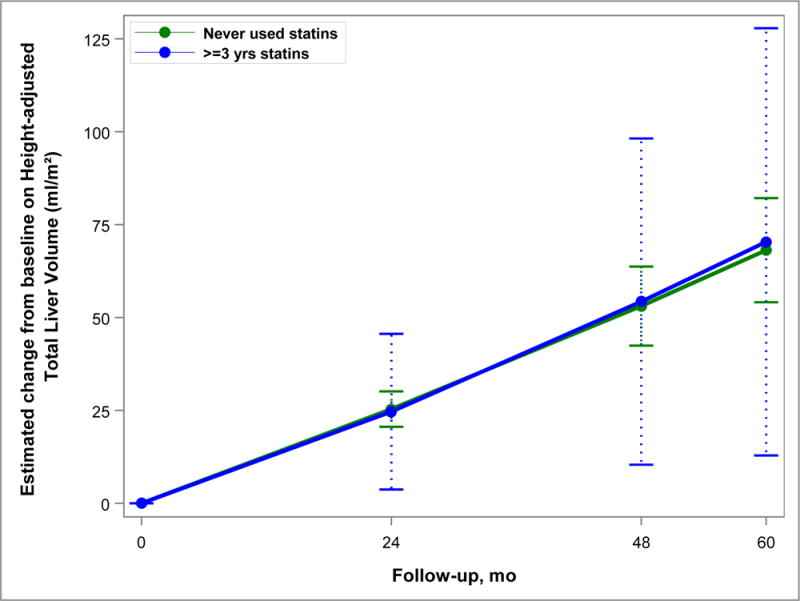
Model-based estimates of change in height-adjusted total liver volume (htTLV) from baseline over 60 months in Study A. Point estimates and 95% confidence intervals derived from linear mixed models accounting for the inverse probability of treatment weights and including predictors for month, statin use group, and their interaction. The difference between statin users (for at least 3 years) and never users was not significant (p=0.54).
Figure 4.
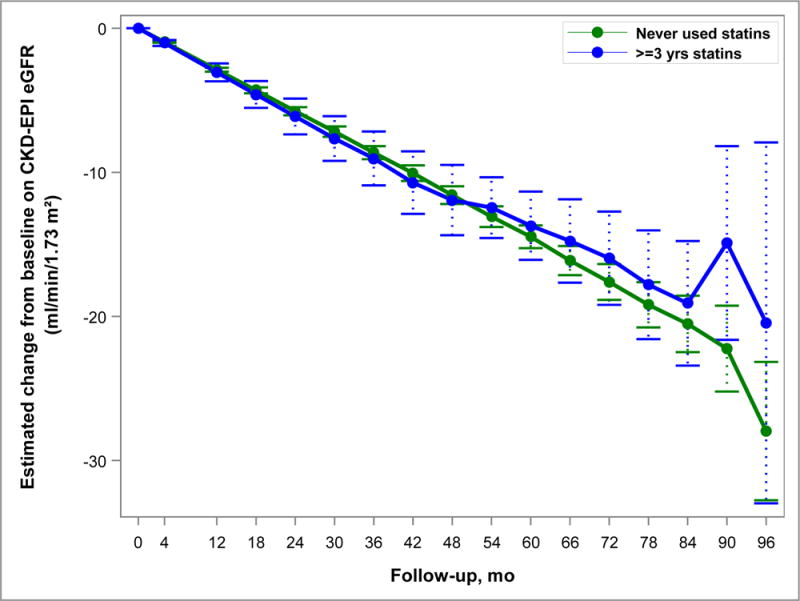
Model-based estimates of change in eGFR over 60 months in Study A. Point estimates and 95% confidence intervals derived from linear mixed models accounting for the inverse probability of treatment weights and including predictors for month, statin use group, and their interaction. The difference between statin users (for at least 3 years) and never users was not significant (p=0.57).
In Study B, 118 of 486 (24%) participants used a statin for at least 3 years (mean 5.0 ± 1.3), and 292 (60%) were in the No Use group. Statin users were more often men and were older than never users (Table 3); after accounting for the propensity weights, standardized mean differences between statin use groups were attenuated with all below 0.20 (Figure 1 b). Average home systolic and diastolic BPs during the trial were similar in the 2 groups (data not shown). Due to missing data on any of the covariates used in the propensity score model, 58 Study B participants were not included in the IPTW analyses. These participants were similar to the 352 who were included (Table 4).
Table 3.
Baseline demographic and clinical characteristics of Study B participants by use of statin drugs, before and after IPTW
| a. Categorical measures: | |||||||
|---|---|---|---|---|---|---|---|
| Before IPTW | After IPTW | ||||||
| Never used (n=292) |
Used 3 or more years (n=118) |
Never used (n=249) |
Used 3 or more years (n=103) |
||||
| Characteristic | Category | n (%) | n (%) | p value | % | % | p value |
| Sex | Male | 133 (45.5%) | 69 (58.5%) | 0.0178 | 50.1% | 50.8% | 0.9112 |
| Female | 159 (54.5%) | 49 (41.5%) | 49.9% | 49.2% | |||
| PKD genotype | NMD | 12 (4.5%) | 10 (8.9%) | 0.1343 | 5.5% | 7.4% | 0.8154 |
| PKD1 | 220 (82.1%) | 83 (74.1%) | 80.2% | 78.2% | |||
| PKD2 | 36 (13.4%) | 19 (17.0%) | 14.3% | 14.4% | |||
| Treatment group | Lisinopril/Telmisartan | 143 (49.0%) | 61 (51.7%) | 0.6177 | 50.7% | 52.8% | 0.7405 |
| Lisinopril/Placebo | 149 (51.0%) | 57 (48.3%) | 49.3% | 47.2% | |||
| CAD or Angina | Yes | 2 (0.7%) | 2 (1.7%) | 0.3462 | 1.2% | 1.3% | 0.9952 |
| No | 290 (99.3%) | 116 (98.3%) | 98.8% | 98.7% | |||
| b. Continuous measures: | ||||||||
|---|---|---|---|---|---|---|---|---|
| Before IPTW | After IPTW | |||||||
| Never used (n=292) |
Used 3 or more years (n=118) |
Never used (n=249) |
Used 3 or more years (n=103) |
|||||
| Measure | n | Mean ± SD | n | Mean ± SD | p value | Mean (SE) | Mean (SE) | p value |
| Age at baseline | 292 | 47.3 ± 8.5 | 118 | 52.2 ± 7.3 | <.0001 | 48.9 (0.6) | 49.6 (0.9) | 0.5425 |
| BMI (kg/m2) | 286 | 27.4 ± 5.2 | 115 | 28.2 ± 5.0 | 0.1856 | 27.6 (0.3) | 28.1 (0.7) | 0.4904 |
| Home average systolic BP (mmHg)* | 281 | 122.9 ± 10.7 | 118 | 122.9 ± 11.0 | 0.9900 | 122.7 (0.7) | 122.9 (1.2) | 0.8889 |
| Home average diastolic BP (mmHg)* | 281 | 81.6 ± 8.4 | 118 | 80.2 ± 7.3 | 0.1181 | 81.0 (0.5) | 80.8 (0.9) | 0.8757 |
| CKD EPI eGFR (mL/min/1.73m2) | 292 | 48.8 ± 11.8 | 118 | 47.9 ± 11.3 | 0.5069 | 48.6 (0.8) | 48.6 (1.3) | 0.9844 |
| Urine albumin (mg/24 hrs) Median (p25, p75) |
277 | 26.8 (16.2, 69.5) | 114 | 31.6 (17.9, 66.0) | 0.5351** | 82.3 (9.5) | 71.1 (11.7) | 0.9013** |
| CKD EPI eGFR at F5 (mL/min/1.73m2) | 274 | 47.3 ± 12.1 | 116 | 45.4 ± 11.4 | 0.1418 | 46.9 (0.8) | 45.6 (1.2) | 0.3729 |
| Statin Use (# years of any use) | 292 | N/A | 118 | 5.0 ± 1.3 | — | — | 4.9 (0.1 | — |
CAD: coronary artery disease; IPTW: inverse probability of treatment weighting; NMD: no mutation detected.
At the visit a participant first reported a home systolic and diastolic BP reading (for 98% of participants, this was at the baseline (78%) or 4 month (19%) visit)
p value from log transformed variable
BMI: body mass index; BP: blood pressure; CKD EPI eGFR: estimated glomerular filtration rate using the Chronic Kidney Disease Epidemiology Collaboration equation; F5: Follow-up visit 5 (after 4 months in the study); IPTW: inverse probability of treatment weighting; p25: 25th percentile.
Table 4.
Baseline demographic and clinical characteristics of Study B participants by included vs not included in IPTW analysis
| a. Categorical measures: | ||||
|---|---|---|---|---|
| Not included (n=57) |
Included (n=352) |
|||
| Characteristic | Category | n (%) | n (%) | p value |
| Sex | Male | 26 (45.6%) | 176 (50.0%) | 0.5389 |
| Female | 31 (54.4%) | 176 (50.0%) | ||
| PKD genotype | NMD | 2 (8.0%) | 19 (5.4%) | 0.8250 |
| PKD1 | 20 (80.0%) | 282 (80.1%) | ||
| PKD2 | 3 (12.0%) | 51 (14.5%) | ||
| Treatment group | Lisinopril/Telmisartan | 26 (45.6%) | 180 (51.1%) | 0.4392 |
| Lisinopril/Placebo | 31 (54.4%) | 172 (48.9%) | ||
| b. Continuous measures: | |||||
|---|---|---|---|---|---|
| Not included (n=57) |
Included (n=352) |
||||
| Measure | n | Mean ± SD | n | Mean ± SD | p value |
| Age at baseline (years) | 57 | 47.4 ± 9.3 | 352 | 48.9 ± 8.3 | 0.2130 |
| BMI (kg/m2) | 48 | 28.4 ± 4.7 | 352 | 27.5 ± 5.2 | 0.2499 |
| Home average systolic BP (mmHg)* | 44 | 124.6 ± 10.4 | 352 | 122.7 ± 10.8 | 0.2637 |
| Home average diastolic BP (mmHg)* | 44 | 82.3 ± 8.2 | 352 | 81.0 ± 8.1 | 0.3316 |
| CKD EPI eGFR (mL/min/1.73 m2) | 57 | 48.9 ± 12.0 | 352 | 48.6 ± 11.7 | 0.8214 |
| Logarithm urine albumin (mg/24 hrs) | 38 | 3.6 ± 1.2 | 352 | 3.6 ± 1.2 | 0.9984 |
At the visit a participant first reported a home systolic and diastolic BP reading (for 98% of participants, this was at the baseline (78%) or 4 month (19%) visit)
BMI: body mass index; BP: blood pressure; CKD EPI eGFR: estimated glomerular filtration rate using the Chronic Kidney Disease Epidemiology Collaboration equation; IPTW: inverse probability of treatment weighting; NMD: no mutation detected
There was no difference in time to the composite endpoint of death, ESRD or 50% decline in eGFR between the 2 groups (HR=0.99; p=0.96) in Study B (Figure 5).
Figure 5.
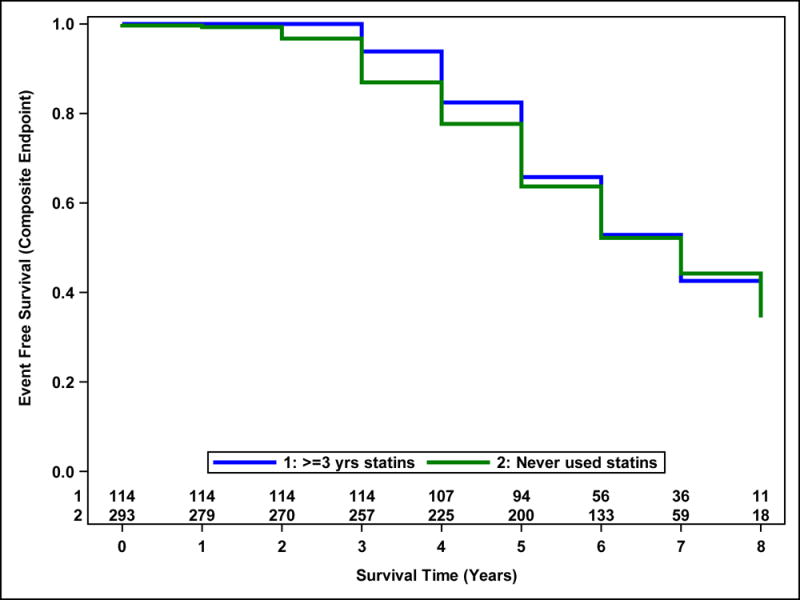
Probability of event-free survival from the composite outcome in Study B. Survival curves estimated from a Cox proportional hazards model accounting for the inverse probability of treatment weights and including statin use group as a predictor. Number of participants at risk are shown above the x-axis. There was no difference between statin users (for at least 3 years) and never users (p=0.96).
DISCUSSION
ADPKD is a common genetic disease with high morbidity and premature mortality, for which a treatment is desperately needed. Due to the slow progression, from birth to the 5th or 6th decade of life when ESRD ensues, treatment will need to be given for many years starting at a young age, requiring a drug with a low side effect profile and no serious toxicity. Statins have been used in millions of people worldwide for lipid lowering and prevention of cardiovascular events, and are generally well tolerated. Statins exert pleiotropic effects besides decreasing serum cholesterol levels25,26. By inhibiting the enzyme hydroxymethylglutaryl-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis, and other mechanisms such as cell cycle arrest and induction of apoptosis, statins inhibit cell proliferation, shown in several cancer cell lines and in clinical studies of malignancies27,28. Tubular epithelial cell proliferation is required for cyst growth, and both simvastatin and pravastatin inhibited proliferation of an immortalized ADPKD cyst cell line in a dose dependent manner [abstract Wang W et al: Statin effect on human ADPKD tubular epithelial cell proliferation. J Am Soc Nephrol 2014; 25: 412A]. In the Han:SPRD rat model of ADPKD, lovastatin treatment resulted in decreased cystic kidney size, decreased volume density of cysts and improved renal function14,15. Most importantly, a randomized controlled trial in pediatric patients with ADPKD showed that treatment with pravastatin (20 mg daily if age 8–12 years and 40 mg daily if age 13–22 years) was associated with slower increase in height-adjusted TKV compared to placebo17. GFR did not change in either group at this early stage of ADPKD.
In contrast, the current post-hoc analysis of the HALT PKD trials does not confirm a beneficial effect of statins on renal volume growth in adults. Although participants in HALT Study A had preserved renal function (baseline eGFR 91.5 ± 17.5 ml/min/1.73 m2), they were significantly older (mean age 36.2 ± 8.3 years)29 than the pediatric patients (mean age 16 ± 4 years), suggesting that antiproliferative treatments may be most effective in early-stage ADPKD. Likewise, in the animal models statins were administered after weaning, a very young age.
Other effects of statins include improvement of endothelial dysfunction by upregulating endothelial and vascular smooth muscle cell production of nitric oxide25,30,31. Endothelial dysfunction is an early feature of ADPKD32–37 and may account for the decreased renal blood flow observed in young people with ADPKD38,39. In fact, a double-blind cross-over study among young (mean age 35 years) normotensive ADPKD patients demonstrated an increase in effective renal plasma flow after 4 weeks of simvastatin treatment (40 mg daily), accompanied by improvement in endothelium-dependent vasodilatation in the forearm40. In contrast, an increase in renal blood flow or GFR was not seen with the same treatment in older (mean age 47 years) patients with more advanced ADPKD41, consistent with the notion that statin benefits may be limited to early stages of this disease42.
Because statins have anti-inflammatory and anti-oxidant effects25,31,43, they have been studied for the prevention of acute kidney injury after cardiac surgery, a state of an intense systemic inflammatory response, with mixed results26,44. Renal interstitial inflammation and oxidative stress are prominent features of ADPKD, resulting in dense fibrosis, tubular atrophy and glomerulosclerosis45–48. Statin treatment was associated with less renal interstitial and systemic inflammation in the Han:SPRD rat model14,16; likewise in the pediatric ADPKD trial, plasma levels of inflammatory and oxidative stress biomarkers declined during pravastatin treatment but not with placebo49. However, any anti-inflammatory effects did not translate into slower decline of eGFR in our post-hoc analysis of either Study A or Study B. Similarly, a small (n = 49) open-label trial of pravastatin 20 mg for 2 years in advanced ADPKD (mean age 51 years) also did not show a benefit50, consistent with the SHARP trial results in very advanced disease18.
Limitations of this study are the small numbers of participants who used a statin for at least 3 years, particularly in Study A, and the nonrandomized allocation to statins. The variety of statin drugs used and the fact that doses were not documented also contribute to uncertainty. Lipid-soluble statins may have better antiproliferative effects than water-soluble ones that do not readily penetrate into epithelial cells. Low-potency statins may not have the same pleiotropic effects as high-potency statins and/or high doses. Clinical trials showing a benefit of pre-angiography statin administration to prevent contrast-induced acute kidney injury typically used high doses of a high-potency statin51–53. A meta-analysis of trials examining statin therapy to prevent progression of chronic kidney disease did not find a benefit of low-intensity statins, but subjects in the high-intensity study arms did have a significantly slower decline in eGFR compared to controls54. Among high-potency statins, atorvastatin may have better renoprotective effects than rosuvastatin, based on a direct comparison in the PLANET (Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease) trials55.
KDIGO (Kidney Disease: Improving Global Outcomes) guidelines recommend the use of statins in all patients with chronic kidney disease who are older than 50 years and not receiving dialysis, regardless of serum cholesterol levels, for prevention of cardiovascular disease56. Therefore, the question whether a high-potency statin drug reduces renal cyst growth and preserves kidney function is relevant mainly for young (age 16–40 years) individuals with ADPKD, who are the most likely to benefit from any intervention. A randomized controlled trial of sufficient size, similar to HALT Study A, is necessary, using a high-potency lipid-soluble statin drug for young people with ADPKD and preserved renal function.
Short summary.
Autosomal dominant polycystic kidney disease (ADPKD) is a common disorder, yet other than blood pressure control, treatment that is well tolerated for long-term use is still lacking. Studies in animal models and a small randomized trial in adolescents have shown benefits of statin therapy for reducing the progression of ADPKD, but no large trial has been undertaken in adults with early to moderately advanced disease. Therefore we performed a secondary analysis of the recently completed HALT PKD trials (1044 participants) to examine whether statin use for at least 3 years, compared to no use, was associated with slower renal and liver cyst growth, or with less decline in renal function.
Acknowledgments
Supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK62402 to Dr. Schrier, DK62411 to Dr. Perrone, DK62410 to Dr. Torres, DK082230 to Dr. Moore, DK62408 to Dr. Chapman, and DK62401 to Washington University in St. Louis) and the National Center for Research Resources General Clinical Research Centers (RR000039 to Emory University, RR000585 to the Mayo Clinic, RR000054 to Tufts Medical Center, RR000051 to the University of Colorado, RR023940 to the University of Kansas Medical Center, and RR001032 to Beth Israel Deaconess Medical Center), National Center for Advancing Translational Sciences Clinical and Translational Science Awards (RR025008 and TR000454 to Emory University, RR024150 and TR00135 to the Mayo Clinic, RR025752 and TR001064 to Tufts University, RR025780 and TR001082 to the University of Colorado, RR025758 and TR001102 to Beth Israel Deaconess Medical Center, RR033179 and TR000001 to the University of Kansas Medical Center, and RR024989 and TR000439 to Cleveland Clinic), by funding from the Zell Family Foundation (to the University of Colorado), and by a grant from the PKD Foundation.
Mutation analysis was supported by DK62410-S1 to Dr. Harris and the Mayo Translational PKD Center (DK090728). Study drugs were donated by Boehringer Ingelheim Pharmaceuticals Inc (telmisartan and matched placebo) and Merck & Co Inc (lisinopril).
Most of all we thank the hundreds of patients who took part in the HALT-PKD trials and the dedicated study coordinators who guided them through the years of participation. We are also indebted to the research program coordinators and program managers at Washington University (Gigi Flynn and Robin Woltman) and the University of Pittsburgh (Susan Spillane and Patty Smith), and the staff at the Image Analysis Center (Johana Schafer and Cheng Tao).
We also thank Diane Comer, Center for Research on Health Data at the University of Pittsburgh, for expert assistance with the statistical analyses.
Footnotes
DISCLOSURES:
Dr. Schrier reports having received fees for serving on advisory boards from Otsuka Pharmaceuticals, Janssen Pharmaceuticals, and Ikaria; Dr. Perrone, consulting fees from Sanofi–Genzyme and Vertex Pharmaceuticals and consulting fees and grant support through his institution from Otsuka Pharmaceuticals; Drs. Torres and Harris, grant support from Otsuka Pharmaceuticals; Dr. Steinman, grant support from Kadmon, Fibrogen and AMAG Pharmaceuticals and a consulting fee from Sanofi-Genzyme; Dr. Rahbari-Oskoui, fees for serving on advisory boards from Otsuka, Kadmon, and Astute medical and also, research support from Otsuka; Dr. Bae, consulting fees from Kadmon Pharmaceuticals; and Dr. Chapman, consulting fees from Kadmon, Otsuka Pharmaceuticals, and Pfizer and grant support from Otsuka Pharmaceuticals. The remaining authors declared no potential conflict of interest.
References
- 1.Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am J Kidney Dis. 1983;2:630–639. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 2.Fick-Brosnahan GM, Ecder T, Schrier RW. Polycystic kidney disease. In: Schrier R, editor. Diseases of the Kidney. 7th. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. pp. 547–588. [Google Scholar]
- 3.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 4.Spithoven EM, Kramer A, Meijer E, et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014;86:1244–1252. doi: 10.1038/ki.2014.120. [DOI] [PubMed] [Google Scholar]
- 5.Reule S, Sexton DJ, Solid CA, Chen SC, Collins AJ, Foley RN. ESRD from autosomal dominant polycystic kidney disease in the United States, 2001–2010. Am J Kidney Dis. 2014;64:592–599. doi: 10.1053/j.ajkd.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw C, Simms RJ, Pitcher D, Sandford R. Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrol Dial Transplant. 2014;29:1910–1918. doi: 10.1093/ndt/gfu087. [DOI] [PubMed] [Google Scholar]
- 7.Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 8.Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 9.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caroli A, Perico N, Perna A, et al. Effect of long-acting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013;382:1485–1495. doi: 10.1016/S0140-6736(13)61407-5. [DOI] [PubMed] [Google Scholar]
- 11.Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol. 2002;13:1733–1739. doi: 10.1097/01.asn.0000018407.60002.b9. [DOI] [PubMed] [Google Scholar]
- 12.Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis. 2011;57:856–862. doi: 10.1053/j.ajkd.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 13.Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev. 2013;9:2–11. doi: 10.2174/1573402111309010002. [DOI] [PubMed] [Google Scholar]
- 14.Gile RD, Cowley BD, Gattone VH, O’Donnell MP, Swan SK, Grantham JJ. Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. Am J Kidney Dis. 1995;26:501–507. doi: 10.1016/0272-6386(95)90497-2. [DOI] [PubMed] [Google Scholar]
- 15.Zafar I, Tao Y, Falk S, McFann K, Schrier RW, Edelstein CL. Effect of statin and angiotensin-converting enzyme inhibition on structural and hemodynamic alterations in autosomal dominant polycystic kidney disease model. Am J Physiol Renal Physiol. 2007;293:F854–859. doi: 10.1152/ajprenal.00059.2007. [DOI] [PubMed] [Google Scholar]
- 16.Klawitter J, Zafar I, Klawitter J, et al. CL. Effects of lovastatin treatment on the metabolic distributions in the Han:SPRD rat model of polycystic kidney disease. BMC Nephrol. 2013;14:165. doi: 10.1186/1471-2369-14-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9:889–896. doi: 10.2215/CJN.08350813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haynes R, Lewis D, Emberson J, et al. Effects of lowering LDL cholesterol on progression of kidney disease. J Am Soc Nephrol. 2014;25:1825–1833. doi: 10.1681/ASN.2013090965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371:2255–2266. doi: 10.1056/NEJMoa1402685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371:2267–2276. doi: 10.1056/NEJMoa1402686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5:102–109. doi: 10.2215/CJN.04310709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The consortium for radiologic imaging studies of polycystic kidney disease (CRISP) cohort. Kidney Int. 2003;64:1035–45. doi: 10.1046/j.1523-1755.2003.00185.x. [DOI] [PubMed] [Google Scholar]
- 23.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009; 150: 604–612. Erratum in: Ann Intern Med. 2011;155:408. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Austin PC, Stuart EA. Moving towards best practice when using inverse probability of treatment weighting (IPTW) using the propensity score to estimate causal treatment effects in observational studies. Stat Med. 2015;34:3661–3679. doi: 10.1002/sim.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002;87:1451–1458. doi: 10.1210/jcem.87.4.8412. [DOI] [PubMed] [Google Scholar]
- 26.Barakat AF, Saad M, Abuzaid A, Mentias A, Mahmoud A, Elgendy IY. Perioperative statin therapy for patients undergoing coronary artery bypass grafting. Ann Thorac Surg. 2016;101:818–825. doi: 10.1016/j.athoracsur.2015.09.070. [DOI] [PubMed] [Google Scholar]
- 27.Horiguchi A, Sumitomo M, Asakuma J, Asano T, Asano T, Hayakawa M. 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitor, fluvastatin, as a novel agent for prophylaxis of renal cancer metastasis. Clin Cancer Res. 2004;10:8648–8655. doi: 10.1158/1078-0432.CCR-04-1568. [DOI] [PubMed] [Google Scholar]
- 28.Kaffenberger SD, Lin-Tsai O, Stratton KL, et al. Statin use is associated with improved survival in patients undergoing surgery for renal cell carcinoma. Urol Oncol. 2015;33:21.e11–7. doi: 10.1016/j.urolonc.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torres VE, Chapman AB, Perrone RD, et al. Analysis of baseline parameters in the HALT polycystic kidney disease trials. Kidney Int. 2012;81:577–585. doi: 10.1038/ki.2011.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perticone F, Ceravolo R, Maio R, et al. Effects of atorvastatin and vitamin C on endothelial function of hypercholesterolemic patients. Atherosclerosis. 2000;152:511–518. doi: 10.1016/s0021-9150(00)00370-1. [DOI] [PubMed] [Google Scholar]
- 31.Palaniswamy C, Selvaraj DR, Selvaraj T, Sukhija R. Mechanisms underlying pleiotropic effects of statins. Am J Ther. 2010;17:75–78. doi: 10.1097/MJT.0b013e31819cdc86. [DOI] [PubMed] [Google Scholar]
- 32.Al-Nimri MA, Komers R, Oyama TT, Subramanya AR, Lindsley JN, Anderson S. Endothelial-derived vasoactive mediators in polycystic kidney disease. Kidney Int. 2003;63:1776–1784. doi: 10.1046/j.1523-1755.2003.00913.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Iversen J, Wilcox CS, Strandgaard S. Endothelial dysfunction and reduced nitric oxide in resistance arteries in autosomal dominant polycystic kidney disease. Kidney Int. 2003;64:1381–1388. doi: 10.1046/j.1523-1755.2003.00236.x. [DOI] [PubMed] [Google Scholar]
- 34.Namli S, Oflaz H, Turgut F, et al. Improvement of endothelial dysfunction with simvastatin in patients with autosomal dominant polycystic kidney disease. Ren Fail. 2007;29:55–59. doi: 10.1080/08860220601038892. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Strandgaard S, Borresen ML, et al. Asymmetric dimethylarginine and lipid peroxidation products in early autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2008;51:184–191. doi: 10.1053/j.ajkd.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: From impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol. 2008;3:986–991. doi: 10.2215/CJN.02330607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klawitter J, Reed-Gitomer BY, McFann K, et al. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am J Physiol Renal Physiol. 2014;307:F1198–1206. doi: 10.1152/ajprenal.00327.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torres VE, King BF, Chapman AB, et al. Magnetic resonance measurements of renal blood flow and disease progression in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2007;2:112–120. doi: 10.2215/CJN.00910306. [DOI] [PubMed] [Google Scholar]
- 39.Meijer E, Rook M, Tent H, et al. Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5:1091–1098. doi: 10.2215/CJN.00360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Dijk MA, Kamper AM, van Veen S, Souverijn JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2001;16:2152–2157. doi: 10.1093/ndt/16.11.2152. [DOI] [PubMed] [Google Scholar]
- 41.Zand L, Torres VE, Larson TS, et al. Renal hemodynamic effects of the HMG-CoA reductase inhibitors in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31:1290–1295. doi: 10.1093/ndt/gfv394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ecder T. Statins in the treatment of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31:1194–1196. doi: 10.1093/ndt/gfv449. [DOI] [PubMed] [Google Scholar]
- 43.Barnett M, Hall S, Dixit M, Arany I. Simvastatin attenuates oleic acid-induced oxidative stress through CREB-dependent induction of heme oxygenase-1 in renal proximal tubule cells. Pediatr Res. 2016;79:243–250. doi: 10.1038/pr.2015.210. [DOI] [PubMed] [Google Scholar]
- 44.Zheng Z, Jayaram R, Jiang L, et al. Perioperative rosuvastatin in cardiac surgery. N Engl J Med. 2016;374:1744–1753. doi: 10.1056/NEJMoa1507750. [DOI] [PubMed] [Google Scholar]
- 45.Zeier M, Fehrenbach P, Geberth S, Möhring K, Waldherr R, Ritz E. Renal histology in polycystic kidney disease with incipient and advanced renal failure. Kidney Int. 1992;42:1259–1265. doi: 10.1038/ki.1992.413. [DOI] [PubMed] [Google Scholar]
- 46.Maser RL, Vassmer D, Magenheimer BS, Calvet JP. Oxidant stress and reduced antioxidant enzyme protection in polycystic kidney disease. J Am Soc Nephrol. 2002;13:991–999. doi: 10.1681/ASN.V134991. [DOI] [PubMed] [Google Scholar]
- 47.Norman J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD) Biochim Biophys Acta. 2011;1812:1327–1336. doi: 10.1016/j.bbadis.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7:556–566. doi: 10.1038/nrneph.2011.109. [DOI] [PubMed] [Google Scholar]
- 49.Klawitter J, McFann K, Pennington AT, et al. Pravastatin therapy and biomarker changes in children and young adults with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2015;10:1534–1541. doi: 10.2215/CJN.11331114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fassett RG, Coombes JS, Packham D, Fairley KF, Kincaid-Smith P. Effect of pravastatin on kidney function and urinary protein excretion in autosomal dominant polycystic kidney disease. Scand J Urol Nephrol. 2010;44:56–61. doi: 10.3109/00365590903359908. [DOI] [PubMed] [Google Scholar]
- 51.Giacoppo D, Capodanno D, Capranzano P, Aruta P, Tamburino C. Meta-analysis of randomized controlled trials of preprocedural statin administration for reducing contrast-induced acute kidney injury in patients undergoing coronary catheterization. Am J Cardiol. 2014;114:541–548. doi: 10.1016/j.amjcard.2014.05.036. [DOI] [PubMed] [Google Scholar]
- 52.Akyuz S, Yaylak B, Altay S, Kasikcioglu H, Cam N. The role of statins in preventing contrast-induced acute kidney injury: A narrative review. Angiology. 2015;66:701–707. doi: 10.1177/0003319714549556. [DOI] [PubMed] [Google Scholar]
- 53.Wang N, Qian P, Yan TD, Phan K. Periprocedural effects of statins on the incidence of contrast-induced acute kidney injury: A systematic review and trial sequential analysis. Int J Cardiol. 2016;206:143–152. doi: 10.1016/j.ijcard.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Sanguankeo A, Upala S, Cheungpasitporn W, Ungprasert P, Knight EL. Effects of statins on renal outcome in chronic kidney disease patients: a systematic review and meta-analysis. PLoS One. 2015;10:e0132970. doi: 10.1371/journal.pone.0132970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeZeeuw D, Anzalone DA, Cain VA, et al. Renal effects of atorvastatin and rosuvastatin in patients with diabetes who have progressive renal disease (PLANET I): a randomised clinical trial. Lancet Diabetes Endocrinol. 2015;3:181–190. doi: 10.1016/S2213-8587(14)70246-3. [DOI] [PubMed] [Google Scholar]
- 56.Kidney Disease: Improving Global Outcomes (KDIGO) Lipid Work Group. KDIGO clinical practice guideline for lipid management in chronic kidney disease. Kidney Int Suppl. 2013;3:259–305. [Google Scholar]