

Abstract
The WNT signaling pathway is involved in cellular and tissue functions that control such diverse processes as body axis patterning, cellular proliferation, differentiation, and lifespan. The long list of molecules that can participate or modify WNT signaling makes this pathway one of the most complex in cell biology. In bone tissue, WNT signaling is required for proper skeletal development, and human mutations in various components of the cascade have revealed insights into pharmacologic targeting that can be harnessed to improve skeletal health. In particular, mutations in genes that code for the WNT signaling inhibitor sclerostin or the WNT co-receptor LRP5 have highlighted the potential therapeutic value of recapitulating those effects in patients with low bone mass. A constant challenge in this area is selectively modifying WNT components in the tissue of interest, as WNT has manifold effects in nearly every tissue.
Keywords: Wnt, Sost, sclerostin, Lrp5, HBM, osteoporosis, bone anabolics
Pharmacologic options for the clinical treatment of low-bone-mass disorders have come a long way since the early 1990s, when hormone replacement therapy and calcitonin were the main choices available to physicians. Since then, numerous FDA-approved anti-resorptive therapies have emerged (the bisphosphonates alendronate, risedronate, ibandronate, and zolendronate; the selective estrogen receptor modulator raloxifene; and biologic denosumab), but the development and approval of new anabolic agents, which can rebuild lost bone, has lagged behind. Currently, only two skeletal anabolics are clinically available (teriparatide and abaloparatide), both of which work by a common mechanism of stimulating the parathyroid hormone receptor (PTHR1). The paucity of skeletal anabolics is not a matter of simple short-sightedness, as it is likely that concerns over patient safety and the promotion of anabolic action in bone potentially affecting tumor development and growth might explain the reluctance to further develop this line of therapy.
The search for biomolecular targets that are useful in drug development is often aided by the identification of genetic mutations in humans, many of which are linked to revealing phenotypes or rare diseases. For some diseases of connective tissue, like skin or muscle, the phenotypes are obvious because they are often immediately visible. For example, mutations that cause an increase in muscle mass are immediately obvious to the patient, friends and family, and the primary physician. Such is the case in animals (McPherron and Lee, 1997, Lee and McPherron, 2001) and humans (Schuelke et al., 2004) with loss-of-function mutations in the GDF-8 gene, which encodes a negative regulator of muscle growth. Loss of GDF-8 significantly increases muscle mass, producing an obvious phenotype, and monoclonal antibodies directed against GDF-8 are currently in clinical trials to treat muscle wasting diseases (Singh et al., 2016). Other unrelated, yet undiscovered rare mutations that cause such an increase in muscle mass would also be unlikely to go unnoticed, and the genetic basis and suitability for targeting could be very quickly revealed and developed. Thus there is a distinct “leg up” advantage, inherent to a tissue like muscle, that facilitates uncovering anabolic targets, based on the visually obvious phenotypic effects of disturbances in normal physiology.
This is not the case for uncovering anabolic targets for bone, where significant changes in bone mass are not obvious and must be fortuitously discovered based on patients that happen to present clinically for other, potentially non-skeletal health issues. As such, the search for skeletal anabolic targets is at distinct disadvantage compared to tissues like muscle and skin, and it is likely that many patients with mutations that cause high bone mass go unnoticed, undocumented, and unexplored for genetic linkage; without genetic linkage, targets are elusive. Consequently, anabolic therapy development for bone is necessarily sluggish, but chance observations have yielded significant leads. A hallmark example of the fortuitous nature of discovering anabolic mechanisms in bone has been described by the Johnson/Recker group at Creighton University in Omaha, NE. A young woman who was involved in a car accident was brought to the local hospital in Omaha and administered routine x-rays to look for fractures. While no fractures were present, the orthopaedist noticed unusually dense bones on x-ray, and referred the patient to a local endocrinologist (Dr. Recker) for further investigation, where she was given a DXA and found to have very high bone mass and density. Genetic analysis of her extended family (Dr. Johnson), in conjunction with dual energy x-ray absorptiometry (DXA) scanning to reveal the presence or absence of the high bone mass (HBM) phenotype, fueled the eventual discovery and localization of the genetic cause of HBM as a missense mutation in the low-density lipoprotein receptor related protein 5 (LRP5), a co-receptor for int/Wingless (WNT; Little et al., 2002). Subsequently, other research groups have identified several other families with HBM that harbored other missense mutations in LRP5 (Van Wesenbeeck et al., 2003), and those LRP5 mutations that have been studied in mouse models all indicate that the receptor potently regulates bone formation (Fig. 1). If not for the vigilant eye of the orthopaedist that initially referred the proband HBM patient, the WNT pathway would not have received the deluge of basic science and translational investigation that it did in the years following the LRP5 discovery; or, at the very least, the revelation of its importance in bone metabolism would have been significantly delayed. This work was a major impetus that brought the WNT field out of Drosophila and Xenopus, and into the mammalian skeleton, where the pathway has undergone major mechanistic clarification. It is obvious, but worth stating, that the lack of outward “visibility” of HBM phenotypes (except for some diagnostic clues like a squared-off or prominent mandible), no matter what their biochemical cause—WNT or otherwise—is a major hurdle to identifying molecular causes of (and consequently, potential solutions to) low bone mass disease. How many other patients are walking around today with high bone mass, but are otherwise asymptomatic, that will never present clinically—or will present to a physician but will be overlooked for study of the genetic cause of their skeletal phenotype since it is not a source of disability?
Figure 1.
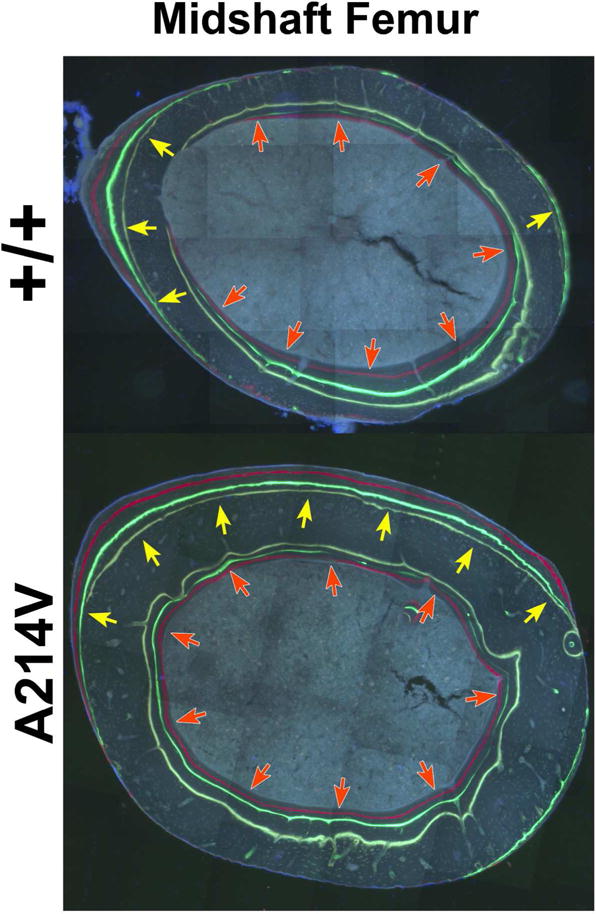
Undecalcified, methyl-methacrylate-embedded transverse histological sections from the midshaft femur of two littermate male mice. The sections are unstained and were imaged under epifluorescent light filtered to excite the oxytetracycline (pale yellow) calcein (green), and alizarin (red) labels that were injected in vivo 8, 6, and 2 weeks before sacrifice, respectively. The mouse in the upper panel is wild-type for Lrp5, whereas the mouse in the lower panel carries a missense mutation in Lrp5 (Arg→Val at amino acid 201, the mouse analog of human amino acid 214) identified in a human family that has HBM. The bone-seeking fluorochrome labels reveal the more rapid and extensive bone modeling activity in the Lrp5 mutant mice (note the extend of both periosteal [gold arrows] and endocortical [red arrows] surface labeling in the mutant), which ultimately leads to a thicker and broader bone cross section in the A214V knockin mice.
While the search for anabolic mechanisms is slow, the foundational LRP5 work described above does highlight the potential for harnessing the WNT pathway for anabolic action in the skeleton. However, targeting is not straightforward because the WNT signaling pathway is one of the most complex biological systems known in cell biology (Nusse and Clevers, 2017). This complexity stems from a multitude of factors, including the number of proteins and accessory proteins involved in signal transduction, the number and diversity of alternative pathways, the tissue expression/selectivity of certain components, and the endogenous inhibitor milieu that suppresses signaling. To illustrate, consider Wnt signaling just at the level of the cell membrane (and momentarily ignore intracellular components involved in Wnt generation, maturation, release, signaling downstream of activated receptors, and nuclear consequences of alterations in the pathway): there are 19 Wnts (the ligand), 10 Frizzleds (one half of the co-receptor complex), and at least 2 LDL-like receptors (the other half of the co-receptor complex) in play in most mammals, and one member from each of these three components is required for a canonical Wnt signal to be transmitted. In light of the number of possible combinations of these components, each of which might have slightly to significantly different effects on the downstream signal, it is not surprising that the pathway is difficult to study experimentally. Moving beyond the cell surface, there are numerous endogenous extracellular modulators of the Wnt signal (e.g., Wnt inhibitors like secreted Frizzled related proteins [sFrp] and Wnt inhibitor factor [Wif], Lrp5/6 inhibitors like dickkopf homolog-1 [Dkk1] and sclerostin, and some of these have tissue-selective expression. An additional layer of complexity comes from the observation that several different classes of Wnt signaling can occur, including the canonical pathway (Wnt-Fzd-Lrp5/6) the noncanonical planar cell polarity (PCP) pathway (Wnt-Fzd-Ror2/Ryk), and the calcium pathway (Wnt-Fzd→PLC). Each of these pathways engages different downstream effectors and has different cellular and transcriptional outcomes. The breadth of WNT signaling, in conjunction with the ubiquitous or near-ubiquitous presence of the pathway in one form or another, across all tissues in the body, positions this system to be a major regulator of cellular activity and disease. WNT affects hair loss, hearing loss, vision loss, cancer, skin disease, liver disease, cognition, fertility, liver disease, and immune function, to name a few. Due to the widespread and diverse role of WNT in these and other processes, targeting WNT for therapeutic purposes has proved to be tricky at best, and to date there are no FDA-approved WNT inhibitors for treatment of disease. For example, WNT signaling is essential for pancreatic ductal adenocarcinoma (PDAC) initiation and progression (Zhang et al., 2013), and so an obvious therapeutic approach to treating PDAC is to inhibit WNT. Several pharmaceutical companies are conducting clinical trials with inhibitors of an enzyme (porcupine) that catalyzes a crucial post-translational modification to WNT—palmitoylation—that is required for WNT secretion. While porcupine inhibitors might turn out to have therapeutic value for patients with PDAC, their bone-wasting effects on the skeleton are devastating (Williams, 2016), as WNT signaling is required for normal bone homeostasis. Thus, the development of WNT inhibitors to treat diseases like colorectal cancer (Novellasdemunt et al., 2015) or type II diabetes (Savic et al., 2011, Tong et al., 2009) have significant challenges ahead, due to the diverse and broad influence WNT signaling has on many organ systems.
A key to properly and safely targeting WNT for therapeutic benefit is finding and exploiting context- or cell type-specific components of the pathway. In the skeletal biology field, such an approach has been taken to stimulate osteoanabolic action based on targeting the WNT pathway inhibitor sclerostin, the protein product of the SOST gene. Sclerostin is a potent LRP5/6 antagonist that serves as a negative regulator of bone formation (Semenov et al., 2005); one can think of sclerostin as the bone equivalent of GDF-8 for muscle. Sclerostin presents a high priority candidate for targeting in the bone field because sclerostin’s expression is highly enriched in a specific cell type within the bone tissue—the osteocyte (Poole et al., 2005; Fig. 2). Sclerostin expression can be detected in a few other tissues, but usually these are transient expression events that occur during development or arise as a result of disease, e.g., plaque formation and calcification of the great vessels. We noted earlier that most patients with high bone mass probably go undetected to the healthcare community, but that is not the case for patients with homozygous loss-of-function mutations in the SOST gene, which codes for sclerostin protein. These patients have even greater bone mineral density (BMD) than LRP5-HBM patients (z-scored upwards of 10–12) and usually present clinically due to symptoms associated with bone overgrowth such as headaches due to increased intracranial pressure from the encroaching inner cranial table, and loss of sensory input (taste, hearing, olfaction, vision) from cranial nerves due to cranial nerve foramina stenosis (Gardner et al., 2005). Beyond the health issues related to bone overgrowth, sclerosteosis patients have remarkably few associated health concerns, and there are no data suggesting that cancer incidence is increased in this patient population (though there are only ~100 identified cases of sclerosteosis so statistical confidence is low).
Figure 2.
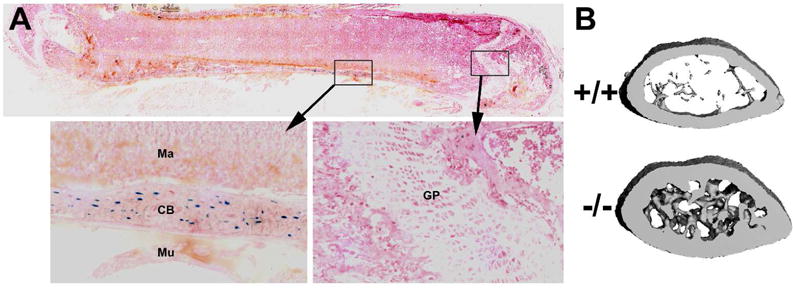
(A) Frozen section through a whole femur from a 4-wk old mouse heterozygous for a LacZ reporter allele knocked into the endogenous Sost coding sequence. The blue staining reports Sost expression. Note the strong Sost expression in osteocytes (cells embedded in the cortical bone [CB]) and lack of staining in the marrow [Ma], muscle [Mu], and growth plate chondrocytes [GP]. The section is counterstained with fast red. (B) μCT reconstructions of a 2-mm slab through the distal femur metaphysis of a 16-wk-old female wild type mouse (upper) and a Sost−/− littermate (lower) illustrating the high bone mass (cortical and cancellous) phenotype in mice with Sost homozygous loss-of-function mutations. These mice model the sclerosteosis phenotype found among patients with homozygous loss-of-function mutations in SOST.
The similarity in phenotype between HBM patients with gain-of-function mutations in LRP5 and those with homozygous loss-of-function mutations in SOST stems from the common mechanisms that is at the root of their conditions, i.e., enhanced WNT signaling in bone. In the case of LRP5 patients, the dozen or so identified missense mutations in LRP5 that cause HBM are all clustered around the central domain of the receptor’s 1st β-propeller, which is also the locale for sclerostin interaction (Holdsworth et al., 2012, Bourhis et al., 2011). It is likely that the amino acid substitutions in this region induce steric changes in the binding pocket for sclerostin such that sclerostin-mediated inhibition of the receptor is compromised, and tonic WNT signaling is enhanced. Thus either the absence of sclerostin (i.e., sclerosteosis patients) or a lack of sclerostin interaction with LRP5 (i.e., LRP5 HBM patients) has the same end result: increased signaling through LRP5 (Niziolek et al., 2011). The observation that sclerosteosis patients have a more severe HBM phenotype than LRP5 HBM patients is likely due to sclerosteosis patients presumably having tonically enhanced signaling of both LRP5 and LRP6 (sclerostin binds and inhibits both receptors) whereas LRP5-HBM patients have tonically enhanced LRP5 signaling alone, with LRP6 being susceptible to sclerostin-mediated inhibition.
The sclerosteosis and LRP5-HBM human phenotypes have prompted several members of the commercial biotech industry to develop sclerostin programs, with the goal of generating sclerostin inhibitors that can improve bone mass and strength in patients with low bone mass disorders. Although the discovery and exploitation of sclerostin as a drug target was more complicated and multifaceted than simply tracking down the genetic cause of sclerosteosis (reviewed in (Paszty et al., 2010), use of sclerostin-neutralizing antibodies in numerous animal models (Jacobsen et al., 2016, Jacobsen et al., 2014, Kedlaya et al., 2013) and in patients (Padhi et al., 2011) with osteoporosis has proven effective. As of this date, the FDA is formally reviewing an application for approval for Romosozumab, a monoclonal antibody directed against sclerostin, for the treatment of postmenopausal osteoporosis. If approved, the skeletal biology field would have its first (and only) anabolic agent outside of the PTH/PTHrP class.
The WNT pathway is a complicated signaling apparatus that controls major components of development, growth, and aging, and has manifold roles in disease initiation and progression. The vast number of proteins involved in WNT signal transduction poses both great challenges and great opportunities for harnessing the pathway to improve quality of life and longevity. Work in the skeletal biology field nicely highlights how select components of the WNT system can be carefully targeted to achieve beneficial effects for the treatment of disease, while potentially leaving other tissues to function normally. Nevertheless, the challenges with targeting WNT are not trivial; even molecules as restricted as sclerostin can be affected in certain pathologic states and manifest in adverse events. A recent press release by Amgen, the company filing for FDA approval of the sclerostin antibody Romosozumab, reported an increase in “cardiovascular serious adverse events” among patients in a phase III trial of the drug. Whether this specific compound gains approval or not, it is clear that potent osteoanabolic targets exist within in the WNT pathway. Designing other strategies to safely target those mechanisms presents a challenge that becomes increasingly within reach as our understanding of this intricate pathway’s nuances are refined by basic, translational, and clinical research.
References
- Bourhis E, Wang W, Tam C, Hwang J, Zhang Y, Spittler D, Huang OW, Gong Y, Estevez A, Zilberleyb I, Rouge L, Chiu C, Wu Y, Costa M, Hannoush RN, Franke Y, Cochran AG. Wnt antagonists bind through a short peptide to the first beta-propeller domain of LRP5/6. Structure. 2011;19:1433–1442. doi: 10.1016/j.str.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Gardner JC, van Bezooijen RL, Mervis B, Hamdy NA, Lowik CW, Hamersma H, Beighton P, Papapoulos SE. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90:6392–6395. doi: 10.1210/jc.2005-1235. [DOI] [PubMed] [Google Scholar]
- Holdsworth G, Slocombe P, Doyle C, Sweeney B, Veverka V, Le Riche K, Franklin RJ, Compson J, Brookings D, Turner J, Kennedy J, Garlish R, Shi J, Newnham L, McMillan D, Muzylak M, Carr MD, Henry AJ, Ceska T, Robinson MK. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of Wnt co-receptors. J Biol Chem. 2012;287:26464–26477. doi: 10.1074/jbc.M112.350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen CM, Barber LA, Ayturk UM, Roberts HJ, Deal LE, Schwartz MA, Weis M, Eyre D, Zurakowski D, Robling AG, Warman ML. Targeting the LRP5 pathway improves bone properties in a mouse model of osteogenesis imperfecta. J Bone Miner Res. 2014;29:2297–2306. doi: 10.1002/jbmr.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen CM, Schwartz MA, Roberts HJ, Lim KE, Spevak L, Boskey AL, Zurakowski D, Robling AG, Warman ML. Enhanced Wnt signaling improves bone mass and strength, but not brittleness, in the Col1a1(+/mov13) mouse model of type I Osteogenesis Imperfecta. Bone. 2016;90:127–132. doi: 10.1016/j.bone.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanis JA, Hans D, Cooper C, Baim S, Bilezikian JP, Binkley N, Cauley JA, Compston JE, Dawson-Hughes B, El-Hajj Fuleihan G, Johansson H, Leslie WD, Lewiecki EM, Luckey M, Oden A, Papapoulos SE, Poiana C, Rizzoli R, Wahl DA, McCloskey EV, Task Force of the, F.I. Interpretation and use of FRAX in clinical practice. Osteoporos Int. 2011;22:2395–2411. doi: 10.1007/s00198-011-1713-z. [DOI] [PubMed] [Google Scholar]
- Kedlaya R, Veera S, Horan DJ, Moss RE, Ayturk UM, Jacobsen CM, Bowen ME, Paszty C, Warman ML, Robling AG. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5:211ra158. doi: 10.1126/scitranslmed.3006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niziolek PJ, Farmer TL, Cui Y, Turner CH, Warman ML, Robling AG. High-bone-mass-producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes. Bone. 2011;49:1010–1019. doi: 10.1016/j.bone.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am J Physiol Cell Physiol. 2015;309:C511–521. doi: 10.1152/ajpcell.00117.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res. 2010;25:1897–1904. doi: 10.1002/jbmr.161. [DOI] [PubMed] [Google Scholar]
- Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. 2011;21:1417–1425. doi: 10.1101/gr.123745.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- Singh P, Rong H, Gordi T, Bosley J, Bhattacharya I. Translational Pharmacokinetic/Pharmacodynamic Analysis of MYO-029 Antibody for Muscular Dystrophy. Clin Transl Sci. 2016;9:302–310. doi: 10.1111/cts.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Lin Y, Zhang Y, Yang J, Zhang Y, Liu H, Zhang B. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: a large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC Med Genet. 2009;10:15. doi: 10.1186/1471-2350-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, Warman ML, De Vernejoul MC, Bollerslev J, Van Hul W. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–771. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BO. Genetically engineered mouse models to evaluate the role of Wnt secretion in bone development and homeostasis. Am J Med Genet C Semin Med Genet. 2016;172C:24–26. doi: 10.1002/ajmg.c.31474. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Morris JPt, Yan W, Schofield HK, Gurney A, Simeone DM, Millar SE, Hoey T, Hebrok M, Pasca di Magliano M. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013;73:4909–4922. doi: 10.1158/0008-5472.CAN-12-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]