

Abstract

ABC triblock copolymers with a poly(ethylene glycol) (PEG) midblock have attractive properties for biomedical applications because of PEG’s favorable properties regarding biocompatibility and hydrophilicity. However, easy strategies to synthesize polymers containing a PEG midblock are limited. In this study, the successful synthesis of a heterofunctional PEG macroinitiator containing both an azoinitiator and an atom transfer radical polymerization (ATRP) initiator is demonstrated. This novel PEG macroinitiator allows the development of elegant synthesis routes for PEG midblock-containing ABC copolymers that does not require protection of initiating sites or polymer end-group postmodification. Polymers with outer blocks composed of different monomers were synthesized to illustrate the versatility of this macroinitiator. N-Isopropylacrylamide (NIPAM) was included to obtain thermosensitive polymers, 2-(dimethylamino)ethyl methacrylate (DMAEMA) provided pH-sensitive properties, and 2-hydroxyethyl acrylate (HEA) functioned as a noncharged hydrophilic block that also allows for postmodifications reactions. This synthesis approach can further contribute to the design of high-precision polymers with tailorable block compositions and polymer topologies, which is highly attractive for applications in nanotechnology.
1. Introduction
Emerging technologies in the nanotechnology field have led to the development of novel polymeric materials with sophisticated properties and smart performances. In particular, block copolymers with well-defined architectures play a central role in polymer science. Control over chemical structures, compositions, and molecular characteristics of block copolymers give rise to innovative self-assembling materials. A rich variety of ordered nano- and macrostructures can be formed when two or more polymeric chains, having different physical properties, are covalently linked to each other. The synthesis of copolymers of the diblock AB type has been extensively reported in the literature. Depending on the copolymer’s composition, different ordered structures can be obtained, including lamellae, cylinders, spheres, and gyroids.1 The shape and size of the achieved supramolecular structure can be varied by tuning the properties of the blocks, such as hydrophobicity, presence of charged groups, or block length. In addition, the environment of the polymer, such as solvent, pH value, or temperature, can influence the self-assembly behavior of these materials.2−4 Besides the diblock AB type materials, multiblock copolymers including ABC triblock copolymers have attracted much attention because of their unique structure with multiple different homopolymer blocks. The addition of another block leads to a much richer variety of phases, and the resulting materials have the potential to generate a wide range of multiphase structures on a mesoscopic length scale.5−7 Consequently, numerous materials for applications ranging from the development of novel surfactants,8 dispersants,9 coatings,10 and adhesives11 to materials for microelectronics12 and biomaterials, such as membranes13 and drug delivery systems,14 became available.
Poly(ethylene glycol) (PEG) is a neutral and hydrophilic polymer and an important component for various biomedical materials.15 This polymer is generally known for its biocompatible properties, and when PEG chains are conjugated to nano- or microparticles, it can prevent aggregation of particles in an aqueous solution.16 In addition, PEGylation of proteins and nanoparticles has shown to reduce the interaction with serum proteins, resulting in longer circulation times.17,18 For these reasons, PEG is often used as building block in AB diblock copolymers for the development of biomedical materials. However, the design and synthesis of PEG-based ABC triblock copolymers, especially as the middle block, is limited. Two strategies are usually followed to obtain ABC triblock copolymers with predetermined number-average molecular weight (Mn) and low polydispersity index (PDI): sequential living polymerizations of the different monomers or selective coupling of two or more functional polymer chains.19,20 The latter one requires the presence of mutually reactive end-groups on the polymer chains. This coupling reaction becomes increasingly less efficient with increasing chain lengths due to steric hindrance, which influences the polymer molecular weight distributions.21 On the other hand, sequential living polymerizations, especially the use of controlled radical polymerizations (CRP), have rapidly increased over the past years.22,23 These polymerization techniques are versatile methods for the synthesis of block copolymers, with their own set of advantages and limitations. One of the limitations of CRP is the difficulty of synthesizing ABC triblock copolymers with a linear PEG midblock. Protection of the initiating sites or polymer end-group modifications is often required for this purpose, which results in a more complex synthesis process. A combination of different polymerization mechanisms can provide an alternative synthetic route to ABC block copolymers and achieve architectures not attainable using only one mechanism. In addition, by using a heterofunctional initiator, a wide range of functionalities can be introduced into a polymer chain.21
In this study, the synthesis of a novel heterofunctional PEG macroinitiator containing both an azoinitiator and an ATRP initiator is shown (Scheme 1). ATRP can be performed at low temperatures, leaving the azoinitiator intact. In the second step, the final block is polymerized at high temperatures with a wide versatility in choice of monomers. This macroinitiator enabled the development of a new strategy for the preparation of high-precision ABC triblock copolymers by two-step radical polymerization. The obtained ABC polymers contain a linear hydrophilic PEG midblock, which can be of particular interest for the design of materials for biomedical applications, and outer blocks having different properties. In addition, no protection of the initiating sites during or polymer end-group modifications after the polymerization process is required.
Scheme 1. Synthesis Route of ABC Triblock Copolymers Using the Heterofunctional PEG Macroinitiator (1); in the First Step, the First Block, Represented by A, Is Synthesized by Atom Transfer Radical Polymerization (ATRP) (2); in the Next Step, the Final Block, Represented by C, Is Synthesized by Free Radical Polymerization (FRP) (3).
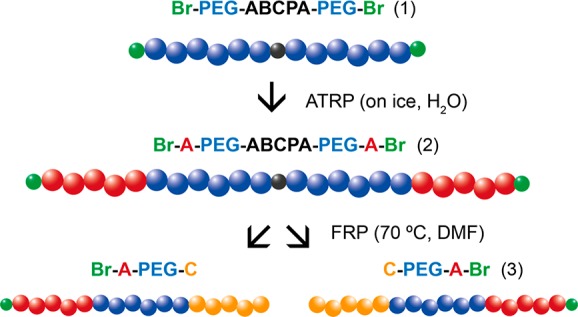
2. Materials and Methods
2.1. Materials
All materials were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands) and used as received unless otherwise noted. α-tert-Butyloxycarbonylamino-ω-hydroxypoly(ethylene glycol) with PEG molecular weight of 5000 Da (Boc-NH-PEG5000-OH) was obtained from Iris Biotech GmbH (Marktredwitz, Germany) and dried overnight in a vacuum oven at room temperature (RT) before use. 4-(Dimethylamino)pyridinium-4-toluenesulfonate (DPTS) was prepared according to a literature procedure.24 Triethylamine (TEA) and 2,4,6-trinitrobenzenesulfonic acid (TNBSA) solution in methanol (5% w/v) were obtained from Thermo Fisher Scientific (Bleiswijk, The Netherlands). Peptide grade dichloromethane (DCM), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), and diethyl ether were purchased from Biosolve (Valkenswaard, The Netherlands). All solvents were dried by molecular sieves for 24 h before use. Prior to use, 2-(dimethylamino)ethyl methacrylate (DMAEMA) was passed through a column of alumina to remove the inhibitor. Slide-A-lyzer dialysis cassettes (Mw cutoff: 3.5 and 10 kDa) were obtained from Thermo Fisher Scientific (Bleiswijk, The Netherlands).
2.2. Synthesis of Heterofunctional PEG Macroinitiator
The synthesis of (Br-NH-PEG5000)2-ABCPA was performed according to a three-step synthesis route as described in detail below.
2.2.1. Characterization of Starting Compound Boc-NH-PEG5000-OH
Prior to the synthesis, the commercially obtained Boc-protected PEG was characterized by 1H NMR by the addition of trichloroacetyl isocyanate (TAIC) to determine the Mn of the polymer. To this end, around 5 mg of the compound was dissolved in 1 mL of deuterated chloroform, and a few drops of TAIC were added. After 15 min incubation at RT, an 1H NMR spectrum was recorded. TAIC reacts with the free hydroxyl group of the Boc-NH-PEG5000-OH, thereby causing a downfield shift to δ = 4.4 ppm of the peak of the methylene group adjacent to the hydroxyl group.25 By comparing the integrals of the peaks at δ = 4.4 ppm and δ = 3.8–3.5 ppm (PEG), the Mn of the polymer was calculated, and this value was used for further calculations in the synthesis steps. Furthermore, the degree of bocylation was determined by comparing the integrals of the peaks at δ = 1.4 ppm (Boc group) and δ = 3.8–3.5 ppm (PEG).
2.2.2. Synthesis of (Boc-NH-PEG5000)2-ABCPA
Boc-NH-PEG5000-OH (1.0 g, 0.20 mmol) was dissolved in 5.0 mL of dry DCM together with 28.0 mg (0.10 mmol) of 4,4′-azobis(4-cyanopentanoic acid) (ABCPA) and 8.8 mg (0.03 mmol) of DPTS. The mixture was cooled on ice, and 5.0 mL of 60 mM (0.3 mmol) of N,N′-dicyclohexylcarbodiimide (DCC) in DCM was added dropwise. The reaction mixture was allowed to react for 24 h at RT under a N2 atmosphere. Afterward, the mixture was filtered through a 0.2 μm nylon filter to remove the formed dicyclohexylurea (DCU), and the product was further purified by precipitation in cold diethyl ether (three times). 1H NMR (CDCl3): δ (ppm) 4.25 (4H, PEG methylene next to ABCPA), 3.8–3.5 (910H, PEG), 2.2–2.6 (8H, ABCPA methylene), 1.71 + 1.64 (6H, ABCPA methyl), 1.4 (18H, Boc).
The amount of unreacted PEG was determined by 1H NMR by the addition of TAIC to the sample as described in section 2.2.1. The amount of monosubstituted Boc-NH-PEG-ABCPA present in the final product was calculated by taking the relative peak area (%) of the GPC peak at 15 min (corresponding to the 5 kDa PEG product) minus the percentage unreacted PEG as determined by 1H NMR spectroscopy.
2.2.3. Deprotection of (Boc-NH-PEG5000)2-ABCPA
The amine groups of the macroinitiator were deprotected with an excess of trifluoroacetic acid (TFA). Therefore, 950 mg (0.19 mmol) of (Boc-NH-PEG5000)2-ABCPA was dissolved in 10 mL of DCM/TFA (1/1, v/v) and stirred overnight at RT. Afterward, the solvent was partially evaporated under reduced pressure, and the product was precipitated in cold diethyl ether (three times). 1H NMR (CDCl3): δ (ppm) 4.25 (4H, PEG methylene next to ABCPA), 3.8–3.5 (910H, PEG), 2.2–2.6 (8H, ABCPA methylene), 1.71 + 1.64 (6H, ABCPA methyl).
2.2.4. Synthesis of (Br-C(CH3)2-CO-NH-PEG5000)2-ABCPA
Functionalization of the macroinitiator with an ATRP initiator was based on a previously described method by de Graaf et al. with some modifications.26 First, 900 mg (0.18 mmol) of (TFA.NH2-PEG5000)2-ABCPA was dissolved in 9 mL of THF, and 100 μL (0.72 mmol) of TEA was added to the solution. Afterward, 89 μL (0.72 mmol) of α-bromoisobutyryl bromide was added, and the mixture was allowed to react overnight at RT under N2 atmosphere. The reaction mixture was filtrated to remove the formed bromide salt, and subsequently the filtrate was concentrated and dissolved in water. The final product, (Br-C(CH3)2-CO-NH-PEG5000)2-ABCPA, was further purified by extensive dialysis against water for 48 h at 4 °C (Mw cutoff: 3500 Da) and subsequently freeze-dried. 1H NMR (CDCl3): δ (ppm) 4.25 (4H, PEG methylene next to ABCPA), 3.8–3.5 (910H, PEG), 2.2–2.6 (8H, ABCPA methylene), 1.71 + 1.64 (6H, ABCPA methyl) 1.94 (12H, methyl bromoisobutyryl).
2.3. Synthesis of ABC Triblock Polymers
The synthesis of ABC triblock polymers followed a two-step procedure, in which the first step involved atom transfer radical polymerization (ATRP) followed by free radical polymerization (FRP).
2.3.1. Synthesis of ABBA Polymers by Atom Transfer Radical Polymerization (ATRP)
In a typical procedure, 1 equiv of the PEG macroinitiator, 290 equiv of N-isopropylacrylamide (NIPAM) or 278 equiv of 2-hydroxyethyl acrylate (HEA), and 8.2 equiv of CuBr were dissolved in water in an airtight screw-cap glass vial. The concentration of the monomer was adjusted to 90 mg/mL. The reaction mixture was flushed with nitrogen for 15 min at RT and subsequently another 15 min on ice. The reaction was started by adding 18 equiv of tris[2-(dimethylamino)ethyl]amine (Me6TREN), which changed the color of the mixture immediately from colorless to blue/green. The polymerization reaction was carried out for 3 h on ice. Next, the polymer solution was transferred to a dialysis cassette and dialyzed against water for 48 h at 4 °C (Mw cutoff: 10 kDa), while changing the dialysate frequently. Finally, the resulting ABBA polymer was recovered by freeze-drying with a typical yield of 80–90% and analyzed by 1H NMR spectroscopy and GPC.
2.3.2. Synthesis of ABC Polymers by Free Radical Polymerization (FRP)
In the next step, the ABBA polymer and a defined amount of the second monomer (NIPAM, DMAEMA, or HEA) for the aimed block length were dissolved in dry DMF in an airtight Schlenk flask. The concentration of the monomer was adjusted to 300 mg/mL. At least three freeze–pump–thaw cycles were applied to degas the solution, after which the reaction was placed in an oil bath at 70 °C and stirred for 24 h under a N2 atmosphere. The polymer solution was transferred to a dialysis cassette and dialyzed against water for 48 h at 4 °C (Mw cutoff: 10 kDa). The final ABC polymer was recovered by freeze-drying with a typical yield of 65–70%. All polymer products were analyzed by 1H NMR spectroscopy and GPC, and the cloud point was determined for thermosensitive polymers.
2.4. 1H NMR Spectroscopy
The macroinitiator and polymers were characterized with 1H NMR spectroscopy on an Agilent 400 MR-NMR spectrometer (Agilent Technologies, Santa Clara, CA). Chemical shifts were referred to the residual solvent peak (δ = 7.26 ppm for CDCl3 and δ = 4.80 ppm for D2O). All data analysis was performed using MestReNova Software version 10.0.1–14719.
2.5. Gel Permeation Chromatography (GPC)
The obtained products were further characterized by GPC using a Waters Alliance System (Waters Corporation, Milford, MA) equipped with a refractive index (RI) detector and a PLgel 5 μM MIXED-D column (Polymer Laboratories) using DMF containing 10 mM LiCl as eluent. The column temperature was set to 65 °C and the flow rate to 1.0 mL/min. Calibration was performed using poly(ethylene glycol) standards of narrow and defined molecular weights. All data analysis was performed using Empower 3 Software 2010.
2.6. Quantification of Primary Amines (TNBSA Assay)
Free primary amine groups of the synthesized products of the PEG macroinitiator were quantified by a TNBSA assay. Samples were dissolved in sodium bicarbonate buffer (0.1 M, pH 8.5) with a concentration of 1–4 mg/mL. Glycine standards were prepared for calibration at concentrations ranging from 0 to 0.15 mM in the same buffer. Next, 250 μL of a freshly prepared 0.01% TNBSA solution in buffer was added to 500 μL of a sample or standard solution, and the solutions were incubated at 37 °C for 2 h. Afterward, 250 μL of 10% SDS and 125 μL of 1 M HCl were added to each solution, and the absorbance at 335 nm was measured in triplicate using a BMG Spectrostar nano wellplate reader. Data analysis was performed using MARS Data analysis software version 2.22 (BMG Labtech, Ortenberg, Germany).
2.7. Determination of Cloud Point (CP) of Thermosensitive Polymers
The CP of thermosensitive polymers was determined by light scattering using a Jasco FP-8300 spectrofluorometer (JASCO Benelux BV., De Meern, The Netherlands). The polymers were dissolved overnight at a concentration of 2 mg/mL in 20 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer, pH 7.4. Next, 1 mL of the polymer solution was transferred to a clean glass cuvette, and the scattering intensity was measured at 550 nm while increasing the temperature from 10 to 60 °C with a heating rate of 1 °C/min. The cloud point was taken as the onset point of increasing scattering intensity.
2.8. Micelle Formation and Characterization by Dynamic Light Scattering (DLS)
Polymeric micelles were formed via the quick heating procedure as described in the literature.27 In brief, the polymers were dissolved overnight at a concentration of 1 mg/mL in 20 mM HEPES buffer, pH 7.4. Subsequently, 1 mL of the polymer solution was transferred to a clean glass vial and characterized by dynamic light scattering (DLS) at 25 °C. To form micelles, the polymer solution was rapidly heated to 50 °C, and after 10 min of incubation the micelles were characterized by DLS at 37 °C. Afterward, the mixtures were cooled down again to 25 °C, and the same procedure to form micelles was repeated. The DLS equipment consisted of a Malvern CGS-3 multiangle goniometer with a JDS Uniphase He–Ne laser (λ = 632.8 nm, 22 mW output power) and a temperature controller (Malvern Instruments Ltd., Malvern, UK). Scattering of the micellar solutions was measured under an angle of 90°.
3. Results and Discussion
3.1. Heterofunctional PEG Macroinitiator Synthesis and Characterization
The PEG macroinitiator was synthesized following a three-step synthesis route, starting with a Boc-protected NH2-PEG-OH (Scheme 2). The first step consisted of a DCC-mediated ABCPA coupling to the free OH group, followed by deprotection of the Boc-protected amine groups and finally end-group functionalization with an ATRP initiator. Boc-NH-PEG5000-OH was commercially obtained, and as expected, no free primary amines could be detected with a TNBSA assay (Table 1). In addition, 1H NMR analysis displayed the characteristic peaks of the Boc-methyl protons at 1.4 ppm, and after addition of TAIC, a number-average molecular weight (Mn) of 5.01 kDa was determined in agreement with the Mn obtained by GPC of 5.5 kDa. In addition, GPC analysis showed a small impurity (2.5%) of high molecular weight PEG in the starting compound.
Scheme 2. Three-Step Synthesis Route of the Heterofunctional PEG Macroinitiator Yielding Both an Atom Transfer Radical Polymerization (ATRP) Initiator and a Free Radical Polymerization (FRP) Initiator.

Table 1. Characteristics of the Starting Compound (Boc-NH-PEG5000-OH), Intermediate Products ((Boc-NH-PEG5000)2-ABCPA and (NH2-PEG5000)2-ABCPA)), and Final PEG Macroinitiator ((Br-C(CH3)2-CO-NH-PEG5000)2-ABCPA)a.
| product | yield (%) | Mn (kDa) | PDI | free amines (%) |
|---|---|---|---|---|
| Boc-NH-PEG5000 | 5.5 | 1.05 | 0 | |
| (Boc-NH-PEG5000)2-ABCPA | 95 | 11.8 | 1.05 | 0 |
| (NH2-PEG5000)2-ABCPA | 95 | 11.8 | 1.05 | 97 |
| (Br-C(CH3)2-CO-NH-PEG5000)2-ABCPA | 80 | 12.2 | 1.07 | 5 |
The molecular weight (Mn) and the polydispersity index (PDI) were determined with GPC. The amount of free amines was determined with a TNBSA assay.
The synthesis of the heterofunctional PEG macroinitiator started with the coupling of the azoinitiator to the amine-protected PEG by a DCC-mediated esterification between the carboxylic acid groups of ABCPA and the hydroxyl groups of PEG (Scheme 2, step 1). 1H NMR analysis showed the appearance of characteristic peaks corresponding to the CH2 and CH3 groups of ABCPA (Figure S1), indicating successful coupling of ABCPA to PEG. GPC analysis showed a major peak at 10 kDa corresponding to the molecular weight of the desired PEG macroinitiator (Figure 1). Overall, 84% of the final product was the desired bifunctional initiator, which corresponds to the typical yield of DCC-mediated esterification reactions.24,28,29 The peak at 5 kDa in the GPC chromatogram is a result of a bimodal molecular weight distribution for the obtained product, which can be ascribed to a mixture of bifunctionalized PEG2-ABCPA (10 kDa) and monofunctionalized PEG-ABCPA and/or unreacted PEG (5 kDa). The amount of unreacted PEG was determined by 1H NMR by the addition of TAIC to the sample. 1H NMR analysis indicated that 11% unreacted Boc-NH-PEG5000-OH was present. GPC showed the presence of 16% of the 5 kDa compound, meaning that the final product contained 5% monofunctionalized Boc-NH-PEG5000-ABCPA. Separation of the 10 and 5 kDa PEG products by commonly used separation techniques, which are based on differences in molecular weight, is rather difficult. For example, to achieve preparative chromatography conditions, a large column would be needed to prevent overloading of the column, resulting in a low-resolution separation. Such large columns would be both impractical to work with and very expensive. In addition, although a dialysis membrane with a 10 kDa Mn cutoff was used, traces of the 5 kDa PEG products were still visible in the GPC chromatogram, indicating that removal of these molecules via dialysis was not efficient.
Figure 1.

GPC chromatograms (RI detection) of the starting compound (Boc-NH-PEG5000-OH), intermediate products ((Boc-NH-PEG5000)2-ABCPA, (NH2-PEG5000)2-ABCPA)), and final PEG macroinitiator ((Br-C(CH3)2-CO-NH-PEG5000)2-ABCPA).
The next step involved the removal of the Boc group with TFA (Scheme 2, step 2), and 1H NMR analysis showed the complete disappearance of the proton signal of the Boc group at δ = 1.4 ppm (Figure S2). In addition, almost full recovery of the free primary amines was found with the TNBSA assay (Table 1), and no changes in the GPC chromatogram were observed. The final step of the synthesis was the functionalization of (NH2-PEG5000)2-ABCPA with α-bromoisobutyryl bromide to obtain a bifunctional ATRP initiator (Scheme 2, step 3). Results from both the 1H NMR analysis and the TNBSA assay indicated a nearly complete Br-functionalization on the PEG macroinitiator. The 1H NMR spectrum showed a sharp peak at δ = 1.94 ppm, which corresponds to the protons of the methyl groups of bromoisobutyryl (Figure S3). Comparing the integral of this peak with the integral of the peak at δ = 3.8–3.5 ppm (PEG) showed 98% Br-functionalization. In addition, results from the TNBSA assay showed the presence of 5% free primary amines, indicating that the final product contained 95% Br-functionalization.
3.2. ABC Triblock Copolymers Synthesis and Characterization
The first polymerization step involved the synthesis of the A block by ATRP, yielding an ABBA polymer. The azoinitiator remains intact during this polymerization step because ATRP reactions can be performed at low temperatures (reactions were performed on ice). After purification, the obtained ABBA polymer was used to synthesize the final block by free radical polymerization. The reaction mixture was heated to 70 °C, at which ABCPA decomposes and generates radicals to start polymerization of the final block. By employing this orthogonal synthesis route, linear ABC triblock copolymers can be polymerized in a two-step process without the requirement of protection of initiating sites. Because of the combination of two types of polymerization, a wide range of monomers can be included, yielding different kinds of polymer compositions. To illustrate the versatility of the heterofunctional PEG macroinitiator, ABC triblock copolymers with different block compositions were synthesized (Table 2). The polymer names are abbreviated according to the block composition, in which the first letter corresponds to the monomer used for the A block, the second letter to the B block, and the third letter to the C block. Three types of monomers with different properties were used for this study: DMAEMA, NIPAM, and HEA. All polymers were characterized by 1H NMR (Figures S4–S8) and GPC to determine the Mn of the polymers and the average lengths of each functional block.
Table 2. Composition of Three Different ABC Triblock Copolymers Synthesized Using the Heterofunctional PEG Macroinitiatora.
| polymer
block composition |
|||
|---|---|---|---|
| name | A | B | C |
| NPD | NIPAM (thermosensitive) | PEG (hydrophilic) | DMAEMA (pH-sensitive) |
| NPH | NIPAM (thermosensitive) | PEG (hydrophilic) | HEA (hydrophilic) |
| HPD | HEA (hydrophilic) | PEG (hydrophilic) | DMAEMA (pH-sensitive) |
N-Isopropylacrylamide (NIPAM) was used to introduce a thermosensitive block in the polymer, 2-(dimethylamino)ethyl methacrylate (DMAEMA) was used to introduce pH-sensitive properties, and 2-hydroxyethyl acrylate (HEA) was used as noncharged hydrophilic monomer.
In the first step, NIPAM was polymerized by ATRP, and the corresponding ABBA polymers (PNIPAM-PEG-PEG-PNIPAM, abbreviated by NPPN) were obtained with a typical monomer conversion of 90% and a typical yield of 85% (Table 3). In addition, GPC analysis confirmed the formation of the ABBA polymer since the peak of the PEG macroinitiator (14 min, 10 kDa) was almost completely shifted to a lower retention time, corresponding to a higher Mn of the polymer product (Figure 2). Small traces of the PEG macroinitiator were visible in the GPC chromatogram, which corresponds to the small fraction of nonfunctionalized PEG macroinitiator. The relative peak area (%) of the GPC peaks at 14 and 15 min (corresponding to the unreacted 10 and 5 kDa PEG macroinitiator, respectively) was 4% for each. In conclusion, the end product contained 92% of the desired ABBA polymer. The Mn obtained by GPC was higher compared to the Mn obtained by 1H NMR analysis, which has been reported in the literature before for NIPAM-based polymers.33 Besides NIPAM, HEA was also polymerized by ATRP to form ABBA polymers (PHEA-PEG-PEG-PHEA, abbreviated by HPPH), and similar results were obtained, with the exception that the monomer yield was slightly lower (65%). After polymerization of the A block, the polymers were extensively dialyzed to remove the copper catalyst to prevent further ATRP upon addition of the second type of monomer.
Table 3. Characteristics of Various ABBA Block Copolymers Synthesized by Atom Transfer Radical Polymerization Using the Heterofunctional PEG Macroinitiatora.
| name | feed PEG:monomer ratio | Mn A blocks (kDa)b | Mn B blocks (kDa)b | total Mn (kDa)b | total Mn (kDa)c | PDIc | yield (%) |
|---|---|---|---|---|---|---|---|
| NPPN_1 | 1:566 | 54 | 10 | 64 | 100 | 1.8 | 81 |
| NPPN_2 | 1:283 | 30 | 10 | 40 | 64 | 1.8 | 90 |
| HPPH | 1:276 | 20 | 10 | 30 | 44 | 2.1 | 65 |
Either NIPAM was used to obtain PNIPAM-PEG-PEG-PNIPAM polymers (abbreviated by NPPN) or HEA to obtain PHEA-PEG-PEG-PHEA polymers (abbreviated by HPPH).
Determined by 1H NMR.
Determined by GPC.
Figure 2.

GPC chromatograms (RI detection) of the ABBA polymer (NPPN_1, 100 kDa) after atom transfer radical polymerization and the corresponding ABC polymer (NPD_1, 83 kDa) after free radical polymerization.
In the second polymerization step, either DMAEMA or HEA was polymerized by FRP to obtain the final ABC triblock copolymers (Table 4). One of the advantages includes the widespread use of FRP and thus the possibility to choose a wide range of monomers suitable for polymerization. The PDI of the ABC triblock copolymers was between 1.8 and 2.5, which is similar to other triblock polymers reported in the literature.34,35 Typically, a monomer conversion of 60–70% was determined by 1H NMR, which can be expected for this type of polymerization.36,37 The GPC chromatogram of the final ABC triblock copolymer was compared with the chromatogram of the ABBA polymer, which acted as initiator for the final polymerization step (Figure 2). The azoinitiator decomposes at 70 °C and initiates the polymerization of the second monomer. The GPC chromatogram of the ABC polymer indicates that, as expected, the overall molecular weight slightly shifted to lower values, indicating the breaking of the starting ABBA polymer resulting into two ABC polymer chains by free radical polymerization. As a control, the ABBA polymer was heated for 24 h at 70 °C in DMF, which represents similar conditions to those used during free radical polymerization, but without the presence of the second monomer. GPC analysis showed indeed a shift in the chromatogram to the lower Mn values of the ABBA polymer after heating corresponding to half the Mn of the ABBA polymer (Figure S9). These results support the fact that the azoinitiator remains intact under ATRP reaction conditions and can be used afterward as an initiator for radical polymerization.
Table 4. Characteristics of Various ABC Triblock Copolymers Synthesized by Radical Polymerization Using the Heterofunctional PEG Macroinitiatora.
| name | feed PEG:monomer ratio (A block) | feed PEG:monomer ratio (C block) | Mn A block (kDa)b | Mn B block (kDa)b | Mn C block (kDa)b | total Mn (kDa)b | total Mn (kDa)c | PDIc | cloud point (°C)d |
|---|---|---|---|---|---|---|---|---|---|
| NPD_1 | 1:566 | 1:789 | 27 | 5 | 43 | 75 | 83 | 1.8 | 34 |
| NPD_2 | 1:283 | 1:1011 | 15 | 5 | 64 | 84 | 62 | 1.8 | 34 |
| NPH | 1:283 | 1:371 | 15 | 5 | 18 | 38 | 28 | 2.1 | 33 |
| HPD | 1:276 | 1:711 | 10 | 5 | 12 | 27 | 26 | 2.5 | n.a.e |
The polymer names are abbreviated according to the block composition, in which the first letter corresponds to the monomer used for the A block, the second letter to the B block, and the third letter to the C block (N = NIPAM, P = PEG, D = DMAEMA, H = HEA).
Determined by 1H NMR.
Determined by GPC.
Determined by light scattering at 550 nm.
n.a. = not applicable.
The tailorable design of the polymers using this PEG macroinitiator was further demonstrated by the synthesis of two different block lengths of NIPAM and DMAEMA (NPD_1 and NPD_2, respectively, Table 4). No significant difference in PDI was observed for these two polymer batches. In addition, PDI values were comparable with those observed for the ABBA polymers. The Mn of NPD_1 obtained by GPC was higher than the Mn obtained by 1H NMR, as was also seen for the ABBA polymers. Interestingly, the other way around was observed for NPD_2, which is composed of a shorter A block (NIPAM) and a longer C block (DMAEMA). The polymer block composition influences the conformation of the polymer in solution (e.g., swollen or condensed state of the polymer chain), which in turn influences the flow rate of the polymer through the GPC column resulting in different retention times. Likely, the ratio between the block lengths of the hydrophilic part of the polymer chain compared to the hydrophobic part plays a major role in the observed hydrodynamic volume of the polymers. Indeed, a lower Mn value was observed by GPC compared to 1H NMR with increasing hydrophilic to hydrophobic ratio. In addition, the same trend was observed for ABC triblock copolymers consisting of a NIPAM and HEA block (Table 4) as well as for other ABC copolymers consisting of NIPAM and DMAEMA (Table S1). To further extend the scope of this synthesis methodology and demonstrate its versatility, HEA was polymerized either as A block or as C block. Well-defined ABC copolymers were obtained when HEA was polymerized in the first step by ATRP as well as by FRP in the final step. The hydroxyl group of HEA is attractive as a functionality in the polymer because they can undergo a wide variety of reactions.38−41 This offers the possibility for postmodification of hydroxylated polymers and thus the design of complex polymeric architectures.
Polymers containing NIPAM units are well-known to respond to changes in temperature, and therefore this monomer was introduced to obtain thermosensitive polymers. Typically, NIPAM-based polymers have a cloud point (CP) of 32 °C in water, which makes them very attractive building blocks for materials in biomedical and pharmaceutical applications. The CPs of the thermosensitive polymers synthesized in this study were 33–34 °C (Table 4 and Figure S10), as expected for these block copolymers.33,42 As a proof-of-principle, the self-assembly behavior of these thermosensitive polymers into well-defined macromolecular micellar structures was demonstrated by DLS (Table 5) in HEPES buffer pH 7.4. At 25 °C, below the CP of the polymers, no particles were detected, indicating complete solubilization of free polymers. However, heating of the polymer solutions above the CP resulted in micellar structures with a size between 73 and 117 nm and a low PDI (0.06–0.17). These results are similar to other thermosensitive polymeric micelles based on NIPAM reported in the literature.24,43,44 The different compositions and Mn’s of the polymer batches led to slight differences in size of the micelles, with the polymer with lowest Mn (NPH, 39 kDa), resulting in the smallest micellar structure (73 ± 7 nm). The reversibility of the thermosensitive behavior was shown by the complete disappearance of the micelles after cooling down the mixture to 25 °C. In addition, when the heating procedure was repeated, micelles with the same size and PDI were obtained as before cooling down. These data support that functional polymers with well-defined properties, such as self-assembly behavior upon a trigger-like change in temperature, can be synthesized with the presented PEG macroinitiator.
Table 5. Particle Size and Polydispersity Index (PDI) of Thermosensitive Micelles Measured by Dynamic Light Scattering (DLS) at 37 °C in 20 mM HEPES Buffer (pH 7.4)a.
| size (nm) |
PDI |
|||
|---|---|---|---|---|
| name | first time heating | second time heating | first time heating | second time heating |
| NPD_1 | 117 ± 6 | 113 ± 7 | 0.17 ± 0.03 | 0.17 ± 0.03 |
| NPD_2 | 101 ± 2 | 102 ± 3 | 0.11 ± 0.04 | 0.11 ± 0.03 |
| NPH | 73 ± 7 | 65 ± 5 | 0.06 ± 0.01 | 0.04 ± 0.02 |
After the micelles were formed (first time heating), the mixtures were cooled down again to 25 °C, and the same procedure to form micelles was repeated (second time heating).
4. Conclusion
This study shows the successful synthesis of a heterofunctional PEG macroinitiator containing both an azoinitiator and an ATRP initiator. Well-defined polymeric products are obtained using a combination of the living polymerization technique ATRP and classical free radical polymerization. The use of this novel heterofunctional PEG macroinitiator allows the synthesis of high-precision ABC copolymers, having a PEG midblock, in an elegant way. In addition, the combination of two types of polymerizations techniques enabled the incorporation of a wide range of monomers into the polymer design. Multiple functionalities were combined in a single polymer chain, including pH-sensitive and thermosensitive properties and functional sites for postmodification reactions. This synthesis approach can further contribute to the design of dual-stimuli responsive polymers with tailorable block compositions and polymer topologies. Different kinds of self-assembled morphologies, depending on specific block properties, make these linear ABC triblock copolymers highly attractive for applications in nanotechnology.
Acknowledgments
The Netherlands Organization for Scientific Research (NWO/VIDI 13457 and NWO/Aspasia 015.009.038) is acknowledged for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.7b01475.
Figures S1–S10 and Table S1 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lazzari M.; Arturo López-Quintela M. Block Copolymers as a Tool for Nanomaterial Fabrication. Adv. Mater. 2003, 15 (19), 1583–1594. 10.1002/adma.200300382. [DOI] [Google Scholar]
- Tai H.; Duvall C. L.; Hoffman A. S.; Stayton P. S.; Wang W. PH-Responsive Hyperbranched Copolymers from One-Pot RAFT Copolymerization. Macromol. Mater. Eng. 2012, 297 (12), 1175–1183. 10.1002/mame.201200227. [DOI] [Google Scholar]
- Liu F.; Urban M. W. Recent Advances and Challenges in Designing Stimuli-Responsive Polymers. Prog. Polym. Sci. 2010, 35, 3–23. 10.1016/j.progpolymsci.2009.10.002. [DOI] [Google Scholar]
- Alarcón C.; Pennadam S.; Alexander C. Stimuli Responsive Polymers for Biomedical Applications. Chem. Soc. Rev. 2005, 34 (3), 276–285. 10.1039/B406727D. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Wang Z.-G. Morphology of ABC Triblock Copolymers. Macromolecules 1995, 28 (21), 7215–7223. 10.1021/ma00125a026. [DOI] [Google Scholar]
- Hadjichristidis N.; Iatrou H.; Pitsikalis M.; Pispas S.; Avgeropoulos A. Linear and Non-Linear Triblock Terpolymers. Synthesis, Self-Assembly in Selective Solvents and in Bulk. Prog. Polym. Sci. 2005, 30 (7), 725–782. 10.1016/j.progpolymsci.2005.04.001. [DOI] [Google Scholar]
- Ruzette A.-V.; Leibler L. Block Copolymers in Tomorrow’s Plastics. Nat. Mater. 2005, 4, 19–31. 10.1038/nmat1295. [DOI] [PubMed] [Google Scholar]
- Li G.; Yates M. Z.; Johnston K. P.; Lim K. T.; Webber S. E. Trifunctional Ambidextrous Surfactants for Latexes in Supercritical CO2 and Water. Macromolecules 2000, 33, 1606–1612. 10.1021/ma9913212. [DOI] [Google Scholar]
- Fujii S.; Cai Y.; Weaver J. V. M.; Armes S. P. Syntheses of Shell Cross-Linked Micelles Using Acidic ABC Triblock Copolymers and Their Application as pH-Responsive Particulate Emulsifiers. J. Am. Chem. Soc. 2005, 127 (20), 7304–7305. 10.1021/ja050049a. [DOI] [PubMed] [Google Scholar]
- Weinman C. J.; Finlay J. A.; Park D.; Paik M. Y.; Krishnan S.; Sundaram H. S.; Dimitriou M.; Sohn K. E.; Callow M. E.; Callow J. A.; Handlin D. L.; Willis C. L.; Kramer E. J.; Ober C. K. ABC Triblock Surface Active Block Copolymer with Grafted Ethoxylated Fluoroalkyl Amphiphilic Side Chains for Marine Antifouling/ Fouling-Release Applications. Langmuir 2009, 25 (20), 12266–12274. 10.1021/la901654q. [DOI] [PubMed] [Google Scholar]
- Knoll K.; Nießner N. Styrolux+ and Styroflex’ - From Transparent High Impact Polystyrene to New Thermoplastic Elastomers. Macromol. Symp. 1998, 132 (1), 231–243. 10.1002/masy.19981320122. [DOI] [Google Scholar]
- Ruiz R.; Kang H.; Detcheverry F. A.; Dobisz E.; Kercher D. S.; Albrecht T. R.; de Pablo J. J.; Nealey P. F. Density Multiplication and Improved Lithography by Directed Block Copolymer Assembly. Science 2008, 321 (5891), 936–939. 10.1126/science.1157626. [DOI] [PubMed] [Google Scholar]
- Stoenescu R.; Graff A.; Meier W. Asymmetric ABC-Triblock Copolymer Membranes Induce a Directed Insertion of Membrane Proteins. Macromol. Biosci. 2004, 4 (10), 930–935. 10.1002/mabi.200400065. [DOI] [PubMed] [Google Scholar]
- Hoare T. R.; Kohane D. S. Hydrogels in Drug Delivery: Progress and Challenges. Polymer 2008, 49 (8), 1993–2007. 10.1016/j.polymer.2008.01.027. [DOI] [Google Scholar]
- Otsuka H.; Nagasaki Y.; Kataoka K. Self-Assembly of Poly(ethylene Glycol)-Based Block Copolymers for Biomedical Applications. Curr. Opin. Colloid Interface Sci. 2001, 6 (1), 3–10. 10.1016/S1359-0294(00)00082-0. [DOI] [Google Scholar]
- Adams M. L.; Lavasanifar A.; Kwon G. S. Amphiphilic Block Copolymers for Drug Delivery. J. Pharm. Sci. 2003, 92 (7), 1343–1355. 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- Peracchia M. T.; Harnisch S.; Pinto-Alphandary H.; Gulik A.; Dedieu J. C.; Desmaële D.; d’Angelo J.; Müller R. H.; Couvreur P. Visualization of in Vitro Protein-Rejecting Properties of PEGylated Stealth? Polycyanoacrylate Nanoparticles. Biomaterials 1999, 20 (14), 1269–1275. 10.1016/S0142-9612(99)00021-6. [DOI] [PubMed] [Google Scholar]
- Bazile D.; Prud’homme C.; Bassoullet M.; Marlard M.; Spenlehauer G.; Veillard M. Stealth Me.PEG-PLA Nanoparticles Avoid Uptake by the Mononuclear Phagocytes System. J. Pharm. Sci. 1995, 84 (4), 493–498. 10.1002/jps.2600840420. [DOI] [PubMed] [Google Scholar]
- Goldmann A. S.; Glassner M.; Inglis A. J.; Barner-Kowollik C. Post-Functionalization of Polymers via Orthogonal Ligation Chemistry. Macromol. Rapid Commun. 2013, 34 (10), 810–849. 10.1002/marc.201300017. [DOI] [PubMed] [Google Scholar]
- Bates F. S.; Hillmyer M. A.; Lodge T. P.; Bates C. M.; Delaney K. T.; Fredrickson G. H. Multiblock Polymers: Panacea or Pandora’s Box?. Science 2012, 336 (6080), 434–440. 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- Bernaerts K. V.; Du Prez F. E. Dual/heterofunctional Initiators for the Combination of Mechanistically Distinct Polymerization Techniques. Prog. Polym. Sci. 2006, 31 (8), 671–722. 10.1016/j.progpolymsci.2006.08.007. [DOI] [Google Scholar]
- Davis K. A.; Matyjaszewski K. ABC Triblock Copolymers Prepared Using Atom Transfer Radical Polymerization Techniques. Macromolecules 2001, 34 (7), 2101–2107. 10.1021/ma002050u. [DOI] [Google Scholar]
- Huang Y.; Yong P.; Chen Y.; Gao Y.; Xu W.; Lv Y.; Yang L.; Reis R. L.; Pirraco R. P.; Chen J.; Kavallaris M.; McCarroll J.; Luzinov I.; Minko S. Micellization and Gelatinization in Aqueous Media of pH- and Thermo-Responsive Amphiphilic ABC (PMMA 82 -B-PDMAEMA 150 -B-PNIPAM 65) Triblock Copolymer Synthesized by Consecutive RAFT Polymerization. RSC Adv. 2017, 7 (46), 28711–28722. 10.1039/C7RA04351A. [DOI] [Google Scholar]
- Neradovic D.; Van Nostrum C. F.; Hennink W. E. Thermoresponsive Polymeric Micelles with Controlled Instability Based on Hydrolytically Sensitive N-Isopropylacrylamide Copolymers. Macromolecules 2001, 34 (22), 7589–7591. 10.1021/ma011198q. [DOI] [Google Scholar]
- Loccufier J.; Van Bos M.; Schacht E. Convenient Method for the Analysis of Primary and Secondary Hydroxyl End Groups in Polyethers. Polym. Bull. 1991, 27 (2), 201–204. 10.1007/BF00296031. [DOI] [Google Scholar]
- De Graaf A. J.; Azevedo Próspero Dos Santos I. I.; Pieters E. H. E.; Rijkers D. T. S.; Van Nostrum C. F.; Vermonden T.; Kok R. J.; Hennink W. E.; Mastrobattista E. A Micelle-Shedding Thermosensitive Hydrogel as Sustained Release Formulation. J. Controlled Release 2012, 162 (3), 582–590. 10.1016/j.jconrel.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Soga O.; Van Nostrum C. F.; Ramzi A.; Visser T.; Soulimani F.; Frederik P. M.; Bomans P. H. H.; Hennink W. E. Physicochemical Characterization of Degradable Thermosensitive Polymeric Micelles. Langmuir 2004, 20 (21), 9388–9395. 10.1021/la048354h. [DOI] [PubMed] [Google Scholar]
- Novo L.; Mastrobattista E.; van Nostrum C. F.; Hennink W. E. Targeted Decationized Polyplexes for Cell Specific Gene Delivery. Bioconjugate Chem. 2014, 25, 802–812. 10.1021/bc500074a. [DOI] [PubMed] [Google Scholar]
- Talelli M.; Rijcken C. J. F.; Oliveira S.; Van Der Meel R.; Van Bergen En Henegouwen P. M. P.; Lammers T.; Van Nostrum C. F.; Storm G.; Hennink W. E. Nanobody - Shell Functionalized Thermosensitive Core-Crosslinked Polymeric Micelles for Active Drug Targeting. J. Controlled Release 2011, 153 (1), 93–102. 10.1016/j.jconrel.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Lima V.; Schoenmakers P. J. Robust Isocratic Liquid Chromatographic Separation of Functional Poly(methyl Methacrylate). J. Chromatogr. A 2003, 1018 (1), 19–27. 10.1016/j.chroma.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Schoenmakers P. J.; Van Dongen J. L. J.; Lou X.; Lima V.; Brokken-Zijp J. Mass Spectrometric Characterization of Functional Poly(methyl Methacrylate) in Combination with Critical Liquid Chromatography. Anal. Chem. 2003, 75 (20), 5517–5524. 10.1021/ac034556r. [DOI] [PubMed] [Google Scholar]
- Berek D. Liquid Chromatography of Macromolecules at the Point of Exclusion - Adsorption Transition. Principle, Experimental Procedures and Queries Concerning Feasibility of Method. Macromol. Symp. 1996, 110 (1), 33–56. 10.1002/masy.19961100104. [DOI] [Google Scholar]
- Boere K. W. M.; Soliman B. G.; Rijkers D. T. S.; Hennink W. E.; Vermonden T. Thermoresponsive Injectable Hydrogels Cross-Linked by Native Chemical Ligation. Macromolecules 2014, 47 (7), 2430–2438. 10.1021/ma5000927. [DOI] [Google Scholar]
- Boere K. W. M.; van den Dikkenberg J.; Gao Y.; Visser J.; Hennink W. E.; Vermonden T. Thermogelling and Chemoselectively Cross-Linked Hydrogels with Controlled Mechanical Properties and Degradation Behavior. Biomacromolecules 2015, 16 (9), 2840–2851. 10.1021/acs.biomac.5b00802. [DOI] [PubMed] [Google Scholar]
- Dubbini A.; Censi R.; Butini M. E.; Sabbieti M. G.; Agas D.; Vermonden T.; Di Martino P. Injectable Hyaluronic acid/PEG-p(HPMAm-Lac)-Based Hydrogels Dually Cross-Linked by Thermal Gelling and Michael Addition. Eur. Polym. J. 2015, 72, 423–437. 10.1016/j.eurpolymj.2015.07.036. [DOI] [Google Scholar]
- Tsai H. C.; Chang C. H.; Chiu Y. C.; Lin S. Y.; Lin C. P.; Hsiue G. H. In Vitro Evaluation of Hexagonal Polymeric Micelles in Macrophage Phagocytosis. Macromol. Rapid Commun. 2011, 32 (18), 1442–1446. 10.1002/marc.201100157. [DOI] [PubMed] [Google Scholar]
- Talelli M.; Morita K.; Rijcken C. J. F.; Aben R. W. M.; Lammers T.; Scheeren H. W.; Van Nostrum C. F.; Storm G.; Hennink W. E. Synthesis and Characterization of Biodegradable and Thermosensitive Polymeric Micelles with Covalently Bound Doxorubicin-Glucuronide Prodrug via Click Chemistry. Bioconjugate Chem. 2011, 22, 2519–2530. 10.1021/bc2003499. [DOI] [PubMed] [Google Scholar]
- Kakwere H.; Perrier S. Design of Complex Polymeric Architectures and Nanostructured Materials/hybrids by Living Radical Polymerization of Hydroxylated Monomers. Polym. Chem. 2011, 2 (2), 270–288. 10.1039/C0PY00160K. [DOI] [Google Scholar]
- Yusa S. I.; Sugahara M.; Endo T.; Morishima Y. Preparation and Characterization of a pH-Responsive Nanogel Based on a Photo-Cross-Linked Micelle Formed from Block Copolymers with Controlled Structure. Langmuir 2009, 25 (9), 5258–5265. 10.1021/la803878s. [DOI] [PubMed] [Google Scholar]
- Gierlich J.; Burley G. A.; Gramlich P. M. E.; Hammond D. M.; Carell T. Click Chemistry as a Reliable Method for the High-Density Postsynthetic Functionalization of Alkyne-Modified DNA. Org. Lett. 2006, 8 (17), 3639–3642. 10.1021/ol0610946. [DOI] [PubMed] [Google Scholar]
- Le Droumaguet B.; Velonia K. Click Chemistry: A Powerful Tool to Create Polymer-Based Macromolecular Chimeras. Macromol. Rapid Commun. 2008, 29 (12–13), 1073–1089. 10.1002/marc.200800155. [DOI] [Google Scholar]
- De Graaf A. J.; Mastrobattista E.; Vermonden T.; Van Nostrum C. F.; Rijkers D. T. S.; Liskamp R. M. J.; Hennink W. E. Thermosensitive Peptide-Hybrid ABC Block Copolymers Obtained by ATRP: Synthesis, Self-Assembly, and Enzymatic Degradation. Macromolecules 2012, 45 (2), 842–851. 10.1021/ma2024667. [DOI] [Google Scholar]
- Chen J.; Liu M.; Gong H.; Cui G.; Lü S.; Gao C.; Huang F.; Chen T.; Zhang X.; Liu Z. Synthesis of Linear Amphiphilic Tetrablock Quaterpolymers with Dual Stimulus Response through the Combination of ATRP and RAFT by a Click Chemistry Site Transformation Approach. Polym. Chem. 2013, 4, 1815–1825. 10.1039/c2py20946b. [DOI] [Google Scholar]
- Topp M. D. C.; Dijkstra P. J.; Talsma H.; Feijen J. Thermosensitive Micelle-Forming Block Copolymers of Poly(ethylene Glycol) and Poly(N-Isopropylacrylamide). Macromolecules 1997, 30, 8518–8520. 10.1021/ma9710803. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.