

Abstract

Anthanthrone and its derivatives are large polycyclic aromatic compounds (PACs) that pose a number of challenges for incorporation into the structure of soluble conjugated polymers. For the first time, this group of PACs was employed as the building blocks for the synthesis of copolymers (P1–P5) based on poly[(arylene ethynylene)-alt-(arylene vinylene)]s backbone (−Ph–C≡C–Anth–C≡C–Ph–CH=CH–Ph–CH=CH−)n. During the synthesis of P1–P5, different alkyloxy side chains were incorporated in order to tune the properties of the polymers. Of the copolymer series only P1 (containing anthanthrone and branched 2-ethylhexyloxy side chains on phenylenes), P2 and P3 (for which the anthanthrones containing carbonyl groups were converted to anthanthrene containing alkyloxy substituents) were soluble. The photophysical, electrochemical, electroluminescent and photovoltaic properties of P1–P3 are reported, compared and discussed with respect to the effects of side chains.
Introduction
Polycyclic aromatic compounds (PACs) have attracted considerable attention as materials with potential applications in supramolecular electronics1−3 and are becoming increasingly popular for optoelectronic applications.4−9 The extensive conjugated π-systems of PACs facilitate electron delocalization, while the rigid flat geometry enables them to stack in well-organized arrays for strong π-stacking interactions. The π-conjugated systems influence the tuning of the optoelectronic properties, while the flat geometry enables good light emitting properties and intermolecular charge transport.10−15 These combined features make PACs appealing building blocks for the preparation of semiconducting polymers. In spite of this, a comparatively low number of conjugated polymers incorporating large PACs building blocks have been reported so far.16−24 The low interest in PAC based polymers (especially for organic photovoltaic cells) is as a result of their relatively high band gap. Although the extension of the effective conjugation of the polymers can be used to overcome this drawback,24−26 it is often accompanied by a significant decrease in solubility, which may hinder their processing into thin films for photovoltaic applications. For example, large hexabenzocoronene PAC was incorporated in the main chain of conjugated polymers.27 These materials, however, were insoluble in common organic solvents and difficult to process. The synthesis of π-conjugated small molecules based on anthanthrone28 is relatively simple, but the preparation of their polymeric analogues is challenging. Its rigidity coupled with its large and flat π surface hinders good solubility in common organic solvents. Only very recently, the synthesis and characterization of the first series of soluble conjugated polymers based on anthanthrone derivative (anthanthrene)29 and 4,10-bis(thiophen-2-yl)anthanthrone (TANT) were reported.30
Herein we report for the first time the preparation of poly[(arylene ethynylene)-alt-(arylene vinylene)]s (PAE-AVs) from anthanthrone and anthanthrene building blocks, respectively, using the Horner-Wadsworth-Emmons (HWE) polycondensation reaction (Scheme 1).
Scheme 1. Reaction Pathway for the Synthesis of the PAE-AVs Polymers.

PAE-AVs are a class of polymeric materials that combine the intrinsic properties of poly(arylene ethynylene) (PAE) and poly(arylene vinylene) (PAV) into a single backbone with additional structure-specific properties.31 The general constitutional unit of the PAE-AV backbones in this study, (−Ph–C≡C–Anth–C≡C–Ph–CH=CH–Ph–CH=CH−)n, contains an anthanthrone or its analogue anthanthrene embedded between two alkyne moieties (Figure 1b). To the best of our knowledge, the use of anthanthrone and its derivative as building blocks for polymer synthesis via the PAE-AV backbone has never been reported. PAE-AV based on other structures such as anthracene,32−41 thiophene and phenylene core31,42,43 have been reported. The aim of the current study was to establish anthanthrone and its derivatives as suitable building blocks for the design of novel PAE-AV backbones with good solubility (through side chain incorporation and variation) and to study their suitability as emitting or absorber materials in organic light-emitting or photovoltaic (OPV) devices, respectively.
Figure 1.

(a) Monomers (M1–M3 and Ma–Mc) and (b) polymers (P1–P5) and a small molecule (SM).
Results and Discussion
Synthesis
The synthesis of new polymers is schematically displayed in Scheme 1. The synthesis of the dialdehydes Ma–Mc and the bisphosphonates M1–M3 follows well-established protocols.32,33,44,45 Starting both with the O-alkylation of hydroquinone, their preparation involves overall 6 and 3 reaction steps, respectively. The reaction sequence for the synthesis of the dialdehyde involves the dibromination of the O-alkylated hydroquinone; the transformation of one bromo substituent by formylation; the substitution of the remaining bromo substituent by trimethylsilylethynyl via palladium-catalyzed Sonogashira cross-coupling reaction; and the cleavage of the trimethylsilane group to yield an alkyne functionalization, which was reacted with the respective dibromo anthanthrone or anthanthrene derivative in the final Sonogashira cross coupling (Scheme 1a).
The chemical structure and purity of the synthesized materials were confirmed by NMR spectroscopy. The 1H NMR, 13C NMR and 31P NMR spectra of the bisphosphonate polycondensation comonomers, M1, M2, and M3, are depicted in the Supporting Information (Figures S1–S9). The NMR spectra of the dialdehyde polycondensation comonomers, Ma–Mc, are displayed in Figures 2 and S10–S14. For the polymers P1, P2, and P3, 1H NMR and FTIR spectroscopy were conducted (Figures 3 and S15–S20), while for SM, only 1H NMR was conducted (Figure S21).
Figure 2.

1H NMR spectra of Ma–Mc.
Figure 3.

1H NMR spectra of P1–P3.
The NMR spectra of the bisphosphonate polycondensation comonomers (M1–M3, Figures S1–S9) are in good agreement with literature.44−50 For Ma–Mc, the 1H signals (Figure 2 and Figures S10–S12) in the range 10.49–10.44 ppm are attributed to the CHO aldehyde protons while the aromatic protons of the anthanthrone/anthanthrene unit and the phenylene group appear in the range 8.94–7.09 ppm. The CH2 protons directly linked to the oxygen in the alkoxy side chain, in the phenyl group, as well as, in the anthanthrene unit, are visible in the 4.92–3.49 ppm region. The CH (ethylhexyloxy side chain) and CH2 (decyloxy side chain) protons following the CH2 groups directly linked to the oxygen of the alkoxy side chains are assigned to the signals in the range 2.36–1.68 ppm. All other CH2 protons present in the compounds show signals in the range 1.68–1.03 ppm, while the signals of all CH3 protons are clearly visible between 1.03 and 0.87 ppm. The 13C spectra of Ma and Mb have been schematically illustrated and explained as shown in Figures S13 and S14, respectively.
While the 1H NMR spectra of the polycondensation comonomers, bisphosphonates, and dialdehydes are well-resolved and show defined signals, the spectra of the HWE polycondensation products are characterized by a distinct signal broadening and low resolution. The depletion or absence of the characteristic signals for the reactive functionalities of the comonomers, CHO (∼10.5 ppm) and CH2–P (3.5 ppm), indicates the successful formation of polymeric species. The signals between 9.25 and 8.41 ppm in P1 are assigned to the protons of the anthanthrone core. Between 8.07 and 6.67 ppm arylene vinylene proton signals can be identified. The signals in the region 4.37–3.20 ppm and 2.49–0.37 ppm are the protons of the alkyloxy side chains (Figure 3). The presence of a residual aldehyde signal in the 1H NMR spectrum of P1 (∼10.5 ppm) suggests that the soluble fraction of P1 consists of low-molecular weight species with aldehyde end groups. Figure 3 shows the 1H NMR signals of P2 and P3. The broad NMR peaks occurring at 9.05–8.22 ppm are the anthanthrenylene protons and that at 7.65–6.92 ppm are due to the arylene vinylene protons. The peaks in the regions 4.61–3.28 ppm and 2.41–0.42 ppm are those of the protons of the alkoxy side chains. In contrast to P1 the 1H NMR spectra of P2 and P3 do not show any sign of a residual aldehyde signal.
Mc was applied in HWE polycondensation reactions with the bisphosphonate M1 and M3 to obtain the polymers P4 and P5, respectively. However, the materials obtained from this reaction showed no reasonable solubility in common solvents, which prevented further characterization. Despite P1, P4, and P5 possessing identical backbones based on anthanthrone units, the presence of only branched 2-ethylhexyloxy side chains apparently granted partial solubility for P1 in common organic solvents. The linear decyloxy side chains in P4 and P5 appear much less efficient in promoting solubility. The significantly better solubility of the polymers P2 and P3 can be apparently attributed to the presence of anthanthrene instead of anthanthrone building blocks in the polymer backbones. The repeating units of P2 and P3 are isomeric. Their structures only differ in the side chains, linear dodecyloxy and branched (3,7-dimethyloctyl)oxy, introduced by the different bisphosphonates, which were used in the respective polycondensation reaction. This was done in order to study the effect of various side chains on the polymer properties.
The characteristic FTIR signals associated with the molecular vibrations of P1, P2, and P3 occur at 3073–3064, 2962–2850, 2198–2180, 1659, 1603–1575, 1500, 1464, 1380, and 1203 cm–1 (Figures S18–S20). The bands between 3073–3064 cm–1 are attributed to the stretching vibrations of aromatic =C–H and unsaturated hydrocarbon (vinylene). Characteristic aliphatic −C–H stretching vibrations of all three polymers show high intensity signals at 2962–2850 cm–1, while the vibrational stretching of the ethynylene linker bond (−C≡C−) gives a weak signal at 2198–2180 cm–1 for all the three polymers. These FTIR spectral signals confirm a disubstituted ethynylene linker bond. The characteristic stretching vibration associated with the ketone group (−C=O) of P1 is observed at 1659 cm–1, and as expected, this ketone signal is completely absent in the spectra of P2 and P3. The signals around 1603–1575 cm–1 are produced by the vinylene linker bond stretching, while those observed at 1500 and 1203–1031 cm–1 are for the stretching vibrations of aromatic −C=C– and aromatic–aliphatic −C=C–O–C– bonds, respectively. The bands observed at 969 and 861–869 cm–1 are attributed to the vinylene double bonds.
Molecular Mass and Thermal Studies
Results obtained from SEC (THF as eluent, polystyrene standards) and thermogravimetric analyses (TGA) are provided in Table 1. The molecular masses of the polymers P1, P2, and P3 are relatively low. The difficulty in the preparation of polymers of high molecular masses that contain anthanthrone or anthanthrene building blocks, is apparent from the results obtained for polymers P1–P3. The low Mn value obtained for P1 can be attributed to the rapid precipitation from the reaction mixture during the polycondensation as a result of the poor solubility of the polymer. The thermogravimetric analysis (TGA) data indicates good thermal stability for all three polymers (P1, P2, and P3). Only a 5% weight loss was observed between 314 and 331 °C (Table 1 and Figure S22).
Table 1. Synthesis and TGA Data of the Copolymersa.
| material | T (°C) | yield (%) | Mwb (g/mol) | Đ | Td (°C) |
|---|---|---|---|---|---|
| P1 | 118 | 95 | 3570 | 2 | 325 |
| P2 | 118 | *95 | 11730 | 3.7 | 314 |
| P3 | 118 | *33 | 7820 | 2.7 | 331 |
| P3 | 111 | *73 | 11800 | 3.2 | – |
T is the reaction temperature, Mw is the weight-average molecular weight, Đ is the dispersity, and Td is the thermal decomposition temperature @ 5% weight loss).
Measured by SEC in tetrahydrofuran as the mobile phase, and polystyrene standards were used for calibration. * Determined for the soluble fraction.
Photophysical Properties
The photophysical properties of the soluble polymers P1, P2, and P3 and the anthanthrene small molecule (SM) (which is used as a model compound) were investigated by UV–vis absorption spectroscopy and photoluminescence spectroscopy, for both the solutions and thin films of the materials. The absorption spectra obtained for measurements taken with dilute solutions and thin films are displayed in Figures 4 and 5, respectively, and their characteristic data are summarized in Tables 2 and 3.
Figure 4.

UV–vis absorption spectra of P1 (black), P2 (red), and P3 (blue) copolymers, and SM (green) model compound measured in dilute toluene (solid curves) and DCB (dashed curves) solutions.
Figure 5.

UV–vis absorption spectra of P1 (black), P2 (red for thicker and magenta for thinner films), P3 (blue for thicker and cyan for thinner films), and SM (green) thin films prepared from toluene (solid) and DCB (dashed) solutions.
Table 2. Photophysical Properties of the Copolymers Measured in Dilute Toluene and DCB Solutions (λabsmax, Absorption Maximum; λPLmax, Emission Maximum; λPLexcmax, Maximum of the Excitation Spectrum; ηPL, Photoluminescence Quantum Yield; Primary Maxima in Bold).
| material | solvent | λabsmax (nm) | λPLmaxa (nm) | λPLexcmaxb (nm) | ηPL |
|---|---|---|---|---|---|
| P1 | toluene | 363sh, 429, 487 sh, 569 | 502, 612 | 349, 550 | – |
| P1 | DCB | 433, 607 | – | – | – |
| P2 | toluene | 333, 451, 478 | 519, 554 | 332, 447, 478 | 0.47c |
| P2 | DCB | 332, 492 (523) | 533, 589 | 337, 484, 530 | 0.54d |
| P3 | toluene | 334, 450, 479 | 520, 551 | 332, 450, 480 | 0.47c |
| P3 | DCB | 334, 455, 483 | 533 | 334, 451, 484 | 0.53c |
| SM | toluene | 327, 400, 447, 477 | 488, 518 | 334, 396, 447, 477 | 0.92c |
| SM | DCB | 332, 405, 450, 480 | 499 | 331, 405, 449, 480 | 0.95c |
Excitation wavelength at λabsmax;
Emission wavelength at λPLmax,
Fluorescein used as the standard.
Rhodamine 6G used as the standard.
Table 3. Photophysical Properties of the Copolymers Measured as Thin Films Prepared from Toluene or DCB (λabsmax, Absorption Maximum; λPLmax, Emission Maximum; λPLexcmax, Maximum of the Excitation Spectrum; d, Film Thickness; Primary Maxima in Bold).
| thin film | prepared from | d (nm) | λabsmax (nm) | λPLmaxa (nm) | λPLexcmaxb (nm) |
|---|---|---|---|---|---|
| P1 | DCB | 10 | 213, 304, 427, 604 | – | – |
| P2 | DCB | 46 | 212, 286, 557, 591 | 625 | 488, 531, 571 |
| P2 | DCB | 87 | 212, 286, 547, 581 | 625 | 487, 529, 570 |
| P2 | toluene | 52 | 213, 336, 488, 590 | 621 | 333, 457, 486, 523, 577 |
| P2 | toluene | 157 | 212, 336, 484, 584 | 623 | 336, 458, 484, 525, 589 |
| P3 | DCB | 47 | 213, 337, 483 | 609 | 335, 454, 485 |
| P3 | DCB | 70 | 213, 335, 479 | 604 | 331, 451, 482 |
| P3 | toluene | 30 | 214, 337, 484 | 605 | 335, 454, 485 |
| P3 | toluene | 111 | 213, 334, 462 | 611 | 334, 456, 485 |
| SM | DCB | 43 | 206, 231, 258, 342, 423, 455 sh, 525 sh | 567 | 339, 421 |
Excitation wavelength at λabsmax;
Emission wavelength at λPLmax
The absorption of P1 covers the whole visible spectral region and its spectra exhibited broad absorption bands in both solutions and as thin films. The long wavelength maxima are located at 429 and 569 nm for measurements done in toluene and red-shifted to 433 and 607 nm in DCB solution spectra. In thin film spectra, the maxima were slightly blue-shifted (427, 604 nm) relative to the DCB solution spectra. The long-wavelength band can be assigned to the π–π* transition of the conjugated polymer backbone and the band at higher energy to the n−π* transition from the carbonyl of the anthanthrone moiety, which is also influenced by conjugation. It is noteworthy that only very thin films of good quality could be prepared by spin-coating due to the limited solubility of P1.
The absorption spectra of the P2 and P3 copolymers with anthanthrene unit significantly differ from the spectra of P1. The corresponding SM model compound for P2 and P3 exhibits well resolved absorption spectrum in toluene and DCB solutions that are typical of fused conjugated systems,51−54 with maxima at 327, 400, 447, and 477 nm in toluene and slightly red-shifted maxima at 332, 405, 450, and 480 nm in DCB. The absorption spectrum of SM thin film is not so well resolved reflecting the intermolecular interactions in solid state. The absorption maxima of the thin film were red-shifted and an absorption extension of up to 550 nm was observed when compared with the spectra obtained for solution. The P2 and P3 exhibit broad-band absorption in solution and thin films. The absorption spectra in their toluene solutions are less resolved but similar for both P2 and P3 copolymers, with the main maximum occurring at 478 and 479 nm, respectively. P2 exhibited more extended shoulder at long wavelengths. Qualitatively different absorption spectrum was observed for P2 when measured in DCB solutions. The P2 absorption spectrum in DCB solution (see Figure 4) shows broad structureless absorption in the visible region from 400 to 600 nm, which is red-shifted compared to its toluene solution spectrum and also the P3 and SM spectra. Broad red-shifted band indicates more planar backbone configuration and also aggregate formation in DCB, whereas the P3 DCB spectra are of similar shape to its absorption spectra measured in toluene with slightly red-shifted maxima. Similar absorption behavior as in P2 toluene solution was observed in its tetrahydrafuran solution, and on the other hand, the aggregation was also observed in P2 chloroform solution. The P3 absorption spectrum is also red-shifted compared to the SM spectrum or its spectrum in toluene, but the red shift is not so pronounced. Two long-wavelength maxima at 455 and 483 nm can be well resolved in its DCB absorption spectra similarly to those of toluene solution. The broad band in P2 absorption spectrum in DCB reflects more planar structure than P3 and the molecular aggregation in dilute solution. In the case of P2, the absorption spectra of their thin films differ when the films are prepared from toluene or DCB solutions, whereas such difference was not observed for P3 thin films. The absorption spectra of P2 thin films prepared from DCB are significantly red-shifted and differ in shape from the absorption spectra of P2 thin films prepared from toluene solution, which are blue-shifted.
New polymers P2 and P3 also exhibit PL emission in both solution and thin films, whereas the PL emission of new polymer P1 was very weak, particularly in DCB solution and thin films. The SM model compound is a highly luminescent material in dilute solution. Therefore, PL properties of P1 were studied in toluene solution and a more detailed PL study was performed for SM, P2, and P3. The PL excitation and emission spectra measured in toluene and DCB solutions are displayed in Figures 6 and 7, respectively, and the characteristic data are summarized in Table 2. In toluene solution (Figure 6), P1 exhibits a PL main maximum at 612 nm when excited at long wavelength absorption maxima. If the excitation wavelength is shorter an additional maximum appeared at about 500 nm originating from anthranthrone-containing segments, which is supported by the different excitation spectra for the emission wavelengths for the 500 nm and the main maximum. In DCB solution the PL emission of P1 is very weak. In toluene solution, P2 and P3 emit yellowish light, which is red-shifted when compared with SM model compound’s emission of greenish light. In DCB solution the P2 emission is significantly red-shifted and the emitted light is of orange-red color whereas PL emission of P3 is similar to its toluene solution. Similarly, as in the absorption study, the PL emission spectra of P2 and P3 measured in toluene solutions are similar, but significantly differ in DCB solutions. In the toluene solution, the emission spectra show vibration structure with main maxima at 519 and 520 nm for P2 and P3, respectively, and the excitation ones are well resolved and follow absorption ones. In the DCB solution PL emission spectra of P2 and P3 (Figure 7, parts a and b) significantly differ. The PL maximum of P2 DCB solution spectrum, located at 589 nm, significantly red-shifted when compared with the maximum of its emission spectrum in toluene and with the 533 nm of the P3 DCB solution spectrum. The maximum at about 530 nm is also observed in the P2 emission spectrum for the shorter excitation wavelength of 482 nm, but the maximum at 589 nm dominates.
Figure 6.

PL emission and excitation spectra of (a) P1, (b) P2, (c) P3, and (d) SM measured in dilute toluene solutions. Normalized absorption spectra are shown in dashed lines.
Figure 7.

PL emission and excitation spectra of (a) P2, (b) P3, and (c) SM measured in dilute DCB solutions. Normalized absorption spectra are shown in dashed lines.
The excitation spectra of P2 in DCB depended on the emission wavelength. For the emission at 531 nm, the PL excitation spectrum is similar to that measured in toluene, whereas for the emission at 589 nm the PL excitation spectrum is broader with a better resolved structure, but it follows the shape of absorption spectrum in DCB. The excitation spectrum of P3 exhibited better-resolved vibration structure when compared with the corresponding absorption spectra and follows well the shape of the absorption one. The PL behavior of P2 supports the results from the absorption study concerning the better planarity and aggregation of P2 molecules in the DCB. The PL quantum yields determined for P2 and P3 in their toluene and DCB solutions were similar for both polymers in the same solution, and both P2 and P3 have slightly higher values in their DCB solutions. PL emission and excitation spectra of the SM model compound are shown in Figures 6 and 7 for comparison. In toluene the PL emission spectrum for SM is better resolved and exhibited a maximum at 488 nm, whereas the maximum of the SM spectrum in DCB is slightly red-shifted at 499 nm. The excitation spectra are well resolved for both solvents and similar to the absorption ones.
The PL spectra measured for the thin films are displayed in Figure 8, and the characteristic data are summarized in Table 3. For the thin films, the emission maxima are red-shifted compared to their solutions. In the PL the emission maxima are at 621–623 nm in the spectra of P2 films prepared from toluene and are slightly red-shifted to 625 nm for P2 prepared from DCB. The shape of emission spectra is similar for both P2 films, but the excitation spectra significantly differ. The excitation spectrum of P2 film prepared from toluene follows a similar pattern to its absorption spectrum, but the excitation spectrum of the P2 film prepared from DCB differs in shape from its absorption one. In the case of P3 films the PL emission with maxima at 605–611 nm is slightly blue-shifted when compared with P2 films and there is nearly no difference for the P3 films prepared from toluene or DCB. The excitation spectra follow a similar behavior to those of the absorption ones and are better resolved. Compared with the toluene solution red shift in the emission maxima of the PL thin film spectra is about 100 nm for P2 and 70 nm for P3 films, which indicates excimer and/or aggregate formation in solid state for both polymers, which is also evident from the large Stokes shifts.
Figure 8.

PL emission and excitation spectra of 52 nm (blue) and 46 nm (black) thick P2 films, 111 nm (blue) and 70 nm (black) thick P3 films prepared from toluene (blue) and DCB (black) solutions, respectively, and 43 nm thick SM prepared from DCB. Normalized absorption spectra are shown in dashed lines.
The PL spectra of SM thin film are also shown
in Figure 8c. A red
shift of
the PL emission spectrum is also observed. The shape of the excitation
spectrum matches well with the absorption spectrum shape. The relative
PL efficiency of a thin film, 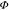 PLrel, was used to compare the
PL efficiencies of the polymers. The
PLrel, was used to compare the
PL efficiencies of the polymers. The 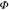 PLrel of
the films prepared from
toluene is similar for both copolymers P2 and P3, whereas
PLrel of
the films prepared from
toluene is similar for both copolymers P2 and P3, whereas 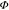 PLrel of P2 films
prepared from DCB is by more than three times higher than the
PLrel of P2 films
prepared from DCB is by more than three times higher than the  PLrel of P3.
PLrel of P3.
Electrochemical Properties
Cyclic voltammetry (CV) measurements were performed to obtain information on the electronic structures of the new polymers. Representative CV curves of P1, P2, and P3 polymer thin films and SM film coated on a Pt wire are displayed in Figure 9. Quasi-reversibility in the oxidation and reduction of all new polymers and also SM model compound was observed. The ionization potential (HOMO level), EIP, and electron affinity (LUMO level), EA, were estimated from the onset potentials, Eonset, of the oxidation and reduction peaks based on the reference energy level of ferrocene (4.8 eV below the vacuum level) using the equation EIP (EA) = |−(Eonset – Eferr) – 4.8| eV, where Eferr is the half-wave potential for ferrocene vs the Ag/Ag+ electrode. The CV curve of SM exhibited a small prepeak in the reduction, which is in good correlation with the shoulder in the thin film absorption spectra. The similar EIP (−EHOMO) values of 5.05, 5.12, and 5.15 eV were evaluated for P2, P3 and SM, respectively, which are lower than the value of 5.36 eV obtained for P1. The EA (−ELUMO) value of 3.87 eV for P1 was higher than 2.92, 2.85, and 2.62/2.84 eV evaluated for P2, P3 and SM, respectively. The EA values reflect the higher electron affinity of the anthanthrone block (which possesses two electron-withdrawing keto groups at the 6- and 12-positions of the anthanthrone unit) than the anthanthrene unit. Electrochemical bandgap values of 1.49, 2.13, and 2.27 eV were determined for P1, P2 and P3, respectively, which were slightly higher values than the optical bandgap values determined from the absorption edge (except for P1), but there was good correlation between the values for the polymers and SM. (see Table 4).
Figure 9.

Representative cyclic voltammograms of (a) P1, (b) P2, (c) P3, and (d) SM thin films coated on a Pt wire, recorded at a scan rate of 50 mV s–1.
Table 4. Electronic Properties of the Copolymers (EIP, Ionization Potential; EA, Electron Affinity; Egelc, Electrochemical Bandgap; Egopt, Optical Bandgap).
| material | EIP (eV) (−EHOMO) | EA (eV) (−ELUMO) | Egelc (eV) | Egopt (eV) |
|---|---|---|---|---|
| P1 | 5.36 | 3.87 | 1.49 | 1.6 |
| P2 | 5.05 | 2.92 | 2.13 | 1.98 |
| P3 | 5.12 | 2.85 | 2.27 | 2.21 |
| SM | 5.15 | 2.62/a2.84 | 2.53/a2.33 | 2.51/2.2 |
Evaluated from the reduction prepeak onset.
Electroluminescent Properties
The new polymers P2 and P3 were used to create active layers in samples of light-emitting diodes (LEDs). LEDs with a hole-injecting electrode formed by indium-tin oxide (ITO) covered with a poly[3,4-(ethylenedioxy)thiophene]/poly(styrenesulfonate) (PEDOT:PSS) layer and an electron-injecting electrode of calcium covered with aluminum (Ca/Al) were prepared and studied. The electroluminescent (EL) spectra of the LEDs are shown in Figure 10. The EL spectrum of the P2 LEDs was similar to the PL thin film spectrum. The EL emission maximum was at 624 nm. The EL spectra of P3 LEDs were slightly blue-shifted to the PL thin film spectra. The EL maximum of the P3 LEDs depended slightly on the applied voltage; a blue shift was observed as voltage increased. Similarity in EL and PL thin-film spectra indicated that similar centers occur in the EL and PL emission processes.
Figure 10.

Normalized EL spectra (solid) of the ITO/PEDOT:PSS/P2/Ca/Al LED (blue) and ITO/PEDOT:PSS/P3/Ca/Al LED (red) measured at 3 V. PL emission spectra of the corresponding thin films (dashed) are displayed for comparison.
The P2 LED exhibited better performance than the P3 LED, which is in good agreement with the relative PL efficiencies of the thin films. An example of the typical dependences of the luminance on the applied voltage measured on the ITO/PEDOT:PSS/polymer/Ca/Al LED is shown in Figure 11. The P2 LED produced higher luminance values (of up to 200 cd m–2, where saturation occurred) and a lower EL onset voltage (of 1.9–2 V) than P3. Lower EL onset voltages for P2 (in comparison with P3) are in good correspondence with the lower bandgap values of P2 compared with P3. The EL results obtained with the nonoptimized simple LEDs gave an interesting comparison of the two new copolymers. P2 seems to be more promising for further polymer or device optimization: for example, by the modification of the injection electrodes, or by using various blends of EL polymers (in the case of polymer-blend LEDs) with appropriate electroluminescent or charge-transporting polymers to make charge injection and transport more balanced.55−62
Figure 11.

Dependence of current density (a) and luminance (b) on the applied voltage measured on the ITO/PEDOT:PSS/P2/Ca/Al (circles) and ITO/PEDOT:PSS/P3/Ca/Al (squares) LEDs.
Photovoltaic Properties
To investigate the performance of the new polymers P1–P3 as absorbers in bulk heterojunction solar cell applications, organic photovoltaic devices (OPVs) with nonoptimized configurations were fabricated and investigated. The OPV devices {ITO/PEI/Polymer:[60]PCBM(1:2)/MoOx/Ag} consisted of active blend layers of P1, P2, and P3 copolymers and the fullerene derivative [6,6]-phenyl-C61-butyric acid methyl ester ([60]PCBM), in an inverted cell architecture where polyethylenimine (PEI) and molybdenum oxide were used as interfacial layers. The current–voltage characteristics of the OPV devices involving P1–P3 are shown in Figure 12, parts a and b. The OPVs based on P2 and P3 copolymers with anthranthrene unit exhibited better performance with power conversion efficiency (PCE) values of 1.17% and 0.61%, respectively, than the OPVs based on P1 with anthranthrone unit having a PCE of 0.18% (Table 5).
Figure 12.

Current–voltage characteristics of ITO/PEI/polymer:[60]PCBM(1:2)/MoOx/Ag OPV devices using (a) P1, (b) P2 (circles), and P3 (squares), which were measured in the dark and under illumination by solar simulator at 100 mW cm–2.
Table 5. Photovoltaic Characteristic Data of ITO/PEI/Polymer:[60]PCBM(1:2)/MoOx/Ag Devices Based on P1, P2, and P3 (Jsc: short-circuit current density; Voc: open-circuit voltage; FF: fill factor; PCE: power conversion efficiency; d: active layer thickness).
| polymer | d (nm) | Jsc (mA cm–2) | Voc (V) | FF | PCE (%) | J @ −2 V (mA cm–2) | J @ + 2 V (mA cm–2) |
|---|---|---|---|---|---|---|---|
| P1 | 92 | 0.58 | 0.93 | 0.33 | 0.18 | –0.94 | 0.05 |
| P2 | 104 | 2.90 | 0.76 | 0.53 | 1.17 | –3.19 | 22.74 |
| P3 | 134 | 2.02 | 0.78 | 0.39 | 0.61 | –2.53 | 0.95 |
The higher efficiency realized with P2-based OPV can be attributed to higher Jsc and FF. The Voc ideally correlates with the energetic difference between the EIP (HOMO) of the donor polymer and the EA (LUMO) of the [60]PCBM acceptor. The OPVs based on P2 and P3 showed resemblance in their Voc values (0.76 and 0.78 V, respectively), which is in good agreement with the similar ionization potentials EIP (HOMO levels) of P2 (5.05 eV) and P3 (5.12 eV). The Voc value for P1 based OPV is higher (Voc = 0.93 V) as expected due to higher EIP value of P1 (5.36 eV). The difference in the EA values of the [60]PCBM and copolymer (LUMO energy offset) is for P1 only 0.03 eV, which is probably too low value for exciton dissociation to be efficient enough, and this could implicate the low JSC value for P1 based OPV. On the other hand, LUMO energy offsets for P2 and P3 (of 1 eV) are large enough for the exciton dissociation. The EA value of 3.9 eV used for [60]PCBM was obtained from our CV measurement. The low FF values of P1 and P3 based OPVs are likely to be attributed to nonsuitable film morphologies. The solubility of conducting polymers plays a major role in the film morphology. Low solubility leads to poor quality of films, which induce low FFs values and invariably lower the efficiency of OPV devices. Low solubility also seems to cause poor miscibility with the electron acceptor [60]PCBM, which might partly explain the limited performance of the devices. Besides other characteristics, generally the solubility, molecular weight, intermolecular interactions, and charge carrier mobilities of the conjugated semiconducting polymers can largely affect their photovoltaic properties.63−68
The external quantum efficiency (EQE), which is the measure of the efficiency of a cell to convert incident photons into free charge carriers, was also investigated. Figure 13 shows EQE curves for the OPVs. As a result, high EQE values can be achieved by the combined effect of efficient exciton generation in the bulk of the active blend layer, the dissociation into free charge carriers, and the charge transport to the respective electrode. The linear correlation between Jsc and EQE can be seen in P1–P3 (Figure 13, Table 5). P2 that has the highest Jsc value exhibits the highest EQE value, while P1, with the lowest Jsc, has the lowest EQE value.
Figure 13.

External quantum efficiencies of ITO/PEI/Polymer:[60]PCBM(1:2)/MoOx/Ag OPV devices using P1 (black), P2 (red), and P3 (blue).
P1–P3 were tested in OPV devices without any optimization and, therefore, the photovoltaic performance is also limited. Optimization of several parameters such as the morphology, variation of the mutual component ratio in blends and utilization of more appropriate device architecture could further improve the OPV devices.63−68 We have recently shown that fullerene aggregation occurs in the blend layers, which can negatively influence the device performance.69 Using blends with a nonfullerene acceptor for the active layer is another way to improve the stability and performance of OPV.70−76
Conclusions
Polycyclic aromatic building blocks based on anthanthrone and anthanthrene, were incorporated for the first time into the backbone of poly[(arylene ethynylene)-alt-(arylene vinylene)]s yielding new copolymers P1–P5. With copolymer P1, which possesses 4,10-linked anthanthrone building blocks and an all-branched 2-ethylhexyloxy side chains, it was possible to demonstrate the preparation of the anthanthrone based PAE-AV polymer with soluble fractions. This was not possible with P4 and P5 with linear decyloxy side chains formed as insoluble materials. Substitution of anthanthrone with anthanthrene units appeared to be an efficient strategy to prepare polymers with appreciable solubility (P2 and P3). P1, P2, and P3 were studied in detail. All three polymers exhibited interesting photophysical properties. The absorption spectrum of P1 in solution and thin films covered the whole visible spectral region. The PL of P1 was weak, particularly in DCB and thin films. The “transformation” of the anthanthrone’s carbonyl groups into anthanthrene’s alkyloxy substituents in the synthesis of P2 and P3 affected the photophysical properties of the resultant copolymers significantly. The properties of P2 and P3 indicated that they were not only dominated by the polycyclic chromophore, but were also influenced by the nature of the side chains on the phenylene–vinylene unit (decyloxy in P2 and 3,7-dimethyloctyloxy in P3). The absorption and PL properties of P2 and P3 were similar in their toluene solution only, whereas in DCB solutions they differ. The absorption spectra of P2 thin films differed whether the thin films were prepared from toluene or from DCB solutions, contrary to P3 thin films for which such a difference was not observed. PL studies also revealed differences in the P2 and P3 behavior in solutions and thin films. The results of the photophysical studies indicate the formation of aggregates in P2 DCB solution, and more pronounced planarity and excimer or aggregate formation in P2 films than in P3 ones. The ionization potential (HOMO level) and electron affinity (LUMO level) values determined for P1 were higher than the corresponding values for P2 and P3, thereby reflecting the higher electron affinity of anthanthrone block than that of anthanthrene unit. The copolymers P2 and P3 were tested as active layers in polymer LEDs. The performance of the LEDs with P2 as an active layer was better than that of the P3 LEDs, which is in good agreement with the relative PL efficiencies of the thin films. P1, P2, and P3 containing [60]PCBM in blend layers were also tested for photovoltaic applications. Similarly, photovoltaic devices based on P2 exhibited better performance than those based on P3. The replacement of decyloxy side chains in P2 with 3,7-dimethyloctyloxy in P3 resulted in lower yield and decrease in photovoltaic efficiency. Optimization of the device composition and architecture might yield significant enhancements of the LED efficiency and the photovoltaic performance.
Experimental Section
Synthesis
All starting materials and solvents were purchased from commercial suppliers (Sigma-Aldrich, Merck and Alfa Aesar and VWR). Most of the details describing the synthesis steps toward the HWE polycondensation monomers, dialdehydes (Ma–Mc) and bisphosphonates (M1–M3) (Scheme 1), are given in the Supporting Information.
Monomer Synthesis
Synthesis of M1
A mixture of 1,4-bis(bromomethyl)-2,5-bis(2-ethylhexyloxy)benzene (15.2 g, 18.97 mmol) and an excess of triethylphosphite (9.47 g, 57.0 mmol) was stirred and heated slowly to 150–160 °C; simultaneously, the evolving ethyl bromide was distilled off. After 4 h, vacuum was applied for 30 min at 180 °C to distill off the excess triethylphosphite. The residual oily product was allowed to cool to room temperature (14.4 g, 83% yield). 1H NMR (300 MHz, CDCl3): δ 6.92 (d, J = 1.6 Hz, 2H), 4.08–3.89 (m, 8H), 3.79 (d, J = 5.6 Hz, 4H), 3.15 (d, J = 20.1 Hz, 4H), 1.76–1.59 (m, 2H), 1.55–1.34 (m, 10H), 1.34–1.11 (m, 18H), 0.99–0.63 (m, 12H). 13C NMR (75 MHz, CDCl3): δ 150.43, 119.30, 114.50, 71.17, 61.8, 39.65, 30.58, 29.09, 27.06, 25.21, 23.89, 23.00, 16.30, 14.01, 11.12.
M2 and M3 were prepared under similar reaction conditions as described for M1
Synthesis of M2
A mixture of 1,4-bis(bromomethyl)-2,5-bis(decyloxy)benzene (3.37 g, 5.85 mmol) and an excess of triethylphosphite (3.0 g, 17.6 mmol) were stirred and heated slowly to 150–160 °C, and the evolving ethyl bromide was distilled off simultaneously. The reaction proceeded for 4 h, after which vacuum was applied for 1 h at 180 °C to distill off any excess of triethylphosphite left in the mixture. The resulting oil was allowed to cool to room temperature to form a white solid, which was recrystallized from diethyl ether (30 mL), yielding a white powder (3.9 g, 97%). 1H NMR (400 MHz, CDCl3): δ 6.89 (s, 2H), 4.00 (m, 8H), 3.90 (t, J = 6.5 Hz, 4H), 3.20 (d, J = 20.3 Hz, 4H), 1.82–1.65 (m, 4H), 1.41–1.15 (m, 40H), 0.86 (t, J = 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 150.31, 119.28, 114.70, 68.89, 61.69, 31.89, 29.61, 29.56, 29.47, 29.44, 29.32, 26.92, 26.13, 25.54, 22.67, 16.38, 16.34, 16.31, 14.10.
Synthesis of M3
A mixture of 1,4-bis(bromomethyl)-2,5-bis(3,7-dimethyloctyloxy)benzene (5.10 g, 8.70 mmol) and an excess of triethylphosphite (4.40 g, 26.1 mmol) was stirred and heated slowly to 150–160 °C, and the evolving ethyl bromide was concurrently distilled off. After 4 h, vacuum was applied for 30 min at 180 °C to distill off any excess of triethylphosphite. The residual yellow oil was allowed to cool to room temperature (5.73 g, 96% yield). 1H NMR (400 MHz, CDCl3): δ 6.85 (s, 2H), 4.07–3.79 (m, 12H), 3.14 (d, J = 20.4 Hz, 4H), 1.86–1.37 (m, 8H), 1.32–1.01 (m, 24H), 0.86 (d, J = Hz, 6H), 0.79 (d, J = 6.6 Hz, 12H). 13C NMR (100 MHz, CDCl3): δ 150.15, 119.18, 114.60, 67.01, 61.61, 38.99, 37.09, 36.23, 29.63, 27.72, 26.71, 25.32, 24.45, 22.45, 22.34, 19.44, 16.09.
Synthesis of Ma
4,10-Dibromoanthanthrone (408 mg, 0.88 mmol) was given to a mixture of diisopropylamine (15 mL) and tetrahydrofuran (40 mL). After deaeration of the reaction for 30 min, Pd(PPh3)4 (45.1 mg, 0.04 mmol) and CuI (7.4 mg, 0.04 mmol) were added. The mixture was allowed to stir and deaerate for another 30 min. Thereafter, 2,5-bis(2-ethylhexyloxy)-4-ethynylbenzaldehyde (750 mg, 2.0 mmol, dissolved in deaerated THF) was added dropwise. The reaction mixture was heated to 60 °C for 72 h in a nitrogen atmosphere. Thin layer chromatography (TLC) was used to monitor the progress of the reaction. Toluene (40 mL) was added and the heating was removed. After cooling to room temperature, the mixture was precipitated in cold methanol and refrigerated for 4 h. The precipitate was filtered off, redissolved in toluene, and purified through column chromatography (silica gel) toluene as eluent to yield 350 mg (37%) of a purplish black substance. 1H NMR (300 MHz, CDCl3): δ 10.47 (s, 2H), 8.87–8.66 (m, 4H), 8.60 (s, 2H), 7.95–7.73 (m, 2H), 7.32 (s, 2H), 7,17 (s, 2H), 4.17–3.82 (m, 8H), 2.03–1.73 (m, 4H), 1.73–1.16 (m, 32H), 1.1–0.8 (m, 24H). 13C NMR (100 MHz, CDCl3): δ 188.98, 182.12, 155.49, 154.07, 134.07, 133.74, 131.28, 129.53, 129.30, 128.63, 127.60, 125.45, 124.11, 119.09, 117.49, 109.36, 94.20, 93.96, 71.72, 71.65, 39.57, 39.48, 30.65, 30.34, 29.71, 29.14, 29.01, 24.04, 23.77, 23.11, 23.08, 14.13, 14.07, 11.26, 11.06.
Synthesis of Mb
A mixture of 4,10-dibromo-6,12-bis(2-ethylhexyloxy)anthanthrene (200 mg, 0.29 mmol), diisopropylamine (10 mL), and toluene (20 mL) was deaerated for 30 min. Pd(PPh3)4 (16.7 mg, 0.015 mmol), and CuI (2.8 mg, 0.02 mmol) were added. After another hour a deaerated solution of 2,5-bis(decyloxy)-4-ethynylbenzaldehyde (320 mg, 0.72 mmol) in toluene (15 mL) was added dropwise into the mixture. The reaction mixture was heated to 70–80 °C for 24 h in a nitrogen atmosphere and monitored with TLC. Toluene (40 mL) was added. After the reaction cooled to room temperature, the precipitated diisopropylammonium bromide was filtered off and the solvent was evaporated under vacuum. The residue was subjected to column chromatography–silica gel with toluene:hexane (4:1) as eluent. 380 mg (93% yield) of a greenish substance was obtained. 1H NMR (300 MHz, CDCl3): δ 10.49 (s, 2H), 8.94 (d, J = 7.4 Hz, 2H), 8.80 (d, J = 8.1 Hz, 2H), 8.73 (s, 2H), 8.19 (m, 2H), 7.38 (s, 2H), 7.22 (s, 2H), 4.29 (d, J = 5.5 Hz, 4H), 4.21–4.07 (m, 8H), 2.23–1.68 (m, 10H), 1.68–1.08 (m, 78H), 1.03 (t, J = 7.0 Hz, 6H), 0.90 (t, J = 6.6 Hz, 6H), 0.77 (t, J = 6.7 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 189.13, 155.51, 153.95, 150.13, 130.72, 127.58, 125.98, 125.61, 124.88, 124.52, 123.95, 121.27, 120.54, 120.22, 119.50, 117.12, 109.28, 96.16, 90.48, 69.26, 59.55, 41.30, 38.17, 31.94, 31.86, 31.26, 30.48, 29.70, 29.62, 29.46, 29.32, 26.12, 24.03, 23.31, 22.72, 22.58, 14.32, 14.15, 14.05, 11.56.
Synthesis of Mc
A mixture of 4,10-dibromoanthanthrone (200 mg, 0.43 mmol), diisopropylamine (12 mL), and toluene (20 mL) was deaerated. Pd(PPh3)4 (20.3 mg, 0.02 mmol) and CuI (3.3 mg, 0.02 mmol) were added. After 30 min, a deaerated solution of 2,5-bis(decyloxy)-4-ethynylbenzaldehyde (390 mg, 0.88 mmol) in toluene was added dropwise. The reaction mixture was heated to 70–80 °C for 24 h in a nitrogen atmosphere. Toluene was added and the mixture was allowed to cool to room temperature. The precipitated diisopropylammonium bromide was filtered off and the solvent was distilled off using a rotary evaporator. The residue was subjected to column chromatography using silica gel, with toluene as eluent to yield 130 mg (26% yield) of a purple substance. The same reaction was conducted in THF at 60 °C for 72 h. After cooling, the mixture was reprecipitated in cold methanol (100 mL). The precipitate was recovered via filtration, and the crude residue was purified by column chromatography (silica gel) with toluene as eluent to afford Mc as a purplish black solid (74% yield). 1H NMR (300 MHz, CDCl3): δ 10.44 (s, 2H), 8.72 (d, d, J = 6.9 Hz, 4H), 8.49 (s, 2H), 7.82–7.68 (m, 2H), 7.24–7.13 (m, 2H), 7.09 (s, 2H), 4.06 (t, t, J = 6.6 Hz, 8H), 2.17 (m, 4H), 1.91 (m, 8H), 1.66–1.08 (m, 56H), 0.87 (t, t, J = 6.2 Hz, 6H, 6H). Acquisition of the 13C NMR spectrum of Mc proved unsuccessful because of the poor solubility and precipitation of the product during the course of the acquisition.
Polymer Synthesis
Synthesis of P1
For the synthesis of P1, a solution of Ma (150 mg, 0.14 mmol) and M1 (89.0 mg, 0.14 mmol) in dry toluene (12 mL) was mechanically stirred and heated under nitrogen to reflux. The polymerization was initiated by the addition of potassium tert-butoxide (63.0 mg, 0.56 mmol) and a spontaneous color change from deep pink to black was observed. The reaction mixture was stirred for 45 min, and toluene (100 mL) was added. The reaction was quenched with aqueous HCl (10 wt %, 10 mL). The organic phase was separated and washed with deionized water until a pH of ∼7 was obtained. Residual water was removed from the mixture by heating to reflux in a Dean–Stark apparatus. The water-free mixture was concentrated with a rotary evaporator, precipitated in cold methanol, and then kept in the refrigerator for 24 h. Afterward the precipitate was filtered off in a Soxhlet thimble, which was transferred into a Soxhlet extractor. Extraction was performed with methanol in order to remove oligomers and any impurity. The extraction continued until the extract appeared colorless. The polymer was redissolved in toluene, reprecipitated in cold methanol and stored in the refrigerator. After 24 h, it was filtered and dried under air to obtain a black, partially soluble polymeric material (190 mg, 95% yield). SEC of the soluble fractions (THF as eluent; polystyrene standards): Mn = 1830 g/mol, Mw = 3570 g/mol, and Đ = 2. 1H NMR of the soluble fractions (300 MHz, CDCl3): δ 10.48 (s), 9.25–8.41 (m, anthanthronylene H’s), 8.07–6.67 (m, arylene vinylene H’s), 4.37–3.20 (m, −CH2O−), 2.49–0.37 (m, alkyl H’s). FTIR, cm–1: 3064 (ν(=C–H)), 2962, 2934, and 2878 (ν(C–H)), 2198 (ν(C≡C)), 1659 (ν(C=O)), 1575 (ν(C=C) vinylene), 1500 (ν(C=C) aromatic ring), 1464 (δ(CH2)), 1380 (δ(CH3)), 1203 (ν(C=C–O–C) aromatic–aliphatic).
P2–P5 were prepared under similar reaction conditions as described for P1
Synthesis of P2
A solution of Mb (170 mg, 0.12 mmol) and M2 (83.0 mg, 0.12 mmol) in dry toluene (12 mL) was mechanically stirred and heated under nitrogen to reflux. Potassium tert-butoxide (54 mg, 0.48 mmol) was added at a stable temperature to initiate the reaction. A color change from bright green to dark green was observed. The reaction mixture was stirred for 80 min. Thereafter, toluene (100 mL) was added, and the reaction was quenched with aqueous HCl (10 wt %, 10 mL). The mixture was cooled to room temperature; the organic phase separated and washed with deionized water until a pH of ∼7 was obtained. Residual water was removed from the mixture by heating to reflux in a Dean–Stark apparatus. The obtained solution was concentrated, precipitated in cold methanol, and kept in the refrigerator for 24 h. Afterward the precipitate was filtered off, transferred into a Soxhlet extractor, and extracted using methanol. The polymer was redissolved in chloroform and reprecipitated in cold methanol. It was filtered and dried for 24 h to obtain a brownish green polymeric material (220 mg, 95% yield). SEC (THF as eluent; polystyrene standards): Mn = 3200 g/mol, Mw = 11 730 g/mol, and Đ = 3.7. 1H NMR (300 MHz, CDCl3): δ 9.05–8.22 (anthanthrenylene H’s), 7.58–6.92 (arylene vinylene H’s), 4.28–3.28 (m, −CH2O−), 2.41–0.42 (m, alkyl H’s). FTIR, cm–1: 3064 (ν(=C–H)), 2962, 2924, and 2850 (ν(C–H)), 2180 (ν(C≡C)), 1594 (ν(C=C) vinylene), 1500 (ν(C=C) aromatic ring), 1464 (δ(CH2)), 1203 (ν(C=C–O–C) aromatic–aliphatic).
Synthesis of P3
P3 was obtained from a solution of Mb (190 mg, 0.14 mmol) and M3 (93 mg, 0.15 mmol) in dry toluene (13 mL). The polymerization was initiated by the addition of potassium tert-butoxide (60 mg, 0.56 mmol). A color change from bright green to dark green was observed. The reaction mixture was stirred for 80 min. Chloroform (100 mL) was thereafter added, and the reaction was quenched with aqueous HCl (10 wt %, 10 mL). Mixture was cooled to room temperature; the organic phase was separated and washed with deionized water until a pH of ∼7 was obtained. Residual water was removed from the mixture by heating to reflux in a Dean–Stark apparatus. The obtained solution was concentrated, precipitated in cold methanol, and kept in the fridge for 24 h. Afterward the precipitate was filtered off using a thimble, transferred into a Soxhlet extractor, and extracted with methanol. The polymer was redissolved in chloroform and reprecipitated in methanol. It was filtered and dried to obtain a brownish green polymeric material. This polymerization was conducted twice using different oil bath temperatures, 111 °C [SEC (THF as eluent; polystyrene standards): Mn = 3650 g/mol, Mw = 11 800 g/mol, Đ = 3.2] and 118 °C [SEC (THF as eluent; polystyrene standards): Mn 2880 g/mol, Mw 7820 g/mol, and Đ = 2.7]. The polycondensation reaction conducted at 111 °C yielded material that is soluble in common organic solvents (180 mg, 73% yield). The reaction performed at higher temperature(118 °C) yielded material only partially soluble at elevated temperatures (33% yield). 1H NMR (300 MHz, CDCl3): δ 8.73 (m, (anthanthreneylene H’s)), 7.65–6.90 (m, arylene vinylene H’s), 4.61–3.48 (m, −CH2O−), 2.38–0.42 (m, alkyl H’s). FTIR, cm–1: 3073 (ν(=C–H)), 2962, 2924, and 2850 (ν(C–H)), 2198 (ν(C≡C)), 1603 (ν(C=C) vinylene), 1500 (ν(C=C) aromatic ring), 1464 (δ(CH2)), 1203 (ν(C=C–O–C) aromatic–aliphatic).
P4 and P5 were synthesized under similar reaction conditions as described for P1–P3. For P4, monomers Mc and M1 were employed while for P5; monomers Mc and M3 were employed. Both polycondensation reactions produced materials that appeared completely insoluble and could not be further characterized.
Synthesis of SM
Mb (190 mg, 0.14 mmol) and diethyl benzylphosphonate (79.0 mg, 0.34 mmol) were dissolved in dried toluene (15 mL) to give a greenish brown color while stirring vigorously under nitrogen and heating under reflux. Potassium-tert-butoxide (62 mg, 0.50 mmol) was added (and the reaction mixture was heated at reflux for 30 min. After 30 min, toluene was added, heat was removed, and aqueous HCl (10 wt %) was added. The purification process yielded a greenish material (170 mg, 79%). 1H NMR (300 MHz, CDCl3): δ 9.08 (d, J = 7.3 Hz, 2H), 8.92–8.78 (m, 2H), 8.27 (t, J = 7.7 Hz, 2H), 7.64 (d, J = 7.6 Hz, 4H), 7.56–7.18 (m, 16H), 4.37 (d, J = 5.2 Hz, 4H), 4.28 (t, J = 6.4 Hz, 4H), 4.15 (t, J = 6.2 Hz, 4H), 2.19 (m, 2H), 1.94–1.03 (m, 80H), 0.94 (d, J = 6.6 Hz, 12H), 0.83 (t, J = 6.2 Hz, 12H).
Instrumentation
NMR spectra were recorded in CDCl3 with a Gemini 300 MHz spectrometer and a Bruker Avance IIIHD 400 MHz Nanobay NMR spectrometer equipped with a 5 mm BBO. Fourier transform infrared (FTIR) spectroscopy was performed with a PerkinElmer ATR TWO FTIR Spectrometer at room temperature. Size exclusion chromatography (SEC) measurements were performed using a Pump Deltachrom (Watrex Comp.) with a Midas autosampler and two columns of MIXED-B LS PL gel, particle size 10 μm. An evaporative light scattering detector (PL-ELS-1000 from Polymer Laboratories) was used; THF was the mobile phase and polystyrene standards were used for calibration. Thermogravimetric analysis (TGA) were performed using PerkinElmer TGA 7 in the range between 20 and 600 °C at a heating rate of 10 K/min in nitrogen atmosphere.
Sample Preparation
Thin films were prepared by spin-coating from toluene and 1,2-dichlorobenzene solutions. Thin films were spin-coated onto fused silica substrates for optical studies or coated on a Pt wire electrode by dipping for electrochemical measurements. For polymer light-emitting devices (LED), polymer layers were prepared on indium–tin oxide (ITO) substrates covered with a thin layer of poly[3,4-(ethylenedioxy)thiophene]/poly(styrenesulfonate) (PEDOT:PSS). All polymer films were dried in a vacuum (10–3 Pa) at 353 K for 2 h. The ITO glass substrates were purchased from Merck (Germany). PEDOT:PSS (CLEVIOS P VP AI 4083) from Heraeus Clevios GmbH (Germany) was used for LED preparation. The 50 nm thick PEDOT:PSS layers were prepared by spin-coating and dried in air at 396 K for 15 min. The calcium (20 nm) and, subsequently, 60–80 nm thick aluminum electrodes were vacuum-evaporated on top of the polymer films to form LEDs. Typical active areas of the LEDs were 4 mm2. Layer thicknesses were measured using a KLA-Tencor P-10 profilometer. All thin film preparations and the device fabrication were carried out in a glovebox under a nitrogen atmosphere. For the fabrication of organic photovoltaic (OPV) devices ITO-coated glass substrates (15 Ω/sq, Xin Yan Tech. LTD) were employed. Polymer blend layers composed of polymer and [60]PCBM (Sigma-Aldrich) were spin-coated on the ITO substrate covered with polyethylenimine (PEI). PEI was treated at 378 K. The MoOx (10 nm) and, subsequently, 100 nm thick silver (Ag) electrodes were vacuum-evaporated on top of the polymer films to finalize OPVs. The thickness of the films was characterized with a Bruker DektakXT profilometer.
Cyclic Voltammetric Measurements
Cyclic voltammetry (CV) was performed with a PA4 polarographic analyzer (Laboratory Instruments, Prague, CZ) with a three-electrode cell. Platinum (Pt) wire electrodes were used as both working and counter electrodes. A nonaqueous Ag/Ag+ electrode (Ag in 0.1 M AgNO3 solution) was used as the reference electrode. CV measurements were made in an electrolyte solution of 0.1 M tetrabutylammonium hexafluorophosphate ((TBA)PF6) in anhydrous acetonitrile under a nitrogen atmosphere in a glovebox. Typical scan rates were 20, 50, and 100 mV s–1.
Photophysical Measurements
UV–vis spectra were measured on a PerkinElmer Lambda 35 UV/vis spectrometer. Solvents of spectroscopic grade were used. The absorption spectra of thin films were also measured in the glovebox using fiber optics connected to the spectrophotometer. Steady-state PL spectra were recorded using a PerkinElmer LS55 Fluorescence spectrophotometer. The PL quantum yield of the polymer in solution was calculated relative to the fluorescein or rhodamine 6G, which were used as standards.77
Electroluminescence Measurements
EL spectra were recorded using an Acton Research Spectrograph with single photon-counting detection (SPEX, RCA C31034 photomultiplier). LEDs were supplied from a Keysight Technologies B2901A Precision Source Measure Unit, which served to simultaneously record the current flowing through the sample. Current–voltage and luminance–voltage characteristics were recorded simultaneously using the source measure unit and a Minolta LS110 luminance meter or a silicon photodiode with integrated amplifier (EG&G HUV-4000B) for the detection of total light output. A voltage signal from the photodiode was recorded with a Hewlett-Packard 34401A multimeter. All measurements were conducted in a glovebox under a nitrogen atmosphere.
Photovoltaic Measurements
Current–voltage characteristics of the devices were recorded in the dark and under illumination (100 mW/cm2) using a Keithley 2401 SourceMeter unit and a LS02821 LOT Quantum Design solar simulator. External quantum efficiencies (EQE) were measured with a setup consisting of EG&G Instruments 7260 DSP lock-in-amplifier, an optical chopper (SCITEC Instruments LTD), a xenon lamp LXH100 (Müller Elektronik-Optik), and a monochromator Acton Spectra Pro150. A calibrated silicon diode S2281 (Hamamatsu) was used as a reference.
Acknowledgments
We thank the Czech Science Foundation (Grant 13-26542S), FWF (Project No. I 1703-N20) and the National Research Foundation (NRF) South African Agency for Science and Technology Advancement (Project No. UID-85102) for financial support. S.V.J. is grateful to the International Centre for Theoretical Physics (ICTP) and the African Network for Solar Energy (ANSOLE), as well as the NRF of South Africa, for financial support.
Supporting Information Available
Supplementary data related to this article are available free of charge via the Internet at The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.7b02136.
Synthesis of precursors, NMR and FTIR spectra, and thermo-gravimetric analysis plots (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Schenning A. P. H. J.; Meijer E. W. Supramolecular electronics; nanowires from self-assembled π-conjugated systems. Chem. Commun. 2005, 3245–3258. 10.1039/b501804h. [DOI] [PubMed] [Google Scholar]
- Hoeben F. J. M.; Jonkheijm P.; Meijer E. W.; Schenning A. P. H. J. About Supramolecular Assemblies of π-Conjugated Systems. Chem. Rev. 2005, 105, 1491–1546. 10.1021/cr030070z. [DOI] [PubMed] [Google Scholar]
- Moulin E.; Cid J.-J.; Giuseppone N. Advances in Supramolecular Electronics – From Randomly Self-assembled Nanostructures to Addressable Self-Organized Interconnects. Adv. Mater. 2013, 25, 477–487. 10.1002/adma.201201949. [DOI] [PubMed] [Google Scholar]
- Pisula W.; Feng X.; Müllen K. Tuning the Columnar Organization of Discotic Polycyclic Aromatic Hydrocarbons. Adv. Mater. 2010, 22, 3634–3649. 10.1002/adma.201000585. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Walker B.; Liu F.; Colella N. S.; Mannsfeld S. C. B.; Watkins J. J.; Nguyen T.-Q.; Briseno A. L. Triisopropylsilylethynyl-functionalized dibenzo[def,mno]chrysene: a solution-processed small molecule for bulk heterojunction solar cells. J. Mater. Chem. 2012, 22, 4266–4268. 10.1039/C2JM14998B. [DOI] [Google Scholar]
- Mei J.; Diao Y.; Appleton A. L.; Fang L.; Bao Z. Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors. J. Am. Chem. Soc. 2013, 135, 6724–6746. 10.1021/ja400881n. [DOI] [PubMed] [Google Scholar]
- Ye Q.; Chi C. Recent Highlights and Perspectives on Acene Based Molecules and Materials. Chem. Mater. 2014, 26, 4046–4056. 10.1021/cm501536p. [DOI] [Google Scholar]
- Ball M.; Zhong Y.; Wu Y.; Schenck C.; Ng F.; Steigerwald M.; Xiao S.; Nuckolls C. Contorted Polycyclic Aromatics. Acc. Chem. Res. 2015, 48, 267–276. 10.1021/ar500355d. [DOI] [PubMed] [Google Scholar]
- Collis G. E. Designing small molecule polyaromatic p- and n-type semiconductor materials for organic electronics. Proc. SPIE 2015, 9668, 96683O–96683O-10. 10.1117/12.2202565. [DOI] [Google Scholar]
- Schmidt-Mende L.; Fechtenkotter A.; Müllen K.; Moons E.; Friend R. H.; MacKenzie J. D. Self-Organized Discotic Liquid Crystals for High-Efficiency Organic Photovoltaics. Science 2001, 293, 1119–1122. 10.1126/science.293.5532.1119. [DOI] [PubMed] [Google Scholar]
- Laschat S.; Baro A.; Steinke N.; Giesselmann F.; Hagele C.; Scalia G.; Judele R.; Kapatsina E.; Sauer S.; Schreivogel A.; Tosoni M. Discotic Liquid Crystals: From Tailor-Made Synthesis to Plastic Electronics. Angew. Chem., Int. Ed. 2007, 46, 4832–4887. 10.1002/anie.200604203. [DOI] [PubMed] [Google Scholar]
- Wong W. W. H.; Singh T. B.; Vak D.; Pisula W.; Yan C.; Feng X.; Williams E. L.; Chan K. L.; Mao Q.; Jones D. J.; Ma C.-Q.; Müllen K.; Bäuerle P.; Holmes A. B. Solution Processable Fluorenyl Hexa-peri-hexabenzocoronenes in Organic Field-Effect Transistors and Solar Cells. Adv. Funct. Mater. 2010, 20, 927–938. 10.1002/adfm.200901827. [DOI] [Google Scholar]
- Pisula W.; Feng X.; Müllen K. Charge-Carrier Transporting Graphene-Type Molecules. Chem. Mater. 2011, 23, 554–567. 10.1021/cm102252w. [DOI] [Google Scholar]
- Zhang Z.; Lei T.; Yan Q.; Pei J.; Zhao D. Electron-transporting PAHs with dual perylenediimides: syntheses and semiconductive characterizations. Chem. Commun. 2013, 49, 2882–2884. 10.1039/c3cc40704g. [DOI] [PubMed] [Google Scholar]
- Xiao S.; Kang S. J.; Zhang S.; Scott A. M.; Moscatelli A.; Turro N. J.; Steigerwald M. L.; Li H.; Nuckolls C.; et al. Controlled Doping in Thin-Film Transistors of Large Contorted Aromatic Compounds. Angew. Chem., Int. Ed. 2013, 52, 4558–4562. 10.1002/anie.201300209. [DOI] [PubMed] [Google Scholar]
- He B.; Tian H.; Geng Y.; Wang F.; Müllen K. Facile Synthesis of 9,10-Diarylphenanthrenes and Poly(9,10-diarylphenanthrene)s. Org. Lett. 2008, 10, 773–776. 10.1021/ol702873z. [DOI] [PubMed] [Google Scholar]
- Kawano S.-I.; Yang C.; Ribas M.; Baluschev S.; Baumgarten M.; Müllen K. Blue-Emitting Poly(2,7-pyrenylene)s: Synthesis and Optical Properties. Macromolecules 2008, 41, 7933–7937. 10.1021/ma8017316. [DOI] [Google Scholar]
- Saleh M.; Park Y.-S.; Baumgarten M.; Kim J.-J.; Müllen K. Conjugated Triphenylene Polymers for Blue OLED Devices. Macromol. Rapid Commun. 2009, 30, 1279–1283. 10.1002/marc.200900332. [DOI] [PubMed] [Google Scholar]
- Chen H.; Hu X.; Ng S.-C. Synthesis and characterization of soluble conjugated polymers having pyrene moiety in the main chain. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5562–5569. 10.1002/pola.24369. [DOI] [Google Scholar]
- Chou C.-E.; Wang D.; Bagui M.; Hsu J.; Chakraborty S.; Peng Z. Syntheses and optical properties of triphenylene-containing conjugated polymers. J. Lumin. 2010, 130, 986–994. 10.1016/j.jlumin.2010.01.010. [DOI] [Google Scholar]
- Lehnherr D.; Tykwinski R. R. Conjugated Oligomers and Polymers Based on Anthracene, Tetracene, Pentacene, Naphthodithiophene, and Anthradithiophene Building Blocks. Aust. J. Chem. 2011, 64, 919–929. 10.1071/CH11169. [DOI] [Google Scholar]
- Kim H.; Schulte N.; Zhou G.; Müllen K.; Laquai F. A High Gain and High Charge Carrier Mobility Indenofluorene-Phenanthrene Copolymer for Light Amplification and Organic Lasing. Adv. Mater. 2011, 23, 894–897. 10.1002/adma.201003797. [DOI] [PubMed] [Google Scholar]
- Yang D. S.; Kim K. H.; Cho M. J.; Jin J.-I.; Choi D. H. Donor–acceptor alternating π-conjugated polymers containing Di(thiophen-2-yl)pyrene and 2,5-Bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione for organic thin-film transistors. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1457–1467. 10.1002/pola.26518. [DOI] [Google Scholar]
- Li G.; Kang C.; Li C.; Lu Z.; Zhang J.; Gong X.; Zhao G.; Dong H.; Hu W.; Bo Z. Planar Conjugated Polymers Containing 9,10-Disubstituted Phenanthrene Units for Efficient Polymer Solar Cells. Macromol. Rapid Commun. 2014, 35, 1142–1147. 10.1002/marc.201400044. [DOI] [PubMed] [Google Scholar]
- Grimsdale A. C.; Müllen K. The Chemistry of Organic Nanomaterials. Angew. Chem., Int. Ed. 2005, 44, 5592–5629. 10.1002/anie.200500805. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Ye Q.; Chi C.; Wu J. Low band gap polycyclic hydrocarbons: from closed-shell near infrared dyes and semiconductors to open-shell radicals. Chem. Soc. Rev. 2012, 41, 7857–7889. 10.1039/c2cs35211g. [DOI] [PubMed] [Google Scholar]
- Hinkel K.; Cho D.; Pisula W.; Baumgarten M.; Müllen K. Alternating Donor–Acceptor Arrays from Hexa-peri-hexabenzocoronene and Benzothiadiazole: Synthesis, Optical Properties, and Self-Assembly. Chem. - Eur. J. 2015, 21, 86–90. 10.1002/chem.201403111. [DOI] [PubMed] [Google Scholar]
- Giguère J.-B.; Sariciftci N. S.; Morin J.-F. Polycyclic anthanthrene small molecules: semiconductors for organic field-effect transistors and solar cells applications. J. Mater. Chem. C 2015, 3, 601–606. 10.1039/C4TC02137A. [DOI] [Google Scholar]
- Lafleur-Lambert A.; Giguère J.-B.; Morin J.-F. Anthanthrene as a large PAH building block for the synthesis of conjugated polymers. Polym. Chem. 2015, 6, 4859–4863. 10.1039/C5PY00603A. [DOI] [Google Scholar]
- Lafleur-Lambert A.; Giguère J.-B.; Morin J.-F. Conjugated Polymers Based on 4,10-Bis(thiophen-2-yl)anthanthrone: Synthesis, Characterization, and Fluoride-Promoted Photoinduced Electron Transfer. Macromolecules 2015, 48, 8376–8381. 10.1021/acs.macromol.5b01449. [DOI] [Google Scholar]
- Egbe D. A. M.; Carbonnier B.; Birckner E.; Grummt U.-W. Arylene-ethynylene/arylene-vinylene copolymers: Synthesis and structure–property relationships. Prog. Polym. Sci. 2009, 34, 1023–1067. 10.1016/j.progpolymsci.2009.03.003. [DOI] [Google Scholar]
- Egbe D. A. M.; Turk S.; Rathgeber S.; Kuhnlenz F.; Jadhav R.; Wild A.; Birckner E.; Adam G.; Pivrikas A.; Cimrova V.; Knor G.; Sariciftci N. S.; Hoppe H. Anthracene Based Conjugated Polymers: Correlation between π–π-Stacking Ability, Photophysical Properties, Charge Carrier Mobility, and Photovoltaic Performance. Macromolecules 2010, 43, 1261–1269. 10.1021/ma902273s. [DOI] [Google Scholar]
- Günes S.; Wild A.; Cevik E.; Pivrikas A.; Schubert U. S.; Egbe D. A. M. Effect of shifting of aromatic rings on charge carrier mobility and photovoltaic response of anthracene and thiophene-containing MEH-PPE-PPVs. Sol. Energy Mater. Sol. Cells 2010, 94, 484–491. 10.1016/j.solmat.2009.11.011. [DOI] [Google Scholar]
- Susarova D. K.; Khakina E. A.; Troshin P. A.; Goryachev A. E.; Sariciftci N. S.; Razumov V. F.; Egbe D. A. M. Photovoltaic performance of PPE-PPV copolymers: effect of the fullerene component. J. Mater. Chem. 2011, 21, 2356–2361. 10.1039/C0JM02681F. [DOI] [Google Scholar]
- Tore N.; Parlak E. A.; Usluer Ö.; Egbe D. A. M.; San S. E.; Aydogan P. Effect of blend ratio on poly(p-phenylene-ethynylene)-alt-poly(p-phenylene-vinylene) polymer solar cells. Sol. Energy Mater. Sol. Cells 2012, 104, 39–44. 10.1016/j.solmat.2012.04.040. [DOI] [Google Scholar]
- Kästner C.; Susarova D. K.; Jadhav R.; Ulbricht C.; Egbe D. A. M.; Rathgeber S.; Troshin P. A.; Hoppe H. Morphology evaluation of a polymer–fullerene bulk heterojunction ensemble generated by the fullerene derivatization. J. Mater. Chem. 2012, 22, 15987–15997. 10.1039/c2jm32629a. [DOI] [Google Scholar]
- Troshin P. A.; Mukhacheva O. A.; Usluer Ö.; Goryachev A. E.; Akkuratov A. V.; Susarova D. K.; Dremova N. N.; Rathgeber S.; Sariciftci N. S.; Razumov V. F.; Egbe D. A. M. Improved Photovoltaic Performance of PPV-Based Copolymers Using Optimized Fullerene-Based Counterparts. Adv. Energy Mater. 2013, 3, 161–166. 10.1002/aenm.201200118. [DOI] [Google Scholar]
- Saidani M. A.; Benfredj A.; Ben Hamed Z.; Romdhane S.; Ulbricht C.; Egbe D. A. M.; Bouchriha H. Franck-Condon analysis of the photoluminescence spectra of a triple-bond containing polymer as a solution and as a thin film. Synth. Met. 2013, 184, 83–85. 10.1016/j.synthmet.2013.09.030. [DOI] [Google Scholar]
- Gasiorowski J.; Boudiba S.; Hingerl K.; Ulbricht C.; Fattori V.; Tinti F.; Camaioni N.; Menon R.; Schlager S.; Boudida L.; Sariciftci N. S.; Egbe D. A. M. Anthracene-containing conjugated polymer showing four optical transitions upon doping: A spectroscopic study. J. Polym. Sci., Part B: Polym. Phys. 2014, 52, 338–346. 10.1002/polb.23419. [DOI] [Google Scholar]
- Kösemen A.; Tore N.; Parlak E. A.; Alpaslan Kösemen Z.; Ulbricht C.; Usluer Ö.; Egbe D. A. M.; Yerli Y.; San S. E. An efficient organic inverted solar cell with AnE-PVstat:PCBM active layer and V2O5/Al anode layer. Sol. Energy 2014, 99, 88–94. 10.1016/j.solener.2013.10.039. [DOI] [Google Scholar]
- Kästner C.; Egbe D. A. M.; Hoppe H. Polymer aggregation control in polymer–fullerene bulk heterojunctions adapted from solution. J. Mater. Chem. A 2015, 3, 395–403. 10.1039/C4TA04736B. [DOI] [Google Scholar]
- Ding L.; Egbe D. A. M.; Karasz F. E. Photophysical and Optoelectronic Properties of Green-Emitting Alkoxy-Substituted PE/PV Hybrid Conjugated Polymers. Macromolecules 2004, 37, 6124–6131. 10.1021/ma040069h. [DOI] [Google Scholar]
- Egbe D. A. M.; Nguyen L. H.; Carbonnier B.; Mühlbacher D.; Sariciftci N. S. Thiophene-containing poly(arylene–ethynylene)-alt-poly(arylene–vinylene)s: Synthesis, characterisation and optical properties. Polymer 2005, 46, 9585–9595. 10.1016/j.polymer.2005.08.008. [DOI] [Google Scholar]
- Wild A.; Egbe D. A. M.; Birckner E.; Cimrova V.; Baumann R.; Grummt U.-W.; Schubert U. S. Anthracene- and thiophene-containing MEH-PPE-PPVs: Synthesis and study of the effect of the aromatic ring position on the photophysical and electrochemical properties. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 2243–2261. 10.1002/pola.23304. [DOI] [Google Scholar]
- Usluer Ö.; Kästner C.; Abbas M.; Ulbricht C.; Cimrova V.; Wild A.; Birckner E.; Tekin N.; Sariciftci N. S.; Hoppe H.; Rathgeber S.; Egbe D. A. M. Charge carrier mobility, photovoltaic, and electroluminescent properties of anthracene-based conjugated polymers bearing randomly distributed side chains. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 3425–3436. 10.1002/pola.26133. [DOI] [Google Scholar]
- Egbe D. A. M.; Roll C. P.; Klemm E. Synthesis, characterisation and properties of highly luminescent and liquid-crystalline alternating PPE/PPV-copolymers. Des. Monomers Polym. 2002, 5, 245–275. 10.1163/156855502760157962. [DOI] [Google Scholar]
- Egbe D. A. M.; Roll C. P.; Birckner E.; Grummt U.-W.; Stockmann R.; Klemm E. Side Chain Effects in Hybrid PPV/PPE Polymers. Macromolecules 2002, 35, 3825–3837. 10.1021/ma012195g. [DOI] [Google Scholar]
- Egbe D. A. M.; Carbonnier B.; Ding L.; Mühlbacher D.; Birckner E.; Pakula T.; Karasz F. E.; Grummt U.-W. Supramolecular Ordering, Thermal Behavior, and Photophysical, Electrochemical, and Electroluminescent Properties of Alkoxy-Substituted Yne-Containing Poly(phenylene–vinylene)s. Macromolecules 2004, 37, 7451–7463. 10.1021/ma0488111. [DOI] [Google Scholar]
- Egbe D. A. M.; Sell S.; Ulbricht C.; Birckner E.; Grummt U.-W. Mixed Alkyl- and Alkoxy-Substituted Poly[(phenylene ethynylene)-alt-(phenylene vinylene)] Hybrid Polymers: Synthesis and Photophysical Properties. Macromol. Chem. Phys. 2004, 205, 2105–2115. 10.1002/macp.200400307. [DOI] [Google Scholar]
- Egbe D. A. M.; Carbonnier B.; Paul E. L.; Muhlbacher D.; Kietzke T.; Birckner E.; Neher D.; Grummt U.-W.; Pakula T. Diyne-Containing PPVs: Solid-State Properties and Comparison of Their Photophysical and Electrochemical Properties with Those of Their Yne-Containing Counterparts. Macromolecules 2005, 38, 6269–6275. 10.1021/ma0507490. [DOI] [Google Scholar]
- Giguere J. B.; Boismenu-Lavoie J.; Morin J. F. Cruciform Alkynylated Anthanthrene Derivatives: A Structure-Properties Relationship Case Study. J. Org. Chem. 2014, 79, 2404–2418. 10.1021/jo402674m. [DOI] [PubMed] [Google Scholar]
- Osuna R. M.; Zhang X.; Matzger A. J.; Hernandez V.; Lopez Navarrete J. T. Combined Quantum Chemical Density Functional Theory and Spectroscopic Raman and UV–vis–NIR Study of Oligothienoacenes with Five and Seven Rings. J. Phys. Chem. A 2006, 110, 5058–5065. 10.1021/jp0607263. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Tao Y.; Cao J.-Z.; Chen R.-F.; Zhao P.; Wu X.-J.; Huang W. The structural, electronic, and optical properties of ladder-type polyheterofluorenes: a theoretical study. J. Mol. Model. 2012, 18, 4929–4939. 10.1007/s00894-012-1483-3. [DOI] [PubMed] [Google Scholar]
- Sghaier T.; Le Liepvre S.; Fiorini C.; Douillard L.; Charra F. Optical absorption signature of a self-assembled dye monolayer on graphene. Beilstein J. Nanotechnol. 2016, 7, 862–868. 10.3762/bjnano.7.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimrová V.; Neher D.; Remmers M.; Kmínek I. Blue Light-Emitting Devices Based on Novel Polymer Blends. Adv. Mater. 1998, 10, 676–680. . [DOI] [Google Scholar]
- Cimrová V.; Výprachtický D.; Hoerhold H.-H. Poly[methyl(phenyl)silanediyl] Modified with Dansyl Fluorophore: Synthesis and Photophysics. J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 2233–2244. 10.1002/pola.24654. [DOI] [Google Scholar]
- Cimrová V.; Výprachtický D. Enhanced Electroluminescence from Light-Emitting Devices Based on Poly(9,9-dihexadecylfluorene-2,7-diyl) and Polysilane Blends. Appl. Phys. Lett. 2003, 82, 642–644. 10.1063/1.1538352. [DOI] [Google Scholar]
- Cimrová V.; Výprachtický D. Electroluminescence and charge photogeneration in poly(9,9-dihexadecylfluorene-2,7-diyl) and its blends. Macromol. Symp. 2004, 212, 281–286. 10.1002/masy.200450831. [DOI] [Google Scholar]
- Mikroyannidis J. A.; Barberis V. P.; Výprachtický D.; Cimrová V. Simple Synthesis, Photophysics and Electroluminescent Properties of Poly[2,7-bis(4-tert-butylstyryl)fluorene-9,9-diyl-alt-alkane-α,ω-diyl]. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 809–821. 10.1002/pola.21842. [DOI] [Google Scholar]
- Chen S.-H.; Li Y.-R.; Yu Ch.-F.; Lin Ch.-F.; Kao P.-Ch. Enhanced luminescence efficiency of Ag nanoparticles dispersed on indium tin oxide for polymer light-emitting diodes. Opt. Express 2013, 21, 26236–26243. 10.1364/OE.21.026236. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Smith G. M.; Loughman E.; Li Y.; Nie W.; Carroll D. L. Effect of multi-walled carbon nanotubes on electron injection and charge generation in AC field-induced polymer electroluminescence. Org. Electron. 2013, 14, 8–18. 10.1016/j.orgel.2012.10.017. [DOI] [Google Scholar]
- Ying L.; Ho Ch.–L.; Wu H.; Cao Y.; Wong W.-Y. White Polymer Light-Emitting Devices for Solid-State Lighting: Materials, Devices, and Recent Progress. Adv. Mater. 2014, 26, 2459–2473. 10.1002/adma.201304784. [DOI] [PubMed] [Google Scholar]
- Beaujuge P. M.; Fréchet J. M. J. Molecular Design and Ordering Effects in π-Functional Materials for Transistor and Solar Cell Applications. J. Am. Chem. Soc. 2011, 133, 20009–20029. 10.1021/ja2073643. [DOI] [PubMed] [Google Scholar]
- Scharber M. C.; Sariciftci N. S. Efficiency of bulk-heterojunction organic solar cells. Prog. Polym. Sci. 2013, 38, 1929–1940. 10.1016/j.progpolymsci.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.; Kim H.-B.; Ko S.-J.; Kim J. Y.; Heeger A. J. An organic surface modifier to produce a high work function transparent electrode for high performance polymer solar cells. Adv. Mater. 2015, 27, 892–896. 10.1002/adma.201404172. [DOI] [PubMed] [Google Scholar]
- Choi H.; Ko S.-J.; Kim T.; Morin P.-O.; Walker B.; Lee B. H.; Leclerc M.; Kim J. Y.; Heeger A. J. Small-bandgap polymer solar cells with unprecedented short-circuit current density and high fill factor. Adv. Mater. 2015, 27, 3318–3324. 10.1002/adma.201501132. [DOI] [PubMed] [Google Scholar]
- Vohra V.; Kawashima K.; Kakara T.; Koganezawa T.; Osaka I.; Takimiya K.; Murata H. Efficient inverted polymer solar cells employing favourable molecular orientation. Nat. Photonics 2015, 9, 403–408. 10.1038/nphoton.2015.84. [DOI] [Google Scholar]
- Yuan J.; Ouyang J.; Cimrová V.; Leclerc M.; Najari A.; Zou Y. Development of quinoxaline based polymers for photovoltaic applications. J. Mater. Chem. C 2017, 5, 1858–1879. 10.1039/C6TC05381E. [DOI] [Google Scholar]
- Cimrová V.; Morávková Z.; Pokorná V.; Výprachtický D. Investigation of donor–acceptor copolymer films and their blends with fullerene in the active layers of bulk heterojunction solar cells by Raman microspectroscopy. Org. Electron. 2017, 47, 194–199. 10.1016/j.orgel.2017.05.016. [DOI] [Google Scholar]
- Liu Y.; Mu C.; Jiang K.; Zhao J.; Li Y.; Zhang L.; Li Z.; Lai J. Y. L.; Hu H.; Ma T.; Hu R.; Yu D.; Huang X.; Tang B. Z.; Yan H. A tetraphenylethylene core-based 3D structure small molecular acceptor enabling efficient non-fullerene organic solar cells. Adv. Mater. 2015, 27, 1015–1020. 10.1002/adma.201404152. [DOI] [PubMed] [Google Scholar]
- Li S.; Liu W.; Shi M.; Mai J.; Lau T.-K.; Wan J.; Lu X.; Li C.-Z.; Chen H. A spirobifluorene and diketopyrrolopyrrole moieties based non-fullerene acceptor for efficient and thermally stable polymer solar cells with high open-circuit voltage. Energy Environ. Sci. 2016, 9, 604–610. 10.1039/C5EE03481G. [DOI] [Google Scholar]
- Li Y.; Liu X.; Wu F.-P.; Zhou Y.; Jiang Z.-Q.; Song B.; Xia Y.; Zhang Z.-G.; Gao F.; Inganäs O.; Li Y.; Liao L.-S. Non-fullerene acceptor with low energy loss and high external quantum efficiency: towards high performance polymer solar cells. J. Mater. Chem. A 2016, 4, 5890–5897. 10.1039/C6TA00612D. [DOI] [Google Scholar]
- Yuan J.; Qiu L.; Zhang Z.-G.; Li Y.; Chen Y.; Zou Y. Tetrafluoroquinoxaline based polymers for non-fullerene polymer solar cells with efficiency over 9%. Nano Energy 2016, 30, 312–320. 10.1016/j.nanoen.2016.10.008. [DOI] [Google Scholar]
- Yuan J.; Qiu L.; Zhang Z.; Li Y.; He Y.; Jiang L.; Zou Y. A simple strategy to the side chain functionalization on the quinoxaline unit for efficient polymer solar cells. Chem. Commun. 2016, 52, 6881–6884. 10.1039/C6CC01771A. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Feng L.; Xu S.; Yuan J.; Zhang Z.-G.; Peng H.; Li Y.; Zou Y. Achieving over 10% efficiency in a new acceptor ITTC and its blends with hexafluoroquinoxaline based polymers. J. Mater. Chem. A 2017, 5, 11286–11293. 10.1039/C7TA02486J. [DOI] [Google Scholar]
- Chen W.; Zhang Q. Recent progress in non-fullerene small molecule acceptors in organic solar cells (OSCs). J. Mater. Chem. C 2017, 5, 1275–1302. 10.1039/C6TC05066B. [DOI] [Google Scholar]
- Brouwer A. M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. 10.1351/PAC-REP-10-09-31. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.