

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) contribute to development of late-onset familial Parkinson's disease (PD), with clinical features of motor and cognitive dysfunction indistinguishable from sporadic PD. Calcium dysregulation plays an important role in PD pathogenesis, but the mechanisms of neurodegeneration remain unclear. Recent reports indicate enhanced excitatory neurotransmission in cortical neurons expressing mutant LRRK2, which occurs before the well-characterized phenotype of dendritic shortening. As mitochondria play a major role in the rapid buffering of cytosolic calcium, we hypothesized that altered mitochondrial calcium handling contributes to dendritic retraction elicited by the LRRK2-G2019S and -R1441C mutations. In primary mouse cortical neurons, we observed increased depolarization-induced mitochondrial calcium uptake. We found that expression of mutant LRRK2 elicited transcriptional upregulation of the mitochondrial calcium uniporter (MCU) and the mitochondrial calcium uptake 1 protein (MICU1) with no change in levels of the mitochondrial calcium antiporter NCLX. Elevated MCU and MICU1 were also observed in LRRK2-mutated patient fibroblasts, along with increased mitochondrial calcium uptake, and in postmortem brains of sporadic PD/PDD patients of both sexes. Transcriptional upregulation of MCU and MICU1 was caused by activation of the ERK1/2 (MAPK3/1) pathway. Inhibiting ERK1/2 conferred protection against mutant LRRK2-induced neurite shortening. Pharmacological inhibitors or RNAi knockdown of MCU attenuated mitochondrial calcium uptake and dendritic/neuritic shortening elicited by mutant LRRK2, whereas expression of a constitutively active mutant of NCLX that enhances calcium export from mitochondria was neuroprotective. These data suggest that an increased susceptibility to mitochondrial calcium dysregulation contributes to dendritic injury in mutant LRRK2 pathogenesis.
SIGNIFICANCE STATEMENT Cognitive dysfunction and dementia are common features of Parkinson's disease (PD), causing significant disability. Mutations in LRRK2 represent the most common known genetic cause of PD. We found that PD-linked LRRK2 mutations increased dendritic and mitochondrial calcium uptake in cortical neurons and familial PD patient fibroblasts, accompanied by increased expression of the mitochondrial calcium transporter MCU. Blocking the ERK1/2-dependent upregulation of MCU conferred protection against mutant LRRK2-elicited dendrite shortening, as did inhibiting MCU-mediated calcium import. Conversely, stimulating the export of calcium from mitochondria was also neuroprotective. These results implicate increased susceptibility to mitochondrial calcium overload in LRRK2-driven neurodegeneration, and suggest possible interventions that may slow the progression of cognitive dysfunction in PD.
Keywords: cortical neuron, dendrite shortening, Lewy body dementia, LRRK2, mitochondrial calcium, mitophagy
Introduction
Mutations in the leucine-rich repeat kinase 2 (LRRK2) represent a common cause of familial Parkinson's disease (PD), the brainstem stage of Lewy body disease (LBD) (Zheng et al., 2010) while also contributing to risk of sporadic PD (Healy et al., 2008). Indeed, in most LRRK2 mutation carriers, the clinical and pathological features are indistinguishable from those of sporadic PD (Dickson et al., 2009). Substantia nigra neurons are vulnerable to Ca2+ dysregulation due to autonomous pacemaking activity, with mitochondrial mechanisms implicated (Guzman et al., 2010). In addition to motor symptoms, PD is characterized quite early in disease by olfactory, autonomic, and cognitive dysfunction, with nearly 80% progressing to dementia (PDD) within 8 years (Aarsland et al., 2003). The symptoms include impairment of concentration, judgment, and visual processing, accompanied by mood disorders and hallucinations. These features are distinct from Alzheimer disease, yet little is known of pathophysiological mechanisms contributing to vulnerability of the cerebral cortex in PD/PDD.
Lewy body pathology in LRRK2-related PD patient brains is highly correlated with clinical patterns of cortical-executive dysfunction characteristic of PD (Kalia et al., 2015). LRRK2, a multidomain protein with GTPase and kinase activities, localizes to membranous structures, including mitochondria and lysosomes (Biskup et al., 2006). LRRK2 phosphorylates Rab GTPases involved in vesicular transport (Steger et al., 2016) and regulates PKA localization in dendritic spines (Parisiadou et al., 2014). PD-associated LRRK2 mutations result in shortening and simplification of the dendritic tree in primary neurons (MacLeod et al., 2006; Plowey et al., 2008; Chan et al., 2011; Ramonet et al., 2011; Cherra et al., 2013; Reinhardt et al., 2013; Sepulveda et al., 2013) and in vivo (MacLeod et al., 2006; Winner et al., 2011). We previously found that LRRK2-G2019S or -R1441C mutations elicit increased excitatory synapse density and increased EPSPs in mouse cortical neurons (Plowey et al., 2014). Enhanced postsynaptic excitatory neurotransmission was also observed in LRRK2-G2019S transgenic hippocampal slices (Sweet et al., 2015). Interestingly, LRRK2-G2019S mice exhibit early hyperactivity and enhanced learning that transform into cognitive deficits with aging (Volta et al., 2015). This suggests that increased excitatory neurotransmission, although tolerated acutely, enhances vulnerability of mutant LRRK2-expressing neurons to neuritic degeneration through undefined mechanisms. We hypothesize that this enhanced vulnerability is mediated through dysregulation of mitochondrial Ca2+ homeostasis.
Mitochondrial dysfunction has emerged as an important factor in LRRK2-associated pathophysiology (Mortiboys et al., 2010; Wang et al., 2012; Cherra et al., 2013), but the specific mechanisms by which mutant LRRK2 promotes mitochondrial injury remain unknown. Mitochondria function to rapidly sequester intracellular Ca2+ released from internal stores or due to sudden influx of extracellular calcium (for review, see Carafoli, 2012). This is achieved through the mitochondrial calcium uniporter (MCU) (Baughman et al., 2011; De Stefani et al., 2011), balanced by release of imported Ca2+ back to the cytosol through a sodium/calcium antiporter (NCLX) (Palty et al., 2010) as other cellular calcium homeostasis mechanisms are engaged. In addition to MCU, regulatory proteins include Mitochondrial Calcium Uptake 1 (MICU1) and Mitochondrial Calcium Uptake 2 (MICU2) proteins that tune the thresholds for mitochondrial calcium uptake through MCU (Perocchi et al., 2010; Plovanich et al., 2013).
In the present study, we discovered that expression of mutant LRRK2 (R1441C and G2019S) increases cytosolic and mitochondrial Ca2+ levels upon KCl stimulation. The increase in mitochondrial Ca2+ uptake is dependent on LRRK2 kinase activity, which upregulates MCU and MICU1 expression through the ERK1/2 (MAPK3/1) pathway. Furthermore, several distinct genetic or pharmacologic strategies to normalize mitochondrial calcium fluxes, including expression of a constitutively active (CA) form of NCLX, conferred protection against mutant LRRK2-mediated dendrite retraction. These results provide a new mechanistic linkage between mitochondrial Ca2+ handling and mutant LRRK2 toxicity in PD/PDD-associated neurodegeneration.
Materials and Methods
Mouse primary neuron culture.
Timed pregnant female C57BL/6 mice were purchased from Charles River Laboratories. All procedures for the derivation of primary neuron cultures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Primary E14–E16 cortical neurons from male or female pups were isolated from cerebral cortices as described previously (Cherra et al., 2013; Dagda et al., 2014). Neurons were plated at 150,000 cells/cm2 in LabTek II coverglass chamber slides coated with poly-L-lysine (0.1 mg/ml). They were maintained in antibiotic-free Neurobasal medium supplemented with 2% B27 and 2 mm Glutamax (Invitrogen). Half of the media was replaced with fresh media every other day.
Cell culture, reagents, and treatments.
SH-SY5Y cells (ATCC catalog #CRL-2266, RRID:CVCL_0019) were purchased from ATCC. Human control fibroblasts (ND34769, 68-year-old female), and two familial LRRK2 patient-derived fibroblast cultures (ND33879, 66-year-old female, PDD with LRRK2-G2019S; and ND32975, 74-year-old female, PD with LRRK2-R1441G) were purchased from the NINDS Cell Repository at RUCDR Infinite Biologics. All cells were maintained in antibiotic-free DMEM (BioWhittaker) supplemented with 10% FBS (Mediatech), 2 mm l-glutamine, and 10 mm HEPES in humidified incubator at 37°C and 5% CO2. Cells were routinely passaged at 1:10 for maintenance. For neurite length analysis, cells were plated at a density of 50,000 cells/cm2 in LabTek II coverglass chamber slides (Thermo Fisher Scientific) in DMEM supplemented with 10 μm retinoic acid for 72 h to induce differentiation before transfection. Pharmacological treatments were added 24 h after transfection. For all other experiments, cells were plated in Biolite 6-well tissue culture plates at the same density (Thermo Fisher Scientific). Cells were transfected the next day for 48 h and treated as indicated in the figure legends. CGP-37157 (catalog #C8874) and U0126 (catalog #U120) were purchased from Sigma-Aldrich. Ru360 (catalog #557440), digitonin (catalog #300410), and ruthenium red (catalog #557450) were purchased from Calbiochem. BAPTA-AM (catalog #B1205) was purchased from Thermo Fisher Scientific.
Calcium and hydrogen peroxide imaging and analysis.
Mouse primary cortical neurons were plated at a density of 250,000 cells/coverslip on poly-L-lysine coated 25 mm glass coverslips (Thermo Fisher Scientific) in a 6-well plate and maintained in Neurobasal medium supplemented with 2% B27 and 2 mm Glutamax (Invitrogen). The following genetically encoded calcium sensors, RCaMP, gift of Loren L. Looger (Howard Hughes Medical Institute Janelia Research Campus, Ashburn, Virginia) (Akerboom et al., 2013) and 2mtGCaMP6m, gift of Diego De Stefani (University of Padova, Padova, Italy) (Patron et al., 2014), were cotransfected with blue fluorescent protein (BFP), gift of Erik Snapp (Howard Hughes Medical Institute Janelia Research Campus, Ashburn, Virginia) (Costantini et al., 2015). For intracellular hydrogen peroxide imaging, neurons were cotransfected with either pHyPer-cyto (catalog #FP941) or pHyPer-dMito (catalog #FP942; Evrogen) and BFP. Neurons were transfected at 7 DIV and imaged 7 d after transfection in imaging buffer containing the following (in mm): 126 NaCl, 5.4 KCl, 0.8 MgCl2, 20 HEPES, 1.8 CaCl2 and 15 glucose, pH 7.4 adjusted with NaOH, supplemented with 0.1% BSA. Neurons were washed 3 times with imaging buffer, and coverslips were mounted into Attoflour cell imaging chamber (catalog #A7816, Invitrogen) and placed on a heated stage (Tokai Hit).
Calcium imaging in digitonin-permeabilized primary neurons or PD patient fibroblasts was conducted in “intracellular-like” imaging buffer as described previously (Brustovetsky et al., 2009; Palty et al., 2010) containing the following (in mm): 125 KCl, 0.5 MgCl2 3 KH2PO4, 3 pyruvate, 1 malate, 0.01 EGTA, 10 HEPES, and 220 sucrose, pH adjusted to 7.4 with KOH. Digitonin concentration and incubation times were optimized to achieve plasma membrane permeabilization without affecting mitochondrial membrane potential. Neurons or fibroblasts were permeabilized by incubating for 10 min at 37°C in imaging buffer containing 2 μm digitonin, washed 2×, and then placed in imaging buffer without digitonin. Time-lapse images were acquired with A1R confocal microscope (Nikon Instruments) every 5 s for neurons or 5 frames/s for fibroblasts in fast scan Galvano mode at 256 × 256 resolution in each channel. Images were analyzed with NIS-Elements Basic Research software (Nikon Instruments).
To stimulate Ca2+ flux in intact neurons, 40 mm KCl was added at the indicated time point. For digitonin-permeabilized neurons or fibroblasts, a bolus of 50 μm CaCl2 was added. In intact neurons, intensities of RCaMP and 2mtGCaMP6m signals in dendrites were normalized to adjacent BFP signal to control for possible focal drift. Similarly, intensity changes in cytosolic HyPer and mitochondrial HyPer were normalized to RFP signal. In fibroblasts, 2mtGCaMP6m intensity was normalized to dsRed targeted to mitochondria (mito-dsRed). Changes in cytosolic and mitochondrial Ca2+ or hydrogen peroxide signals were calculated using the following equation: (F1/Fo), where F1 is the fluorescence intensity of the image and Fo is the intensity of the first image. Maximum response in intensity from baseline (ΔF) was determined as maximum intensity after stimulation minus baseline intensity before stimulation, expressed as percentage increase.
Plasmid and siRNA transfections.
Mouse primary cortical neurons were cotransfected at 7 DIV with HA-tagged LRRK2 and the indicated calcium or hydrogen peroxide sensors at a 15:1 mass ratio (∼4:1 molar ratio) using Lipofectamine 2000 as described previously (Cherra et al., 2013). These conditions routinely yielded >90% cotransfection. For intracellular Ca2+ or ROS analysis, cells were imaged at 7 d after transfection (14 DIV). For dendritic length analysis, cells were fixed and immunostained 14 d after transfection (21 DIV). Patient fibroblasts were transfected with 500 ng total plasmid/well with 2mtGCaMP6m and mito-dsRed at a 4:1 mass ratio. Cells expressing both plasmids were imaged 48 h after transfection.
For neurite length analysis, RA-differentiated SH-SY5Y cells were cotransfected as described previously (Plowey et al., 2008). Generation and details of GFP-tagged ERK plasmids (wild-type [WT], CA, and kinase dead [KD]) have been previously described (Dagda et al., 2008). Cells were fixed with 4% PFA and processed for immunofluorescence 48 h after transfection. For biochemical analyses, the 4D-Nucleofector system (Lonza) was used according to the manufacturer's instructions. Cells were harvested for Western blot 48 h after nucleofection.
Small interfering RNA (siRNA) transfections were performed as described previously (Zhu et al., 2007). siRNA targeting human MCU (5′-CAGUUCACACUCAAGCCUCdTdT-3′; 50 nm, Sigma-Aldrich) was cotransfected with LRRK2 plasmids using 0.1% Lipofectamine 2000 reagent. Cotransfection efficiency was >95% as determined by using Cy3-conjugated scrambled oligonucleotide (20 nm; Dharmacon). A random nontargeting RNA oligonucleotide pool was used as control. Knockdown efficiency was determined by quantitative RT-PCR and immunoblot analysis.
Immunoblot analysis.
SH-SY5Y cells or PD patient fibroblasts were lysed in buffer containing the following (in mm): 150 NaCl, 5 EDTA, 25 HEPES, 10% glycerol, and 1% Triton X-100 supplemented with protease inhibitors as described previously (Cherra et al., 2013). Equal amounts of protein, as determined by Coomassie Plus Protein Assay (Thermo Scientific), were resolved on a 10% gel by SDS-PAGE and transferred to PVDF membrane. Membranes were blocked with 5% nonfat milk and probed with rabbit anti-MCU (catalog #HPA016480, 1:500, Sigma-Aldrich; RRID:AB_1233699), rabbit anti-MICU1 (catalog #HPA037480, 1:500, Sigma-Aldrich; RRID:AB_106969334), mouse anti-HA (catalog #16B12, 1:1000, Biolegend), rabbit anti-EFHA1/MICU2 (catalog #ab101645, 1:1000, Abcam; RRID:AB_10711219), rabbit-anti p-ERK1/2 (catalog #E7028, 1:1000, Sigma-Aldrich; RRID:AB_259347), rabbit anti-LRRK2 (catalog #C41-2, 1:1000, MJFF), rabbit total-ERK (catalog #06-182, 1:10,000, Millipore), a custom rabbit anti-NCLX produced in the laboratory of Dr. Seklar (Palty et al., 2010), rabbit anti-GAPDH (catalog #ab37168, 1:5000, Abcam;RRID:AB_732652), and rabbit anti-β-tubulin (catalog #ab6046, 1:2000, Abcam; RRID:AB_2210370) overnight with gentle agitation at 4°C. Immunoreactive bands were detected using anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (GE Healthcare) followed by exposure to ECL solution. Images were acquired using the Odyssey Fc imaging system (Li-Cor) for densitometry using Image Studio software (Li-Cor).
Flash frozen mid-frontal cortical specimens were obtained from the University of Pittsburgh Brain Bank, following standardized premortem neurological and postmortem neuropathological assessment. Diagnoses were confirmed and Braak tangle and Braak PD staging performed by the study neuropathologist (C.T.C.) as previously described (Chu et al., 2000; Alafuzoff et al., 2009). The study design was reviewed and approved by the University of Pittsburgh Committee for Oversight of Research Involving the Dead. Mid-frontal cortex from 8 PDD patients and 6 control subjects, matched for age, sex, and postmortem intervals (Table 1), was used for analysis.
Table 1.
Clinical and pathological characteristics of human postmortem subjects
| Age (yr) | Male:female | Postmortem time (h) | Braak AD stage | Braak PD stage | |
|---|---|---|---|---|---|
| Control | 77.3 ± 10.3 | 3:3 | 9.3 ± 3.5 | 1.8 ± 0.3 | 0 ± 0 |
| PDD | 77.0 ± 8.7 | 4:4 | 7 ± 3.5 | 2 ± 0.5 | 5.0 ± 0.8 |
| Statistics | p = 0.95 (t test) | NA | p = 0.24 (t test) | p = 0.69 (χ2) | NA |
Indirect immunofluorescence and neurite/dendrite length analysis.
Mouse primary cortical neurons or SH-SY5Y cells were fixed with 4% PFA and permeabilized with 0.1% Triton X-100 in PBS. Cells were blocked with Superblock Buffer (catalog #37515, Thermo Scientific). Transfected cells were identified by staining with rabbit anti-GFP (catalog #A6455, 1:1000, Invitrogen, RRID:AB_221570) and AlexaFlour-488-conjugated goat anti-rabbit IgG (catalog #A-11008, Thermo Fisher Scientific). Cells were imaged on an Olympus IX71 microscope with Microsuite Five imaging software (Olympus America). Color images were extracted to 8-bit greyscale images for quantification using NIH Image J supplemented with the Neuron J plug-in. Mean neurite length/cell (SH-SY5Y cells) and total dendritic length/neuron (mouse primary cortical neuron) were measured as described previously (Plowey et al., 2008; Cherra et al., 2013), using the intensity-tracing algorithm in ImageJ (Chu et al., 2009).
To assess autophagy or mitophagy, SH-SY5Y cells were cotransfected with GFP-LC3 and pcDNA or LRRK2 constructs. Twenty-four hours after transfection, Ru360 (10 μm) or vehicle was added for another 24 h, and then cells were fixed and immunostained for the mitochondrial protein HSP60 (catalog #611563, BD Biosciences, RRID:AB_399099). Images were obtained with Nikon A1 confocal microscope at 60× magnification. Two-dimensional projections were created from Z stacks (12-μm-thick sections from 0.25-μm-thick slices) using Nikon Elements software. Percentage mitophagy was quantified using the “mitophagy” ImageJ plug-in (Zhu et al., 2011). Numbers of GFP-LC3 puncta/cell and numbers of HSP60-colocalized GFP-LC3 puncta were quantified as previously described (Chu et al., 2013), analyzing at least 20 cells/condition from two independent experiments.
Paraffin-embedded midbrain sections (5 μm thick) were immunostained as previously described (Zhu et al., 2003), using rabbit anti-phospho ERK (Sigma-Aldrich) at 1:2500 followed by tyramide signal amplification (PerkinElmer). Immunoreactivity was visualized using the Nova Red chromagen (Vector Labs).
RNA isolation and quantitative RT-PCR.
Total RNA was isolated from SH-SY5Y cells or fibroblasts using RNeasy kit (QIAGEN). A total of 1 μg of RNA was used to generate cDNA using the GeneAmp RNA PCR kit (Invitrogen). MCU expression was analyzed using SYBR green (Sigma-Aldrich) primers (forward, 5′-GGTACGAATTGAGATTAGCAG-3′; reverse, 5′-GTGATGAAGTATGTTACTGGC-3′) normalized to GAPDH (forward, 5′-ACAGTTGCCATGTAGACC-3′; reverse, 5′-TTTTTGGTTGAGCACAGG-3′). NCLX (probe ID:Hs00227951_m1), MICU1 (probe ID:Hs00246104_m1), MICU2 (probe ID:Hs00381563_m1) expression was determined using TaqMan probe and normalized to GAPDH (4333764F) or ACTB (β-actin; 4333762F), all purchased from Thermo Fisher Scientific.
Statistics.
Data are mean ± SEM, except for mutant LRRK2 patient materials, which were not analyzed statistically (N < 3). Parametric data were analyzed by Student's t test for two-group comparisons or ANOVA followed by post hoc t test with Bonferroni correction for multigroup comparisons. p values shown were multiplied by the Bonferroni correction factor. Nonparametric data were analyzed using (χ2).
Results
Mutant LRRK2 modulates MCU expression
Mitochondrial calcium uptake is mediated by activity of MCU (Raffaello et al., 2013), whereas calcium antiporters, such as NCLX, mediate mitochondrial calcium release (Palty et al., 2010). To determine whether or not LRRK2 affected expression of MCU or NCLX, we performed quantitative RT-PCR on SH-SY5Y cells transfected with HA-tagged LRRK2 constructs (Fig. 1A). SH-SY5Y cells transfected with PD-associated LRRK2 mutants (-G2019S and -R1441C) 48 h before analysis showed increased MCU mRNA expression compared with cells transfected with pcDNA (vector control), whereas no changes were observed for NCLX mRNA (Fig. 1A). Significant elevations of MCU mRNA were blocked by the LRRK2-In-1 inhibitor and also observed at 24 and 72 h after transfection (data not shown). Similar results were observed whether GAPDH or ACTB was used as reference gene (see Fig. 6). Increases in MCU protein expression were observed in SH-SY5Y cells expressing LRRK2-G2019S or -R1441C (Fig. 1B,C), along with an increase in MICU1, another component of the MCU complex (Perocchi et al., 2010) (Fig. 1B,D). There were no changes in expression of MICU2 protein, which plays a gatekeeper role limiting calcium uptake by MCU (Patron et al., 2014) (Fig. 1B). These results suggested that PD-linked LRRK2 mutants may regulate MCU complex activity.
Figure 1.

PD-associated LRRK2 mutants modulate MCU, but not NCLX, expression, and inhibiting MCU prevents mutant LRRK2-mediated neurite/dendrite retraction. A, PD-associated LRRK2 mutants, G2019S (GS) and R1441C (RC), elicit increased MCU mRNA expression in SH-SY5Y cells at 48 h after transfection, which is not seen in cells expressing vector (pcDNA), LRRK2-WT, or the kinase inactive K1906M (KM) mutant. No change in NCLX mRNA expression was observed in the same samples. *p = 0.009, MCU (GS vs pcDNA); p = 0.014, MCU (RC vs pcDNA). B, Representative Western blot reveals increased MCU protein expression in SH-SY5Y cells expressing the LRRK2-GS and -RC mutants compared with pcDNA, LRRK2-WT, or -KM-transfected cells. MICU1 expression was also increased, but there were no changes in MICU2. Densitometry normalized to GAPDH is shown for (C) MCU (*p = 0.013, GS vs pcDNA; p = 0.008, RC vs pcDNA) and (D) MICU1 (*p = 0.0035, GS vs pcDNA; p = 0.012, RC vs pcDNA). E, RA-differentiated SH-SY5Y cells were transfected with pcDNA vector or the indicated forms of LRRK2. The inhibitors Ru360, cyclosporine A, CGP, or vehicle control were added for the last 24 h before measurement of mean neurite length/cell as described in Materials and Methods. *p = 0.003, Veh (GS/RC vs pcDNA). &p = 0.043, pcDNA (CGP vs Veh). #p = 0.045, GS (Ru360 vs Veh); p = 0.026, RC (Ru360 vs Veh). F, SH-SY5Y cells were mock-transfected or transfected with control siRNA (siCtrl) or siRNA targeting human MCU (siMCU). After 48 h, cells were harvested and knockdown was verified by quantitative RT-PCR (top left, †p = 0.013) and by immunoblot (top right). Middle, RA-differentiated SH-SY5Y cells were cotransfected with pcDNA and siCtrl/siMCU or with the indicated forms of LRRK2 and siCtrl/siMCU. Mean neurite length/cell was measured as described in Materials and Methods. *p = 0.0452, siCtrl (GS vs pcDNA); p = 0.0439, siCtrl (RC vs pcDNA). #p = 0.0419, GS (siMCU vs siCtrl); p = 0.0428, RC (siMCU vs siCtrl). Bottom, Representative images of RA-differentiated SH-SY5Y cells show restoration of neurite length by siRNA knockdown of MCU in cells expressing LRRK2-GS and -RC. Scale bar, 25 μm. G, Mouse primary cortical neurons were cotransfected with GFP and either pcDNA or LRRK2 mutants at 7 DIV. The indicated inhibitors were added at 8 DIV and total dendritic length/neuron measured at 15 DIV. *p = 0.0045, Veh (GS/RC vs pcDNA). &p = 0.047, pcDNA (CGP vs Veh). #p = 0.005, GS/RC (Ru360 vs Veh). A, C–G, ANOVA followed by post hoc Bonferroni-corrected t test.
Figure 6.

ERK1/2 inhibition prevents increased MCU and MICU1 expression and protects against mutant LRRK2-mediated neurite retraction. A–D, SH-SY5Y cells were transfected with pcDNA and LRRK2 constructs using nucleofection and treated with vehicle (DMSO) or the MEK inhibitor U0126 (10 μm). After 48 h, mRNA was studied using quantitative RT-PCR and normalized to β-actin (ACTB). A, MCU mRNA expression. *p = 0.0006, Veh (GS vs pcDNA); p = 0.0017, Veh (RC vs pcDNA). #p = 0.0091, GS (U0126 vs Veh); p = 0.002, RC (U0126 vs Veh). B, MICU1 mRNA expression. *p = 0.044, Veh (GS vs pcDNA); p = 0.034, Veh (RC vs pcDNA). #p = 0.041, GS (U0126 vs Veh); p = 0.03, RC (U0126 vs Veh). C, D, MICU2 and NCLX mRNA expression normalized to β-actin. E, In parallel experiments, SH-SY5Y cells were lysed and proteins resolved using 10% SDS-PAGE for immunoblot studies. Representative blot from two independent experiments. F, Mean neurite length/cell in RA-differentiated SH-SY5Y cells transfected with pcDNA or LRRK2 plasmids, after treatment with DMSO (Veh) or 10 μm U0126. *p = 0.041, Veh (GS vs pcDNA); p = 0.045, Veh (RC vs pcDNA). #p = 0.00969, GS (U0126 vs Veh); p = 0.01593, RC (U0126 vs Veh). G, MCU mRNA expression in SH-SY5Y transfected with GFP or the indicated GFP tagged ERK2 constructs, after treatment with Veh or 10 μm U0126. *p = 0.037, Veh (ERK-WT vs GFP); p = 0.0001, Veh (ERK-CA vs GFP). #p = 0.03, ERK-WT (U0126 vs Veh); p = 0.000246, ERK-CA (U0126 vs Veh). H, Mean neurite length/cell in RA-differentiated SH-SY5Y cells transfected with GFP or the indicated constructs of ERK2-GFP. *p = 0.0018, CA-ERK2-GFP versus GFP; p = 0.047, WT-ERK2-GFP versus GFP. A, B, F–H, ANOVA followed by post hoc Bonferroni-corrected t test.
Inhibiting mitochondrial calcium uptake prevents LRRK2 mutant-induced neurite shortening
Whereas the G2019S and R1441C/G mutations differ with regard to effects on kinase activity (West et al., 2005; Jaleel et al., 2007) or protein aggregation (Nichols et al., 2010; Kett et al., 2012), kinase domain and Roc-COR mutations of LRRK2 converge on a common phenotype of dendritic/neuritic shortening and simplification (MacLeod et al., 2006; Cherra et al., 2010, 2013; Plowey et al., 2014). This dendrite shortening may be due to either degeneration (Yao et al., 2010) or to reduced outgrowth during development (Parisiadou et al., 2009), although the delayed retraction that occurs in the current system supports a degenerative process (Plowey et al., 2014). Given that NMDA receptor antagonists and L-type calcium channel inhibitors can reverse mutant LRRK2-induced autophagy, mitophagy, and dendritic/neuritic shortening (Cherra et al., 2013; Plowey et al., 2014), calcium dysregulation plays a causal role. To determine whether the uptake of calcium by mitochondria represents a protective or a pathogenic response, we treated differentiated SH-SY5Y cells expressing mutant LRRK2 with a panel of inhibitors that modulate mitochondrial calcium transport. Ru360, a cell-permeable inhibitor of MCU, prevented neurite shortening in differentiated SH-SY5Y cells (Fig. 1E). In contrast, treatment with cyclosporine A to prevent the mitochondrial permeability transition did not protect against mutant LRRK2-mediated neurite retraction. Inhibition of mitochondrial calcium export using the NCLX inhibitor CGP37157 (CGP) not only failed to protect against mutant LRRK2-induced neurite shortening, but also elicited a small, but significant, degree of neurite shortening in vector control transfected cells relative to vehicle. Together, these data indicate that increased mitochondrial calcium levels contribute to the pathogenesis of LRRK2-induced neurite shortening.
To control for possible off-target effects of Ru360, we used siRNA to knockdown MCU mRNA (Fig. 1F, top left) and protein (Fig. 1F, top right). Similar to Ru360, knockdown of MCU prevented mutant LRRK2-induced neurite retraction in differentiated SH-SY 5Y cells (Fig. 1F, graph and representative images). In mouse primary cortical neurons, inhibition of MCU with Ru360 was also neuroprotective (Fig. 1G), whereas neither inhibitors of mitochondrial permeability transition nor inhibiting calcium efflux through NCLX prevented mutant LRRK2-mediated dendrite retraction. These results suggest that mutant LRRK2 not only modulates cytosolic calcium homeostasis (Cherra et al., 2013), but also mitochondrial calcium handling through regulation of MCU.
Mutant LRRK2 mediates enhanced mitochondrial calcium uptake through MCU
To directly visualize dendritic mitochondrial calcium uptake, we conducted live cell calcium imaging in mouse primary cortical neurons cotransfected with LRRK2 mutants or controls, two genetically encoded cytosolic and mitochondrial calcium sensors with spectrally distinct emissions (RCaMP and 2mt-GCaMP6m, respectively) and BFP. Live time-lapse confocal imaging shows that upon neuronal stimulation with KCl (black arrows), the increased calcium influx into the dendrites of neurons expressing LRRK2 mutants (G2019S and R1441C) (Fig. 2A,E) is accompanied by a greater degree of calcium uptake by dendritic mitochondria (Fig. 2C,F) than in pcDNA- or WT LRRK2- transfected controls. As expected, Ru360 treatment did not block the increased cytosolic calcium uptake in neurons expressing mutant LRRK2 (Fig. 2B,E) but reversed the increases in mitochondrial calcium uptake induced by mutant LRRK2 (Fig. 2D,F).
Figure 2.
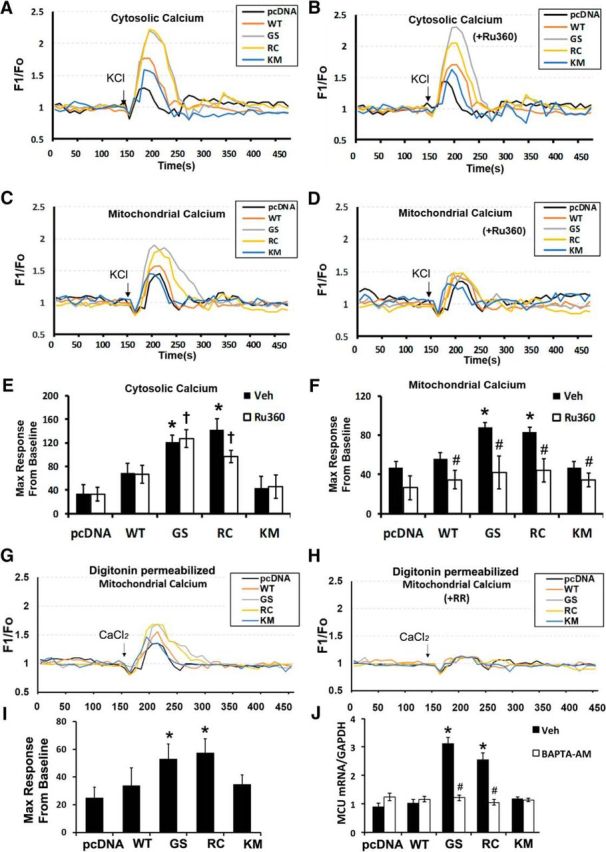
Mutant LRRK2 enhances MCU-mediated mitochondrial calcium uptake in primary cortical neurons. A–F, Mouse primary cortical neurons were cotransfected with the indicated LRRK2 constructs and cytosolic and mitochondrial calcium sensors. PD-associated LRRK2 mutants showed altered cytosolic (A) and mitochondrial (C) calcium levels upon stimulation with 40 mm KCl (black arrow) added after 150 s of baseline recording. Changes in cytosolic and mitochondrial calcium levels were recorded without Ru360 (A, C) or in the presence of Ru360 (B, D) as described in Materials and Methods. Each trace represents an average intensity of calcium recording from 8 to 10 different neurons. E, F, Data were quantified as the percentage increase over baseline of the maximum F1/FO, and averaged across 8–10 neurons. Error bars indicate SEM. *p = 0.002, pcDNA versus GS/RC in the presence of vehicle (black bars). †p = 0.01, pcDNA versus GS/RC in the presence of Ru360 (white bars). #p = 0.0489/p = 0.0298/p = 0.0402/p = 0.047, WT/GS/RC/KM, respectively (Ru360 vs Veh). G, H, Mitochondrial calcium levels were measured in digitonin-permeabilized mouse primary cortical neurons expressing GCaMP6m in the absence (G) or presence of ruthenium red (H, RR, 2 μm). Calcium (CaCl2, 50 μm) was added to increase the calcium concentration in the imaging chamber (black arrow). I, Quantification of GCaMP6m traces in digitonin-permeabilized neurons in G. *p = 0.013, GS versus pcDNA; p = 0.0083, RC versus pcDNA. J, Chelation of intracellular calcium with 5 μm BAPTA-AM in SH-SY5Y cells transfected with pcDNA or the indicated constructs of LRRK2 prevents MCU mRNA upregulation. Error bars indicate SEM from three independent experiments. *p = 0.02, Veh (GS vs pcDNA); p = 0.0202, Veh (RC vs pcDNA). #p = 0.0011, GS (BAPTA-AM vs Veh); p = 0.003, RC (BAPTA-AM vs Veh). E, F, I, J, ANOVA followed by post hoc Bonferroni-corrected t test.
To further determine whether increased expression of MCU directly modulated mitochondrial calcium uptake, digitonin treatment of mouse primary cortical neurons was used to eliminate differences in “cytosolic” calcium levels between mutant and control neurons. Upon addition of exogenous calcium (CaCl2, 50 μm), digitonin-permeabilized neurons expressing mutant LRRK2 showed significant increases in mitochondrial calcium uptake (Fig. 2G,I), which were inhibited by pretreatment with ruthenium red (Fig. 2H). To determine whether increased mutant LRRK2-mediated intracellular calcium levels were involved in triggering increased MCU expression, pcDNA- or LRRK2-transfected SH-SY5Y cells were treated with DMSO or 5 μm of BAPTA-AM, and mRNA levels determined by quantitative RT-PCR. The increase in MCU mRNA expression in mutant LRRK2-expressing cells was reversed by BAPTA-AM treatment (Fig. 2J). These results confirm that the neuroprotective effect of Ru360 were not due to off-target effects on cytosolic calcium uptake, and further support a causative role for mitochondrial calcium dysregulation due in part to enhanced expression of MCU in the mutant LRRK2 phenotype.
Mutant LRRK2-mediated mitophagy is attenuated by inhibition of mitochondrial calcium uptake without significant changes in intracellular ROS generation
An increase in kinase activity of LRRK2 has been associated with increases in ROS generation (Heo et al., 2010). Similar to previous results, we found a significant increase in cytosolic ROS in LRRK2-G2019S-transfected neurons, with nonsignificant trends in LRRK2-R1441C and LRRK2-WT-transfected neurons (Fig. 3A, top traces, B; GS > RC > WT), upon stimulation with 40 mm KCl. There were no changes in mitochondrial ROS generation upon KCl stimulation (Fig. 3A, bottom traces, C).
Figure 3.

Mutant LRRK2-mediated mitophagy is attenuated by inhibition of mitochondrial calcium uptake without significant changes in intracellular ROS generation. A, Mouse primary cortical neurons were transfected with the indicated LRRK2 constructs and either cytosolic or mitochondrial hydrogen peroxide sensors and monitored after addition of KCl. Each trace represents an average intensity of calcium recording from 8 to 10 different neurons. B, The LRRK2–G2019S mutant showed a moderate, but significant, increase in cytosolic ROS levels upon stimulation with 40 mm KCl. *p = 0.026, GS versus pcDNA. Not significant, p = 0.056, RC versus pcDNA. C, There were no significant changes in signal from the mitochondrially targeted sensor. D, The LRRK2-stimulated autophagy, as monitored by increase in numbers of GFP-LC3 puncta/cell, was not modulated by MCU inhibition. *p = 0.0064, Veh (WT vs pcDNA); p = 0.0043, Veh (GS vs pcDNA); p = 0.0053, Veh (RC vs pcDNA). Mutant LRRK2-mediated mitophagy was significantly attenuated by inhibition of MCU, whether monitored by the percentage of GFP-LC3 puncta colocalizing with HSP60-stained mitochondria (E) or the increase in numbers mitochondrially colocalized GFP-LC3 puncta (F). E, *p = 0.0045, Veh (GS vs pcDNA); p = 0.047, Veh (RC vs pcDNA). #p = 0.017, GS (Ru360 vs Veh); p = 0.0028, RC (Ru360 vs Veh). F, *p = 0.0044, Veh (GS vs pcDNA); p = 0.0026, Veh (RC vs pcDNA). #p = 0.0093, GS (Ru360 vs Veh); p = 0.0215, RC (Ru360 vs Veh). Data are mean ± SEM from three independent experiments. B, D–F, ANOVA followed by post hoc Bonferroni-corrected t test.
WT and mutant LRRK2 have been previously linked to changes in autophagy and mitophagy (Plowey et al., 2008; Alegre-Abarrategui et al., 2009; Gómez-Suaga et al., 2012a; Bravo-San Pedro et al., 2013; Cherra et al., 2013; Schapansky et al., 2014). The LRRK2-mediated increase in autophagosome numbers was not affected by Ru360 (Fig. 3D). However, LRRK2-mediated increases in mitophagy, as evidenced by targeting of mitochondria to GFP-LC3 puncta, were significantly attenuated upon inhibition of MCU (Fig. 3E,F). As inhibition of autophagy, or mitochondrial clearance by autophagy, has previously been shown to protect against mutant LRRK2-induced neuritic/dendritic shortening (Cherra et al., 2010), these data suggest that MCU-mediated mitochondrial calcium uptake plays an upstream role in triggering mitophagy, which contributes to neuritic/dendritic shortening.
Fibroblasts from PD patients with mutations in LRRK2 show increased mitochondrial calcium uptake accompanied by increased MCU and MICU1 expression
We obtained PD patient-derived fibroblasts harboring LRRK2-R1441G and -G2019S mutations to study whether similar changes are observed in the presence of endogenous levels of mutant LRRK2 (Fig. 4A–C). These cells also showed increased mitochondrial calcium uptake (Fig. 4A,B), accompanied by increases in MCU and MICU1 protein expression, but not MICU2 or NCLX (Fig. 4C).
Figure 4.
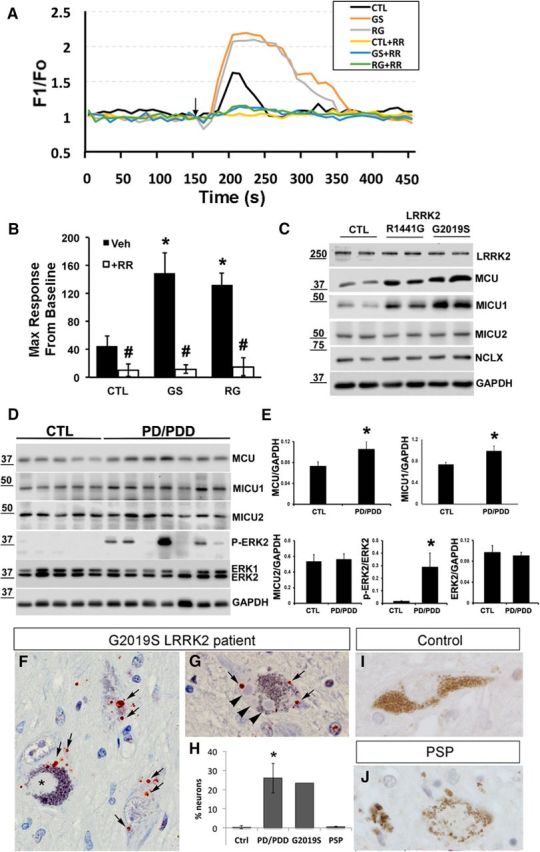
Patient-derived LRRK2-mutated fibroblasts and human PD/PDD brains show upregulation of MCU and MICU1 in conjunction with increased mitochondrial calcium uptake and elevations in phospho-ERK2. A, Human fibroblasts were cotransfected with 2mt-GCaMP6m and mito-dsRed. Mitochondrial calcium uptake was measured in digitonin-permeabilized fibroblast in the absence or presence of ruthenium red (RR, 10 μm). A bolus of CaCl2 was added at the indicated time point (black arrow). Each trace represents an average intensity of 8–10 fibroblasts. B, Data were quantified as the percentage increase over baseline of the maximum F1/FO, and averaged across 8–10 fibroblasts. Error bars indicate SEM. *p = 8.89257 × 10−9 Veh (CTL vs GS); p = 5.19185 × 10−11 Veh (CTL vs RG). #p = 6.56837 × 10−7 CTL (Veh vs RR); p = 1.73223 × 10−9 GS (Veh vs RR); p = 7.66794 × 10−13 RG (Veh vs RR). C, Western blot of patient-derived fibroblasts carrying LRRK2 mutations (-R1441G and -G2019S) shows increased MCU and MICU1 expression, with no changes in MICU2 and NCLX. D, Representative Western blot of human mid-frontal cortices showing increased expression of MCU, MICU1, and ERK2 phosphorylated at its activation motif (p-ERK2). E, Densitometric quantification of MCU (p = 0.0084 vs CTL), MICU1 (p = 0.044 vs CTL), MICU2 and ERK2 expression normalized to GAPDH, and p-ERK2 normalized to ERK2 (p = 0.001 vs CTL). F–J, Immunohistochemistry for phosphorylated ERK1/2 (arrows, red chromagen) in midbrain neurons of a G2019S PD/PDD patient compared with healthy control and progressive supranuclear palsy (PSP) midbrain neurons. As previously reported in sporadic PD/PDD (Zhu et al., 2003), cytoplasmic phospho-ERK1/2 was observed in neurons without synuclein pathology as well as those with pale bodies (F, asterisk) and or mature Lewy bodies (G, arrowheads). In contrast, this pattern of staining was not prominent in age-matched control brains (I) or in tangle-bearing midbrain neurons of patients with PSP (J). H, Quantification of the percentage of SNc neurons exhibiting ERK phosphorylation in Control (Ctrl, n = 6), PD/PDD (n = 9), G2019S (n = 1), and PSP (n = 3) cases. *p = 1.5 × 10−5, PD/PDD versus Ctrl. E, Student's t test. B, H, ANOVA followed by post hoc Bonferroni-corrected t test.
Human PD/PDD brains show upregulation of MCU and MICU1 in conjunction with elevations in phosphorylated ERK2
To determine whether elevated MCU and MICU1 protein are also observed in sporadic human PD/PDD brain tissues, we performed Western blot analysis on frozen mid-frontal cortical samples. Similar to cells expressing mutant LRRK2, we observed a significant increase in MCU and MICU1 protein levels in PD/PDD human brain samples compared with age-matched control cases (Fig. 4D,E). As observed previously in PD/PDD cases (Zhu et al., 2003), there were also increases in phosphorylation of extracellular signal-regulated protein kinases, particularly ERK2, in the majority of the PD/PDD samples with no difference in expression of total ERK1/2 (Fig. 4D,E). Although no frozen brain tissue was available from patients with LRRK2 mutations, we were able to examine paraffin-embedded sections from a single transitional LBD patient with the G2019S mutation in LRRK2 (courtesy of Dr. Dennis Dickson). This revealed increases in phosphorylated ERK1/2 in degenerating neurons (Fig. 4F,G) in a punctate, cytoplasmic staining pattern similar to that previously reported in sporadic PD/PDD patients (Zhu et al., 2003), which are not observed in age-matched controls (Fig. 4H,I). Although ERK1/2 phosphorylation was observed in PD/PDD patients in neurons undergoing neurodegeneration, as evidenced by the presence of Lewy bodies, it was not observed in midbrain neurons with tau aggregates in patients with progressive supranuclear palsy, a different neurodegenerative disease characterized by degeneration of the same dopaminergic neuronal population (Fig. 4H,J). Moreover, previous studies of early stage “incidental” PD revealed a similar pattern of ERK1/2 phosphorylation in neurons lacking visible signs of pathology (Zhu et al., 2003). These observations suggest that ERK1/2 activation occurs early as well as late in LBDs, and is not simply a nonspecific consequence of neurodegeneration. Together, these pathological data further support a role for ERK1/2 and increased MCU expression in the pathogenesis of mutant LRRK2, and suggest that similar changes may occur in sporadic PD/PDD.
ERK1/2 inhibition prevents MCU and MICU1 upregulation and protects against mutant LRRK2-mediated neurite retraction
To further explore the role of ERK1/2 signaling in regulation of the MCU complex, mRNA expression of MCU, MICU1, MICU2, and NCLX was determined in LRRK2-mutated patient fibroblasts (Fig. 5) and in pcDNA or mutant LRRK2-transfected SH-SY5Y cells (Fig. 6). The mutant LRRK2-mediated increases in MCU (Figs. 5A, 6A) and MICU1 (Figs. 5B, 6B) expression were reversed in cells treated with the MEK inhibitor U0126, which prevents ERK1/2 activation. Consistent with the cell culture and human tissue Western blots discussed above, mutant LRRK2 expression did not increase MICU2 (Figs. 5C, 6C) or NCLX (Figs. 5D, 6D) mRNA expression.
Figure 5.

ERK1/2 inhibition prevents increased MCU and MICU1 expression in PD patient-derived fibroblasts. A–D, Control or PD patient-derived fibroblasts were treated with vehicle (DMSO) or the MEK inhibitor U0126 (10 μm) for 48 h. mRNA levels were studied using quantitative RT-PCR and normalized to β-actin (ACTB). A, MCU mRNA expression. *p = 0.004, Veh (GS vs CTL); p = 0.004, Veh (RG vs CTL). #p = 0.007, GS (U0126 vs Veh); p = 0.012, RG (U0126 vs Veh). B, MICU1 mRNA expression. *p = 0.0003, Veh (GS vs CTL); p = 0.0011, Veh (RG vs CTL). #p = 0.0004, GS (U0126 vs Veh); p = 0.0012, RG (U0126 vs Veh). C, D, MICU2 and NCLX mRNA expression normalized to β-actin. E, Left, Representative Western blots of human fibroblasts treated with vehicle or U0126 for 48 h. Right, Gel densitometry from 3 to 6 independent experiments. p-ERK2: *p = 0.027, Veh (RG vs CTL); p = 2.287 × 10−6 Veh (GS vs CTL). #p = 0.012, RG (Veh vs U0126); p = 8.518 × 10−8 GS (Veh vs U0126). MCU: *p = 0.009, Veh (RG vs CTL); p = 0.005, Veh (GS vs CTL). #p = 0.006, RG (Veh vs U0126); p = 0.004, GS (Veh vs U0126). MICU1: *p = 0.042, Veh (RG vs CTL); p = 0.0107, Veh (GS vs CTL). #p = 0.0495, RG (Veh vs U0126); p = 0.0086, GS (Veh vs U0126).
Previous studies have demonstrated that the ERK1/2 signaling pathway mediates several effects of mutant LRRK2 in differentiated SH-SY5Y cells, HEK cells, and PD-patient induced pluripotent stem cells (Plowey et al., 2008; Carballo-Carbajal et al., 2010; Reinhardt et al., 2013). In agreement with these previous studies, we found that, in transfected SH-SY5Y cells, LRRK2-G2019S and R1441C mutants increased ERK1/2 phosphorylation (Fig. 6E), whereas LRRK2-WT had a moderate effect. PD patient fibroblasts bearing endogenous G2019S and R1441G mutations also exhibited increased phospho-ERK (Fig. 5E). Furthermore, the increase in MCU expression could be reversed by treatment with U0126 (Figs. 5E, 6E), which also protected against the neurite-shortening phenotype (Fig. 6F). Interestingly, MCU mRNA expression was significantly upregulated in SH-SY5Y cells transfected with a ERK-CA construct, which was reversed by U0126 treatment (Fig. 6G). Moreover, ERK-CA was sufficient to significantly reduce neurite length (Fig. 6H).
Restoring mitochondrial calcium homeostasis is neuroprotective
To further investigate the possible role of mitochondrial calcium overload in the mutant LRRK2 phenotype, we sought another mechanism to alleviate increased mitochondrial calcium levels. In addition to inhibiting or reducing mitochondrial calcium uptake by inhibition or RNAi knockdown of MCU, we reasoned that, if the increased mitochondrial calcium was harmful, we should be able to achieve neuroprotection by other mechanisms to decrease mitochondrial calcium levels. NCLX plays an important role in the efflux of calcium out of mitochondria. We recently found that NCLX is activated by phosphorylation by protein kinase A (Kostic et al., 2015). To determine whether NCLX-WT or a phosphomimetic mutant NCLX (S258D) could rescue mutant LRRK2-mediated calcium homeostasis and neurite length, we co-overexpressed different forms of LRRK2 and NCLX in differentiated SH-SY5Y cells (Fig. 7A) and in primary cortical neurons (Fig. 7B). In both systems, expression of the CA NCLX-S258D significantly protected against LRRK2-G2019S and LRRK2-R1441C mediated neurite/dendrite shortening (Fig. 7A–C). As predicted by the CGP results, downregulation of NCLX activity by overexpression of the nonphosphorylatable NCLX mutant (S258A) or by shRNA-mediated knockdown of endogenous NCLX did not protect against the neurite/dendrite shortening phenotype. These results further establish that increased mitochondrial calcium flux contributes to pathogenic responses to mutant LRRK2, highlighting the neuroprotective potential of interventions that enhance mitochondrial calcium efflux in neurons.
Figure 7.

Restoring mitochondrial calcium homeostasis is neuroprotective. A, RA-differentiated SH-SY5Y cells were cotransfected with pcDNA or LRRK2 with the indicated NCLX constructs. Knockdown efficiency for shRNA targeting NCLX was 50 ± 5% (mean ± SD). *p = 0.0105 Vec (GS vs pcDNA); p = 0.0141 Vec (RC vs pcDNA). &p = 0.0153 pcDNA (shNCLX vs vector). #p = 0.0045 GS (NCLX-S258D vs vector); p = 0.0258 RC (NCLX-S258D vs vector). B, Mouse primary cortical neurons were cotransfected with the indicated plasmids at 7 DIV. Total dendritic length/neuron was quantified at 15 DIV. N = 30–35 neurons/condition from three independent experiments. *p = 0.0142 Vec (GS vs pcDNA); p = 0.0244 Vec (RC vs pcDNA). #p = 0.01 GS (NCLX-S258D vs vector); p = 0.01 RC (NCLX-S258D vs vector). A, B, ANOVA followed by post hoc Bonferroni-corrected t test. C, Representative images of neurons quantified in B. Scale bar, 100 μm.
Discussion
Mutations in LRRK2 have been linked to cytosolic and lysosomal Ca2+ dysregulation, with changes in mitochondrial calcium handling implicated (Gómez-Suaga et al., 2012b; Cherra et al., 2013; Bedford et al., 2016). Here, we show that the PD-associated G2019S and R1441C mutations in LRRK2 elicit ERK1/2-dependent transcriptional upregulation of MCU and MICU1. Similar ERK1/2-dependent increases in MCU and MICU1 expression were observed in PD patient fibroblasts with LRRK2-G2019S and -R1441G mutations. Elevated ERK1/2 phosphorylation and MCU/MICU1 expression were also observed in human PD/PDD postmortem brains. Cotransfection of mouse primary cortical neurons or PD patient fibroblasts with genetically encoded calcium sensors confirmed increases in MCU-mediated mitochondrial calcium uptake. Inhibiting ERK1/2 activation not only reversed changes in MCU mRNA but also prevented mutant LRRK2-mediated neurite retraction, suggesting a pathogenic role for increased mitochondrial calcium. To test this directly, we used several strategies to oppose mutant LRRK2-elicited increases in activity-triggered mitochondrial Ca2+. Pharmacological inhibition and RNAi knockdown of MCU were neuroprotective against mutant LRRK2-mediated neurite/dendrite retraction. Moreover, expression of CA NCLX-S258D, which promotes mitochondrial Ca2+ efflux (Kostic et al., 2015), protected against mutant LRRK2-mediated neurodegeneration. These data provide a new mechanistic linkage implicating LRRK2 and mitochondrial Ca2+ dyshomeostasis in the pathogenesis of PD/LBD.
Dysregulation of intracellular Ca2+ homeostasis plays a critical role in PD pathogenesis (Calì et al., 2014). SN neurons exhibit spontaneous pacemaking activity (Puopolo et al., 2007), and cortical neurons degenerate as patients progress from the executive dysfunction that is prevalent in PD to frank PDD (Aarsland et al., 2003). Both SN and cortical neurons express L-type voltage-gated channels, which contribute to Ca2+ influx during an action potential (Guzman et al., 2009). Moreover, L-type calcium channel inhibitors protect both neuron types in toxic (Ilijic et al., 2011) and genetic (Cherra et al., 2013) models of PD. Enhanced cytosolic calcium is a shared feature of mutant LRRK2 (Cherra et al., 2013; Hockey et al., 2015) and α-synuclein models (Danzer et al., 2007).
Earlier studies found that inhibiting Ca2+ uptake or enhancing chelation of intracellular Ca2+ protects against mutant LRRK2-mediated pathology (Gómez-Suaga et al., 2012b; Cherra et al., 2013). Increased calcium uptake by mitochondria in mutant LRRK2-expressing neurons could represent either an adaptive response or part of the pathogenic sequence. Together with prior studies showing increased excitatory neurotransmission in mutant LRRK2 cortical and hippocampal neurons (Beccano-Kelly et al., 2014; Plowey et al., 2014; Sweet et al., 2015), our data indicate that the pathogenic consequences of increased calcium on neuritic/dendritic structures are mediated by elevations in mitochondrial calcium. This fits with observations that increased mitochondrial Ca2+ uptake during excitatory stimuli promotes cell death (Pivovarova et al., 2004), whereas inhibition of mitochondrial Ca2+ uptake prevents Ca2+ overload and is neuroprotective in other injury systems (Peng et al., 1998; Qiu et al., 2013). Interestingly, the BAPTA studies indicate that increased cytosolic calcium, which is known to activate ERK1/2 (Lahne and Gale, 2008; Doyle et al., 2010), was essential for mutant LRRK2-mediated upregulation of MCU. In turn, MCU upregulation may decrease calcium-mediated inhibition of L-type calcium channels (Nita et al., 2014), potentially contributing to amplification of calcium dysregulation in mutant LRRK2 cells. Taken together, we propose that signaling pathways leading to upregulation of MCU are activated to compensate for increased cytosolic calcium but may represent a maladaptive response in the mutant LRRK2 setting.
It is important to note that we did not monitor cytosolic calcium in the soma, or the entire dendrite, but focused on cytosolic regions immediately adjacent to dendritic mitochondria. Also, although there were no changes in NCLX expression level, possible post-translational effects cannot be excluded. KCl-induced cytosolic calcium traces in the soma of mutant LRRK2-expressing neurons (Cherra et al., 2013) suggest that somatic calcium handling may also be affected by other mechanisms, such as lysosomal calcium dysregulation (Gómez-Suaga et al., 2012b). Neurons in particular exhibit compartmentalized signaling responses, and mutant LRRK2 affects the localization, and therefore substrate accessibility, of PKA in spines (Parisiadou et al., 2014). Interestingly, NCLX represents the second PKA phospho-target that protects against mutant LRRK2-mediated dendrite shortening, in addition to the autophagy protein LC3 (Cherra et al., 2010).
We previously found that increased excitatory synaptic density/activity occurs within the first few days after transfection with mutant LRRK2 (Plowey et al., 2014), in a similar time frame to upregulation of MCU mRNA observed here. This is followed by loss of dendritic mitochondria, which occurs before dendritic shortening elicited by mutant LRRK2 in primary cortical neurons (Cherra et al., 2013). Although there were no significant changes in mitochondrial ROS, mitochondrial calcium uptake through MCU was necessary for the induction of mitophagy by mutant LRRK2. Mitochondrial calcium overload may thus represent another trigger for selective mitophagy, in addition to ROS, depolarization and cardiolipin externalization (Ding et al., 2010; Matsuda et al., 2010; Chu et al., 2013). Together, these data suggest that alterations in mitochondrial calcium homeostasis can trigger mitophagy. The resulting loss in dendritic mitochondria contributes to dendritic remodeling, although additional mechanisms involving cytoskeletal dysregulation may also play a role (Jaleel et al., 2007; Parisiadou et al., 2009; Kett et al., 2012).
In further support of this mechanism are observations that preventing excess extracellular uptake or chelating intracellular calcium reverses effects of mutant LRRK2 on mitochondrial depolarization in dendrites and their loss via mitophagy (Cherra et al., 2013). Interestingly, LRRK2-G2019S acts to promote basal autophagy in human patient-derived fibroblasts (Bravo-San Pedro et al., 2013), accentuating autophagy in perisynaptic regions in Drosophila (Soukup et al., 2016). Inhibition of autophagy in turn prevents mutant LRRK2-mediated neurite/dendrite shortening (Plowey et al., 2008; Cherra et al., 2013). Both the G2019S and R1441C mutations selectively reduce mitochondrial content in dendrites, although neither affects axonal mitochondrial density (Cherra et al., 2013). This correlates with relatively selective effects on dendritic morphology, implicating enhanced neuronal excitability as a pathogenic factor for mutant LRRK2. As mitochondrial content is important for supporting dendrite extension and maintaining dendritic arbors, multiple mechanisms may account for enhanced vulnerability to calcium-driven synaptic-dendritic pathology.
MCU is the low-affinity mitochondrial Ca2+ uptake channel (Kirichok et al., 2004; Baughman et al., 2011; De Stefani et al., 2011) required for mitochondrial Ca2+ uptake (Pan et al., 2013). Its activity is regulated by MICU1 (Perocchi et al., 2010), MICU2 (Plovanich et al., 2013), EMRE (Sancak et al., 2013), MCUR1 (Mallilankaraman et al., 2012), and MCUb (Raffaello et al., 2013). Fine-tuning of mitochondrial Ca2+ uptake is regulated by MICU1 and MICU2 (Patron et al., 2014), with the former acting to stimulate MCU activity at high calcium concentrations whereas MICU2 prevents channel opening at lower calcium levels. Although decreased MICU2 expression could also result in enhanced mitochondrial calcium uptake, mutant LRRK2 did not affect MICU2 expression.
The ERK1/2 signaling pathway has been implicated as a mediator of mutant LRRK2 effects (Plowey et al., 2008; Reinhardt et al., 2013). Activated ERK1/2 may translocate to the nucleus (Chen et al., 1992) to phosphorylate and activate transcription factors (Lu and Xu, 2006) or to mitochondria as observed in degenerating PD neurons (Zhu et al., 2003). Although an elevation in MCU mRNA was not observed in a microarray study of induced pluripotent stem cell-differentiated DA neurons from 2 patients with G2019S-linked PD, this difference may relate to observations that mutant LRRK2 suppresses dopaminergic neurotransmission (Li et al., 2010; Melrose et al., 2010), whereas it stimulates glutamatergic neurotransmission (Plowey et al., 2014; Sweet et al., 2015). Interestingly, fibroblasts from PD/PDD patients with either G2019S or R1441G mutations also showed increased MCU and MICU1, but not MICU2 and NCLX, expression. Moreover, paraffin sections from a PD patient with the LRRK2-G2019S mutation reveal a similar pattern and degree of phospho-ERK1/2 staining as observed in several distinct cohorts of sporadic PD/PDD patients. Given the high degree of clinical and pathological similarity between dominant genetic forms of PD and sporadic PD, the ability of ERK1/2 to modulate mitochondrial calcium uptake through transcriptional regulation of MCU may be applicable to other forms of PD as well.
Indeed, our neuroprotection data suggest a convergence of mechanisms between dominant LRRK2-linked and recessive PINK1-linked forms of PD. Mutations in LRRK2 and loss of PINK1 function share a common phenotype of dendritic shortening and simplification (Abeliovich and Flint Beal, 2006; Cherra et al., 2009; Dagda et al., 2014). Loss of PINK1 function inhibits mitochondrial calcium extrusion through NCLX, leading to enhanced cell death that could be rescued by expression of CA NCLX-S258D (Kostic et al., 2015). In the current study of mutant LRRK2, both increased cytosolic calcium and increased MCU expression contribute to increased mitochondrial calcium uptake. Nevertheless, alleviation of mitochondrial calcium overload through expression of the phosphomimetic NCLX-S258D was protective in either PINK1-deficient or mutant LRRK2 models. Together, these data indicate that excess levels of activity-dependent mitochondrial calcium flux may represent a shared mechanism in PD/PDD triggered by recessive and dominant etiologies. Strategies targeting either MCU or NCLX may serve to normalize activity-dependent mitochondrial calcium flux to protect against neurodegeneration.
Footnotes
This work was supported by National Institutes of Health Grants R01-AG026389, R01-NS065789, R01-NS101628, and the P01-NS059806 Neuropathology Core to C.T.C., R21-MH107966 to Z.P.W., and University of Pittsburgh Medical Center Stimulating Pittsburgh Research in Geroscience Initiative Pilot Grant. The University of Pittsburgh Brain Bank (supported in part by P50-AG05133) provided frozen and paraffin embedded brain tissues. Dennis Dickson (Mayo Clinic Jacksonville and CurePSP: Society for PSP Brain Bank, supported in part by P50-NS40256) provided unstained slides from the G2019S LBD patient. We thank Diego De Stefani, Loren L. Looger, and Erik Snapp for plasmid reagents; and Simon Watkins and the Center for Biological Imaging for imaging and software support.
The authors declare no competing financial interests.
References
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sørensen P (2003) Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 60:387–392. 10.1001/archneur.60.3.387 [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Flint Beal M (2006) Parkinsonism genes: culprits and clues. J Neurochem 99:1062–1072. 10.1111/j.1471-4159.2006.04102.x [DOI] [PubMed] [Google Scholar]
- Akerboom J, Carreras Calderón N, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schüler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, et al. (2013) Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6:2. 10.3389/fnmol.2013.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, Bogdanovic N, Bugiani O, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovács GG, Meyronet D, Monoranu C, Parchi P, et al. (2009) Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 117:635–652. 10.1007/s00401-009-0523-2 [DOI] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Lourenco Venda L, Ansorge O, Wade-Martins R (2009) LRRK2 regulates autophagic activity and localises to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 18:4022–4034. 10.1093/hmg/ddp346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ, Milnerwood AJ (2014) Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front Cell Neurosci 8:301. 10.3389/fncel.2014.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford C, Sears C, Perez-Carrion M, Piccoli G, Condliffe SB (2016) LRRK2 regulates voltage-gated calcium channel function. Front Mol Neurosci 9:35. 10.3389/fnmol.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 60:557–569. 10.1002/ana.21019 [DOI] [PubMed] [Google Scholar]
- Bravo-San Pedro JM, Niso-Santano M, Gómez-Sánchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, Climent V, López de Maturana R, Sanchez-Pernaute R, López de Munain A, Fuentes JM, González-Polo RA (2013) The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci 70:121–136. 10.1007/s00018-012-1061-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky T, Li V, Brustovetsky N (2009) Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium 46:18–29. 10.1016/j.ceca.2009.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calì T, Ottolini D, Brini M (2014) Calcium signaling in Parkinson's disease. Cell Tissue Res 357:439–454. 10.1007/s00441-014-1866-0 [DOI] [PubMed] [Google Scholar]
- Carafoli E. (2012) The interplay of mitochondria with calcium: an historical appraisal. Cell Calcium 52:1–8. 10.1016/j.ceca.2012.02.007 [DOI] [PubMed] [Google Scholar]
- Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ (2010) Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal 22:821–827. 10.1016/j.cellsig.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Citro A, Cordy JM, Shen GC, Wolozin B (2011) Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2). J Biol Chem 286:16140–16149. 10.1074/jbc.M111.234005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Sarnecki C, Blenis J (1992) Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol 12:915–927. 10.1128/MCB.12.3.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ 3rd, Dagda RK, Tandon A, Chu CT (2009) Mitochondrial autophagy as a compensatory response to PINK1 deficiency. Autophagy 5:1213–1214. 10.4161/auto.5.8.10050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT (2010) Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 190:533–539. 10.1083/jcb.201002108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182:474–484. 10.1016/j.ajpath.2012.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, et al. (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15:1197–1205. 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM (2000) Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Mod Pathol 13:420–426. 10.1038/modpathol.3880072 [DOI] [PubMed] [Google Scholar]
- Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ 3rd,Clark RS (2009) Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol 453:217–249. 10.1016/S0076-6879(08)04011-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini LM, Baloban M, Markwardt ML, Rizzo M, Guo F, Verkhusha VV, Snapp EL (2015) A palette of fluorescent proteins optimized for diverse cellular environments. Nat Commun 6:7670. 10.1038/ncomms8670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Zhu J, Kulich SM, Chu CT (2008) Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy 4:770–782. 10.4161/auto.6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 128:864–877. 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M (2007) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 27:9220–9232. 10.1523/JNEUROSCI.2617-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340. 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157. 10.1016/S1474-4422(09)70238-8 [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285:27879–27890. 10.1074/jbc.M110.119537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle S, Pyndiah S, De Gois S, Erickson JD (2010) Excitation-transcription coupling via calcium/calmodulin-dependent protein kinase/ERK1/2 signaling mediates the coordinate induction of VGLUT2 and Narp triggered by a prolonged increase in glutamatergic synaptic activity. J Biol Chem 285:14366–14376. 10.1074/jbc.M109.080069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Suaga P, Churchill GC, Patel S, Hilfiker S (2012a) A link between LRRK2, autophagy and NAADP-mediated endolysosomal calcium signalling. Biochem Soc Trans 40:1140–1146. 10.1042/BST20120138 [DOI] [PubMed] [Google Scholar]
- Gómez-Suaga P, Luzón-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S (2012b) Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 21:511–525. 10.1093/hmg/ddr481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ (2009) Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29:11011–11019. 10.1523/JNEUROSCI.2519-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468:696–700. 10.1038/nature09536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7:583–590. 10.1016/S1474-4422(08)70117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W (2010) LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res 316:649–656. 10.1016/j.yexcr.2009.09.014 [DOI] [PubMed] [Google Scholar]
- Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, Futter CE, Schapira AH, Marchant JS, Patel S (2015) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128:232–238. 10.1242/jcs.164152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 43:364–371. 10.1016/j.nbd.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J 405:307–317. 10.1042/BJ20070209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, Van Deerlin VM, Trojanowski JQ, Hurtig HI, Alcalay RN, Marder KS, Clark LN, Gaig C, Tolosa E, Ruiz-Martínez J, Marti-Masso JF, Ferrer I, López de Munain A, Goldman SM, et al. (2015) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72:100–105. 10.1001/jamaneurol.2014.2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, Ellisman M, Dauer WT (2012) LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet 21:890–899. 10.1093/hmg/ddr526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360–364. 10.1038/nature02246 [DOI] [PubMed] [Google Scholar]
- Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu C, Abramov AY, Sekler I (2015) PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep 13:376–386. 10.1016/j.celrep.2015.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Gale JE (2008) Damage-induced activation of ERK1/2 in cochlear supporting cells is a hair cell death-promoting signal that depends on extracellular ATP and calcium. J Neurosci 28:4918–4928. 10.1523/JNEUROSCI.4914-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 30:1788–1797. 10.1523/JNEUROSCI.5604-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Xu S (2006) ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 58:621–631. 10.1080/15216540600957438 [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52:587–593. 10.1016/j.neuron.2006.10.008 [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14:1336–1343. 10.1038/ncb2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221. 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose HL, Dächsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L, Liang YQ, Beevers JE, Boules M, Dugger BN, Serna VA, Gaukhman A, Yu X, Castanedes-Casey M, Braithwaite AT, Ogholikhan S, et al. (2010) Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis 40:503–517. 10.1016/j.nbd.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75:2017–2020. 10.1212/WNL.0b013e3181ff9685 [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR (2010) 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem J 430:393–404. 10.1042/BJ20100483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, Sekler I (2014) Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J 28:3301–3312. 10.1096/fj.13-248161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A 107:436–441. 10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15:1464–1472. 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29:13971–13980. 10.1523/JNEUROSCI.3799-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H (2014) LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17:367–376. 10.1038/nn.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53:726–737. 10.1016/j.molcel.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TI, Jou MJ, Sheu SS, Greenamyre JT (1998) Visualization of NMDA receptor-induced mitochondrial calcium accumulation in striatal neurons. Exp Neurol 149:1–12. 10.1006/exnr.1997.6599 [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467:291–296. 10.1038/nature09358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB (2004) Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci 24:5611–5622. 10.1523/JNEUROSCI.0531-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8:e55785. 10.1371/journal.pone.0055785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT (2008) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105:1048–1056. 10.1111/j.1471-4159.2008.05217.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Johnson JW, Steer E, Zhu W, Eisenberg DA, Valentino NM, Liu YJ, Chu CT (2014) Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim Biophys Acta 1842:1596–1603. 10.1016/j.bbadis.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27:645–656. 10.1523/JNEUROSCI.4341-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4:2034. 10.1038/ncomms3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32:2362–2376. 10.1038/emboj.2013.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, et al. (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6:e18568. 10.1371/journal.pone.0018568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P, Schmid B, Burbulla LF, Schöndorf DC, Wagner L, Glatza M, Höing S, Hargus G, Heck SA, Dhingra A, Wu G, Müller S, Brockmann K, Kluba T, Maisel M, Krüger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki OE, et al. (2013) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12:354–367. 10.1016/j.stem.2013.01.008 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382. 10.1126/science.1242993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet 23:4201–4214. 10.1093/hmg/ddu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda B, Mesias R, Li X, Yue Z, Benson DL (2013) Short- and long-term effects of LRRK2 on axon and dendrite growth. PLoS One 8:e61986. 10.1371/journal.pone.0061986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernández-Díaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, Geens A, De Strooper B, Verstreken P (2016) A LRRK2-dependent endophilinA Phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92:829–844. 10.1016/j.neuron.2016.09.037 [DOI] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M (2016) Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5:e12813. 10.7554/eLife.12813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet ES, Saunier-Rebori B, Yue Z, Blitzer RD (2015) The Parkinson's disease-associated mutation LRRK2–G2019S impairs synaptic plasticity in mouse hippocampus. J Neurosci 35:11190–11195. 10.1523/JNEUROSCI.0040-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta M, Cataldi S, Beccano-Kelly D, Munsie L, Tatarnikov I, Chou P, Bergeron S, Mitchell E, Lim R, Khinda J, Lloret A, Bennett CF, Paradiso C, Morari M, Farrer MJ, Milnerwood AJ (2015) Chronic and acute LRRK2 silencing has no long-term behavioral effects, whereas wild-type and mutant LRRK2 overexpression induce motor and cognitive deficits and altered regulation of dopamine release. Parkinsonism Relat Disord 21:1156–1163. 10.1016/j.parkreldis.2015.07.025 [DOI] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X (2012) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 21:1931–1944. 10.1093/hmg/dds003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102:16842–16847. 10.1073/pnas.0507360102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH (2011) Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis 41:706–716. 10.1016/j.nbd.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, El Khoury R, Wang W, Byrd TA, Pehek EA, Thacker C, Zhu X, Smith MA, Wilson-Delfosse AL, Chen SG (2010) LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson's disease. Neurobiol Dis 40:73–81. 10.1016/j.nbd.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, et al. (2010) PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med 2:52ra73. 10.1126/scitranslmed.3001059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT (2003) Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 13:473–481. 10.1111/j.1750-3639.2003.tb00478.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT (2007) Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 170:75–86. 10.2353/ajpath.2007.060524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Dagda RK, Chu CT (2011) Monitoring mitophagy in neuronal cell cultures. Methods Mol Biol 793:325–339. 10.1007/978-1-61779-328-8_21 [DOI] [PMC free article] [PubMed] [Google Scholar]