

Abstract
Background
Expression of caveolin-1 (Cav-1) is frequently altered in many human cancers and both tumor suppression and promotion functions of Cav-1 have been suggested based on its expression status. However, it remains unanswered how Cav-1 provokes opposite effects in different cancers or different phases of tumor progression.
Methods
To explore the implication of Cav-1 alteration in gastric tumorigenesis, the expression and mutational status of Cav-1 and its effects on tumor cell growth were characterized.
Results
A substantial fraction of primary tumors and cell lines displayed abnormally low or high Cav-1 mRNA expression, indicating the bidirectional alteration of Cav-1 in gastric cancers. While allelic imbalance and mutational alterations of the Cav-1 gene were rarely detected, aberrant promoter hyper- or hypo-methylation showed a tight correlation with bidirectional alteration of its expression. Abnormally low and high Cav-1 expression was more frequently observed in early and advanced cancers, respectively, suggesting the oncogenic switch of its function in tumor progression. Cell cycle progression, DNA synthesis, and colony forming ability were markedly decreased by Cav-1 transfection in low-expressing tumor cells but by its depletion in high-expressing cells. Interestingly, Cav-1 exerted opposite effects on MEK-ERK signaling in these two cell types through the reciprocal regulation of the RAF-ERK negative feedback loop. A feedback inhibition of RAF by ERK was stimulated by restoration of Cav-1 expression in low-expressing cells but by it depletion in high-expressing cells. As predicted, the opposite effects of Cav-1 on both tumor cell growth and inhibitory RAF phosphorylation were abolished if ERK is depleted.
Conclusion
Bidirectional alteration of Cav-1 is linked to its opposite effects on gastric tumor cell growth, which stem from the reciprocal control on the RAF-ERK negative feedback loop.
Keywords: Caveolin, Promoter methylation, Gastric cancer, RAF, Erk
Background
Caveolae are flask-shaped vesicular invaginations of the plasma membrane characterized by the existence of integral membrane proteins termed caveolins. Caveolae is implicated in many cellular functions, including membrane trafficking, endocytosis, lipid metabolism, cell adhesion, signal transduction in cellular proliferation and apoptosis [1]. Caveolins are a family of proteins composed of three isoforms, Caveolin (Cav)-1, −2, and −3. Among the three caveolins, Cav-1 is a principal structural component of caveolae and forms a high molecular complex of homo-oligomer or hetero-oligomer with Cav-2. A scaffolding domain within Cav-1 allows this protein to interact with signaling molecules, including growth factor receptors, G-protein coupled receptors, small GTPases, Src kinases, nitric oxide synthases, and integrins [2]. Integrations and complex formation of Cav-1 with signaling molecules functionally affect the activity of these molecules.
Despite a growing body of evidence on Cav-1 implication in tumorigenesis, its role in tumor growth and underlying molecular mechanisms remain largely undefined. Both tumor suppression and promotion roles of Cav-1 have been proposed on the basis of its expression status detected in cancers. Cav-1 expression is frequently down-regulated in many human cancers mainly due to promoter hypermethylation whereas its elevation correlates with enhanced progression, multidrug resistance, and metastatic potentials of certain tumors [3–6]. Furthermore, Cav-1 gene amplification and mutation were reported in a subgroup of breast cancers [7, 8]. These findings demonstrate that Cav-1 has differential functions in tumorigenesis depending on the types, origins, or genetic contexts of tumors.
Caveolae have been proposed to be the site of epidermal growth factor receptor (EGFR) signaling, including EGFR autophosphorylation [9]. EGF-induced tumor cell proliferation and migration is suppressed when Cav-1 binds to the EGFR, suggesting that Cav-1 may play a role in maintaining the EGFR in an inactive state, with dissociation from Cav-1 promoting EGFR activation [10]. It was also shown that many components of Ras signaling, including RAF, MEK, and ERK appear to be compartmentalized within caveolin-rich membrane domains and that Cav-1 downregulation results in constitutive activation of ERK signaling while activation of Ras-ERK signaling causes Cav-1 reduction [11, 12]. In contrast, Cav-1 appears to promote metastasis of Ewing sarcoma and the proliferation of metastatic lung cancer cells through activation of the MAPK-ERK pathway [13, 14]. A recent study also showed that Cav-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated ERK1/2 activation, Ras-induced senescence, and transformation [15]. These findings thus indicate that Cav-1 functions as an endogenous inhibitor or stimulator of the Ras-ERK cascade. Nevertheless, the molecular basis for the opposite effects of Cav-1 on EGFR and Ras-MAPK signaling and its implication in tumorigenesis remains largely undefined.
Gastric cancer is one of the most commonly diagnosed malignancies worldwide and a leading cause of cancer mortality in certain areas such as Korea, Japan, South America, and Eastern Europe [16, 17]. Although a number of study indicates that genetic and/or epigenetic alterations of multiple genes, such as p53, K-Ras, and E-Cadherin are associated with the development and progression of gastric cancers, molecular events that drive the neoplastic process remain to be characterized [18]. In this study, we found that Cav-1 is abnormally down- and up-regulated in a considerable fraction of gastric cancers due to promoter hyper- and hypo-methylation, respectively. In low- and high-expressing tumor cells, Cav-1 evokes the opposite effects on cell proliferation and colony formation through the reciprocal control on the RAF-ERK negative feedback loop. Therefore, our study demonstrates that Cav-1 acts as a positive or negative regulator of the RAF-ERK feedback loop and that the mitogenic switch of Cav-1 function is highly associated with bidirectional alteration of its expression in tumor progression.
Methods
Tissues specimens and cell lines
Total 180 gastric tissues including 100 primary carcinomas, 4 adenomas, 6 hamartomas, 6 hyperplastic polyps, and 64 normal gastric tissues were obtained were obtained from 100 gastric cancer patients and 80 noncancer patients by surgical resection in the Kyung Hee University Medical Center (Seoul, Korea). Signed informed consent was obtained from each patient. Tissue specimens were snap-frozen in liquid N2 and stored at −70 °C until used. Tissue slices were subjected to histopathological review and tumor specimens composed of at least 70% carcinoma cells and adjacent tissues found not to contain tumor cells were chosen for molecular analysis. Fourteen human gastric cancer cell lines (SNU5, SNU16, SNU216, SNU484, SNU601, SNU620, SNU638, SNU719, MKN1, MKN28, MKN45, MKN74, AGS, and KATO-III) were obtained from Korea Cell Line Bank (Seoul, Korea) or American Type Culture Collection (Rockville, MD).
Quantitative RT- and genomic PCR
RNA extraction and cDNA synthesis were performed as described previously [19]. Briefly, 1 μg of total cellular RNA was converted to cDNA using random hexamer primers and M-MLV reverse transcriptase (Invitrogen Corporation, Carlsbad, CA). PCR was initially carried out over 24–40 cycles and 12.5 ng cDNA (50 μl PCR reaction) undergoing 30–36 cycles showed logarithmic amplification with primers Cav-1S/Cav-1AS for Cav-1, C1αA/C1αAS for Cav-1α, C1β/C1βAS for Cav-1β, Cav-2S/Cav-2AS for Cav-2, C2αA/C2αAS for Cav-2α, C2βA/C2βAS for Cav-2β, and G2/G3 for an endogenous expression standard gene GAPDH (Table 1). PCR was done in 1.5 mM MgCl2-containing reaction buffer (PCR buffer II) (Perkin Elmer, Branchburg, NJ) and 10 μl of PCR products were resolved on 2% agarose gels. Quantitation was achieved by densitometric scanning of the ethidium bromide-stained gels. Integration and analysis was performed using Molecular Analyst software program (Bio-Rad, Richmond, CA). For genomic PCR, intron 2 regions of Cav-1 and Cav-2 and intron 5 region of GAPDH were amplified with intron-specific primers RF2S/RF2AS and G3/G5, respectively (Table 1). Quantitative PCR was repeated at least three times for each specimen and the mean was obtained.
Table 1.
Primers used for PCR, LOH and bisulfite sequencing analysis
| Gene | Primer | Sequence (5′ to 3′) |
|---|---|---|
| Cav1 | Cav-1S | TCTGGGGCGTCGTGCGCAAA |
| Cav-1AS | GAACCTTGATGAAGCCTGTG | |
| C1⍺A | AGTTTTCATCCAGCCACGGG | |
| C1⍺AS | TCTTGACCACGTCATCGTTGAG | |
| C1βA | CATTTTTCCTCCCACCGCCGTT | |
| C1βAS | AAAACTGTGTGTCCCTTCTG | |
| RF2S | ATGTATATGTACATCAGGGA | |
| RF2AS | CAGGCACATAGCTGGGTACC | |
| SNP-1 | GGCTCAACATTGTGTTCCCATTTCAGC | |
| SNP-2 | GTGTCAGGAAGACTGGAAGAGGCA | |
| P1 | TGTGTATTTTGTAAATATGGTATA | |
| P2 | AAGTTAAAGATTTTTATTTTTTATT | |
| Cav2 | Cav-2S | ATCTGCAGCCATGCCCTCTTTG |
| Cav-2AS | GGGTCCAAGTATTCAATCCTGG | |
| C2⍺A | ATGGGGCTGGAGACGGAGAA | |
| C2⍺AS | ACTGAAGGCAGAACCATTAGGCA | |
| C2βA | TGCGTCCTGTCTCCTCAGCT | |
| C2βAS | ACTGAAGGCAGAACCATTAGGCA | |
| GAPDH | G2 | CATGTGGGCCATGAGGTCCACCAC |
| G3 | AACCATGAGAAGTATGACAACAGC | |
| G5 | GAGTCCTTCCACGATACCAAAG |
Loss of heterozygosity (LOH) analysis
LOH of the Cav-1 gene was determined using an intraexonic SNP (5′-AGCATCC/T-3′) located at +2061 nucleotide (exon 3) from the transcription start site. PCR was performed on each tumor and normal DNA sample pair obtained from 50 patients using primers SNP-1/SNP-2 (Table 1). Five μl of the PCR products were used for cutting with the endonuclease BtsCI (NEB, Beverly, MA) and enzyme-digested PCR products were electrophoresed on 2% agarose gels. Signal intensity of fragments and the relative ratio of tumor and normal allele intensities were determined by scanning densitometry.
RT-PCR-single strand conformation polymorphism (SSCP) analysis
To screen the presence of somatic mutations, RT-PCR-SSCP analysis of Cav-1 and Cav-2 was performed using 3 sets of primers that were designed to cover the entire coding region of the genes. Twenty μl of PCR products mixed with 10 μl of 0.5 N NaOH, 10 mM EDTA, and 15 μl of denaturing loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue and 0.15% xylene cyanol). After heating at 95 °C for 5 min, samples were loaded in wells pre-cooled to 4 °C and run using 8% nondenaturing acrylamide gels containing 10% glycerol at 4–8 °C and 18–22 °C.
5-Aza-dC treatment and bisulfite DNA sequencing analysis
To assess re-activation of Cav-1 expression, cells were treated with 5 μM of 5-Aza-dC (Sigma, St. Louis, MO) for 4 days. For bisulfite sequencing analysis, 1 μg of genomic DNA was incubated with 3 M sodium bisulfite (pH 5.0) and DNA samples were purified as described previously [20]. Fifty ng of bisulfite-modified DNA were subjected to PCR amplification of the 37 CpG sites within the promoter and exon 1 using primers P1/P2 (Table 1). The PCR products were cloned into pCRII vectors (Invitrogen Corporation, Alemeda, CA) and 5 clones of each specimen were subjected to DNA sequencing analysis to determine the methylation status.
Immunoblotting assay
Cells were lysed in a lysis buffer containing 60 mM octylglucoside, 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium phosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin and 1 mM PMSF. Twenty μg of total protein were supplemented with Laemmli buffer and loaded on a 10% SDS-polyacrylamide gel for electrophoresis. Western analyses were performed using antibodies specific for Cav-1 (sc-894, Santa Cruz, CA), Cav-2 (610,685, BD bioscience, CA), EGFR (#4267, Cell Signaling, Danvers, MA), RAF (#2330, Cell Signaling), MEK1/2 (#9911, Cell Signaling), ERK (#9101, Cell Signaling), AKT (#4060, Cell Signaling), JNK (#4668, Cell Signaling), and β-tubulin (T8328, Sigma). Antibody binding was detected by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) using a secondary antibody conjugated to horseradish peroxidase.
Immunofluorescence and immunohistochemistry (IHC) assay
For immunofluorescence assay, cells were fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100 and blocked with 2% bovine serum albumin-PBS. Slides were incubated with anti-GFP antibody and fluorescent imaging was obtained with a confocal laser scanning microscope (Carl Zeiss, Jena, Germany). IHC study was carried out using tissue arrays (SuperBioChips Laboratory, Seoul, Korea) and Vectastain ABC (avidin-biotin-peroxidase) kit (Vector Laboratories) as described previously [21]. Briefly, slides were incubated with Cav-1 antibody overnight using biotin-free polymeric horseradish peroxidase-linker antibody conjugate system. Slides were counterstained with hematoxylin, dehydrated and visualized using an Olympus CK40 microscopy (Tokyo, Japan). For the immunoreactive score, we established a 1- to 12-point system by multiplying the percentage of positive cells by the intensity of the staining score. Two pathologists performed the assessment of immunostaining sections. Immunoreactive scores of 0–5 were classified as negative, and scores of 6–12 were regarded as positive [22].
Ras activity assay
Cells were lysed with Mg-containing lysis buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, 1% Igeal CA-630, 10% Glycerol, 25 mM NaF, 10 mM MgCl2, 1 mM EDTA, 1 mM Sodium orthovanadate, 10 μg/ml Leupeptin, 10 μg/ml Aprotinin, and 1 mM PMSF. Cell lysates were mixed with RAF-1 RBD agarose (Millipore, Billerica, MA) and the reaction mixture were rocked gently at 4 °C for 30 min. Agarose beads were collected by centrifugation, washed 3 times with lysis buffer, and resuspended in 2X Laemmli sample buffer. Samples were electrophoresed on SDS-PAGE and immunoblotted.
Expression plasmids, siRNA, shRNA, and transfection
GFP or Flag-tagged Cav-1 gene was cloned into the pcDNA3.1-V5-His (Invitrogen Corporation) and the pEGFP-N3 vector (Clontech, Mennheim Germany) using the Expand High Fidelity PCR system (Roche Molecular Biochemicals, Palo Alto, CA). siRNA against Cav-1 (5′-AACCAGAAGGGACACACAGUU-3′) and ERK2 (5′-CACCAUUCAAGUUCGACAUUU-3′) were synthesized by Dharmacon Research (Lafayette, CO). shRNA plasmid for Cav-1 (5′-caccACCTTCACTGTGACGAAATACTGGTTtctcAACCAGTATTTCGTCACAGTGAAGG-3′) was constructed by Genolution (Seoul, Korea). Transfection was performed using FuGENE 6 (Roche Molecular Biochemicals) or Oligofectamine (Invitrogen Corporation).
Cell proliferation, DNA synthesis, and colony formation assay
To measure in vitro cellular growth, cells were seeded at the density of 4 × 104 cells per well in triplicate and cell numbers were counted using a hemocytometer at 24 h intervals. For flow cytometry analysis, cells were fixed with 70% ethanol and resuspended in PBS containing 50 mg/ml RNase and 50 mg/ml propidium iodide (Sigma). The assay was performed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and analyzed using Modfit software (Becton Dickinson). For DNA synthesis assay, cells were pulse-labeled for 4 h with 1 μCi/ml [3H] thymidine and harvested with lysis buffer (0.1 N NaOH, 1% SDS). The cell lysates were mixed with the liquid scintillation cocktail (ICN Inc., Irvine, CA) and [3H]thymidine incorporation was counted with Scintillation Counter (Wallac, Milton Keynes, UK). For colony formation assay, 1 × 105 cells per dish were maintained in the presence of G418 (1600 μg/ml) for 4–6 weeks. Selection medium were replaced every 2 days. Colonies were fixed with methanol for 15 min and stained with 0.05% crystal violet in 20% ethanol.
Statistical analysis
The results of cell growth, apoptosis and colony forming assays were expressed as mean ± SD. A student’s t-test was used to determine the statistical significance of the difference. The Chi-square test was used to determine the statistical significance of expression and methylation levels between tumor and normal tissues. A P value of less than 0.05 was considered significant.
Results
Cav-1 expression is commonly down- or up-regulated in gastric cancers
To explore the implication of caveolin alteration in gastric tumorigenesis, we initially investigated its mRNA expression status in 14 cancer cell lines and 180 gastric tissues, including 100 matched sets of primary tumors and adjacent noncancerous tissues. While all 64 normal and 16 benign tumor tissues we examined showed easily detectable mRNA levels of α and β isoforms of Cav-1 and -2, a substantial fraction of cell lines and primary tumors displayed abnormally low or high expression of the transcripts (Fig. 1a-c). An immunoblot assay revealed that both Cav-1 and -2 protein levels are well consistent with their mRNA levels, indicating that caveolin expression is controlled mainly at the transcript level (Fig. 1c). However, MKN45 and MKN74 cells showed relatively low Cav-1 protein levels compared to their mRNA levels. Except for SNU216 cells which show no Cav-1α but high Cav-1β expression, all cell lines and tissue specimens we examined displayed comparable expression patterns of the isoforms (Fig. 1c). Cav-1 expression levels (Cav-1/GAPDH) were detected in a range of 0.66–1.89 (mean 1.27), 0.07–3.43 (mean 1.09), and 0.00–3.40 (mean 1.41) in normal tissues, primary tumors, and cancer cell lines, respectively (Fig. 1d). Based on expression ranges of normal tissues, 30 (30%) and 16 (16%) of 100 primary tumors were classified as abnormally low and high Cav-1 expressors, respectively and 6 (42.9%) and 6 (42.9%) of 14 cancer cell lines were classified as abnormally low and high expressors, respectively. Abnormal reduction of Cav-1 was significantly more frequent in early (16 of 39, 41%) versus advanced tumors (14 of 61, 23%) whereas abnormal elevation was more frequent in advanced (14 of 61, 23%) versus early (2 of 39, 5.1%) and high (12 of 52, 23.1%) versus low (4 of 48, 8.3%) grade tumors (Fig. 1e). Meanwhile, both abnormal reduction and elevation of Cav-1 were more frequently observed in diffuse versus intestinal type tumors.
Fig. 1.
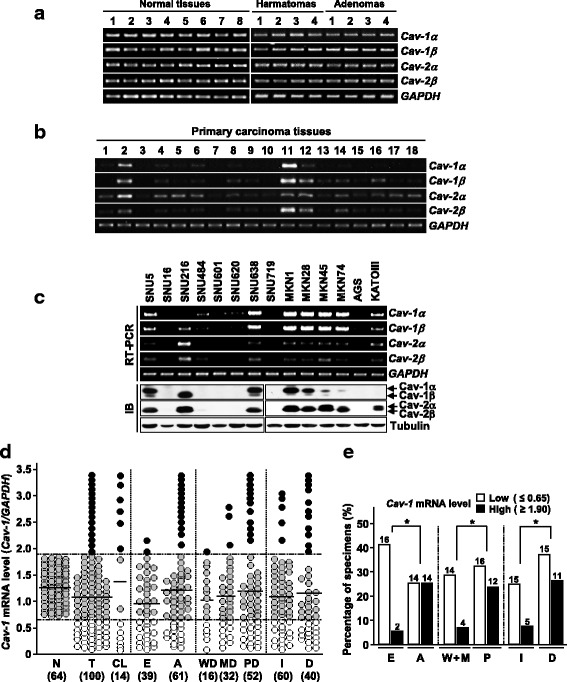
Expression status of Cav-1 in gastric tissues and cell lines. a Quantitative RT-PCR analysis of caveolin expression in normal and benign tumor tissues. b, c Caveolin expression in gastric tissues and cell lines. IB, immunoblot. d, e Association of Cav-1 expression levels with tumor stages, grades, and types. N, normal tissues; T, primary tumors; CL, cell lines; E, early; A, advanced; WD; well-differentiated; MD, moderately differentiated; PD, poorly differentiated; I, intestinal; D, diffused. Bar indicates the mean levels of each specimen group
Next we compared Cav-1 expression in normal and tumor tissues obtained from 100 cancer patients. Compared to adjacent noncancerous tissues, 46 and 30 cancers tissues showed down-and up-regulation of Cav-1 mRNA, respectively (Fig. 2a, b). An immunoblot assay for eight representative tissue sets showed that mRNA levels analyzed by RT-PCR correlate well with protein levels in both normal and cancer tissues (Fig. 2a). Cav-1 reduction in cancerous lesion was more frequent in early (22 of 39, 56.4%) versus advanced (24 of 61, 39.3) tumors while its elevation was more common in advanced (22 of 61, 36.1%) versus early (8 of 39, 20.5%) tumors, further supporting the biphasic alteration of Cav-1 expression during gastric tumor progression (Fig. 2c). To further confirm the finding, we performed an IHC study using additional 40 matched tissue sets. As predicted, a substantial decrease and increase in Cav-1 immunopositivity were observed in 19 (47.5%) and 11 (27.5%) cancer tissues, respectively (Fig. 2d). Collectively, these results indicate that Cav-1 is commonly down- and up-regulated in early and advanced gastric cancers, respectively, suggesting the oncogenic conversion of its function during tumor progression.
Fig. 2.
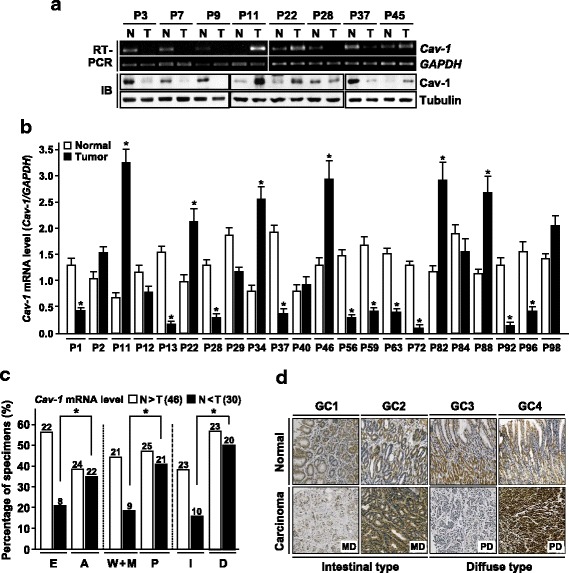
Comparison of Cav-1 expression in matched tissue sets. a RT-PCR and immunoblot analysis of Cav-1 expression in cancer and adjacent noncancerous tissues obtained from the same cancer patients. P, patient. b Relative Cav-1 mRNA levels in cancer and adjacent noncancerous tissues. c Comparison of Cav-1 expression in matched sets and its association with tumor stages, grades and types. P, patient. d Immunohistochemical analysis of Cav-1 in tumor and matched normal tissues. GC, gastric cancer
Bidirectional alteration of Cav-1 is associated with promoter hyper- and hypo-methylation
To define whether altered expression of Cav-1 in cancers is caused by gene deletion or amplification, genomic status of Cav-1 was analyzed. Semi-quantitative DNA-PCR assay revealed that all cancer cell lines and primary tumors we tested have Cav-1 gene levels comparable to those of normal cells (Fig. 3a). An allelotyping assay of 100 matched sets using an intragenic single nucleotide polymorphism (5′-AGCATCC/T-3′) in exon 3 and the endonuclease BtsCI digestion identified 49 informative cases, but none of these showed detectable allelic imbalance between normal and cancer tissues (Fig. 3b). Interestingly, however, PCR-SSCP analysis of 14 cell lines and 50 tumors detected 3 missence and 1 silent sequence alterations in the Cav-1 gene from SNU638 cells (Y97N, TAC to AAC) and three primary tumors (K57R, AAA to AGA; D8G, GAC to GGC; A31A, GCC to GCT) (Fig. 3c). An immunofluorescence microscopic assay revealed that all of these mutant Cav-1 proteins exhibited both perinuclear and plasma membrane localization with a punctuated manner (Fig. 3d).
Fig. 3.
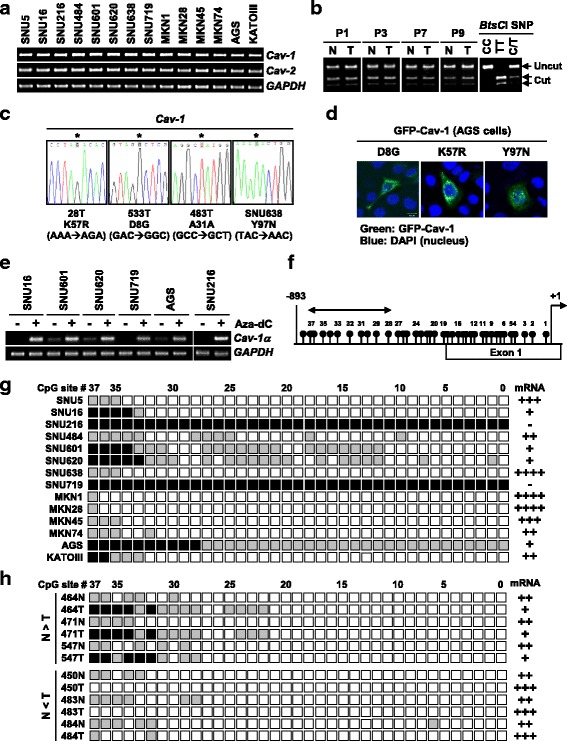
Mutation and promoter methylation analysis of Cav-1 in gastric cancers. a Genomic levels of Cav-1 in 14 cancer cell lines. b LOH analysis of the Cav-1 gene. Exon 3 region containing a SNP (5′-AGCATCC/T-3′) was amplified by PCR and digested with endonuclease BtsCI. P, patient; N, normal; T, tumor. c Sequence alterations of Cav-1 in cell lines and tumors. d Immunofluorescence assay for expression and subcellular localization of mutant Cav-1 proteins in AGS cells. e Quantitative RT-PCR analysis showing re-activation of Cav-1 expression after 5-Aza-dC treatment. f A map of the 37 CpG sites in the promoter and exon 1 region of Cav-1. The first nucleotide of ATG start codon is indicated by an arrow at +1. g Methylation status of the 37 CpGs in 14 cell lines. The gene region comprised of 37 CpGs was amplified by PCR and the PCR products were cloned. Five plasmid clones were sequenced for each cell line. Black, gray, and white circles represent complete methylation (4–5 clones), partial methylation (1–3 clones), and unmethylation, respectively. h Methylation status of the CpGs in primary tumors. N, adjacent noncancerous tissue; T, tumor tissue
Next, we tested whether differential expression of Cav-1 is due to the epigenetic alteration of transcription. In 6 cell lines with no or low expression, Cav-1 transcript level was markedly increased after treatment with the demethylating agent 5-Aza-dC, suggesting that abnormal Cav-1 down-regulation in these cells might be associated with promoter hypermethylation (Fig. 3e). On this basis, we performed bisulfite DNA sequencing analysis of 37 CpG sites within the promoter and exon 1 region (Fig. 3f). Five PCR clones from each specimen were sequenced to determine methylation frequency at individual CpG sites. Among 37 CpGs, 21–37 sites were partially or completely methylated in 6 cell lines with no or low Cav-1 expression, whereas only 1–4 sites showed methylation in 6 cell lines with high Cav-1 expression (Fig. 3g). In particular, methylation status of 10 CpGs (numbered 28–37 in Fig. 3f) within nucleotides −446 to −772 was most tightly associated with mRNA expression status. While all of these 10 CpGs were completely methylated in 3 cell lines (SNU216, SNU719, and AGS) with extremely low expression, partial methylation only at 1–4 sites was found in 6 high expressor cell lines. We next compared methylation status in tumors and adjacent noncancerous tissues using 5 low Cav-1 and 5 high Cav-1 tumors. Low and high Cav-1 tumors showed methylation at 9–14 and 0–2 sites, respectively while noncancerous tissues showed 4–6 sites methylation (Fig. 3h). Consistent with cell lines, methylation at the 10 distant CpG sites showed a tight association with mRNA expression status, supporting that methylation status of this region is crucial for the transcriptional control of the gene. These results indicate that aberrant promoter hyper- or hypo-methylation of Cav-1 is a common event in gastric tumorigenesis and tightly correlates with its bidirectional expression.
Cav-1 exerts opposite effects on growth of low- and high-expressing tumor cells
To address the biological significance of bidirectional alteration of Cav-1 in gastric tumor progression, we examined its effects on tumor cell growth. Ectopic overexpression of wild-type (WT) Cav-1 in AGS and SNU601 cells (low Cav-1) resulted in 36–42% inhibition of cellular growth, whereas siRNA-mediated depletion of endogenously expressed Cav-1 in MKN1 and KNK28 cells (high Cav-1) caused 41–60% reduction in cellular growth (Fig. 4a), indicating that Cav-1 has opposite roles in the growth regulation of low and high expressor cells. Consistently, Cav-1-expressing stable sublines of AGS and SNU601 showed 71–78% decrease in colony formation whereas shCav-1 sublines of MKN1 and MKN28 showed 83–85% decrease in colony formation (Fig. 4b). Flow cytometric analysis of the cell cycle showed that AGS/Cav-1 and MKN1/shCav-1 sublines showed higher fractions of the G1 phase cells compared to control sublines (Fig. 4c). [3H]Thymidine uptake assay also revealed that DNA synthesis is inhibited by Cav-1 expression in AGS cells but by its depletion in MKN1 cells (Fig. 4d). However, Cav-1 did not affect both basal and genotoxic stress-induced apoptosis (Fig. 4e, f). Next, we utilized a dominant negative mutant form of Cav-1 (P132L) to further test the opposite effects of Cav-1 in these cells [23]. As reported, Cav-1/P132L proteins were localized predominantly in the perinuclear region, indicating the loss of its caveolae-scaffolding protein (Fig. 4g). Both the growth-inhibiting (AGS) and growth-promoting (MKN1) effects of Cav-1 were significantly suppressed when Cav-1/P132L was co-transfected while apoptotic response to 5-FU was not affected by Cav-1/P132L (Fig. 4h-j). Collectively, these findings indicate that up- and down-regulated Cav-1 in gastric cancers has opposite roles in the regulation of tumor cell growth.
Fig. 4.
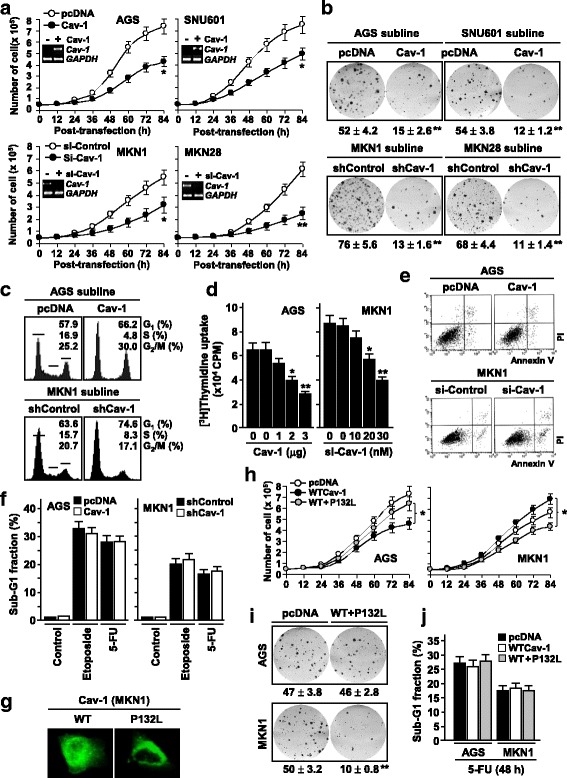
Cav-1 effect on tumor cell growth. a Opposite effects of Cav-1 on growth of low-and high-expressing tumor cells. AGS and SNU601 were transfected with 2 μg of WT-Cav-1 expression or empty vector (pcDNA), and MKN1 and MKN28 were transfected with 30 nM of si-Cav-1 or si-Control. Data represent means of triplicate assays (Bars, SD) (*, P < 0.05; **, P < 0.01). b Cav-1 effect on colony forming ability of tumor cells. Cav-1-expressing sublines of AGS and SNU601 and Cav-1-depleted sublines of MKN1 and MKN28 were maintained in the presence of G418 (1600 μg) for 4–6 weeks. c Flow cytometric analysis of Cav-1 effect on cell cycle progression. d [3H]thymidine uptake assay showing opposite effects of Cav-1 on DNA synthesis in AGS and MKN1 cells. e Flow cytometric analysis of Annexin V-positive cells showing no significant effect of Cav-1 on apoptosis. f Flow cytometric analysis of sub-G1 fraction. Cells were exposed to etoposide (15 μM) or 5-FU (15 μM) for 48 h. g Subcellular distribution of WT- and MT-Cav-1 (P132L) proteins. (h-j) Effect of a dominant negative Cav-1 mutant (P132L) on tumor cell growth, colony formation, and apoptosis
Opposite functions of Cav-1 stems from reciprocal regulation of ERK phosphorylation
To address the molecular basis for the opposite roles for Cav-1 in the regulation of tumor cell proliferation, we initially defined its effect on growth-regulating signaling components. As shown in Fig. 5a, phospho-p38 and phospho-JNK levels were not affected by Cav-1 in both AGS and MKN1 cells while phospho-AKT level was greatly decreased by Cav-1 expression in AGS but not affected by Cav-1 depletion in MKN1 cells. This result suggests that Cav-1 down-regulation but not its up-regulation is associated with AKT activation in gastric tumor cells. Intriguingly, we found that phospho-ERK1/2 level is decreased by Cav-1 expression in AGS cells and by Cav-1 depletion in MKN1 cells (Fig. 5a, b). Likewise, EGF-induced ERK1/2 phosphorylation was blocked by Cav-1 expression in AGS cells but by Cav-1 depletion in MKN1 cells in a dose-associated manner (Fig. 5c). Consistently, the mitogenic effect of EGF was profoundly attenuated by Cav-1 expression and depletion in AGS and MKN1 cells, respectively (Fig. 5d). These support that opposite effects of Cav-1 on cell growth are associated with its reciprocal regulation of ERK signaling.
Fig. 5.
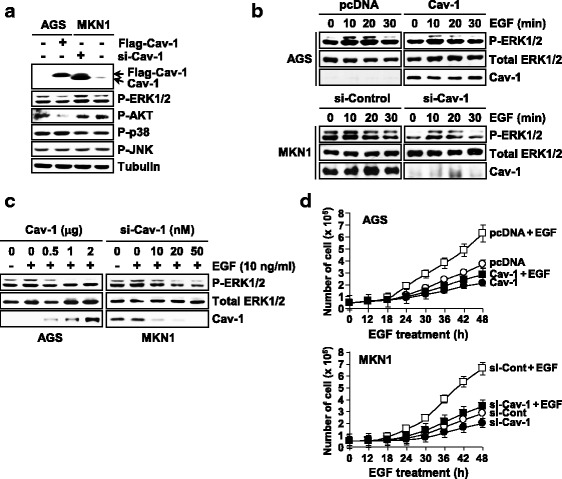
Cav-1 regulation of ERK signaling. a Opposite effects of Cav-1 on ERK1/2 phosphorylation. AGS and MKN1 cells were transfected with WT-Cav-1 or si-Cav-1 and its effect on phosphorylation levels of AKT, ERK1/2, p38, and JNK were examined by an immunoblot assay. b Effect of Cav-1 on EGF-induced phosphorylation of ERK1/2. Cells transfected with WT-Cav-1 or si-Cav-1 were exposed to EGF (10 ng/ml) and phopho-ERK1/2 level was determined. c A dose-associated effect of Cav-1 on EGF-induced ERK1/2 phosphorylation. d Opposite effects of Cav-1 on EGF-induced cell growth. WT-Cav-1- or si-Cav-1-transfected cells were incubated with EGF (10 ng/ml). Data represent means of triplicate assays (Bars, SD)
Cav-1 evokes opposite effects on RAF inhibitory phosphorylation in low- and high-expressing cells
To further dissect the opposite roles for Cav-1 in ERK signaling, we compared its regulation of the EGFR-Ras-RAF-MEK-ERK signaling cascade in AGS and MKN1 cells. Upon ligand stimulation, EGFR is internalized and its signaling is attenuated by ubiquitin-mediated degradation [24]. Compared with controls, Cav-1-transfected AGS cells showed more rapid degradation of EGFR whereas siCav-1-transfected MKN1 cells exhibited more delayed degradation, indicating that EGFR degradation is promoted by Cav-1 in both cells (Fig. 6a). A Ras kinase activity assay also revealed that Cav-1 represses Ras activity in both cells (Fig. 6b). However, EGF-induced phosphorylation of MEK1/2 was attenuated by Cav-1 in AGS cells but by siCav-1 in MKN1 cells, suggesting that kinase(s) upstream of MEK and downstream of Ras might be a target for the reciprocal regulation of Cav-1 (Fig. 6c, d).
Fig. 6.
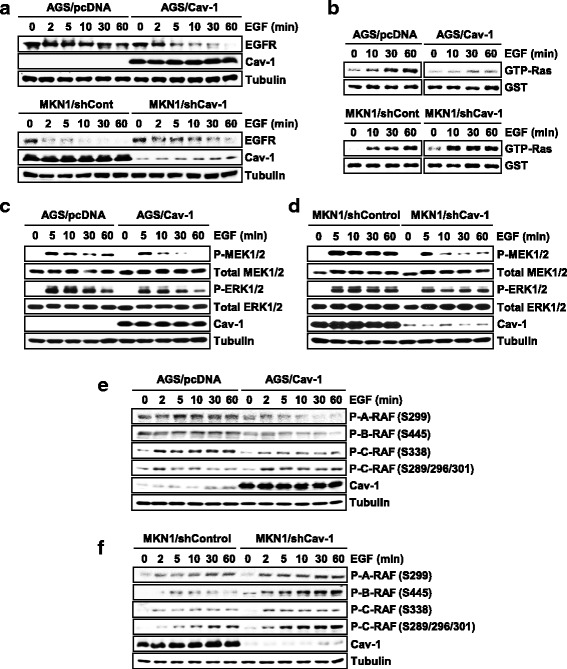
Opposite effects of low and high Cav-1 on inhibitory phosphorylation of RAF. a Cav-1 inhibition of EGFR stability. WT-Cav-1- or shCav-1-expressing cells were exposed to EGF (10 ng/ml) and EGFR level was determined using an immunoblot assay. b Effect of Cav-1 on Ras activity. GTP-Ras levels were measured using Ras activity assay. c, d Opposite effects of Cav-1 on EGF-induced MEK1/2 phosphorylation. e, f Effect of Cav-1 on EGF-induced RAF phosphorylation. Stimulatory phosphorylation of A-RAF (P-S299), B-RAF (P-S445) and C-RAF (P-S339) and inhibitory phosphorylation of C-RAF (P-S289/296/301) were detected using antibodies specific to phospho-RAF isoforms
The C-RAF kinase is a crucial molecule to transmit Ras signals to MEK and its activity is precisely controlled by interplays with isoforms (A-RAF and B-RAF) and differential phosphorylation at multiple sites. In particular, C-RAF has been known to be regulated by ERK1/2 via direct feedback phosphorylation [25]. Activated ERK1/2 phosphorylates C-RAF at multiple serine residues (S29, S289, S296, S301, and S642) and thus blocks the MEK-activating function of active C-RAF (P-S338). On this basis, we characterized Cav-1 effect on RAF isoforms and their phosphorylation status. In both AGS and MKN1 cells, Cav-1 was found to down-regulate active phosphorylation of three RAF isoforms (A-RAF/P-S299, B-RAF/P-S445, and C-RAF/P-S338) (Fig. 6e, f). Intriguingly, however, the inhibitory phospho-RAF (P-S289/S296/S301) was increased by Cav-1 expression in AGS cells but by Cav-1 depletion in MKN1 cells. Therefore, these results strongly suggest that ERK inhibition of C-RAF by feedback phosphorylation might be a critical target that is reciprocally regulated by Cav-1 in low- and high-expressor cancer cells.
Overexpressed Cav-1 blocks the RAF-ERK negative feedback loop
To elicit whether the opposite effects of Cav-1 on cell growth is associated with its reciprocal regulation of the ERK-RAF feedback loop, we tested Cav-1 effect on inhibitory phosphorylation of RAF under ERK-blocked conditions. EGF treatment induced inhibitory phosphorylation of RAF (P-S289/S296/S301) in both AGS and MKN1 cells and this effect was attenuated by the MEK inhibitor UO126, supporting the ERK inhibition of RAF by feedback phosphorylation (Fig. 7a). We next tested whether the reciprocal control of MEK1/2 phosphorylation by Cav-1 is abolished if ERK is depleted. ERK depletion led to an increase in phospho-MEK1/2 level, possibly due to increased RAF kinase activity by loss of the RAF-ERK feedback loop (Fig. 7b, c). As predicted, down-regulation of MEK1/2 phosphorylation by Cav-1 expression in AGS cells and by its depletion in MKN1 cells was not detected when ERK2 expression is knock-downed (Fig. 7b). Furthermore, the opposite effects of Cav-1 on EGF-induced cell proliferation were substantially debilitated in ERK-depleted cells (Fig. 7d, e). Collectively, these results indicate that the bidirectional alteration of Cav-1 expression in gastric cancers is linked to its opposite effects on tumor cell proliferation, which stems from the reciprocal control of the RAF-ERK negative feedback loop (Fig. 7f).
Fig. 7.
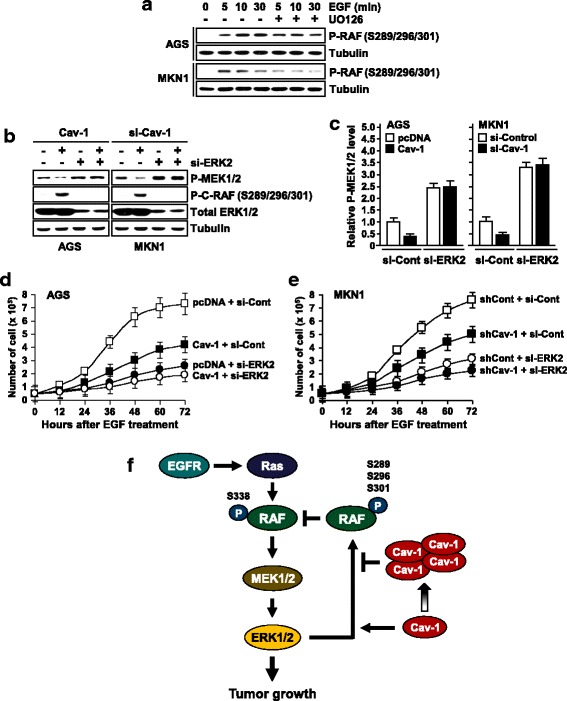
Reciprocal regulation of the RAF-ERK feedback loop by Cav-1. a Validation of the RAF-ERK feedback regulation. Cells were treated with the MEK inhibitor UO126 and its effect on the inhibitory phosphorylation level of C-RAF was determined. b, c Effect of ERK depletion on Cav-1 regulation of RAF, MEK, and ERK. Cells were co-transfected with si-ERK2 and WT-Cav-1 or si-Cav-1. d, e Effect of ERK depletion on Cav-1 regulation of EGF-induced cell growth. WT-Cav-1- or shCav-1-expressing sublines were transfected with si-ERK2 and incubated with EGF (10 ng/ml). Data represent means of triplicate assays (Bars, SD). f Schematic representation of the opposite functions of Cav-1 in the regulation of the RAF-ERK feedback phosphorylation loop in gastric cancer cells
Discussion
Cav-1 has opposite functions in tumorigenesis depending on the cellular contexts [26]. However, the molecular mechanism underlying the differential effects of Cav-1 on tumor growth has been poorly defined. In the present study, we observed a bidirectional alteration of Cav-1 expression in gastric cancers, which is linked to the mitogenic conversion of its function. Our study provides evidence that the mitogenic conversion of Cav-1 function is associated with the switch of its role for the RAF-ERK feedback phosphorylation loop.
Previous IHC studies reported contrasting results on Cav-1 expression in gastric cancers [27–30]. A study using frozen tissues and a monoclonal antibody revealed that Cav-1 is expressed in only a small fraction of intestinal type cancers [28]. Meanwhile, a study using formalin-fixed specimens and a polyclonal antibody showed that Cav-1 is expressed in both diffuse and intestinal types at variable levels [30]. In the present study, we identified that both intestinal and diffuse types of cancers express Cav-1 at highly variable levels. However, Cav-1 down-regulation was significantly more frequent in early versus advanced tumors while its up-regulation was more common in advanced versus early tumors and high versus low grade tumors, supporting that alteration of Cav-1 expression is associated with the oncogenic switch of its function [29, 31]. Our data thus suggest that Cav-1 may act as a stage-specific growth modulator in gastric cancer, which is inactivated during the early stages of tumorigenesis and its subsequent elevation confers growth advantages and malignant progression [26, 32].
It is becoming clear that altered expression of Cav-1 in tumor stroma, particularly in cancer-associated fibroblasts (CAFs), is linked to the malignant progression of various types of human cancers [33, 34]. A study showed that loss of CAFs Cav-1 promotes tumor microenvironment remodeling and tumor development [35]. However it was also reported that stromal Cav-1 favors tumor invasion and metastasis [36]. Therefore, the role for CAFs Cav-1 in tumorigenesis remains largely undefined. It was demonstrated that Cav-1 is not expressed in the epithelial compartment in normal gastric mucosa and in the metaplastic intestinal epithelium while its expression is significantly higher in advanced versus early cancers and an independent prognostic factor of poor survival [29]. In contrast, a recent study using quantum dots immunofluorescence histochemistry identified that epithelial Cav-1 expression gradually decreases with the progression of gastric cancer [37]. Interestingly, this study also showed that low Cav-1 expression in CAFs rather than in tumor cells predicts recurrence and survival in cancer patients, suggesting that loss of stromal Cav-1 heralds poor prognosis of gastric cancer patients, which is consistent with the finding in breast and prostate cancer [38, 39]. Although we did not characterize CAFs Cav-1 expression status in the current study, it was recognized that compared to normal gastric mucosa, noncancerous tissues adjacent to cancerous tissues exhibit much variable levels of Cav-1 mRNA. Further studies will be required to address whether Cav-1 gene expression in CAFs also shows a bidirectional alteration due to promoter hypo- and hyper-methylation during gastric tumor progression.
Mutational alteration of the Cav-1 gene has been rarely found in human cancers. However, Cav-1 mutations were reported in certain tumor types [8]. A mutant Cav-1 (P132L) found in scirrhous breast cancer was identified to exert a dominant negative function by cytoplasmic retention [8]. Interestingly, we detected three missence and one silent sequence alterations in Cav-1 from 3 of 50 primary tumors and 1 of 14 cancer cell lines. Our preliminary data suggest that all of these mutants are expressed. The central α-helical region of Cav-1 protein interacts with the catalytic subunit of protein kinase A (PKAcat) through a hydrogen bond between its Y97 residue and W196 residue of PKAcat [38]. Our finding of Y97N mutation in the SNU638 cancer cell line thus raises the possibility that Cav-1 regulation of PKA signaling might be altered in these gastric cancer cells.
Cav-1 null mice exhibit increases in tumor incidence, tumor area, and tumor number compared with wild-type counterparts [39]. In contrast, prostate cancer cells secrete Cav-1, which stimulates clonal growth of tumor cells, and high Cav-1 expression exerts anti-apoptotic effect under clinically relevant circumstances [40, 41]. Our previous study showed that high Cav-1 expression enhances the metastatic potential of gastric tumor cells by increasing the adhesion ability of the cells to endothelium through the regulation of cell surface VCAM [42]. In the present study, we observed that Cav-1 provokes either a growth-inhibiting or growth-promoting effect in gastric cancers, and this property of Cav-1 is associated with its reciprocal regulation of ERK. Signaling components, including Ras, RAF, MEK, and ERK, are known to be compartmentalized within caveolin-rich membrane domains [2]. However, there is a disagreement in the data concerning whether Cav-1 plays an inhibitory or stimulatory role in Ras-ERK signaling in cancer cells. We found that Cav-1 reciprocally regulates MEK and ERK in low- and high-expressing tumor cells. RAF is a crucial factor to transmit growth factor-induced Ras signals to MEK, and its activity is precisely regulated by differential phosphorylation of RAF isoforms and a negative feedback loop between RAF and ERK [25]. We found that Cav-1 evokes opposite effects on the inhibitory phosphorylation of RAF in low- and high-expressing cells through the reciprocal control for the ERK-mediated inhibitory phosphorylation of RAF. We validated that in EGF-treated gastric tumor cells, RAF is inhibited by ERK feedback phosphorylation and this feedback loop is differentially regulated by Cav-1 in these two cell types. The opposite effects of Cav-1 on tumor cell growth were disrupted if ERK expression is depleted, supporting that the mitogenic conversion of Cav-1 effect is linked to its reciprocal regulation of the ERK feedback phosphorylation of RAF. Although further studies are required to understand the molecular mechanism underlying the Cav-1 regulation of the RAF-ERK negative feedback loop, studies suggest that Cav-1 may affect ERK feedback regulation of signaling components, including RAF, MEK, and KSR1 [15, 43, 44]. It is thus conceivable that MEK-ERK signaling is activated through Cav-1 up- and down-regulation in early and advanced tumors, respectively.
Conclusions
Cav-1 acts as a positive or negative regulator of tumor cell growth via the reciprocal control for the RAF-ERK feedback loop, and the mitogenic switch of Cav-1 function is tightly linked to bidirectional alteration of its expression in tumor progression. Therefore, Cav-1 represents one critical modulator of the RAF-ERK negative feedback loop, adding a new mechanism by which Cav-1 functions as a regulator of tumor growth.
Acknowledgements
Not applicable
Funding
This work was supported in part by grants from National Research Foundation of Korea (NRF-2015R1A2A1A01005389) and a Korea University Intramural Grant-in-Aid (2016). Funding bodies did not have any influence in the design of the study and collection, analysis and interpretation of data or in writing the manuscript.
Availability of data and materials
All relevant data and materials are presented in the manuscript. Additional datasets analyzed during the current study are available from the corresponding author.
Abbreviations
- EGF
epidermal growth factor
- ERK
extracellular signal-related kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEK1
MAPK/ERK kinase 1
- RT-PCR
reverse transcription-polymerase chain reaction
Authors’ contributions
BKR and MGL carried out gene and protein expression, cell growth, apoptosis, colony formation assays, and statistical analysis. BKR carried out methylation studies. SJO and JRM carried out immunohistochemical study. NHK, KYL, and HJK provided human tissue specimens and participated in the design of the study. SGC obtained funding, conceived of the study, and participated in its design and coordination. All authors analyzed data and approved the final manuscript.
Ethics approval and consent to participate
The study protocol was approved by The Regional Committee of Medical Ethics of The Industry-Academic Cooperation Foundation of Kyung Hee University. Written informed agreements were obtained from the patients and clinicians for the samples usage.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Byung-Kyu Ryu, Email: bkryu@korea.ac.kr.
Min-Goo Lee, Email: mglee@korea.ac.kr.
Nam-Hoon Kim, Email: n-hkim@paik.ac.kr.
Kil Yeon Lee, Email: isaac34@korea.com.
Shin-Ju Oh, Email: linus204@naver.com.
Jung-Rock Moon, Email: ukino816@naver.com.
Hyo Jong Kim, Email: hjkim@khmc.or.kr.
Sung-Gil Chi, Phone: +82-2-3290-3443, Email: chi6302@korea.ac.kr.
References
- 1.Parton RG, Simons K. The multiple faces of caveolae. Nature Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 2.de Laurentiis A, Donovan L, Arcaro A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem J. 2007;1:12–32. doi: 10.2174/1874091X00701010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiechen K, Diatchenko L, Agoulnik A, Scharff KM, Schober H, Arlt K, Zhumabayeva B, Siebert PD, Dietel M, Schäfer R, Sers C. Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. Am J Pathol. 2001;159:1635–1643. doi: 10.1016/S0002-9440(10)63010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao X, Evans J, Chae H, Pilrose J, Kim S, Yan P, Huang RL, Lai HC, Lin H, Liu Y, Miller D, Rhee JK, Huang YW, Gu F, Gray JW, Huang TM, Nephew KP. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene. 2013;32:4519–4928. doi: 10.1038/onc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang G, Truong LD, Timme TL, Ren C, Wheeler TM, Park SH, Nasu Y, Bangma CH, Kattan MW, Scardino PT, Thompson TC. Elevated expression of caveolin-1 is associated with prostate and breast cancer. Clin Cancer Res. 1998;4:1873–1880. [PubMed] [Google Scholar]
- 6.Van den Eynden GG, Van Laere SJ, Van der Auwera I, Merajver SD, Van Marck EA, van Dam P, Vermeulen PB, Dirix LY, van Golen KL. Overexpression of caveolin-1 and -2 in cell lines and in human samples of inflammatory breast cancer. Breast Cancer Res Treat. 2006;95:219–228. doi: 10.1007/s10549-005-9002-1. [DOI] [PubMed] [Google Scholar]
- 7.Savage K, Lambros MB, Robertson D, Jones RL, Jones C, Mackay A, James M, Hornick JL, Pereira EM, Milanezi F, Fletcher CD, Schmitt FC, Ashworth A, Reis-Filho JS. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: a morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clin Cancer Res. 2007;13:90–101. doi: 10.1158/1078-0432.CCR-06-1371. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y, Hayakawa T, Hamaguchi M. Invasion activation caveolin-1 mutation in human scirrhous breast cancers. Cancer Res. 2001;61:2361–2364. [PubMed] [Google Scholar]
- 9.Balbis A, Posner BI. Compartmentalization of EGFR in cellular membranes: role of membrane rafts. J Cell Biochem. 2010;109:1103–1108. doi: 10.1002/jcb.22505. [DOI] [PubMed] [Google Scholar]
- 10.Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;271:30429–30438. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 11.Galbiati F, Volonte D, Engelman JA, Watanabe G, Burk R, Pestell RG, Lisanti MP. Targeted downregulation of caveolin-1 is sufficient to derive cell transformation and hyperactivates the p42/44 MAP kinase cascade. EMBO J. 1998;17:6633–6648. doi: 10.1093/emboj/17.22.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, Pereira de Souza A, Kitsis RN, Russell RG, Weiss LM, Tang B, Jelicks LA, Factor SM, Shtutin V, Tanowitz HB, Lisanti MP. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:C457–C474. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 13.Pancotti F, Roncuzzi L, Maggiolini M, Gasperi-Campani A. Caveolin-1 silencing arrests the proliferation of metastatic lung cancer cells through the inhibition of STAT3 signaling. Cell Signal. 2012;24:1390–1397. doi: 10.1016/j.cellsig.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Lagares-Tena L, García-Monclús S, López-Alemany R, Almacellas-Rabaiget O, Huertas-Martínez J, Sáinz-Jaspeado M, Mateo-Lozano S, Rodríguez-Galindo C, Rello-Varona S, Herrero-Martín D, Tirado OM. Caveolin-1 promotes Ewing sarcoma metastasis regulating MMP-9 expression through MAPK/ERK pathway. Oncotarget. 2016;7:56889–56903. doi: 10.18632/oncotarget.10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kortum RL, Fernandez MR, Costanzo-Garvey DL, Johnson HJ, Fisher KW, Volle DJ, Lewis RE. Caveolin-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated extracellular signal-regulated kinase 1/2 activation, H-RasV12-induced senescence, and transformation. Mol Cell Biol. 2014;34:3461–3472. doi: 10.1128/MCB.01633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Hoed CM, Kuipers EJ. Gastric cancer: how can we reduce the incidence of this disease? Curr Gastroenterol Rep. 2016;18:34–42. doi: 10.1007/s11894-016-0506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh CM, Won YJ, Jung KW, Kong HJ, Cho H, Lee JK, Lee DH, Lee KH. Community of Population-Based Regional Cancer Registries: cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2013. Cancer Res Treat. 2016;48:436–450. doi: 10.4143/crt.2016.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, Yu W, Wu Y, Lee M, Wu J, Poh D, Wan WK, Rha SY, So J, Salto-Tellez M, Yeoh KG, Wong WK, Zhu YJ, Futreal PA, Pang B, Ruan Y, Hillmer AM, Bertrand D, Nagarajan N, Rozen S, The BT. Tan P. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 19.Lee MG, Huh JS, Chung SK, Lee JH, Byun DS, Ryu BK, Kang MJ, Chae KS, Lee SJ, Lee CH, Kim JI, Chang SG, Chi SG. Promoter CpG hypermethylation and downregulation of XAF1 expression in human urogenital malignancies: implication for attenuated p53 response to apoptotic stresses. Oncogene. 2006;25:5807–5822. doi: 10.1038/sj.onc.1209867. [DOI] [PubMed] [Google Scholar]
- 20.Chung SK, Lee MG, Ryu BK, Lee JH, Han J, Byun DS, Chae KS, Lee KY, Jang JY, Kim HJ, Chi SG. Frequent alteration of XAF1 in human colorectal cancers: implication for tumor cell resistance to apoptotic stresses. Gastroenterology. 2007;132:2459–2477. doi: 10.1053/j.gastro.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 21.Han J, Kim YL, Lee KW, Her NG, Ha TK, Yoon S, Jeong SI, Lee JH, Kang MJ, Lee MG, Ryu BK, Baik JH, Chi SG. ZNF313 is a novel cell cycle activator with an E3 ligase activity inhibiting cellular senescence by destabilizing p21WAF1. Cell Death Differ. 2013;20:1055–1067. doi: 10.1038/cdd.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang MJ, Ryu BK, Lee MG, Han J, Lee JH, Ha TK, Byun DS, Chae KS, Lee BH, Chun HS, Lee KY, Kim HJ, Chi SG. NF-κB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology. 2008;135:2030–2042. doi: 10.1053/j.gastro.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Bonuccelli G, Casimiro MC, Sotgia F, Wang C, Liu M, Katiyar S, Zhou J, Dew E, Capozza F, Daumer KM, Minetti C, Milliman JN, Alpy F, Rio MC, Tomasetto C, Mercier I, Flomenberg N, Frank PG, Pestell RG, Lisanti MP. Caveolin-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am J Pathol. 2009;174:1650–1662. doi: 10.2353/ajpath.2009.080648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim ES, Khuri FR, Herbst RS. Epidermal growth factor receptor biology (IMC-C225) Curr Opin Oncol. 2001;13:506–513. doi: 10.1097/00001622-200111000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 26.Goetz JG, Lajoie P, Wiseman SM, Nabi IR. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–735. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- 27.Juhász M, Chen J, Tulassay Z, Malfertheiner P, Ebert MP. Expression of caveolin-1 in gastrointestinal and extraintestinal cancers. J Cancer Res Clin Oncol. 2003;129:493–497. doi: 10.1007/s00432-003-0468-0. [DOI] [PubMed] [Google Scholar]
- 28.Burgermeister E, Xing X, Röcken C, Juhasz M, Chen J, Hiber M, Mair K, Shatz M, Liscovitch M, Schmid RM, Ebert MP. Differential expression and function of caveolin-1 in human gastric cancer progression. Cancer Res. 2007;67:8519–8526. doi: 10.1158/0008-5472.CAN-07-1125. [DOI] [PubMed] [Google Scholar]
- 29.Nam KH, Lee BL, Park JH, Kim J, Han N, Lee HE, Kim MA, Lee HS, Kim WH. Caveolin 1 expression correlates with poor prognosis and focal adhesion kinase expression in gastric cancer. Pathobiology. 2013;80:87–94. doi: 10.1159/000341685. [DOI] [PubMed] [Google Scholar]
- 30.Barresi V. Giuffre' G, Vitarelli E, Todaro P, Tuccari G: Caveolin-1 immuno-expression in human gastric cancer: histopathogenetic hypotheses. Virchows Arch. 2008;453:571–578. doi: 10.1007/s00428-008-0681-y. [DOI] [PubMed] [Google Scholar]
- 31.Hung KF, Lin SC, Liu CJ, Chang CS, Chang KW, Kao SY. The biphasic differential expression of the cellular membrane protein, caveolin-1 in oral carcinogenesis. J Oral Pathol Med. 2003;32:461–467. doi: 10.1034/j.1600-0714.2003.00185.x. [DOI] [PubMed] [Google Scholar]
- 32.Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288:C494–C506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 33.Simpkins SA, Hanby AM, Holliday DL, Speirs V. Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J Pathol. 2012;227:490–498. doi: 10.1002/path.4034. [DOI] [PubMed] [Google Scholar]
- 34.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle. 2009;8:2420–2424. doi: 10.4161/cc.8.15.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–467. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 36.Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibáñez T, Pellinen T, Echarri A, Cerezo A, Klein-Szanto AJ, Garcia R, Keely PJ, Sánchez-Mateos P, Cukierman E, Del Pozo MA. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, He Y, Gao J, Fan L. Li z, Yang G, Chen H: Caveolin-1 expression level in cancer associated fibroblasts predicts outcome in gastric cancer. PLoS One. 2013;8:e59102. doi: 10.1371/journal.pone.0059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levin AM, Coroneus JG, Cocco MJ, Weiss GA. Exploring the interaction between the protein kinase a catalytic subunit and caveolin-1 scaffolding domain with shotgun scanning, oligomer complementation, NMR, and docking. Protein Sci. 2006;15:478–486. doi: 10.1110/ps.051911706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, Lisanti MP. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol. 2003;162:2029–2039. doi: 10.1016/S0002-9440(10)64335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tahir SA, Yang G, Ebara S, Timme TL, Satoh T, Li L, Goltsov A, Ittmann M, Morrisett JD, Thompson TC. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res. 2001;61:3882–3885. [PubMed] [Google Scholar]
- 41.Timme TL, Goltsov A, Tahir S, Li L, Wang J, Ren C, Johnston RN, Thompson TC. Caveolin-1 is regulated by c-Myc and suppresses c-Myc-induced apoptosis. Oncogene. 2000;19:3256–3265. doi: 10.1038/sj.onc.1203654. [DOI] [PubMed] [Google Scholar]
- 42.Shin J, Kim J, Ryu BK, Chi SG, Park H. Caveolin-1 is associated with VCAM-1 dependent adhesion of gastric cancer cells to endothelial cells. Cell Physiol Biochem. 2006;17:211–220. doi: 10.1159/000094126. [DOI] [PubMed] [Google Scholar]
- 43.Eblen ST, Slack-Davis JK, Tarcsafalvi A, Parsons JT, Weber MJ, Catling AD. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol Cell Biol. 2004;24:2308–2317. doi: 10.1128/MCB.24.6.2308-2317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009;106:11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data and materials are presented in the manuscript. Additional datasets analyzed during the current study are available from the corresponding author.