

Abstract
Idiopathic macular holes (IMH) are full-thickness defects of retinal tissue that cause severe vision loss due to disruption of the anatomic fovea. Abnormal vitreous traction is involved in the formation of macular holes. Both glial cells and hyalocytes contribute to epiretinal membrane formation in IMH. In order to gain further insight into the pathophysiology of IMH, we conducted a discovery phase investigation of the vitreous proteome in four patients with macular holes and six controls using one-dimensional gel fractionation and liquid chromatography–tandem mass spectrometry analyses on an Orbitrap Elite mass spectrometer. Of a total of 5912 vitreous proteins, 32 proteins had increased and 39 proteins had decreased expression in IMH compared with controls, using a false discovery rate approach with p value < 0.001 and q value < 0.05. IMH was associated with increased expression of proteins in the complement pathway, α-2-macroglobulin, a major inducer of Müller glial cell migration, fibrinogen, and extracellular matrix proteins, and decreased expression of proteins involved in protein folding and actin filament binding. A proteomic approach revealed proteins and biological pathways that may be involved in the pathogenesis of IMH and could be targeted for future studies.
Electronic supplementary material
The online version of this article (10.1186/s12014-017-9172-y) contains supplementary material, which is available to authorized users.
Keywords: Eye, Idiopathic macular hole, Proteomics, Retina, Vitreous
Background
Idiopathic macular holes (IMH) are full-thickness defects of retinal tissue that are an important cause of severe, unilateral vision loss due to mechanical disruption of the anatomic fovea (Fig. 1). Most common in the sixth through eighth decades of life, macular holes are three times more likely to affect women than man [1, 2] and have up to a 14% risk of developing in the fellow eye [1]. An analysis of U.S. data recently showed that Asian-Americans are at higher risk of IMH [3].
Fig. 1.
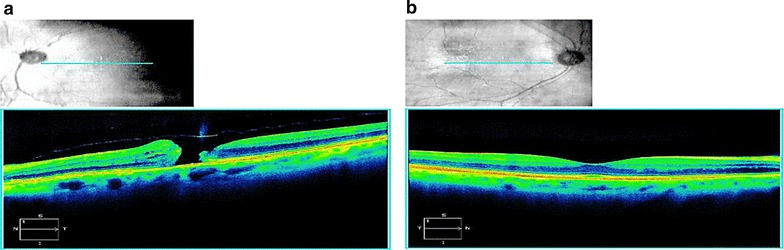
Zeiss spectral-domain image showing a full-thickness macular hole in the left eye (a) and a normal macula with good foveal contour in the right eye (b). Blue line in fundus image (above) denotes the plane of the cross-sectional image of the macula (below)
Vitreoretinal traction is a central mechanism implicated in macular hole formation [4–6]. The role of epiretinal cell proliferation at the internal limiting membrane (ILM) remains controversial [7]. Activated glial cells, such as Müller cells and astrocytes, are considered a source of cells that migrate from the retina to the vitreous [8]. Conversely, it has been hypothesized that hyalocytes, macrophage-like cells in the vitreous, become activated and proliferate at the ILM and form epiretinal membranes [4]. Morphological criteria for identifying the originating cell types in epiretinal membranes has been difficult since cells undergo phenotypic transdifferentiation [9]. Recently, immunocytochemical and ultrastructural studies showed that epiretinal cell proliferation of both glial cells and hyalocytes occurs in the ILM at all stages of IMH [7]. Both cell migration and proliferation play a dominant role in the development of macular holes [7]. In patients with small full-thickness macular holes, single glial cells without proliferation or contraction were found on the ILM [10]. Larger clusters of cells which are positive for both glial and hyalocyte markers were also present on the ILM. These clusters may have greater potential for proliferation and exertion of tangential traction on the retina [10].
While treatment options exist for IMH, none are without significant associated morbidity. Surgical intervention with pars plana vitrectomy, possible internal limiting membrane peeling, and often intraocular gas tamponade may be associated with morbidity from prolonged face-down positioning, cataract progression, elevated eye pressure, retinal tear, and retinal detachment [11]. Ocriplasmin, a recombinant protease which has activity against fibronectin and laminin, is sometimes used for outpatient treatment of IMH, but treatment may be associated with decreased vision and abnormalities on electroretinogram testing [12].
Previous case series, including the Eye Disease Case–Control Study (EDCCS), have attempted to identify physiologic factors related to the risk of developing macular hole. The EDCSS observed a higher plasma fibrinogen concentrations in those affected with IMH compared to those without IMH [12]. Identification of the molecular abnormalities of the vitreous that predispose to IMH formation may lay the groundwork for the development of targeted therapies that could preclude macular hole formation at the earliest stages.
Proteomic analysis is a powerful tool for characterizing the diversity and abundance of proteins in both health and disease in tissues and cells and may provide new insights into the pathogenesis of IMH [13, 14]. We used a proteomic approach to identify proteins that are differentially expressed in IMH and to identify potential pathogenic mechanisms involved in IMH formation.
Experimental procedures
We compared the vitreous proteome in four patients with IMH and six control patients with dislocated intraocular lens or epiretinal membrane. The mean age of cases and controls was 66.0 and 67.5 years, respectively. Adults in the study underwent standard of care pars plana vitrectomy for IMH, dislocated intraocular lens, or epiretinal membrane, and had no previous history of vitrectomy, Ocriplasmin injection, trauma, uveitis, or non-clearing vitreous hemorrhage in the eye undergoing surgery. Undiluted vitreous samples were collected from all patients.
Preparation of vitreous samples
Vitreous samples were immediately snap frozen and stored at − 80 °C until processing. Vitreous was suspended in 1 ml of lysis buffer (10 mM HEPES, 42 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1.0 mM dithiothreitol [DTT], 1 × phosphatase inhibitor (Thermo Scientific, Waltham, MA, USA), 1 × protease inhibitor (Sigma Aldrich, St. Louis, MO, USA), and homogenized using a motorized handheld Eppendorf mortar/pestle. Samples were sonicated three times for 10 s and stored on ice to prevent overheating between sonications. Sodium dodecyl sulfate was added for a final concentration of 2% (w/v). Samples were incubated at room temperature (RT) for 10 min to extract the proteins. The samples were then spun for 45 min, 14,000×g, at RT. Protein concentration of the supernatant was determined using the Micro BCA Protein Assay kit (Thermo Scientific).
Protein separation and fractionation
One-dimensional SDS-PAGE 4–12% gradient gels were used to separate proteins before double distilled H2O was added to the samples, resulting in 50 µg of protein in a 45 µl volume in a low retention Eppendorf tube. Forty-five µl 4 × lithium dodecyl sulfate SE + 100 mM DDT were added to tube, which was then vortexed, spun at 1000 rpm for 30 s at RT, and then placed on a 106 °C heat block for 10 min to denature the proteins. Tubes were again vortexed and spun twice at 1000 rpm for 30 s at RT. Fifty µg of sample were run on each lane of a 1D SDS gel, using NuPAGE 4–12% Bis–Tris gel 1.5 mm, 200 V. Coomassie blue (250 ml methanol, 50 ml acetic acid, 200 ml dH2O, 0.5 g Coomassie brilliant blue) was used to stain gels at RT for 30 min. Gels were then destained (50 ml methanol, 75 ml acetic acid, 875 ml d H2O) overnight. Each gel was cut into 12 bands. Gel bands were placed in separate vials and disrupted with forceps. Gel bands were then destained, dehydrated with acetonitrile, and dried down with a Savant SPD2010 Speedvac concentrator (Thermo Scientific) before samples were reduced with 10 mM DTT for 1 h at 55 °C and then alkylated with 55 mM iodoacetamide in 25 mM ammonium bicarbonate for 30 min at RT.
Sample in-gel digestion and cleanup by C18 reverse column
The samples were then washed with ultrapure water, dehydrated with 100% acetonitrile, and dried by Speedvac concentrator before undergoing digestion with trypsin/LysC (V5073; ratio of protein to enzyme = 50:1, Promega, Madison, WI, USA) at 37 °C overnight with constant shaking. Supernatant with peptides was then collected into a new Eppendorf tube. The gel slices were incubated with 1% TFA v/v in 80% acetonitrile for extracting peptides twice and then combined for drying by Speedvac concentrator. Peptides were resuspended in solvent 0.1% v/v formic acid in water (Optima™ LC/MS, Thermo Scientific), desalted by the column (Spin-Tip, Thermo Scientific), spun, eluted with 0.1% v/v formic acid in 80% acetonitrile (Optima™ LC/MS, Thermo Scientific), and then dried by Speedvac concentrator. Finally, samples were resuspended in 0.1% formic acid in water and spiked with 5 fmol/µl calibration peptides (Peptide Retention Time Calibration Mixture, Thermo Scientific).
Mass spectrometry analysis
Samples were run LC–MS/MS using a 1-h gradient of 2–30% acetonitrile (Fisher Scientific) with 0.1% formic acid (Sigma Aldrich) using an EASY-Spray source coupled with an Orbitrap Elite (Thermo Scientific) mass spectrometer. EASY-Spray source was run at 35 °C using a 25 cm × 75 μm integrated spray tip column. Peptides were trapped at 980 bar on a 2 cm × 75 μm trapping column. The trap was a 3 μm particle, and the column was 2 μm Acclaim PepMap C18. The Orbitrap instrument was operated in a positive ion mode, and the data-dependent product-ion mode was applied for all analyses. MS1 data were acquired with top 10 most intense spectra, at a resolution of 120,000, survey scans of m/z 400–2000 Th, and a target value of 1 × 106. Peptide precursors with charge state 2–6 were sampled for MS2. Ion trap CID spectra were performed with an isolation width of 2.0 Th, 35% NCE, target value of 1 × 104, and the max injection time was 100 ms. Dynamic exclusion was enabled with the following settings: repeat count = 1; repeat duration = 60 s; exclusion duration = 60 s; mass tolerance, ± 7 ppm. All individual samples were run with two technical replicates.
Protein identification and quantification
MS raw files were batch processed using search engines of Mascot and X!Tandem and peak lists were searched against the UniProt Human Sequence database with 20,197 reviewed proteins. Mascot and X!Tandem were searched with a fragment ion mass tolerance of 0.10 Da and a parent ion tolerance of 50 PPM, allowing for 2 missed tryptic cleavages. Carbamidomethylation of cysteine was specified in Mascot and X!Tandem as a fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine, acetylation of the n-terminus, carbamylation of lysine and phosphorylation of serine, threonine and tyrosine were specified as variable modifications.
Peptide identifications were accepted if they were established at > 95.0% probability by the Peptide Prophet algorithm with Scaffold Δ-mass correction. Protein identifications were accepted if they were established at > 99.0% probability and contained at least 2 identified peptides (minimum length of 7 amino acids). Protein Prophet algorithm was used to assign protein probabilities using a false discovery rate (FDR) of 0.2% for peptides and 1% for proteins against the target-decoy database. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Scaffold 4.8.4 (Proteome Software Inc., Portland, OR, USA) was used for FDR filtering and to validate MS/MS-based peptide and protein identifications. The number of proteins and peptides that overlapped in the technical replicates were averaged to provide a mean for each sample. PANTHER analysis was used to classify protein function and to conduct pathway analyses. Relative quantification was performed using weighted spectral counting. Data log (2) transformed values were normalized based on total spectral counts per sample and compared using the tools described below.
Bioinformatics analysis
An R package, DEGseq [15] was used to identify the differentially expressed proteins between samples from IMH and controls. p Values and q values were calculated using a FDR approach as described by Benjamini and Cohen [16] and Storey and Tibshirani [17]. FDR was used to control the proportion of false positives among potentially differentially expressed proteins, and the extended “q values” were defined as the minimum FDR that can be attained as p values have for type-I error control and calculated from the observed p values after estimating the proportion of differentially expressed proteins. An MA plot was used for visualization of proteomic data in significance analyses [18]. Using a p value < 0.001 and a cutoff of q value < 0.05, 71 differentially expressed proteins were identified. GO analyses were performed on these 71 proteins with DAVID (Database for Annotation, Visualization and Integrated Discovery). Protein–protein interaction networks were studied Ingenuity® Pathway Analysis (QIAGEN Bioinformatics, Redwood City, CA, USA).
Multiplexed selected reaction monitoring analysis
To validate for quantitative analysis by multiplexed selected reaction monitoring (SRM) assays, representative proteins with > 1.5-fold change were randomly selected and at least 2–3 of their tryptic peptides and optimization of transitions were selected following the guideline of Skyline (Seattle Proteome Center, Seattle, WA, USA.) (Additional file 1: Table S4). The heavy isotopically labeled tryptic fragment peptides were obtained from New England Peptide (Gardner, MA, USA) and prepared by Fmoc-based solid-phase peptide synthesis using per-15N, 13C-labeled (> 99% isotopic purity) Arg or Lys as the C-terminal residue attached to the resin. Peptides were purified by reversed phase chromatography (C18 stationary phase using water-acetonitrile gradients, ion-pairing agent ~ 0.1% TFA). The purity of the synthetic heavy peptides was ≥ 95% for each by the confirmation of analytical HPLC. Purified peptide solutions were prepared and the concentration of the solution was determined by amino acid analysis. Vitreous samples were carried out in-solution digestion in brief: 0.1 mg of proteins was aliquoted in 0.1% (w/v) RapiGest buffer containing 100 mM Tris–HCl, pH 8.0, and 100 mM dithiothreitol and incubated at 55 °C for 1 h for denaturation and reduction. The samples were then alkylated for 30 min at room temperature in dark with a final concentration of 50 mM iodoacetamide, digested by trypsin/LysC mix (Promega) at an enzyme-to-substrate ratio of 1:50 for 18 h at 37 °C. The digestion was terminated with 10% TFA to a final concentration of 1%, followed by centrifugation at 14,000×g for 15 min to remove RapiGest, and the sample was spiked with a heavy labeled peptide mixture at a concentration of 100 fmol/µl and cleaned up with a 96-well extraction plate vacuum manifold (Waters, Milford, MA, USA) according to manufacturer’s instruction.
SRM assays in triplicates were run on a 5500 QTrap (Sciex, Framingham, MA, USA) mass spectrometer equipped with an electrospray ionization source, a CBM-20A command module, LC-20ADXR pump, and a CTO-10Ac column oven heater (all Shimadzu, Kyoto, Japan). A sample volume of 10 µL was injected onto the column via a Shimadzu SIL-20AXR autosampler set to 4 °C at a flow rate of 0.2 ml/min, with an instrument run time of 18 min/sample including the 5 min column regeneration step. Chromatographic separations were conducted using an XBridge BEH C18 Column, 130 Å, 3.5 µm, 2.1 mm × 100 mm (Waters), with a linear gradient starting from 5% phase B increasing to 36% phase B within 10 min with the column oven set at 37 °C. Mobile phases consisted of water containing 0.1% formic acid (phase A) and 98% acetonitrile containing 0.1% formic acid (phase B). The mass spectrometry analysis was performed in positive ion mode and performed at unit resolution in both Q1 and Q3 quadrupoles. All sample data were collected using Analyst 1.5 software and processed using MultiQuant software version 3.02 (Sciex) with the parameters: Gaussian smooth width = 0, minimum peak width of points = 3, noise percentage = 40.0%, baseline subtraction window = 2.00 min, and peak splitting points = 2. All raw peak area ratios were calculated by contrasting against the heavy standard peptides and normalized by albumin. ANOVA post hoc tests were used for statistical analysis and validation of protein changes between cases (n = 12) and controls (n = 18).
Results
In vitreous, the primary searching generated 6054 clusters and 6393 proteins. There were 5912 non-redundant proteins and 326,434 spectra (Additional file 2: Table S1), after removing immunoglobulins (Ig fragments) and redundant proteins. The change in an MA plot of IMH versus control proteins is shown in Fig. 1, with red points identified as differentially expressed. There were 32 proteins with increased expression and 39 proteins with decreased expression in IMH compared with controls. The 1.5-fold change in expression between IMH and controls are shown in Additional file 3: Table S2 and Additional file 4: Table 3.
Proteins with increased expression in the vitreous of IMH compared with controls (Table 1) were involved in the complement pathway (complement factor H, complement factor B, complement C3, complement C4-A), extracellular matrix (ECM) (brevican core protein, spondin-1, retinol-binding protein 3, versican core protein, collagen α-1(II) chain, target of Nesh-SH3), hyaluronan binding (inter-α-trypsin inhibitor heavy chain H1, inter-α-trypsin inhibitor heavy chain H2, inter-α-trypsin inhibitor heavy chain H4), and fibrinogen (Fig. 2).
Table 1.
Thirty-two proteins with increased expression in the vitreous of idiopathic macular holes compared with controls
| Protein name | Gene | neXtProt ID | Function |
|---|---|---|---|
| Melanotransferrin | MELTF | P08582 | Iron ion binding |
| Brevican core protein | PGCB | Q96GW7 | Hyaluronan binding; ECM component; involved in cell adhesion |
| Neurexin-2 | NRXN2 | Q9P2S2 | Neuronal cell surface protein involved in cell recognition and cell adhesion |
| Ubiquitin-associated protein 2 | UBAP2 | Q5T6F2 | Cadherin binding involved in cell–cell adhesion |
| Spondin-1 | SPON1 | Q9HCB6 | Cell adhesion protein; found in ECM |
| Apolipoprotein B-100 | APOB | P04114 | Phospholipid/cholesterol transporter activity |
| Metallothionein-1A | MT1A | P04731 | Zinc ion binding; negative regulation of growth |
| Retinol-binding protein 3 | RBP3 | P10745 | Serine-type peptidase activity; found in ECM between retina and RPE |
| Fibrinogen gamma chain | FGG | P02679 | Together with fibrinogen α and β, polymerizes to form an insoluble fibrin matrix |
| Complement factor H | CFH | P08603 | Acts as cofactor in the inactivation of the alternative complement pathway |
| Collagen α-2(IX)chain | COL92A | Q14055 | Structural component of the vitreous |
| Versican core protein | VCAN | P13611 | Intracellular signaling and connecting cells with ECM; binds hyaluronan |
| Amyloid-like protein 2 | APLP2 | Q06481 | Serine-type endopeptidase inhibitor activity |
| Inter-α-trypsin inhibitor heavy chain H2 | ITIH2 | P19823 | Involved as binding protein between hyaluronan and other matrix proteins |
| Inter-α-trypsin inhibitor heavy chain H4 | ITIH4 | Q14624 | Type II acute phase response protein; serine-type endopeptidase inhibitor activity |
| Collagen α-1(II) chain | COL2A1 | P02458 | Extracellular matrix structural constituent conferring tensile strength |
| Target of Nesh-SH3 | ABI3BP | Q7Z7G0 | Collagen binding; ECM organization; positive regulator of cell-substrate adhesion |
| Complement factor B | CFB | P00751 | Role in alternative complement pathway; cleaved by factor D into Ba and Bb fragments |
| Apolipoprotein A-IV | APOA4 | P06727 | Component of chylomicrons, HDL; cholesterol and lipid transport |
| Inter-α-trypsin inhibitor heavy chain H1 | ITIH | P19827 | Carrier of hyaluronan or as binding protein between hyaluronan and other matrix proteins |
| Haptoglobin | HP | P00738 | Antioxidant activity |
| α-2-Macroglobulin | A2 M | P01023 | Inhibits all four classes of proteinases |
| Fibrinogen α chain | FGA | P02671 | Polymerizes with fibrinogen β and γ to form an insoluble fibrin matrix |
| Calsyntenin-1 | CLSTN1 | O94985 | Interacts with amyloid precursor protein, kinesin-1 |
| Apolipoprotein A-I | APOA1 | P02647 | Cholesterol transport |
| Serotransferrin | TF | P02787 | Iron binding transport protein |
| Complement C3 | C3 | P01024 | Central role in activation of the complement system |
| Ceruloplasmin | CP | P00450 | Copper-binding glycoprotein; ferroxidase activity in oxidizing Fe2+ to Fe3+ |
| Gelsolin | GSN | P06396 | Role in actin filament capping and polymerization |
| Pigment epithelium-derived factor | SERPINF1 | P36955 | Inhibitor of angiogenesis; neurotrophic |
| Complement C4-A | C4A | P0C0L4 | Nonenzymatic component of C3 and C5 convertases; essential for propagation of classical complement pathway |
| Serum albumin | ALB | P02768 | Main plasma protein; binds water, Ca2+, Na+, K+, zinc, fatty acids, etc. |
Differentially expressed proteins with the normalized log2 fold change at p < 0.001 and q value < 0.05
Fig. 2.
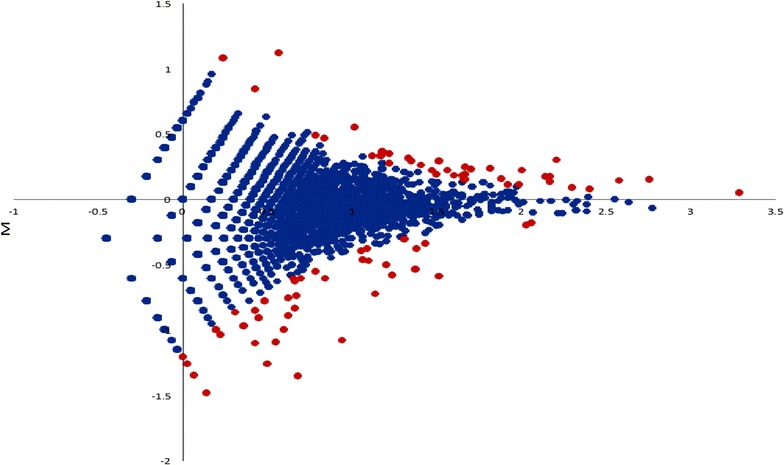
Significance analysis of vitreous proteins in IMH versus control. MA plot of human vitreous proteins [15, 18]. X-axis shows the average protein expression level, and Y-axis shows the fold change between IMH and control (in log2 scale). Each point represents a protein detected in the MS assay. The differentially expressed proteins were highlighted in red
Proteins with decreased expression in the vitreous of IMH compared with controls (Table 2) included those involved in protein folding (T-complex protein 1 subunit ζ, heat shock protein β-1, heat shock 70 kDa protein 1A, UDP-glucose:glycoprotein), actin filament binding (filamin-A, vinculin), cell adhesion (transforming growth factor-β-induced protein ig-h3, transgelin-2, myocilin), crystallin proteins (α-crystallin B chain, α-crystallin A chain, β-crystallin A3, and β-crystallin B2), and glycolytic enzymes (phosphoglycerate kinase 1, α-enolase).
Table 2.
Thirty-nine proteins with decreased expression in the vitreous of idiopathic macular holes compared with controls
| Protein name | Gene | neXtProt ID | Function |
|---|---|---|---|
| Stromelysin-1 | MMP3 | P08254 | Degrades fibronectin; involved in breakdown and remodeling of ECM proteins |
| Transforming growth factor-β-induced protein ig-h3 | TGFBI | Q15582 | Plays a role in cell adhesion; involved in cell–collagen interactions |
| T-complex protein 1 subunit zeta | CCT6A | P40227 | Molecular chaperone; assists in the folding of proteins, including actin and tubulin |
| RNA-binding protein 43 | RBM43 | Q6ZSC3 | Nucleotide and RNA binding |
| Vimentin | VIM | P08670 | Class-III intermediate filament; normal component of vitreous |
| Pro-neuregulin-1, membrane-bound isoform | NRG1 | Q02297 | Tyrosine kinase activator activity; ligand for integrins |
| α-Crystallin B chain | CRYAB | P02511 | Normal component of vitreous, lens, cornea; molecular chaperone |
| α-Crystallin A chain | CRYAA | P02489 | Normal component of vitreous, lens, cornea; molecular chaperone |
| Mixed lineage kinase domain-like protein | MLKL | Q8NB16 | Pseudokinase with role in TNF-induced necroptosis, a programmed cell death process |
| Retinal dehydrogenase 1 | ALDH1A1 | P00352 | Converts retinaldehyde to retinoic acid; binds retinal |
| Heat shock protein β-1 | HSPB1 | P04792 | Involved in stress resistance and actin organization |
| Lipocalin-1 | LCN1 | P31025 | Cysteine-type endopeptidase inhibitor activity |
| Phosphoglycerate kinase 1 | PGK1 | P00558 | Glycolytic enzyme |
| CLOCK-interacting pacemaker | CIPC | Q9C0C6 | Transcriptional repressor |
| Glucoside xylosyltransferase 2 | GXYLT2 | A0PJZ3 | Glycosyltransferase which elongates the O-linked glucose attached to EGF-like repeats in the extracellular domain of Notch proteins by catalyzing the addition of xylose |
| Carbonic anhydrase 1 | CA1 | P00915 | Reversible hydration of carbon dioxide |
| Leucine-rich repeat and IQ domain-containing protein 3 | LRRIQ3 | A6PVS8 | Protein binding |
| Transgelin-2 | TAGLN2 | P37802 | Regulates cell migration; cadherin binding involved in cell–cell adhesion |
| β-Crystallin A3 | CRYBA1 | P05813 | Normal component of vitreous, lens, cornea |
| Decorin | DCN | P07585 | Extracellular matrix binding; glycosaminoglycan binding; may affect rate of fibrils formation |
| Tetraspanin-16 | TSPAN16 | Q9UKR8 | Cell surface receptor signaling pathway |
| Taste receptor type 2 member 9 | TAS2R9 | Q9NYW1 | Taste receptor activity |
| SLIT and NTRK-like protein 3 | SLITRK3 | O94933 | Suppresses neurite outgrowth; axonogenesis |
| β-Crystallin B2 | CRYBB2 | P43320 | Normal component of vitreous, lens, cornea |
| DNA-directed RNA polymerase III subunit RPC2 | POLR3B | Q9NW08 | Contributes to RNA polymerase III activity |
| 55 kDa erythrocyte membrane protein | MPP1 | Q00013 | Regulates neutrophil polarity |
| α-Enolase | ENO1 | P06733 | Plays a role in glycolysis; role in fibrinolytic system as receptor and activator of plasminogen; cadherin binding involved in cell–cell adhesion |
| Heat shock 70 kDa protein 1A | HSPA1A | P0DMV8 | Stabilizes preexistent proteins against aggregation in cooperation with other chaperones |
| Apolipoprotein D | APOD | P05090 | Lipid transport |
| Filamin-A | FLNA | P21333 | Involved in actin filament binding; promotes orthogonal branching of actin filaments; links actin filaments to membrane glycoproteins |
| Myocilin | MYOC | Q99972 | Secreted glycoprotein that regulates activation of different signaling pathways involved in cell adhesion, cell–matrix adhesion, cytoskeleton organization, and cell migration; binds with fibronectin |
| UDP-glucose:glycoprotein glucosyltransferase 2 | UGGT2 | Q9NYU1 | Recognizes glycoproteins with minor folding defects; provide quality control for protein folding in the endoplasmic reticulum |
| Uncharacterized protein C1orf167 | C1orf167 | Q5SNV9 | Unknown |
| Vinculin | VCL | P18206 | Actin filament binding protein involved in cell matrix adhesion and cell–cell adhesion; regulates cell-surface E-cadherin expression |
| Dynein heavy chain 1, axonemal | DNAH1 | Q9P2D7 | Cilium movement involved in cell motility |
| β-Actin-like protein 2 | ACTBL2 | Q562R1 | Major constituent of the contractile apparatus |
| Tectonic-1 | TCTN1 | Q2MV58 | Cilium assembly |
| Hemoglobin subunit β | HBB | P68871 | Oxygen transport; contributes to haptoglobin binding |
| Potassium voltage-gated channel subfamily G member 1 | KCNG1 | Q9IUX4 | Potassium channel activity |
Differentially expressed proteins with the normalized log2 fold change at p < 0.001 and q value < 0.05
We used SRM to validate differentially expressed proteins. Thirteen proteins with 1.5-fold changes were assayed and validated with significant differential expressions between cases (n = 12) and controls (n = 18) (Additional file 5: Figures S1–S25).
Protein–protein interactions among vitreous proteins that are differentially expressed between IMH and controls are shown in Fig. 3. GO processes that were identified include metabolism of chondroitin sulfate and dermatan sulfate (versican, brevican), platelet degranulation (fibrinogen γ chain, gelsolin, metallothionein-1A), complement cascade (complement factor H, complement factor B, complement C3, complement C4-A), scavenging of heme from plasma (haptoglobin, hemoglobin beta), lipoprotein metabolism (apolipoprotein A-I, apolipoprotein A-IV, apolipoprotein B-100, apolipoprotein D).
Fig. 3.
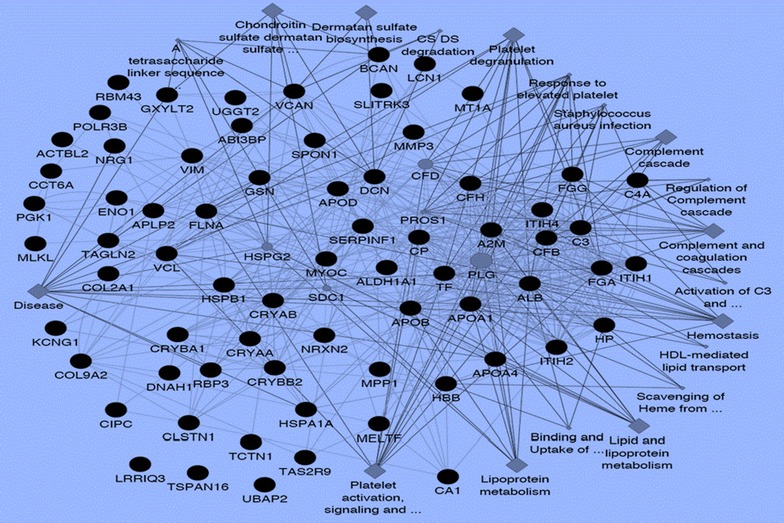
Protein–protein interaction network. The network weighting method is based on GO biological process. Black nodes represent genes associated with the biological process, diamond represents the biological process and the edge represents the type and strength of gene interaction. Biological processes include: binding and uptake of ligands by scavenger receptors, scavenging of heme from plasma, platelet activation, signaling and aggregation, activation of C3 and C5, chondroitin sulfate dermatan sulfate metabolism, and A tetrasaccharide linker sequence is required for GAG synthesis
Discussion
The present proteomic investigation of the vitreous identifies proteins, pathways, and biological processes that could be involved in the pathogenesis of IMH, including the complement pathway, induced migration of Müller glial cells, fibrinogen, ECM proteins, protein folding, and actin filament binding, as presented in greater detail below.
The complement system of innate immunity is involved in the recognition of diseased or damaged host cells, regulation of cellular immune responses, and interaction with the coagulation cascade [19]. Complement—considered a “master of sensing”—can discriminate between foreign, altered, and healthy surfaces, but excessive activation or insufficient control of the complement system can lead to an imbalance that exacerbates the pathology of disease. Two proteins of the alternative complement pathway, complement factors H and B, were upregulated in the vitreous from eyes with IMH compared with controls. In addition, complement C3, which plays a central role in the activation of the complement system, was also upregulated. The increased expression of complement C4-A in IMH also suggests involvement of the classical complement pathway.
The migration of Müller glial cells from the retina to the vitreal surface has been implicated in the pathogenesis of IMH formation [8]. α-2-macroglobulin (α2M), a major antiproteinase in plasma and fluids, was increased in the vitreous of patients with IMH. α2M inhibits a broad spectrum of proteases [20]. α2M consists of two noncovalently bound dimers of disulfide-linked identical subunits. Near the middle of the polypeptide chain is a unique sequence of amino acids, known as the “bait” region, which is susceptible to cleavage by proteinases. Cleavage by proteinases introduces a conformational change and activation of α2M [20]. Activated α2M can specifically bind to low-density lipoprotein receptor-related protein 1 (LRP1), which induces Müller glial cell migration [21]. Increased expression of α2M is a possible mechanism by which Müller glial cell may acquire the ability to migrate to the vitreous in the formation of IMH.
A multicenter epidemiological study from the EDCCS involving 198 cases of IMH and 1023 controls showed that elevated circulating fibrinogen concentrations were a strong independent risk factor for IMH [13]. The underlying basis for this unexpected epidemiological association was unclear. It was speculated that elevated fibrinogen could possibly compromise blood flow in the macular region or act upon the vitreous in an unknown way that increases the susceptibility of the macula to vitreous traction [13]. In the present study, there was increased expression of fibrinogen α and γ chains in the vitreous of patients with IMH compared with controls. These observations appear to corroborate and extend the findings from the EDCCS. In an animal model, fibrinogen induced the formation of vitreous membranes when injected into the vitreous [22]. Fibrinogen may be transformed into a long fibrin polymer that forms a matrix that allows surrounding cells to proliferate in the vitreous [22].
Five protein components of the extracellular matrix (brevican core protein, spondin-1, retinol-binding protein 3, versican core protein, collagen α-1(II) chain, target of Nesh-SH3) and three proteins involved in binding with hyaluronan, a major constituent of the extracellular matrix (inter-α-trypsin inhibitor heavy chain H1, inter-α-trypsin inhibitor heavy chain H2, inter-α-trypsin inhibitor heavy chain H4), were upregulated in IMH compared with controls. The ECM undergoes extensive remodeling during cell proliferation, differentiation, and migration, and such changes can determine the course of disease pathogenesis [23]. Versican and fibrinogen bind with hyaluronan in the ECM [23]. Members of the inter-α-trypsin inhibitor proteoglycan family covalently bind to hyaluronan and stabilize the ECM [24]. These findings suggest that the ECM undergoes active remodeling during the formation of IMH.
Six molecular chaperone proteins involved in the proper folding of proteins were downregulated in IMH: T-complex protein 1 subunit ζ, heat shock protein β-1, heat shock 70 kDa protein 1A, UDP-glucose:glycoprotein, α-crystalline A chain, and α-crystalline B chain. T-complex protein 1 subunit ζ folds various proteins, including actin [25]. Heat shock protein β-1 plays a role in proper assembly of actin filaments [26]. Heat shock 70 kDa protein 1A prevents the aggregation of misfolded proteins and can convert misfolded proteins into active conformation [27]. UDP-glucose: glycoprotein recognizes glycoproteins that have minor folding defects [28]. α-crystalline A chain and α-crystalline B chain have chaperone-like activity and may prevent aggregation of various proteins under conditions of stress. The downregulation of these molecular chaperone proteins suggests that surveillance for misfolded proteins may be compromised in IMH. Two actin filament binding proteins, filamin-A and vinculin, had lower expression in IMH compared with controls. Vinculin plays a role in binding and rearranging the actin cytoskeleton [29]. Filamin-A plays a role in cross-linking and stabilizing the F-actin cytoskeleton [30].
Conclusions
This study showed that the complement pathway, α2M, an inducer of Müller glial cell migration, extracellular matrix remodeling, fibrinogen, molecular chaperones involved in protein folding, and actin filament binding proteins may play a role in the pathogenesis of IMH.
Additional files
Additional file 1: Table 4. SRM assay for protein changes related to the pathogenesis of human idiopathic macular hole.
Additional file 2: Table 1. Proteins identified in the vitreous.
Additional file 3: Table 2. Differentially expressed proteins in the vitreous between idiopathic macular hole and controls.
Additional file 4: Table 3. The 1.5-fold change in protein expression between IMH and controls.
Additional file 5. Figures S1–S25. SRM assay to validate differentially expressed proteins.
Authors’ contributions
PZ performed analysis on all samples, interpreted data, edited manuscript and acted as corresponding author. MZ made contributions to acquisition and interpretation of SRM data. YZ and JQ performed statistics analysis and helped in data interpretation and manuscript evaluation. CD performed mass spectrum analysis on all samples. RT completed sample preparation. RDS and SDS supervised development of work and wrote manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported in part by the Wilmer Eye Institute Pooled Professors Research Fund, the National Institutes of Health Grants R01 EY024596, R01 AG027012, the Edward N. & Della L. Thome Memorial Foundation, and Research to Prevent Blindness. The authors are grateful to the following Wilmer surgeons who contributed to surgical specimen collections as part of this research: Dr. Yannis Paulus, Dr. Ian Han, Dr. Connie Chen, Dr. Syed Shah, Dr. Ingrid Zimmer-Galler, Dr. Adrienne Scott, Dr. Adam Wenick, Dr. Peter Campochiaro, Dr. Howard Ying, Dr. Akrit Sodhi, Dr. David Poon, Dr. Lisa He, Dr. James Handa, Dr. Alvin Liu.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD006687.
Consent for publication
The authors have signed the agreement with for publication.
Ethics approval and consent to participate
The protocol was approved by the Johns Hopkins University School of Medicine Institutional Review Board. Patients provided written, informed consent to participate. The study adhered to the tenets of the Declaration of Helsinki and was conducted in compliance with the regulations set forth by the Health Insurance Portability and Accountability Act. Consecutive patients were enrolled from September 2013 to July 2014.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12014-017-9172-y) contains supplementary material, which is available to authorized users.
Contributor Information
Pingbo Zhang, Phone: (410) 955-2571, Email: pzhang7@jhmi.edu.
Min Zhu, Email: zhumi@grc.nia.nih.gov.
Yuming Zhao, Email: zymyoyo@hotmail.com.
Jiang Qian, Email: jiang.qian@jhmi.edu.
Craig Dufresne, Email: craig.dufresne@thermofisher.com.
Randi Turner, Email: rturner6@umd.edu.
Richard D. Semba, Email: rdsemba@jhmi.edu
Sharon D. Solomon, Email: ssolomon1@jhmi.edu
References
- 1.McCannel CA, Ensminger JL, Diehl NN, Hodge DN. Population-based incidence of macular holes. Ophthalmology. 2009;116:1366–1369. doi: 10.1016/j.ophtha.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chew EY, Sperduto RD, Hiller R, Nowroozi L, Seigel D, Yanuzzi LA, Burton TC, Seddon JM, Gragoudas ES, Haller JA, Blair NP, Farber M. Clinical course of macular holes: the eye disease case–control study. Arch Ophthalmol. 1999;117:242–246. doi: 10.1001/archopht.117.2.242. [DOI] [PubMed] [Google Scholar]
- 3.Ali FS, Stein JD, Blachley TS, Ackley S, Stewart JM. Incidence of and risk factors for developing idiopathic macular hole among a diverse group of patients throughout the United States. JAMA Ophthalmol. 2017;135:299–305. doi: 10.1001/jamaophthalmol.2016.5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sebag J. Anomalous posterior vitreous detachment: a unifying concept in vitreo-retinal disease. Graefe’s archive for clinical and experimental ophthalmology. Graefes Arch Clin Exp Ophthalmol. 2004;242:690–698. doi: 10.1007/s00417-004-0980-1. [DOI] [PubMed] [Google Scholar]
- 5.Green WR. The macular hole: histopathologic studies. Arch Ophthalmol. 2006;124:317–321. doi: 10.1001/archopht.124.3.317. [DOI] [PubMed] [Google Scholar]
- 6.Jackson TL, Nicod E, Simpson A, Angelis A, Grimaccia F, Kanavos P. Symptomatic vitreomacular adhesion. Retina. 2013;33:1503–1511. doi: 10.1097/IAE.0b013e31829232fd. [DOI] [PubMed] [Google Scholar]
- 7.Schumann RG, Eibl KH, Zhao F, Scheerbaum M, Scheler R, Schaumberger MM, Wehnes H, Walch AK, Haritoglou C, Kampik A, Gandorfer A. Immunocytochemical and ultrastructural evidence of glial cells and hyalocytes in internal limiting membrane specimens of idiopathic macular holes. Invest Ophthalmol Vis Sci. 2011;52:7822–7834. doi: 10.1167/iovs.11-7514. [DOI] [PubMed] [Google Scholar]
- 8.Bringmann A, Wiedemann P. Involvement of Muller glial cells in epiretinal membrane formation. Graefe’s archive for clinical and experimental ophthalmology. Graefes Arch Clin Exp Ophthalmol. 2009;247:865–883. doi: 10.1007/s00417-009-1082-x. [DOI] [PubMed] [Google Scholar]
- 9.Vinores SA, Campochiaro PA, McGehee R, Orman W, Hackett SF, Hjelmeland LM. Ultrastructural and immunocytochemical changes in retinal pigment epithelium, retinal glia, and fibroblasts in vitreous culture. Invest Ophthalmol Vis Sci. 1990;31:2529–2545. [PubMed] [Google Scholar]
- 10.Schumann RG, Hagenau F, Haritoglou C, Wolf A, Schaumberger MM, Kampik A, Gandorfer A. Cells at the vitreoretinal interface in small full-thickness macular holes. Retina. 2015;35:1158–1165. doi: 10.1097/IAE.0000000000000441. [DOI] [PubMed] [Google Scholar]
- 11.Jackson TL, Donachie PH, Sparrow JM, Johnston RL. United Kingdom National Ophthalmology Database study of vitreoretinal surgery: report 2, macular hole. Ophthalmology. 2013;120:629–634. doi: 10.1016/j.ophtha.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Khan MA, Haller JA. Ocriplasmin for treatment of vitreomacular traction: an update. Ophthalmol Ther. 2016;5:147–159. doi: 10.1007/s40123-016-0062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Eye Disease Case-Control Study Group Risk factors for idiopathic macular holes. Am J Ophthalmol. 1994;118:754–761. doi: 10.1016/S0002-9394(14)72555-3. [DOI] [PubMed] [Google Scholar]
- 14.Semba RD, Enghild JJ, Venkatraman V, Dyrlund TF, Van Eyk JE. The Human Eye Proteome Project: perspectives on an emerging proteome. Proteomics. 2013;13:2500–2511. doi: 10.1002/pmic.201300075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 16.Benjamini Y, Cohen R. Weighted false discovery rate controlling procedures for clinical trials. Biostatistics. 2017;18:91–104. doi: 10.1093/biostatistics/kxw030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gatto L, Breckels LM, Naake T, Gibb S. Visualization of proteomics data using R and bioconductor. Proteomics. 2015;15:1375–1389. doi: 10.1002/pmic.201400392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehman AA, Ahsan H, Khan FH. alpha-2-Macroglobulin: a physiological guardian. J Cell Physiol. 2013;228:1665–1675. doi: 10.1002/jcp.24266. [DOI] [PubMed] [Google Scholar]
- 21.Barcelona PF, Jaldin-Fincati JR, Sanchez MC, Chiabrando GA. Activated alpha2-macroglobulin induces Muller glial cell migration by regulating MT1-MMP activity through LRP1. FASEB J. 2013;27:3181–3197. doi: 10.1096/fj.12-221598. [DOI] [PubMed] [Google Scholar]
- 22.Mansour AM, Chess J, Henkind P. Fibrinogen-induced vitreous membranes. Ophthalmic Res. 1987;19:164–169. doi: 10.1159/000265489. [DOI] [PubMed] [Google Scholar]
- 23.Wight TN. Provisional matrix: a role for versican and hyaluronan. Matrix Biol. 2017;61:38–56. doi: 10.1016/j.matbio.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bost F, Diarra-Mehrpour M, Martin JP. Inter-alpha-trypsin inhibitor proteoglycan family—a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem. 1998;252:339–346. doi: 10.1046/j.1432-1327.1998.2520339.x. [DOI] [PubMed] [Google Scholar]
- 25.Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, Artandi SE. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell. 2014;159:1389–1403. doi: 10.1016/j.cell.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doshi BM, Hightower LE, Lee J. HSPB1, actin filament dynamics, and aging cells. Ann N Y Acad Sci. 2010;1197:76–84. doi: 10.1111/j.1749-6632.2010.05191.x. [DOI] [PubMed] [Google Scholar]
- 27.Finka A, Sharma SK, Goloubinoff P. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front Mol Biosci. 2015;2:29. doi: 10.3389/fmolb.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda Y, Seko A, Fujikawa K, Izumi M, Kajihara Y, Ito Y. Effects of domain composition on catalytic activity of human UDP-glucose:glycoprotein glucosyltransferases. Glycobiology. 2016;26:999–1006. doi: 10.1093/glycob/cww069. [DOI] [PubMed] [Google Scholar]
- 29.Bays JL, DeMali KA. Vinculin in cell–cell and cell–matrix adhesions. Cell Mol Life Sci. 2017;74:2999–3009. doi: 10.1007/s00018-017-2511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutherland-Smith AJ. Filamin structure, function and mechanics: are altered filamin-mediated force responses associated with human disease? Biophys Rev. 2011;3:15–23. doi: 10.1007/s12551-011-0042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table 4. SRM assay for protein changes related to the pathogenesis of human idiopathic macular hole.
Additional file 2: Table 1. Proteins identified in the vitreous.
Additional file 3: Table 2. Differentially expressed proteins in the vitreous between idiopathic macular hole and controls.
Additional file 4: Table 3. The 1.5-fold change in protein expression between IMH and controls.
Additional file 5. Figures S1–S25. SRM assay to validate differentially expressed proteins.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD006687.