

Abstract
Background
The non-small cell lung cancer (NSCLC) is the leading cause of cancer death worldwide. In NSCLC, the oncogenic AKT-mTOR, ERK and STAT3 pathways are commonly dysregulated and have emerged as attractive targets for therapeutic developments. In a relatively limited subset of NSCLC, these pathways driven by mutant EGFR can be treated by the tyrosine kinase inhibitors (TKIs)-mediated targeted therapy. However, for the most NSCLC, more novel targeted agents are imperatively needed. Therefore, we investigated the inhibitory effects of the active fraction HS7 from Taiwanofungus camphoratus, a unique medicinal fungus in Taiwan, on these pathways in CL1-0 EGFR wild-type human NSCLC cells.
Methods
The active fraction HS7 was prepared by n-hexane extraction of T. camphoratus followed by silica gel chromatography. Its effects on the cell viabilities were determined by sulforhodamine B colorimetric assay. Flow cytometry was used to analyze cell-cycle regulation and apoptosis induction. The changes in cellular protein levels were examined by Western blot.
Results
The active fraction HS7 vigorously inhibits AKT-mTOR, ERK and STAT3 signaling pathways in CL1-0 cells. At dose of 25 μg/mL, these signaling pathways were almost completely inhibited by HS7, accompanied with induction of cyclin-dependent kinase inhibitors such as p15, p21 and p27. Accordingly, the AKT-mTOR downstream targets p-p70S6K and HIF-1α were also suppressed as well. At this dose, the cell proliferation was profoundly suppressed to 23.4% of control and apoptosis induction was observed.
Conclusions
The active fraction HS7 from n-hexane extract of T. camphoratus exerts multi-targeting activity on the suppression of AKT-mTOR, ERK and STAT3 pathways and induction of p15, p21 and p27 in EGFR wild-type NSCLC cells. This multi-targeting activity of HS7 suggests its potential as an alternative medicine for the treatment of EGFR TKIs resistant NSCLC.
Electronic supplementary material
The online version of this article (10.1186/s13020-017-0154-9) contains supplementary material, which is available to authorized users.
Keywords: Non-small cell lung cancer, HS7, Taiwanofungus camphoratus, AKT-mTOR, ERK, STAT3
Background
Lung cancer is the leading cause of cancer death worldwide and its incidence is still on the increase [1]. About 89% of lung cancer patients are non-small cell lung cancer (NSCLC) [2] and 20–30% of these patients were diagnosed at a locally advanced stage [3]. In most NSCLC patients, the oncogenic AKT, ERK and STAT3 signaling pathways are constitutively activated. It has been reported that 50–70% overexpression of phosphorylated AKT [4], 70% expression of activated ERK [5] and over 50% high levels of activated STAT3 [6] were observed in NSCLC. Aberrant activation of these three signaling pathways results in uncontrolled proliferation, apoptosis resistance and other oncogenic cascades in lung cancer cells [4, 6–8]. Therefore, there has been increasing research interest in identifying novel therapeutics to target these oncogenic signaling pathways for effectively treating NSCLC patients [4, 9, 10].
PI3K-AKT-mTOR and RAS-RAF-MEK-ERK are two main downstream signaling pathways driven by epidermal growth factor receptor (EGFR) mutations or abnormal fusion of echinoderm microtubule-associated protein-like 4 and anaplastic lymphoma kinase (EML4-ALK) genes [4]. STAT3 (signal transducer and activator of transcription 3), an important point of convergence for various signaling pathways [6] is also required for the oncogenic effects of NSCLC-associated EGFR mutations [11]. The relatively limited subset of NSCLC carrying the above genetic mutations can be effectively treated by the tyrosine kinase inhibitors (TKIs)-mediated targeted therapy [4, 12, 13]. However, most NSCLC patients do not harbor these genomic events and the 5-year survival rate remains dismal [13]. More novel targeted agents to suppress these oncogenic pathways are imperatively needed.
Taiwanofungus camphoratus (syn. Antrodia camphorata) is a widely used medicinal polypore fungi, which belongs to the Polyporaceae, Basidiomycotine family, and grows in a unique host, the endemic perennial tree Cinnamomun kanehirai (Bull camphor tree) in Taiwan [14, 15]. It is a well-known folk medicine and has been used as a local remedy to treat abdominal pain, diarrhea, drug intoxication, hypertension, and skin itching as well as to improve immune system and liver function [15, 16]. On the other hand, many studies have demonstrated its anticancer effects in the aspects of anti-proliferation, apoptosis induction and anti-invasion [14, 16–18]. Instead of the commonly used ethanol, we use n-hexane to extract the T. camphoratus and further separate the extract into eight fractions (HS1–HS8) by silica gel chromatography. We have isolated the most potent active fraction (HS7) according to its anti-proliferative activities against a panel of human cancer cell lines, including, lung (CL1-0), prostate cancer (PC3) and hepatocellular carcinoma (HepG2, Hep3B and Huh7) cells (see Additional file 1). Our previous study had demonstrated the effects of HS7 on the apoptosis induction and Wnt/β-catenin signaling inhibition in human colon cancer cells [14]. In the present study, we explore its effects on the aforementioned AKT-mTOR, ERK and STAT3 signaling pathways in a human NSCLC cell line CL1-0, which harbors wild-type EGFR and is resistant to EGFR TKIs [19]. The results show that HS7 vigorously suppresses the signaling pathways described above and arrests the cell growth, accompanied with induction of cyclin-dependent kinase (CDK) inhibitors such as p15, p21 and p27. Our findings suggest the potential of HS7 as an alternative medicine for the treatment of NSCLC.
Methods
Information of experimental design and resources
The information of experimental design, statistics, and resources used in this study are attached in Minimum standards of reporting checklist (Additional file 2).
Cell culture
The CL1-0 human lung adenocarcinoma cell line was kindly provided by Dr. Shine-Gwo Shiah (NHRI, Miaoli, Taiwan) and the MRC-5 normal fetal human lung fibroblasts were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan). CL1-0 cells were maintained in RPMI1640 (Gibco, CA, USA) and MRC-5 cells were in MEM (Gibco, CA, USA) medium supplemented with 10% fetal bovine serum and 1× penicillin–streptomycin–glutamine (Gibco, CA, USA). Cells were cultured at 37 °C in a water-jacketed 5% CO2 incubator.
Preparation of ethanol and n-hexane extracts of T. camphoratus and the active fraction HS7
As shown in our previous study [14], the fruiting body-like T. camphoratus (Voucher Number TC-2004-09-001) was cultivated and provided by Well Shine Biotechnology Development Co. (Taipei, Taiwan). Briefly, air-dried ground powder of cultivated T. camphoratus was extracted exhaustively with n-hexane or ethanol. The n-hexane extract was then further separated to eight fractions (HS1–HS8) by silica gel chromatography, and the seventh fraction (HS7) exerted the most potent effect on the growth inhibition of a screening panel of cancer cell lines (CL1-0, PC3, HepG2, Hep3B and Huh7). After lyophilization, the stock solutions of ethanol and n-hexane extracts and HS7 dissolved in dimethyl sulfoxide (DMSO) at concentration of 100 mg/mL were made. They were diluted in phosphate-buffered saline (PBS) prior to use. The final concentrations of DMSO were all below 0.2%.
Reagents
The sulforhodamine B (SRB) dye for cell viability assay, propidium iodide for cell cycle analysis and the EGFR-TKI gefitinib (Iressa) were from Sigma-Aldrich (St. Louis, MO, USA). The MEK-ERK pathway inhibitor U0126 and PI3K-AKT pathway inhibitor LY294002 were from Cell Signaling Technology (Danvers, MA, USA). The JAK-STAT3 pathway inhibitor AG490 was from Calbiochem (La Jolla, CA, USA).
Cell viability assay by sulforhodamine B (SRB) staining
According to the method described by Vichai and Kirtikara [20], SRB dye-binding assay was used to determine the viability of cancer cells. CL1-0 and MRC-5 cells were seeded in a 96-well plate at a density of 2 × 103 cells/well in 10% FBS-RPMI or MEM medium. After 24 h of incubation, cells were treated with agents as indicated or PBS only for another 72 h. Cells were then harvested and fixed by 10% trichloroacetic acid (TCA). After fixing, cells were washed by distilled water and stained viable cells by 0.4% (w/v) SRB dye dissolved in 1% acetic acid. After staining for 30 min, the unbound dye was then washed away by 1% acetic acid and the plate was air-dried. The cell-bound SRB dye was then dissolved in 200 μL of 10 mM Tris base and the absorbance was read on a microplate reader (Molecular Devices, CA, USA) at a wavelength of 562 nm. The absorbance was directly proportional to the cell number over a wide range.
Cell cycle analysis
Propidium iodide (PI) staining and flow cytometry were used to determine the cell cycle distribution. One day after being seeded in a six-well plate (105 cells/2 mL/well), CL1-0 cells were treated with different doses of HS7 for 72 h. At harvest, cells were washed with PBS, incubated with 0.25% Trypsin EDTA at 37 °C for 5–10 min and then suspended in medium at a concentration of 1 × 106 cells/tube. After being washed with PBS and centrifuged at 1200 rpm at 4 °C for 5 min, cells were resuspended in 500 μL PBS and fixed with 70% ethanol followed by gentle vortexing. Cells were allowed to stand overnight at − 20 °C. Fixed cells were spun down and washed with PBS. The cells were suspended in 500 μL PI (2 μg/mL)/Triton X-100 (0.1% v/v) staining solution with RNase A (200 μg/mL) at room temperature for 20 min and then analyzed by a flow cytometer (FACSCalibur™, BD Bioscience, CA, USA). Approximately 10,000 counts were made for each sample. The percentages of cell-cycle distribution were calculated by CellQuest software (BD Bioscience, CA, USA).
Western blotting
A total of 4 × 105 CL1-0 cells/10 cm dish were incubated for 24 h after seeding and then treated as indicated in figures for 72 h. On the day of harvest, the whole-cell lysates were prepared with radioimmunoprecipitation (RIPA) lysis buffer containing 1× tyrosine phosphatase inhibitor cocktail (FC0020-0001, BIONOVAS, Toronto, Canada), 1× protease inhibitor cocktail-full range (FC0070-0001, BIONOVAS, Toronto, Canada), and 1× serine/threonine phosphatase inhibitor cocktail (FC0030-0001, BIONOVAS, Toronto, Canada). Samples of protein extract were size fractionated electrophoretically by polyacrylamide SDS-PAGE gel and transferred onto a PVDF membrane using the BioRad Mini Protean electrotransfer system (CA, USA). The membranes blots were incubated with 5% milk in PBST for 1 h to block nonspecific binding and then were incubated with primary antibodies overnight at 4 °C. The membranes were detected with an appropriate peroxidase-conjugated secondary antibody incubated at room temperature for 1 h. Intensive PBS washing was performed after each time of incubation. The immune complexes were visualized using an enhanced chemiluminescence detection system (ECL, Perkin Elmer, Waltham, MA, USA) according to the manufacturer’s instructions. Primary antibodies against ERK (sc-94), p-ERK (sc-7383), STAT3 (sc-8019), p-STAT3 (sc-7993), p-p70S6K (sc-7984-R), p15 (sc-613), tubulin (sc-5286) and GAPDH (sc-47724) were purchased from Santa Cruz Biotechnology (San Diego, CA, USA). Primary antibodies for p-AKT (#4051), p-mTOR (#2971), mTOR (#2972) and p-pRB (#9308) were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies for p21 (#05-345) and HIF-1α (07-628) were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Primary antibody for p27 (#610241) was purchased from BD Transduction Laboratories (San Jose, CA, USA).
Photograph of the cells
At harvest, cells were examined under a Nikon inverted microscope. The photomicrographs were captured by a CCD camera (Nikon, Mito, Japan) adapted to the microscope.
Statistical analysis
Cell viability data are expressed as mean ± SE. Differences between the cell viabilities of control and treated groups were evaluated by one-way ANOVA followed by Dunnett’s t test. Probability value of p < 0.05 was considered statistically significant. Single asterisk (*) indicate p < 0.05; double asterisks (**) indicate p < 0.01; triple asterisks (***) indicate p < 0.001.
Results
The n-hexane extract of T. camphoratus exerts more potent effect than the ethanol extract on the growth inhibition of CL1-0 cells
To further investigate the anticancer activities of T. camphoratus, we extracted the T. camphoratus with ethanol and n-hexane, respectively, and compared their activities on the growth of CL1-0 cells. The result shows that both the two extracts inhibit the growth of CL1-0 cells in a dose-dependent manner and the n-hexane extract appears to have higher potency. As shown in Fig. 1a, the proliferation of CL1-0 cells was reduced to 30.7 and 31.2% of control by ethanol and n-hexane extracts at dose of 200 and 100 μg/mL, respectively. The n-hexane extract is at least two times more potent than the ethanol extract. We thus further separated the n-hexane extract to eight fractions (HS1–HS8) by silica gel chromatography according their polarity displayed in TLC (thin-layer chromatography) assay (Fig. 1b). These eight fractions were then examined for their activities on the growth inhibition of CL1-0 cells. The result shows that at dose of 50 μg/mL, the seventh fraction (HS7, the 7th fraction of silica gel chromatography after n-hexane extraction) exerts the highest potency (Fig. 1c). Similar results were also observed in PC3, HepG2, Hep3B and Huh7 cells (see Additional file 1). We thus chose the most potent HS7 for the subsequent experiments.
Fig. 1.
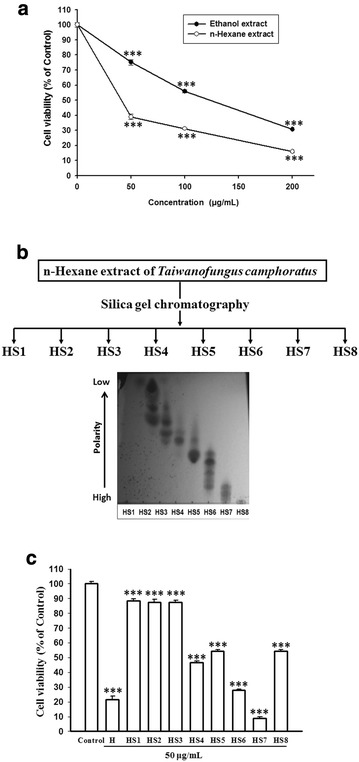
The activities of ethanol and n-hexane extracts of T. camphoratus and the eight separated fractions from n-hexane extract on the growth inhibition of CL1-0 lung cancer cells. a The cell viabilities of CL1-0 cells were measured after treatment with the ethanol or n-hexane extract of T. camphoratus for 72 h. b Eight fractions (HS1–HS8) were separated from the n-hexane extract by silica gel chromatography according to their polarities displayed in thin-layer chromatography. c The cell viabilities of CL1-0 cells were measured after treatment with n-hexane extract (H) or the separated fraction (HS1–HS8) at dose of 50 μg/mL as indicated for 72 h. Cell viability data (mean ± SE) are expressed as a percentage compared to the control and analyzed as described in “Statistical analysis”. ***p < 0.001 compared to the control group
Active fraction HS7 extracted from T. camphoratus profoundly inhibits the proliferation of CL1-0 cells
At first, we examined the effect of gefitinib (Iressa), a typical EGFR-TKI, on the proliferation of CL1-0 cells within its clinically achievable concentration (1 μM) [21]. As shown in Fig. 2a, consistent with the previous study by Lin et al. [19], gefitinib had no significant effect on the cell viability of EGFR wild-type CL1-0 cells. We then examined the effect of HS7 and found that it dose-dependently decreased the proliferation of CL1-0 cells after 72 h of treatment with IC50 of 11.4 μg/mL (Fig. 2b). Significant decrease of cell growth to 68.9% of control by HS7 could be found at dose of 6.25 μg/mL (Fig. 2b). When the dose was increased to 25 μg/mL, the proliferation of CL1-0 cells was suppressed to 23.4% of control (Fig. 2b). The inhibitory effect of HS7 was also confirmed by microscopic observation. As shown in Fig. 2c, significant decrease of CL1-0 cell number by HS7 could be observed at dose of 6.25 μg/mL and the effect was even more profound when the dose was increased to 25 μg/mL. By contrast, HS7 had no significant effect on the viability of MRC-5 cells, the normal fetal human lung fibroblasts, at doses within 25 μg/mL (Fig. 3a). The microscopic observation also showed the same result (Fig. 3b).
Fig. 2.
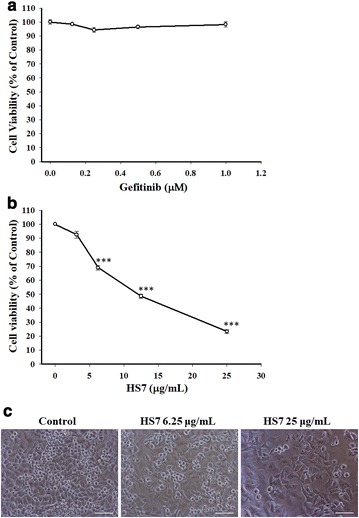
The effects of gefitinib (Iressa) and HS7 on the proliferation of CL1-0 lung cancer cells. a The cell viability of CL1-0 cells was measured after treatment with gefitinib for 72 h. b The cell viability of CL1-0 cells was measured after treatment with HS7 for 72 h. c The effect of HS7 on CL1-0 cells was examined by phase-contrast microscopy after 72 h of treatment, scale bar = 100 μm. Cell viability data (mean ± SE) are expressed as a percentage compared to the control and analyzed as described in “Statistical analysis”. ***p < 0.001 compared to the control group
Fig. 3.
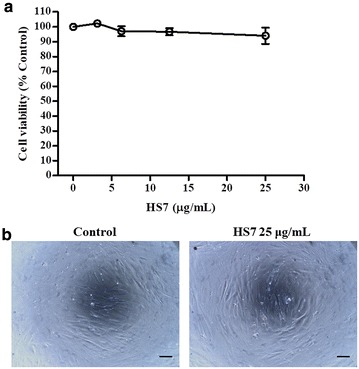
The effect of HS7 on the cell viability of MRC-5 normal fetal human lung fibroblasts. a The cell viability of MRC-5 cells was measured after treatment with HS7 for 72 h. b The effect of HS7 on MRC-5 cells was examined by phase-contrast microscopy after 72 h of treatment, scale bar = 100 μm. Cell viability data (mean ± SE) are expressed as a percentage compared to the control and analyzed as described in “Statistical analysis”
HS7 induces apoptotic sub-G1 fraction in CL1-0 cells
After treatment with HS7 for 72 h, the CL1-0 cells were stained with PI and analyzed by flow cytometry to measure the change in cell-cycle distribution. As shown in Fig. 4 and Table 1, HS7 increased apoptotic sub-G1 fraction from 1.4% in control group to 10% at dose of 25 μg/mL but did not significantly change the distribution of cell populations in G0/G1, S and G2/M cell-cycle phases within this dose. Regarding the profound growth inhibition by HS7 shown in Fig. 2b, c, the progression of cell-cycle might be suppressed by HS7 through inhibition of the proliferative signaling pathways and the subsequent induction of CDK inhibitors that broadly inhibit each phase of the cell-cycle progression.
Fig. 4.
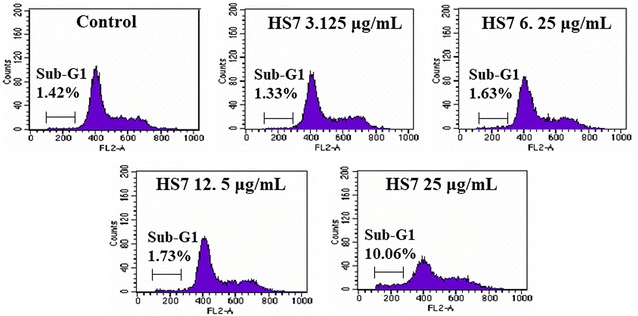
Representative flow cytometry histograms of CL1-0 lung cancer cells after treatment with HS7 for 72 h. HS7 increased the apoptotic sub-G1 fraction in CL1-0 cells at dose of 25 μg/mL but did not significant change the distribution of cells in the G0/G1, S and G2/M phases of the cell cycle within this dose. The percentage of cells in different phases of the cell cycle is shown in Table 1
Table 1.
Cell-cycle phase distribution (%) of 72 h HS7-treated CL1-0 cells
| Treatment | Sub-G1 (%)a | Cell-cycle distribution | ||
|---|---|---|---|---|
| G0/G1 (%) | S (%) | G2/M (%) | ||
| Control | 1.42 | 69.8 | 14.6 | 15.6 |
| HS7 3.125 μg/mL | 1.33 | 65.9 | 13.2 | 20.9 |
| HS7 6.25 μg/mL | 1.63 | 68.3 | 13.6 | 18.1 |
| HS7 12.5 μg/mL | 1.73 | 68 | 14.4 | 17.6 |
| HS7 25 μg/mL | 10.06 | 63.1 | 18.6 | 18.3 |
aSub-G1 proportion is excluded from the percentage calculation of cell-cycle distribution
HS7 inhibits AKT-mTOR signaling cascades in CL1-0 cells
The AKT-mTOR pathway is commonly dysregulated in NSCLC and plays a crucial role in tumourigenesis [4]. To investigate the molecular events underlying the inhibitory effect of HS7 on CL1-0 cell growth, we examined the AKT-mTOR signaling in HS7-treated cells. As shown in Fig. 5a, the constitutively activated/phosphorylated AKT protein (p-AKT) in CL1-0 cells was decreased in a dose-dependent manner after 72 h treatment with HS7. Marked decrease of activated/phosphorylated mTOR protein (p-mTOR) could be observed when the dose of HS7 was reached to 6.25 μg/mL (Fig. 5a).
Fig. 5.
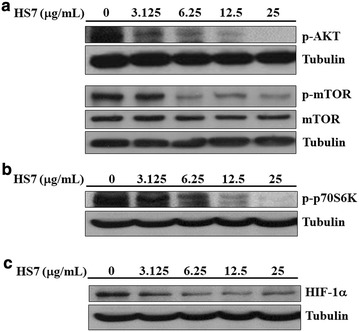
Inhibitory effects of HS7 on the AKT-mTOR signaling pathway and its downstream effectors in CL1-0 lung cancer cells after 72 h of treatment. a HS7 decreased the protein levels of phosphorylated AKT (p-AKT) and phosphorylated mTOR (p-mTOR) in a dose-dependent manner. b HS7 dose-dependently decreased the phosphorylated p70S6K protein (p-p70S6K). c HS7 dose-dependently decreased the HIF-1α protein. Cell lysates were analyzed by Western blot, using tubulin as loading control
The activated mTOR promotes protein translation and cell growth predominantly through activating p70S6K (ribosomal p70S6 kinase) [4, 22]. We thus examined the effect of HS7 on this downstream effector of AKT-mTOR signaling. In accordance with the result of Fig. 2b, HS7 significantly reduced the activated/phosphorylated p70S6K protein (p-p70S6K) in CL1-0 cells at the dose of 6.25 μg/mL and almost diminished the protein level when the dose was increased to 25 μg/mL (Fig. 5b).
In addition to p70S6K, the pivotal transcription factor HIF-1α (hypoxia-inducible transcription factor-1α) that regulates angiogenesis by inducing the expression of vascular endothelial growth factor (VEGF) is also a downstream target of mTOR [23, 24]. We next examined the change of HIF-1α in CL1-0 cells during mTOR inhibition by HS7. As shown in (Fig. 5c), the protein level of HIF-1α was also significantly downregulated when the dose of HS7 was reached to 6.25 μg/mL.
The suppression of these two important downstream effectors p70S6K and HIF-1α demonstrates the substantial inhibitory effects of HS7 on the AKT-mTOR signaling in CL1-0 cells.
HS7 inhibits ERK and STAT3 signaling pathways in CL1-0 cells
In addition to the AKT-mTOR pathway, the MEK-ERK pathway is also constitutively active in the majority of NSCLC cell lines [7] and patients [5]. It is considered to have a key role in the pathogenesis of lung cancer [25]. Intriguingly, HS7 also dose-dependently reduced the activated/phosphorylated ERK protein (p-ERK) in CL1-0 cells without significantly affecting the total ERK protein level (Fig. 6a).
Fig. 6.
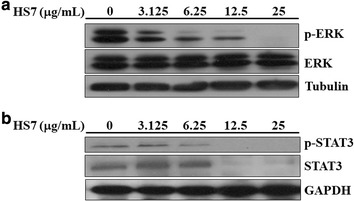
Inhibitory effects of HS7 on the ERK and STAT3 signaling pathways in CL1-0 lung cancer cells after 72 h of treatment. a HS7 decreased the protein levels of phosphorylated ERK (p-ERK) in a dose-dependent manner. b HS7 dose-dependently decreased both the protein levels of total (STAT3) and phosphorylated STAT3 (p-STAT3). Cell lysates were analyzed by Western blot, using tubulin or GAPDH as loading control
The signal transducer and activator of transcription 3 (STAT3) is one of the three major downstream pathways (AKT-mTOR, ERK and STAT3) activated by EGFR phosphorylation, which promote proliferation and survival of NSCLC cells [9, 26]. Constitutive activation of STAT3 is also a common feature in NSCLC and it can be activated by JAK2 independent of known driver mutations [27]. We were interested to investigate if HS7 also inhibits this pro-survival and proliferative signaling in CL1-0 cells. As expected, HS7 significantly reduced the activated/phosphorylated STAT3 protein (p-STAT3) at dose of 6.25 μg/mL and almost diminished both the p-STAT3 and total STAT3 protein levels when the dose was increased to 12.5 μg/mL (Fig. 6b).
HS7 is more effective than the synthetic inhibitors on the inhibition of signaling pathways in CL1-0 cells
To compare the effects of HS7 with the synthetic inhibitors of these three signaling pathways, we treated CL1-0 cells with LY294002 (PI3K-AKT inhibitor), U0126 (MEK-ERK inhibitor), AG490 (JAK-STAT3 inhibitor) for 72 h at the doses which significantly reduced the proliferation of CL1-0 cells (Fig. 7a). At this 72 h treatment time point, LY294002 did not show inhibitory effect on the p-AKT protein but reduced the p-ERK protein as U0126 did (Fig. 7b). On the other hand, probably due to the feedback or compensatory activation at this time point, the reduction of p-ERK by U0126 or LY294002 was also accompanied with induction of p-AKT and p-STAT3 (Fig. 7b). The reduction of p-STAT3 by AG490 also was not observed at this time point, however, the compensatory induction of p-AKT was shown (Fig. 7b). Consistent with these feedback or compensatory inductions of p-AKT by these synthetic inhibitors, the protein levels of downstream component HIF-1α were also increased accordingly (Fig. 7b). In comparison, HS7 vigorously inhibits these three signaling pathways and HIF-1α without the feedback activation of pathway components or compensatory activation of parallel circuits induced by the synthetic inhibitors. This result implies that the multi-targeting HS7 might exert more effective anticancer activity than the single use of each synthetic signaling pathway inhibitor.
Fig. 7.
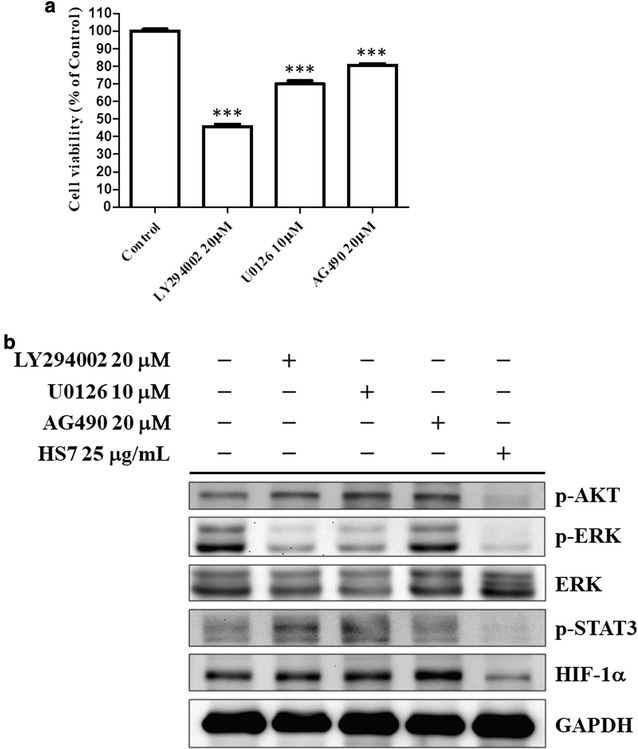
Effects of synthetic inhibitors and HS7 on the AKT ERK and STAT3 signaling pathways in CL1-0 lung cancer cells after 72 h of treatment. a The cell viabilities of CL1-0 cells were measured after treatment with LY294002 (PI3K-AKT inhibitor), U0126 (MEK-ERK inhibitor) or AG490 (JAK-STAT3 inhibitor) at indicated doses for 72 h. Cell viability data (mean ± SE) are expressed as a percentage compared to the control and analyzed as described in “Statistical analysis”. ***p < 0.001 compared to the control group. b The effects of LY294002, U0126, AG490 and HS7 on the protein levels of phosphorylated AKT (p-AKT), phosphorylated ERK (p-ERK), ERK, phosphorylated STAT3 (p-STAT3) and HIF-1α in CL1-0 cells. Cell lysates were analyzed by Western blot, using GAPDH as loading control
HS7 induces CDK inhibitors in CL1-0 cells
It has been shown that inhibition of AKT and ERK signaling by the typical EGFR-TKI gefitinib is accompanied with induction of CDK inhibitors such as p15, p21 and p27, and hypophosphorylation of retinoblastoma protein (pRb) [28, 29]. We thus examined the changes of these cell-cycle regulating components in HS7-treated cells. As expected, treatment with HS7 for 72 h dose-dependently increased the protein levels of p15, p21 and p27 (Fig. 8). As shown in the right-down side of Fig. 9, induction of these CDK inhibitors can prevent the pRb inactivation (phosphorylation) by CDK2/4/6 and the subsequent release of E2F to promote the G1/S transition of cell cycle [30]. Accordingly, the phosphorylation of pRb protein (p-pRb) was dose-dependently inhibited by HS7 while the CDK inhibitors (p15, p21 and p27) were induced (Fig. 8).
Fig. 8.

HS7 induces CDK inhibitors and inhibits phosphorylation of pRb protein in CL1-0 lung cancer cells after 72 h of treatment. HS7 dose-dependently increased protein levels of p15, p21 and p27, and decreased phosphorylated pRb protein (p-pRb). Cell lysates were analyzed by Western blot, using tubulin or GAPDH as loading control
Fig. 9.
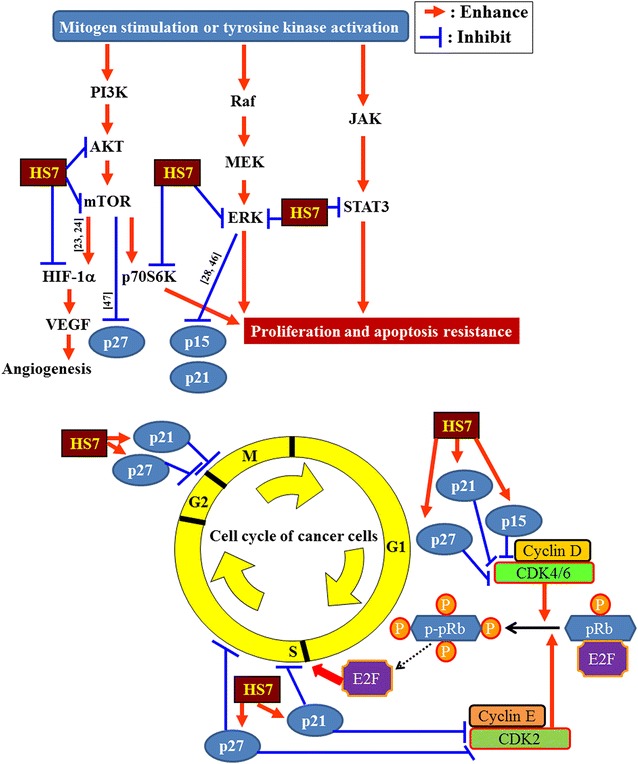
Schematic diagram displays the proposed mechanisms of action of HS7 in CL1-0 lung cancer cells. Through inhibition of AKT-mTOR, ERK and STAT3 signaling pathways, the active fraction HS7 from T. camphoratus increases the CDK inhibitors (p15, p21 and p27) and leads to cell cycle suppression and apoptosis induction. References for the proposed inhibition or enhancement are indicated along the arrow or T-bar. ERK extracellular signal-regulated kinases, HIF-α hypoxia-inducible factor 1-alpha, JAK Janus kinase, MEK mitogen-activated protein/extracellular signal-regulated kinase, mTOR mammalian target of rapamycin, p70S6K ribosomal p70 S6 kinase, PI3K phosphoinositide 3-kinase, Raf rapidly accelerated fibrosarcoma, STAT3 signal transducer and activator of transcription 3, VEGF vascular endothelial growth factor
Significant induction of CDK inhibitors and inhibition of p-pRb were also observed when the dose of HS7 was reached to 6.25 μg/mL. This effective dose of HS7 is consistent with the aforementioned results shown in Figs. 2, 5 and 6, indicating the close correlation between the HS7-mediated events in CL1-0 cells. As proposed in Fig. 9, inhibition of these signaling pathways by HS7 might increase the CDK inhibitors (p15, p21 and p27) and lead to the suppression of cell cycle progression.
Discussion
The oncogenic EGFR signaling in NSCLC engages the activation of downstream effectors such as AKT-mTOR, ERK and STAT3 to promote cell proliferation, cell survival, and tumor growth [4, 9, 31, 32]. Blockade of EGFR activation by TKIs targeted therapy has significantly changed the treatment paradigm in NSCLC [4]. However, only a small proportion of NSCLC patients can respond to clinically used TKIs [4, 31]. Most of the NSCLC patients do not carry the genetic alterations for the effectiveness of EGFR-TKIs treatment [13]. Tumor samples derived from NSCLC patients can show robust activation of AKT, ERK, and STAT3 while EGFR is not activated [31]. Alternative novel targeted therapeutics other than the EGFR-TKIs is imperatively needed. Nowadays, lots of ongoing efforts have been conducted in identifying potential therapeutics targeting the above-mentioned effectors (ERK, AKT-mTOR and STAT3), which are frequently dysregulated in NSCLC.
The anticancer activities of T. camphoratus (syn. A. camphorata) have been studied in numerous types of cancer cells including NSCLC [14, 16–18, 33, 34]. Its potential for the treatment of NSCLC has been shown in a preclinical evaluation in which the significant tumor suppression and apoptosis induction were observed [33]. To further develop the application of T. camphoratus for NSCLC treatment, we isolate a more potent active fraction (HS7) from its n-hexane extract. For the demanding of novel targeted therapeutics mentioned above, we investigate and demonstrate the substantial inhibition of ERK, AKT-mTOR and STAT3 signaling pathways by HS7 in EGFR-TKI resistant CL1-0 human NSCLC cells (EGFR wild-type).
The interplay between these pathways is complex in CL1-0 cells. Our results show that treatments with synthetic inhibitors of these pathways such as LY294002, U0126 and AG490 induce feedback or compensatory activation of parallel circuits in CL1-0 cells. This phenomenon is similar to that reported in other previous studies of synthetic inhibitors. The feedback activation of AKT plays an important role in the unsatisfactory clinical results of mTOR inhibitor in cancer treatment [35, 36]. Combining with AKT inhibitor to enhance the therapeutic effects of mTOR inhibitor for NSCLC treatment was thus suggested [35]. Our results show the simultaneous inhibition of p-AKT and mTOR signaling cascade (p-mTOR, p-p70S6K and HIF-1α) by HS7, implying its potential for this therapeutic strategy. On the other hand, targeting components of ERK signaling (RAS-RAF-MEK-ERK) also has been proposed for NSCLC treatment [37]. However, activation of alternative signaling pathways almost always occurs after inhibition of ERK [37]. Like that reported in the study by Hayashi et al. [38], inhibition of p-ERK by U0126 accompanied with increased p-AKT is also observed in our result. Combination with inhibitor of another parallel signaling tract such as AKT pathway has been studied in animal model [39] and clinical trial [37] for NSCLC treatment. Regarding the simultaneous suppression of both ERK and AKT-mTOR pathways by HS7, it might have the advantage over the individual use of synthetic ERK signaling inhibitors for NSCLC therapy.
Inhibition of STAT3 signaling may be effective for treatment of NSCLC irrespective of the EGFR mutation status [9]. In addition, activation of STAT3 also has been shown to participate in the resistance of NSCLC cells to erlotinib (EGFR-TKI) [9, 40] and radiation [9, 41]. An old FDA-approved anthelmintic drug niclosamide was recently found to overcome acquired erlotinib resistance and reverse radioresistance through suppression of STAT3 in NSCLC xenografts [40, 41]. In this perspective, the significant inhibition of STAT3 pathway by HS7 might also have the potential to reduce the resistance of NSCLC cells to EGFR-TKIs or radiotherapy. Further future investigation is warranted.
The success of targeted therapy can be limited by the ultimately developed resistance of cancer cells through mutation of the target kinase, signaling redundancy, feedback activation of pathway components, compensatory activation of parallel circuits and so forth [32]. One strategy to improve the efficacy is combination therapy [5, 22]. Therefore, simultaneously co-targeting the signaling pathways of such as AKT and ERK [5, 22, 39], mTOR and ERK [42], STAT3 and mTOR [43] have been proposed to improve the success of NSCLC targeted therapy. According to this strategy, the aforementioned multi-targeting activity of HS7 is worth of further development for its integrative use in NSCLC targeted therapy. The point of convergence targeted by HS7 to exert its multi-targeting effects is remained to be elucidated. It is also possible that the multi-components in HS7 are responsible for its multi-targeting activity. Compounds including polysaccharides, ergostan-type triterpenoids, a sesquiterpene, and phenyl and biphenyl derivatives had been isolated from T. camphoratus (syn. A. camphorata) [44, 45]. It is worthy to identify and quantify the active components in the HS7 fraction for the future development.
Like that observed in gefitinib (iressa)-treated cells [28, 29], inhibition of proliferative signaling cascades by HS7 is also accompanied with induction of p15, p21 and p27, and decrease of pRb phosphorylation. It is reported that suppression of proliferative signaling like ERK and AKT-mTOR could inhibit the degradation of p21 and p27, respectively [46, 47], and up-regulate the p15 mRNA level [28]. HS7 might also modulate the proteolysis or transcription of these CDK inhibitors by the similar ways, resulting in inhibition of pRb phosphorylation. On the other hand, in addition to constrain the cell-cycle progression, HS7 also induced apoptosis at dose of 25 μg/mL, and massive apoptosis induction (46% sub-G1 fraction) could be observed at higher dose up to 50 μg/mL (see Additional file 3). The profound inhibition of these targeted signaling effectors by HS7 did have apoptotic killing effect on CL1-0 cells.
Conclusions
From T. camphoratus, we isolate an active fraction (HS7) that simultaneously inhibits the signaling pathways (AKT-mTOR, ERK and STAT3) and induces CDK inhibitors (p15, p21 and p27) in EGFR TKI resistant CL1-0 NSCLC cells, resulting in growth inhibition and apoptosis induction (Fig. 9). Combinations of synthetic signaling pathway inhibitors for NSCLC targeted therapy have been proposed by many studies; however, none of them are clinically available at present. This multi-targeting activity of HS7 implies its potential as an alternative medicine for the treatment of EGFR TKIs resistant NSCLC.
Additional files
Additional file 1. The activities of eight separated fractions (HS1-HS8) from the n-hexane extract of Taiwanofungus camphoratus on the growth inhibition of four cancer cell lines.
Additional file 2. Minimum standards of reporting checklist-R.
Additional file 3. Apoptosis induction in CL1-0 lung cancer cells by HS7 at dose of 50 μg/mL.
Authors’ contributions
ICL, GML, CJY and KJB conceived the study and wrote the manuscript. CFY, CHL and JLY performed the experiments and data analysis. JMC and HLL reviewed literature and interpreted the results. SEC and JWP revised the manuscript. KJB and CJY equally contributed to the paper. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Well Shine Biotechnology Development Co. (Taipei, Taiwan) for providing the fruiting body-like Taiwanofungus camphoratus powder. This work was supported in part by Health and welfare surcharge of tobacco products (MOHW106-TDU-B-212-144001) and Ministry of Science and Technology, Taiwan (MOST 104-2314-B-038-077-MY3).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The information is included in the Additional file 2.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported in part by Health and welfare surcharge of tobacco products (MOHW106-TDU-B-212-144001) and Ministry of Science and Technology, Taiwan (MOST 104-2314-B-038-077-MY3).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CDK
cyclin-dependent kinase
- ERK
extracellular signal-regulated kinase
- EGFR
epidermal growth factor receptor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HIF-1α
hypoxia-inducible factor 1α
- JAK
Janus kinase
- MEK
mitogen-activated protein/extracellular signal-regulated kinase
- mTOR
mammalian target of rapamycin
- p70S6K
ribosomal p70 S6 kinase
- PBS
phosphate-buffered saline
- PI3K
phosphoinositide 3-kinase
- pRb
retinoblastoma protein
- Raf
rapidly accelerated fibrosarcoma
- SRB
sulforhodamine B
- STAT3
signal transducer and activator of transcription 3
- TKI
tyrosine kinase inhibitor
- VEGF
vascular endothelial growth factor
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13020-017-0154-9) contains supplementary material, which is available to authorized users.
Contributor Information
I-Chun Lai, Email: littlelai0114@gmail.com.
Gi-Ming Lai, Email: gminlai@nhri.org.tw.
Jyh-Ming Chow, Email: chow0803@yahoo.com.tw.
Hsin-Lun Lee, Email: b001089024@tmu.edu.tw.
Chuan-Feng Yeh, Email: 950308@nhri.org.tw.
Chi-Han Li, Email: han@nhri.org.tw.
Jiann-Long Yan, Email: 000405@nhri.org.tw.
Shuang-En Chuang, Email: sechuang@nhri.org.tw.
Jacqueline Whang-Peng, Email: jqwpeng@nhri.org.tw.
Kuan-Jen Bai, Phone: 886-2-29307930, Email: kjbai@w.tmu.edu.tw.
Chih-Jung Yao, Phone: 886-2-29307930, Email: yao0928@tmu.edu.tw.
References
- 1.Tsiambas E, Lefas AY, Georgiannos SN, Ragos V, Fotiades PP, Grapsa D, Stamatelopoulos A, Kavantzas N, Patsouris E, Syrigos K. EGFR gene deregulation mechanisms in lung adenocarcinoma: a molecular review. Pathol Res Pract. 2016;212(8):672–677. doi: 10.1016/j.prp.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Abacioglu U, Yumuk PF, Caglar H, Sengoz M, Turhal NS. Concurrent chemoradiotherapy with low dose weekly gemcitabine in stage III non-small cell lung cancer. BMC Cancer. 2005;5:71. doi: 10.1186/1471-2407-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4(2):165–176. doi: 10.3978/j.issn.2218-6751.2015.01.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heavey S, O’Byrne KJ, Gately K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat Rev. 2014;40(3):445–456. doi: 10.1016/j.ctrv.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Dutta P, Sabri N, Li J, Li WX. Role of STAT3 in lung cancer. JAKSTAT. 2014;3(4):e999503. doi: 10.1080/21623996.2014.999503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brognard J, Dennis PA. Variable apoptotic response of NSCLC cells to inhibition of the MEK/ERK pathway by small molecules or dominant negative mutants. Cell Death Differ. 2002;9(9):893–904. doi: 10.1038/sj.cdd.4401054. [DOI] [PubMed] [Google Scholar]
- 8.Papadimitrakopoulou V, Adjei AA. The Akt/mTOR and mitogen-activated protein kinase pathways in lung cancer therapy. J Thorac Oncol. 2006;1(7):749–751. [PubMed] [Google Scholar]
- 9.Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers. 2014;6(2):708–722. doi: 10.3390/cancers6020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stinchcombe TE, Johnson GL. MEK inhibition in non-small cell lung cancer. Lung Cancer. 2014;86(2):121–125. doi: 10.1016/j.lungcan.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66(6):3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 12.Mello RA, Liu DJ, Aguiar PN, Tadokoro H. EGFR and EML4-ALK updated therapies in non-small cell lung cancer. Recent Pat Anticancer Drug Discov. 2016;11(4):393–400. doi: 10.2174/1574892811666160803090944. [DOI] [PubMed] [Google Scholar]
- 13.Richer AL, Friel JM, Carson VM, Inge LJ, Whitsett TG. Genomic profiling toward precision medicine in non-small cell lung cancer: getting beyond EGFR. Pharmgenomics Pers Med. 2015;8:63–79. doi: 10.2147/PGPM.S52845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeh CT, Yao CJ, Yan JL, Chuang SE, Lee LM, Chen CM, Yeh CF, Li CH, Lai GM. Apoptotic cell death and inhibition of Wnt/beta-catenin signaling pathway in human colon cancer cells by an active fraction (HS7) from Taiwanofungus camphoratus. Evid Based Complement Alternat Med. 2011;2011:750230. doi: 10.1155/2011/750230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu DZ, Liang HJ, Chen CH, Su CH, Lee TH, Huang CT, Hou WC, Lin SY, Zhong WB, Lin PJ, et al. Comparative anti-inflammatory characterization of wild fruiting body, liquid-state fermentation, and solid-state culture of Taiwanofungus camphoratus in microglia and the mechanism of its action. J Ethnopharmacol. 2007;113(1):45–53. doi: 10.1016/j.jep.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Chen LY, Sheu MT, Liu DZ, Liao CK, Ho HO, Kao WY, Ho YS, Lee WS, Su CH. Pretreatment with an ethanolic extract of Taiwanofungus camphoratus (Antrodia camphorata) enhances the cytotoxic effects of amphotericin B. J Agric Food Chem. 2011;59(20):11255–11263. doi: 10.1021/jf2024215. [DOI] [PubMed] [Google Scholar]
- 17.Lin LT, Tai CJ, Su CH, Chang FM, Choong CY, Wang CK. The ethanolic extract of Taiwanofungus camphoratus (Antrodia camphorata) induces cell cycle arrest and enhances cytotoxicity of cisplatin and doxorubicin on human hepatocellular carcinoma cells. Biomed Res Int. 2015;2015:415269. doi: 10.1155/2015/415269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee WT, Lee TH, Cheng CH, Chen KC, Chen YC, Lin CW. Antroquinonol from Antrodia camphorata suppresses breast tumor migration/invasion through inhibiting ERK-AP-1- and AKT-NF-kappaB-dependent MMP-9 and epithelial–mesenchymal transition expressions. Food Chem Toxicol. 2015;78:33–41. doi: 10.1016/j.fct.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 19.Lin YC, Shih JY, Huang WJ, Chao SW, Chang YL, Chen CC. DUSP1 expression induced by HDAC1 inhibition mediates gefitinib sensitivity in non-small cell lung cancers. Clin Cancer Res. 2015;21(2):428–438. doi: 10.1158/1078-0432.CCR-14-1150. [DOI] [PubMed] [Google Scholar]
- 20.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 21.Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, Szabo K, Homolya L, Varadi A, Buday L, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65(5):1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 22.Cheng H, Shcherba M, Pendurti G, Liang Y, Piperdi B, Perez-Soler R. Targeting the PI3K/AKT/mTOR pathway: potential for lung cancer treatment. Lung Cancer Manag. 2014;3(1):67–75. doi: 10.2217/lmt.13.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37(1):19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 24.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24(50):7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 25.Sarris EG, Saif MW, Syrigos KN. The biological role of PI3K pathway in lung cancer. Pharmaceuticals. 2012;5(11):1236–1264. doi: 10.3390/ph5111236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Osta H, Shackelford R. Personalized treatment options for ALK-positive metastatic non-small-cell lung cancer: potential role for ceritinib. Pharmgenomics Pers Med. 2015;8:145–154. doi: 10.2147/PGPM.S71100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE. 2012;7(2):e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koyama M, Matsuzaki Y, Yogosawa S, Hitomi T, Kawanaka M, Sakai T. ZD1839 induces p15INK4b and causes G1 arrest by inhibiting the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway. Mol Cancer Ther. 2007;6(5):1579–1587. doi: 10.1158/1535-7163.MCT-06-0814. [DOI] [PubMed] [Google Scholar]
- 29.Di Gennaro E, Barbarino M, Bruzzese F, De Lorenzo S, Caraglia M, Abbruzzese A, Avallone A, Comella P, Caponigro F, Pepe S, et al. Critical role of both p27KIP1 and p21CIP1/WAF1 in the antiproliferative effect of ZD1839 (‘Iressa’), an epidermal growth factor receptor tyrosine kinase inhibitor, in head and neck squamous carcinoma cells. J Cell Physiol. 2003;195(1):139–150. doi: 10.1002/jcp.10239. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8(9):714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorgisen G, Pehlivanoglu S, Ozes D, Ozes ON. Personalized treatment options in non-small cell lung cancer. Recept Clin Investig. 2014;1:e152. [Google Scholar]
- 32.Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26(7):641–650. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiou JF, Wu AT, Wang WT, Kuo TH, Gelovani JG, Lin IH, Wu CH, Chiu WT, Deng WP. A preclinical evaluation of Antrodia camphorata alcohol extracts in the treatment of non-small cell lung cancer using non-invasive molecular imaging. Evid Based Complement Alternat Med. 2011;2011:914561. doi: 10.1093/ecam/nep228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh CT, Huang WC, Rao YK, Ye M, Lee WH, Wang LS, Tzeng DT, Wu CH, Shieh YS, Huang CY, et al. A sesquiterpene lactone antrocin from Antrodia camphorata negatively modulates JAK2/STAT3 signaling via microRNA let-7c and induces apoptosis in lung cancer cells. Carcinogenesis. 2013;34(12):2918–2928. doi: 10.1093/carcin/bgt255. [DOI] [PubMed] [Google Scholar]
- 35.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65(16):7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 36.Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci. 2015;16(2):3267–3282. doi: 10.3390/ijms16023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heigener DF, Gandara DR, Reck M. Targeting of MEK in lung cancer therapeutics. Lancet Respir Med. 2015;3(4):319–327. doi: 10.1016/S2213-2600(15)00026-0. [DOI] [PubMed] [Google Scholar]
- 38.Hayashi H, Tsuchiya Y, Nakayama K, Satoh T, Nishida E. Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells. 2008;13(9):941–947. doi: 10.1111/j.1365-2443.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- 39.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, Peng Z, Herbst RS, Papadimitrakopoulou V, Minna JD, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS ONE. 2010;5(11):e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, Ramalingam SS, Curran WJ, Khuri FR, Deng X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol Cancer Ther. 2013;12(10):2200–2212. doi: 10.1158/1535-7163.MCT-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You S, Li R, Park D, Xie M, Sica GL, Cao Y, Xiao ZQ, Deng X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol Cancer Ther. 2014;13(3):606–616. doi: 10.1158/1535-7163.MCT-13-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Legrier ME, Yang CP, Yan HG, Lopez-Barcons L, Keller SM, Perez-Soler R, Horwitz SB, McDaid HM. Targeting protein translation in human non small cell lung cancer via combined MEK and mammalian target of rapamycin suppression. Cancer Res. 2007;67(23):11300–11308. doi: 10.1158/0008-5472.CAN-07-0702. [DOI] [PubMed] [Google Scholar]
- 43.Jin HO, Lee YH, Park JA, Kim JH, Hong SE, Kim HA, Kim EK, Noh WC, Kim BH, Ye SK, et al. Blockage of Stat3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem Biophys Res Commun. 2014;444(4):502–508. doi: 10.1016/j.bbrc.2014.01.086. [DOI] [PubMed] [Google Scholar]
- 44.Yang HL, Hseu YC, Chen JY, Yech YJ, Lu FJ, Wang HH, Lin PS, Wang BC. Antrodia camphorata in submerged culture protects low density lipoproteins against oxidative modification. Am J Chin Med. 2006;34(2):217–231. doi: 10.1142/S0192415X06003783. [DOI] [PubMed] [Google Scholar]
- 45.Chen CH, Yang SW, Shen YC. New steroid acids from Antrodia cinnamomea, a fungal parasite of Cinnamomum micranthum. J Nat Prod. 1995;58(11):1655–1661. doi: 10.1021/np50125a002. [DOI] [PubMed] [Google Scholar]
- 46.Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. 2013;5(3):a008904. doi: 10.1101/cshperspect.a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alkarain A, Slingerland J. Deregulation of p27 by oncogenic signaling and its prognostic significance in breast cancer. Breast Cancer Res. 2004;6(1):13–21. doi: 10.1186/bcr722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. The activities of eight separated fractions (HS1-HS8) from the n-hexane extract of Taiwanofungus camphoratus on the growth inhibition of four cancer cell lines.
Additional file 2. Minimum standards of reporting checklist-R.
Additional file 3. Apoptosis induction in CL1-0 lung cancer cells by HS7 at dose of 50 μg/mL.
Data Availability Statement
The information is included in the Additional file 2.