

Abstract
MicroRNAs contribute to the maintenance of optimal cellular functions by fine‐tuning protein expression levels. In the pancreatic β‐cells, imbalances in the exocytotic machinery components lead to impaired insulin secretion and type 2 diabetes (T2D). We hypothesize that dysregulated miRNA expression exacerbates β‐cell dysfunction, and have earlier shown that islets from the diabetic GK‐rat model have increased expression of miRNAs, including miR‐335‐5p (miR‐335). Here, we aim to determine the specific role of miR‐335 during development of T2D, and the influence of this miRNA on glucose‐stimulated insulin secretion and Ca2+‐dependent exocytosis. We found that the expression of miR‐335 negatively correlated with secretion index in human islets of individuals with prediabetes. Overexpression of miR‐335 in human EndoC‐βH1 and in rat INS‐1 832/13 cells (OE335) resulted in decreased glucose‐stimulated insulin secretion, and OE335 cells showed concomitant reduction in three exocytotic proteins: SNAP25, Syntaxin‐binding protein 1 (STXBP1), and synaptotagmin 11 (SYT11). Single‐cell capacitance measurements, complemented with TIRF microscopy of the granule marker NPY‐mEGFP demonstrated a significant reduction in exocytosis in OE335 cells. The reduction was not associated with defective docking or decreased Ca2+ current. More likely, it is a direct consequence of impaired priming of already docked granules. Earlier reports have proposed reduced granular priming as the cause of reduced first‐phase insulin secretion during prediabetes. Here, we show a specific role of miR‐335 in regulating insulin secretion during this transition period. Moreover, we can conclude that miR‐335 has the capacity to modulate insulin secretion and Ca2+‐dependent exocytosis through effects on granular priming.
Keywords: Beta cell, exocytosis, insulin secretion, microRNA, patch‐clamp, SNAP25, STXBP1, TIRF, Type 2 Diabetes
Introduction
Hyperglycemia and the development of type 2 diabetes (T2D) depend on environmental components together with genetic factors resulting in insulin resistance in the target tissues and reduced capacity of the pancreatic β‐cells to secrete enough insulin. The release of insulin is biphasic and evidence have put forward that impaired insulin secretion precedes insulin resistance (Gerich 2002) and that first‐phase insulin secretion is lost already when the patient has impaired glucose tolerance (IGT) or prediabetes (del Prato and Tiengo 2001). In the β‐cells, exocytosis serves to release insulin from large dense‐core vesicles in response to elevated cytosolic [Ca2+]i (Ammala et al. 1993, Eliasson et al., 2008; Wang and Thurmond 2009). Prior to fusion with the plasma membrane, the granules need to dock to the release site and undergo the process of granular priming to make them ready for release of their insulin cargo. Impaired priming has been suggested to underlie the absence of first‐phase insulin secretion in individuals with pre‐ and full‐blown diabetes (Eliasson et al. 2008).
As in neuronal cells, the assembly of the SNARE (Soluble NSF Attachment protein Receptor)‐proteins, including VAMP2 (vesicle‐associated‐membrane‐protein2), STX1A (Syntaxin1A), and SNAP25 (Synaptosomal‐Associated‐Protein‐of‐25‐kDa), is a key process in insulin exocytosis (Eliasson et al., 2008, Wang and Thurmond 2009; Pang and Sudhof 2010). In addition, several other proteins are required for the specific targeting of secretory granules to the release sites and the regulation of exocytosis (Wang and Thurmond 2009; Pang and Sudhof 2010). These include the Sec1/Munc18 (or syntaxin‐binding protein 1 (STXBP1)) proteins, thought to guide SNARE complex assembly (Gulyas‐Kovacs et al. 2007; Tomas et al. 2008; Rizo and Sudhof 2012), and the synaptotagmins (SYTs) the primary Ca2+ sensors of exocytosis (Gauthier and Wollheim 2008; Pang and Sudhof 2010). The expression of many exocytotic genes is down‐regulated in islets from donors with T2D (Ostenson et al. 2006; Andersson et al. 2012) as well as in the spontaneous diabetes model the Goto‐Kakizaki (GK) rat (Gaisano et al. 2002; Zhang et al. 2002).
MiRNAs are small (~20‐nt), nonprotein coding, endogenously expressed RNAs that regulate gene expression (Bartel 2004). MiRNAs guide the RNA‐induced silencing complex (RISC) to target the mRNAs by direct base‐pairing, which may include wobble base‐pairing in mammalian systems, between the miRNA 5′ seed region (~7‐nt long) and their target mRNA sequence usually in the 3′UTR (Brennecke et al. 2005). The regulation of protein output by miRNAs has been suggested to function either as a classical binary off‐switch or as a rheostat, in which miRNAs fine‐tune optimal protein output (Bartel 2009).
In the context of T2D several miRNAs have been shown to directly or indirectly regulate crucial components of glucose‐stimulated insulin secretion and exocytosis in β‐cells (Lovis et al. 2008; Eliasson and Esguerra 2014). The exocytotic process involves a plethora of proteins (Eliasson et al., 2008, Wang and Thurmond 2009), several of which are regulated by miRNAs (Lovis et al. 2008; Eliasson and Esguerra 2014; Salunkhe et al. 2015). We have earlier demonstrated up‐regulation of at least 24 miRNAs (Esguerra et al. 2011) in the islets of the GK‐rat, whose hallmark phenotype is impaired glucose‐stimulated insulin secretion (Portha et al. 2009). We found enrichment of transport and secretory genes among the putative targets of the most upregulated miRNAs in GK rat islets. Specifically, we demonstrated that miR‐335 downregulates Stxbp1 mRNA (syntaxin‐binding protein 1 or Munc18‐1) by direct interaction (Esguerra et al. 2011).
Here we investigated if modulation of miR‐335 expression correlates to altered insulin secretion output in human islets. In addition, we performed a detailed analysis on how miR‐335 overexpression influences the exocytotic process using capacitance measurements, live‐TIRF imaging, and western blot analyses of potential targets. The data highlight the important regulatory role of miRNAs during T2D development.
Methods
Ethical approval
The use of human islets from deceased donors was approved by the ethics committees in Malmö and Uppsala, Sweden.
Cell culture
Rat insulinoma INS‐1 832/13 cells (Hohmeier et al. 2000) was maintained in RPMI 1640 medium containing 11.1 mmol/L glucose (HyClone, UT, USA) as previously described (Salunkhe et al. 2015). EndoC‐βH1 cells (EndoCells, Paris, France) (Ravassard et al. 2011; Andersson et al. 2015) were maintained in a culture medium containing: DMEM (5.6 mmol/L glucose), 2% BSA fraction V (Roche Diagnostics, Mannheim, Germany), 10 mmol/L nicotinamide (Merck Millipore, Darmstadt, Germany), 50 μM 2‐mercaptoethanol, 5.5 μg/mL transferrin, 6.7 ng/mL sodium selenite (Sigma‐Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin (PAA Laboratories, Pasching, Austria). Cells were tested for mycoplasma regularly.
Human islets
Islets from 28 human donors (F/M 16/12, age 58.2 ± 1.63, BMI 26.8 ± 0.6 kg/m2, HbA1c 5.82 ± 0.11, days in culture 2.8 ± 0.29) were provided by the Nordic Network for Clinical Islet Transplantation (Uppsala, Sweden). Human islets were hand‐picked under microscope to ensure high purity.
Transfection
One day prior to transfection cells were seeded in antibiotic‐free RPMI 1640 media in a 24‐well‐plate. For transient overexpression, a final concentration of 25 nmol/L chemically modified double‐stranded mature miRNA miR‐335 Pre‐miR™ miRNA Precursor (PM10063, Life Technologies, CA) or Pre‐miR™ miRNA Precursor Negative Control #1 (AM17110, Life Technologies) were used. For down‐regulation, a final concentration of 50 nmol/L of the following was used: miRcury anti‐miR LNA335 (#410201‐00, Exiqon, Denmark), LNA Negative Control B (#199005‐00, Exiqon, Denmark) Silencer ® Select Pre‐Designed siRNA against Syt11 (s133472, Life Technologies), and Silencer ® Select Negative Control No. 2 siRNA (Life Technologies) was used. Transfection was performed according to the manufacturer's protocol using lipofectamine® RNAiMAX Reagent (Invitrogen, CA). Cells were transfected 72 h prior to experiments.
Glucose‐stimulated insulin secretion
For glucose‐stimulated insulin secretion assay, cells were plated in triplicate for each condition and the assay was performed as described (Salunkhe et al. 2015). For K+‐induced insulin secretion the secretion buffer contained 50 mmol/L KCl (adjusted by reducing equimolar amount of NaCl). Insulin secretion measurements were normalized to total protein/well. Protein extraction was performed using RIPA buffer as previously described (Salunkhe et al. 2015). Protein content in cell homogenates was analyzed using BCA assay (Pierce®BCA Protein Assay Kit #23227, IL). Secreted and total insulin were measured using Coat‐a‐Count RIA (Millipore Corporation, MA) and Mercodia insulin ELISA (Mercodia, Uppsala, Sweden).
RNA extraction, RT‐PCR, and qPCR
RNA extraction, RT‐PCR for total RNA, and stem‐loop RT‐qPCR for microRNA was performed as previously described (Esguerra et al. 2011; Salunkhe et al. 2015) using pooled stem‐loop primers from TaqMan®MiRNA Assays: hsa‐miR‐335‐5p (#RT_000546), U6 (#RT_001973), and U87 (#RT_001712). The human miR‐335 (miRBase ID: hsa‐miR‐335‐5p/Acc. no. MIMAT0000765) and rat miR‐335 (rno‐miR‐335/Acc.no. MIMAT0000575) have identical mature sequences and were detected by the same Taqman miRNA assay. qPCR for protein coding genes was performed using primers from TaqMan®Gene Expression Assays; Syt11 (Rn00581475_m1) and endogenous controls Hprt1 (Rn01527840_m1) and Ppia (Rn00690933_m1). Relative expressions were calculated using the ΔΔCt method.
Western blot analysis
Protein extraction and measurement of protein content was performed ~72 h after transfection as described above. Protein samples were separated on 4–15% precast gradient polyacrylamide gels (Bio‐Rad Laboratories, CA) and then transferred to PVDF membranes. The membranes were blocked (at 4°C) with 5% milk and 1% BSA in a buffer consisting of 20 mmol/L Tris, 150 mmol/L NaCl and 0.1% (v/v) Tween‐20 (pH 7.5) for 1 h. Proteins were probed with antibodies for SNAP25 (1:500; #111011, Synaptic Systems, Germany), STXBP1 (1:500; #116002, Synaptic Systems, Germany), SYT11 (1:500; #WH0023208M3 Sigma‐Aldrich, Germany), Beta‐actin (1:1000; #A5441, Sigma‐Aldrich, Germany), and Cyclophilin B (1:2000; #ab16045 Abcam, UK), and incubated overnight at 4°C. The primary antibodies were detected using HRP‐conjugated goat anti‐rabbit/anti‐mouse secondary antibody (1:10,000; #7074S, Cell Signaling Technology) and anti‐mouse immunoglobulins/HRP antibody (1:1000; #P0448, Dako, Denmark). Bands were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (#34096; Thermo Scientific, MA) and AlphaImager (ProteinSimple, CA). Quantification was made using FluorChem SP software (ProteinSimple).
Electrophysiology
To measure ion channel currents and exocytosis (as changes in membrane capacitance) whole‐cell patch clamp experiments on single cells were performed as previously described (Salunkhe et al. 2015), and with a pipette solution containing (mmol/L): 125 Cs‐Glutamate, 10 NaCl, 10 CsCl, 1 MgCl2, 0.05 EGTA, 3 Mg‐ATP, 5 HEPES, and 0.1 cAMP (pH 7.15 using CsOH) and an extracellular solution with (mmol/L): 118 NaCl, 20 TEA‐Cl, 5.6 KCl, 2.6 CaCl2, 1.2 MgCl2, 5 glucose, and 5 HEPES (pH 7.4 using NaOH). The recordings were performed using patch master software (version 2–73) and EPC‐10 amplifier (Heka Elektronik, Lambrecht, Germany). Exocytosis was measured as changes in cell membrane capacitance, and it was evoked by a train of ten 500‐msec depolarizations from −70 mV to 0 mV applied at 1 Hz. Voltage‐dependent currents were investigated using an IV‐protocol, in which the membrane was depolarized from −70 mV to voltages between −40 mV and +40 mV during 50 msec. All experiments were carried out with constant buffer perfusion at 32°C. The measured voltage‐dependent current consists of Na+‐ and Ca2+‐current components. The rapid peak‐current (Ip) represents the Na+ current and the sustained current (Isus), measured during the latter 20 msec of the depolarizations, reflects the Ca2+‐current. Charge (Q) was measured ~ 2 msec after the onset of the pulse to exclude the Na+‐current and is therefore representative of the Ca2+‐influx.
TIRF microscopy
INS‐1 832/13 cells were plated on coverslips coated with poly‐D‐lysine and immediately cotransfected with mature miR‐335 and the granule marker NPY‐EGFP. Cells were imaged 36 h after plating in a solution containing (in mmol/L) 138 NaCl, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 d‐glucose, 5 Hepes HEPES (pH 7.4 with NaOH), supplemented with 200 μmol/L Diazoxide and 2 μmol/L forskolin. Exocytosis was evoked by timed local application of high K+ (75 mmol/L KCl equimolarly replacing NaCl) for 1 min through a pressurized glass electrode, similar to those used for patch clamp experiments. All experiments were carried out with constant buffer perfusion at 32°C.
Cells were imaged using a custom‐built lens‐type total internal reflection (TIRF) microscope based on an Axiovert 135 microscope with a 100x/1.45 objective (Carl Zeiss). Excitation was from a DPSS laser at 491 (Cobolt, Stockholm, Sweden), controlled with an acousto‐optical tunable filter (AA‐Opto, France) and using dichroic Di01‐R488/561 (Semrock) and emission filter FF01‐523/610 (Semrock). Scaling was 160 nm per pixel and exposure time 100 msec per frame at 10 frames/sec.
Data analysis
In the TIRF microscopy imaging experiments exocytosis events were found by eye. The moment of exocytosis was defined as the first significant change (2 SD) from the preexocytosis baseline. This definition applied both types of event, with or without preceding flash. The decay time was then defined as the time from exocytosis until the signal reached less than one‐third of the amplitude of the event. Traces were read out as ΔF, defined as average fluorescence in a 0.5 μm circle minus the average fluorescence in a surrounding annulus of 0.8 μm. The point of exocytosis was calculated by fitting the granule fluorescence during exocytosis with a discontinuous function, which assumes constant fluorescence before fusion, an inverted exponential decay just after fusion, and finally exponential decay during content release.
| (1) |
for t < t 1
for t 2 > t ≥ t 1
for t ≥ t 2,where t is time; c is average fluorescence in a 0.48‐μm‐wide circle at the granule site; A 1, A 2, and A 3 are the fluorescence values at the plateaus; τ 1 and τ 2 are the decay constants for the fluorescence increase after fusion and content release; and t 1 and t 2 are the times of fusion and release, respectively.
Statistics
Data are presented as mean ± SEM and statistical differences between groups were tested using two‐tailed Student's t‐test unless specified otherwise.
For the human islets, a simple linear regression analysis was performed on the miR‐335 expression and stimulatory index data (a measure of insulin secretion capacity in the human islets). F‐test was used to determine significance at P < 0.05 as implemented in IBM SPSS v.22.
Results
Correlation of miR‐335 expression and insulin secretion in human islets
We first investigated miR‐335 expression in human islets. For this we used miR‐335 expression data from 18 donors with HbA1c within the normal glucose tolerance levels (NGT; HbA1c = 5.50 ± 0.06), and 10 donors with high HbA1c corresponding to impaired glucose tolerance levels (IGT; HbA1c = 6.41 ± 0.15, P = 7.69E‐06 vs. NGT). MiR‐335 expression negatively correlated with insulin secretion, in islets from donors with IGT, whereas there was no correlation in islets from NGT donors (Fig. 1A–B). In line with these data, the human β‐cells EndoC‐βH1 overexpressing miR‐335 had reduced glucose‐stimulated insulin secretion (Fig. 1C). There was also a tendency of reduced K+‐induced secretion in these cells although this did not reach significance (Fig 1D).
Figure 1.
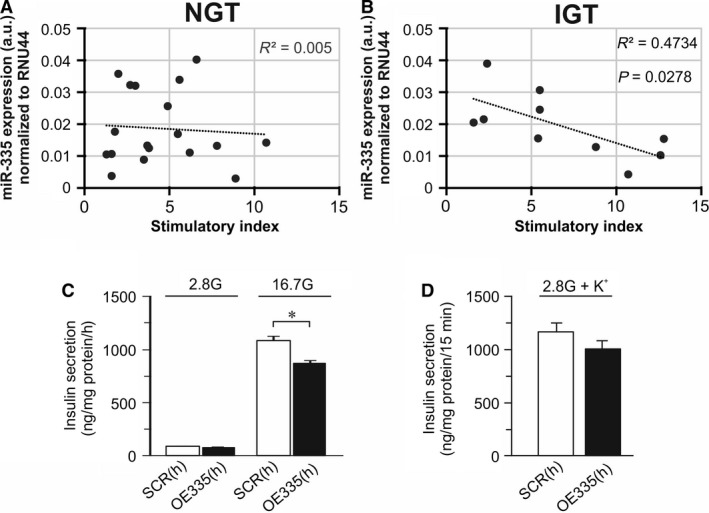
miR‐335 and insulin secretion in human islets and EndoC‐βH1 cells. (A) miR‐335 expression in human islets NGT donors against stimulation index, (B) As in A, but data are measured in human islets from IGT donors. (C) Insulin secretion in EndoC‐βH1 cells overexpressing miR‐335 (OE335(h); black bars) and in control cells (SCR(h); white bars) cells after stimulation for 1 h in 2.8 mmol/L or 16.7 mmol/L glucose (2.8G and 16.7 G) as indicated. n = 3; *P < 0.05. (D) As in C, but insulin secretion was measured after 15 min stimulation in 2.8 mmol/L glucose (2.8G) in the absence and presence of 50 mmol/L KCl (K+).
Decreased expression of STXBP1, SNAP25, and SYT11 after overexpression of miR‐335
To investigate the functional role of miR‐335 in β‐cells we overexpressed mature miR‐335 in the rat INS‐1 832/13 β‐cell line (Fig. 2A). Hereafter these cells are referred to as OE335 cells and the controls are referred to as SCR (scrambled) cells.
Figure 2.
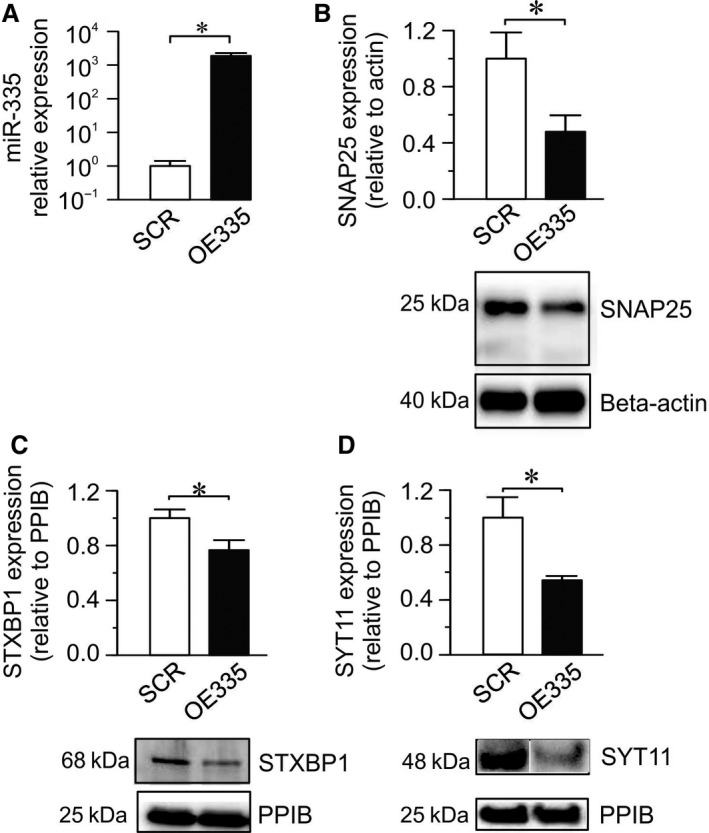
Overexpression of miR‐335 and its effect on the expression of three of its putative protein targets. (A) Expression of miR‐335 using mature miR‐335 mimic (OE335; black bar) relative to its expression using scramble control (SCR; white bar). Mir‐335 expression was normalized to endogenous controls U6 and U87. (n = 4; *P < 0.05). (B, C, D). Average protein levels of SNAP25, STXBP1 and SYT11 in OE335 (black bar) compared to SCR (white bar) cells (n = 4 each group; *P < 0.05). Representative western blots are shown for each protein. Expression levels were normalized to beta‐actin or cyclophilin B (PPIB).
Using TargetScan prediction tool (http://www.targetscan.org/) we found several exocytosis genes to be putative targets of miR‐335, both in rodents and/or humans, including the exocytosis genes Snap25, Syt11, and Stxbp1, which we decided to investigate further. Overexpression of miR‐335 significantly reduced protein levels of all these miR‐335 targets, SNAP25, STXBP1, and SYT11 (Fig. 2B–D).
Overexpression of miR‐335 leads to reduced glucose‐stimulated insulin secretion and exocytosis in INS‐1 832/13 cells
As in the human EndoC‐βH1 cells, OE335 cells displayed decreased glucose‐stimulated insulin secretion compared to SCR cells (Fig. 3A), without any differences in insulin content (Fig 3B). K+‐induced insulin secretion was likewise reduced in OE335 cells (Fig. 3C), indicating effects on exocytosis. We used the patch clamp technique to measure depolarization‐induced exocytosis in OE335 cells (Fig. 3D–E). We found a significantly reduced increase in membrane capacitance elicited by the train of depolarizations (∑Depolall) in OE335 cells as compared to SCR (Fig. 3E). The increase in membrane capacitance evoked by the first two depolarizations (∑Depol1‐2), was not affected, but the capacitance increase due to the latter depolarizations (∑Depol3‐10) was reduced by ~60% in OE335 cells (Fig. 3E). The average cell size did not differ significantly between OE335 and SCR cells (5.91 ± 0.38 pF vs. 5.44 ± 0.27 pF; n = 13–16). The close link between exocytosis and influx of Ca2+ through voltage‐gated Ca2+‐channels (Ammala et al. 1993) made us investigate the effect of miR‐335 modulation on the current–voltage relationships of the voltage‐dependent Ca2+‐currents in OE335 cells (Fig. 3F–G). We observed no differences in the Ca2+‐current at any of the voltages tested.
Figure 3.
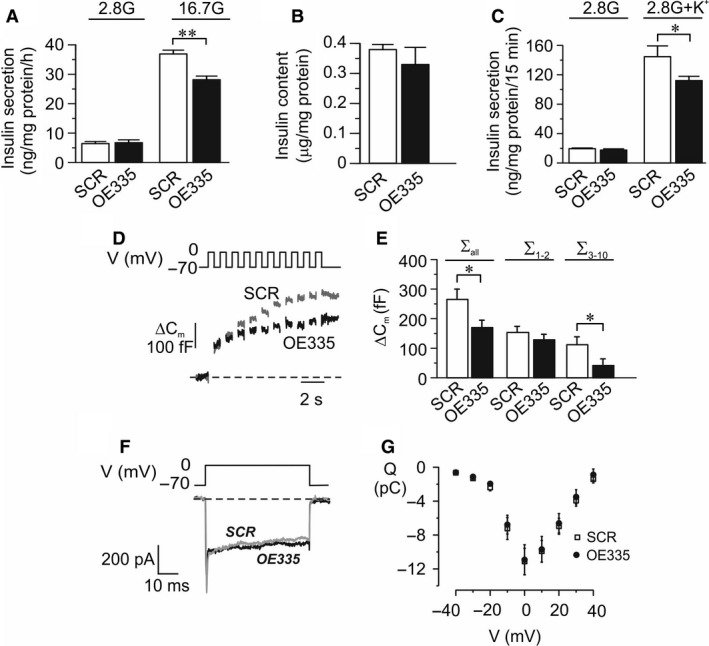
Consequence on insulin secretion and exocytosis in INS‐1 832/13 cells by miR‐335 overexpression. (A) Insulin secretion in OE335 (black bars) and SCR (white bars) cells after stimulation for 1 h in 2.8 mmol/L or 16.7 mmol/L glucose as indicated. n = 3; **P < 0.01. Secreted insulin is measured using human insulin‐RIA.(B) Insulin content in OE335 (black bar) and SCR (white bars) cells. n = 3. (C) Insulin secretion in OE335 (black bars) and SCR (white bars) cells after stimulation for 15 min. 2.8 mmol/L glucose with or without 50 mmol/L K+ as indicated. n = 5; *P < 0.05. (D) Representative traces of depolarization‐induced increases of membrane capacitance in OE335 (black trace) and SCR (gray trace) cells. (E) Summary of capacitance changes presented as the summed increased in membrane capacitance during all ten depolarizations (ΣDepolall), increase evoked by the first two depolarizations (ΣDepol1‐2) or the latter eight depolarizations (ΣDepol3‐10). n = 13 for OE335 (black bar) and n = 16 for SCR (white bar) cells; *P < 0.05. (F) Representative traces of a voltage‐dependent Ca2+ currents evoked by a depolarization from −70 mV to 0 mV in a control cell (SCR) and a cell overexpressing miR‐335 (OE335), respectively. (G) Summary of the charge (Q)‐voltage (V) relationship. n = 15 for SCR and n = 17 for OE335 cells.
We also reduced expression of miR‐335 using locked nucleic acid (LNA) against miR‐335 (LNA‐335; Fig 4A) and investigated the effects on insulin secretion and content (Fig. 4B–D), and on Ca2+‐induced exocytosis (Fig. 4E–F). LNA335 reduced insulin content (Fig. 4C), but had no effect on insulin secretion in INS‐1 832/13 cells (Fig 4B and D). However, LNA‐335 significantly increased membrane capacitance elicited by the train of depolarizations (∑Depolall). Reduction in miR‐335 expression significantly increased both ∑Depol1‐2 and ∑Depol3‐10 as compared to control (Fig. 4E–F).
Figure 4.
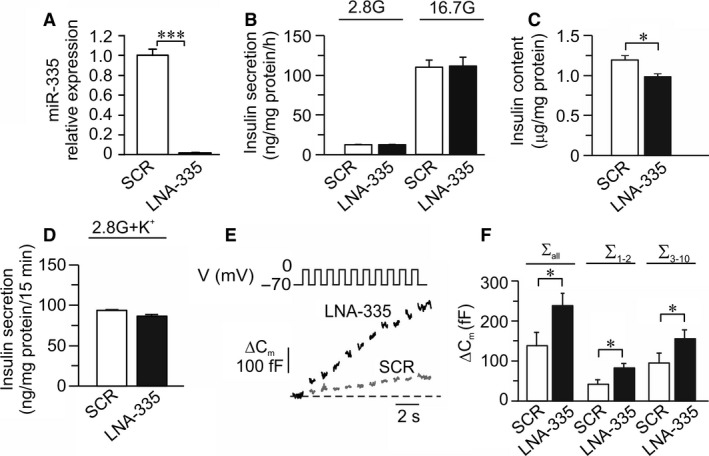
Influence of miR‐335 knockdown on insulin secretion and exocytosis in INS‐1 832/13 cells. (A) Expression of miR‐335 in cells after knockdown of miR‐335 by LNA against miR‐335 (LNA335; black bar) relative to expression in scramble control cells (SCR; white bar). n = 4; ***P < 0.001. (B) Insulin secretion in LNA335 (black bars) and SCR (white bars) cells after stimulation for 1 h in 2.8 mmol/L or 16.7 mmol/L glucose as indicated (n = 4). The amount of released insulin is measured using human/rat insulin‐ELISA. (C) Insulin content in LNA335 (black bars) and SCR (white bars) cells. n = 8; *P < 0.05. (D) Insulin secretion in OE335 (black bars) and SCR (white bars) cells after stimulation for 15 min. in 2.8 mmol/L glucose with 50 mmol/L K+. n = 4. (E) Representative traces of depolarization‐induced increases of membrane capacitance in LNA335 (black trace) and SCR (gray trace) cells. (F) Summary of capacitance changes presented as the summed increased in membrane capacitance during all ten depolarizations (ΣDepolall), increase evoked by the first two depolarizations (ΣDepol1‐2) or the latter eight depolarizations (ΣDepol3‐10). n = 10 for LNA335 (black bar) and n = 9 for SCR (white bar) cells; *P < 0.05
Overexpression of miR‐335 reduces exocytosis and accelerates content release from individual granules in INS‐1 832/13 cells
To study in detail the effects of elevated miR‐335 levels on insulin granule docking and release, OE335 and SCR cells were cotransfected with granular marker neuropeptide‐Y (NPY)‐mEGFP, and imaged by TIRF microscopy. Docked granules were present in both groups and their density was similar to previous estimates in these cells (Gandasi and Barg 2014), with 0.59 ± 0.02 μm‐2 (n = 31) in OE335 and 0.64 ± 0.02 μm‐2 (n = 53) in SCR (Fig. 5A–B).
Figure 5.
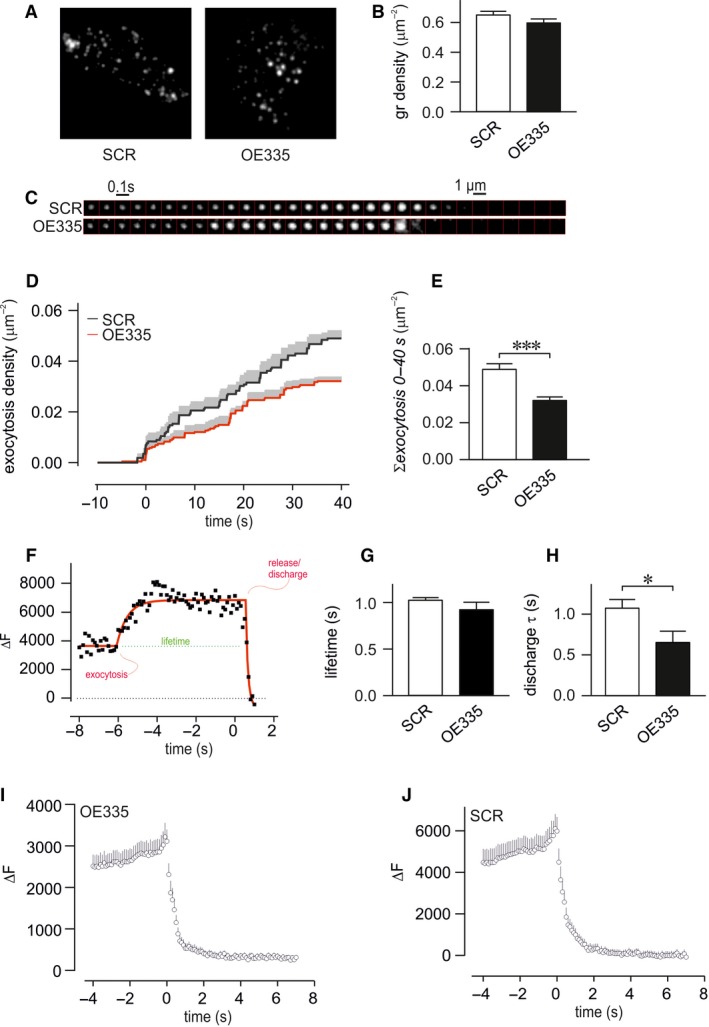
Function of miR‐335 on density of granules and exocytosis measured by TIRF microscopy in INS‐1 832/13 cells. (A) Representative TIRF images of OE335 and SCR cells co‐transfected with NPY‐mEGFP. (B) Density of granules in OE335 (black bar; n = 31) and SCR (white bar; n = 53) cells. (C) Examples of single exocytosis event measured in OE335 and SCR cells, frames are 100 msec apart. Exocytosis was stimulated through depolarization using elevated K+. (D, E) Cumulative number of exocytosis events per area in OE335 and SCR cells (n = 8 each group; ***P < 0.001). (F) Representative image of fluorescence time course during a single exocytosis event (black) with fit overlaid (red) illustrating the numerical analysis. The interval between the moments of exocytosis/fusion and release is taken as fusion pore lifetime (green). (G, H) Summary of fitted exocytosis and decay constants of individual events as described in (F) The lifetime of the events (*P=n.S.) and decay constant from individual granules during content release (*P < 0.05) was measured in OE335 and SCR cells (n = 8 each group). (I,J) Average fluorescence signal of granule fluorescence for exocytosis events in OE335 and SCR cells as in C.
We stimulated exocytosis by local application of elevated K+, which induces rapid exocytosis of docked and primed granules seen as rapid disappearance of granule fluorescence (Fig. 5C). Two thirds of the exocytotic events were preceded by a temporary increase in granule fluorescence (Fig. 5C, I–J) due to unquenching of EGFP‐fluorescence that occurs when the granule lumen makes aqueous contact with the extracellular medium. Lack of this feature indicates rapid release and implies instantaneous widening of the fusion pore (Barg et al. 2002). The duration of the transient increase, which reflects fusion pore lifetime, was estimated by nonlinear fit of a discontinuous function to the fluorescence time course (Fig. 5F and methods); no significant difference between OE335 cells and SCR cells was detected (Fig. 5G). The kinetics of content release was analyzed by fitting a single exponential function to the fluorescence decay of individual events (Fig. 5F–H). The decay of vesicular fluorescence was twice as fast in OE335 cells compared to SCR cells (Fig. 5H). The total number of exocytosis events in OE335 was significantly less than in SCR cells (Fig. 5D–E).
Discussion
The contribution of miRNAs in the regulation of pancreatic β‐cell function has been a topic of great interest in diabetes research. Consequently, mechanistic details on how dysregulated miRNA expression in the β‐cell contributes to the pathophysiology of T2D are beginning to emerge. We have previously shown that miR‐335 is abnormally overexpressed in the pancreatic islets of the GK rat (Esguerra et al. 2011), and we have validated Stxbp1 mRNA as a direct target of miR‐335. Here we show a negative correlation between miR‐335 expression and insulin secretion in human islets from donors with IGT and provide evidence that overexpression of miR‐335 results in (1) downregulation of three exocytosis protein targets: STXBP1, SNAP25, and SYT11, and (2) impaired exocytosis of insulin granules and decreased insulin secretion.
Although it is known that the defective insulin secretory capacity can be due to defects in the exocytotic machinery, for example, through reduced expression of exocytosis proteins in the GK‐rat (Zhang et al. 2002), it remains unclear how β‐cell exocytosis in general is influenced by dysregulated expression of specific miRNAs. Our data support the hypothesis that the main function of miR‐335 is in the regulation of the final stages of insulin secretion. Indeed, both single‐cell capacitance measurements (Fig. 3D–E) and TIRF microscopy data (Fig. 5) confirmed defective priming of already docked granules and deficiencies in postpriming processes of exocytosis after overexpression of miR‐335. The expression of miR‐335 is >1000 times the endogenous levels, prompting us to perform experiments in which the endogenous levels of miR‐335 were silenced (Fig. 4). In these experiments exocytosis was instead increased confirming that miR‐335 is indeed involved in the regulation of β‐cell exocytosis. However, while LNA‐335 increased exocytosis, it simultaneously reduced insulin content. The reduced insulin content after miR‐335 knockdown was to some extent surprising and shows that the knock‐down of miR‐335 needs to be adjusted if it should be used therapeutically. The summed outcome of reduced insulin content and increased exocytosis is unchanged insulin secretion in LNA‐335 cells. Our results demonstrate the inherent complexity by which a single miRNA can influence the regulation of multiple targets and hence the overall targeted cellular process. Classically miRNAs has been thought to act as binary off‐switches to repress mRNA expression; models that are more recent suggest that miRNAs can act as rheostats to control protein levels enabling customized expression in different cells and cellular processes (Bartel 2009). Thus, it seems like miR‐335 differentially regulate targets within processes controlling insulin content and insulin exocytosis. Altogether, we can still conclude that the overexpression of miR‐335 reduce insulin secretion mainly through an effect on exocytosis.
We asked if we could identify potential targets of miR‐335 within the exocytosis pathway and if the overexpression reduced the protein level of these targets. Indeed, we found reduced protein expression of SNAP25, STXBP1, and SYT11 in OE335 cells. SYT11 is one of six Ca2+‐insensitive isoforms of the synaptotagmin family (Sudhof 2004; Gauthier and Wollheim 2008; Milochau et al. 2014). The cellular function of SYT11 is beginning to be understood (Andersson et al. 2012; Arango Duque et al. 2013; Fadista et al. 2014; Milochau et al. 2014), but details regarding its role in exocytosis remain unclear. Recently SYT11 was also shown to interact with components of the RNA‐induced silencing complex (RISC) and proteins involved in endoplasmic reticulum/Golgi derived‐granule transport in rat beta (INS‐1E) cells implicating SYT11 in both gene regulation by miRNAs and membrane trafficking (Milochau et al. 2014). Although less is known about SYT11 our data presented here are in agreement with the established roles of SNAP25 and STXBP1 as essential components in priming and fusion of insulin‐containing granules (Zhang et al. 2000, 2002; Kang et al. 2002; Ohara‐Imaizumi et al. 2004b, Gulyas‐Kovacs et al. 2007; Eliasson et al., 2008; Tomas et al. 2008). STXBP1 binds to the folded conformation of STX1A during docking (Han et al. 2011), and subsequently dissociates from STX1A upon Ca2+ influx to allow formation of the SNARE complex (Tomas et al. 2008) in the priming step (Gulyas‐Kovacs et al. 2007). In addition, studies have implicated STXBP1, as well as SNAP25 and the synaptotagmin isoforms 1 and 7, in the recruitment of granules to the membrane (Gulyas‐Kovacs et al. 2007; Ohara‐Imaizumi et al. 2007; Tomas et al. 2008; de Wit et al. 2009; Dolai et al. 2016). Indeed, granule docking coincides with the formation of clusters containing STXBP1 (Gandasi and Barg 2014) and the physiological importance of docking in insulin secretion was shown in the diabetic GK‐rat (Ohara‐Imaizumi et al. 2004a). Although STXBP1, SNAP25, and SYT11 were down‐regulated in the OE335 cells, we did not observe a reduced number of docked granules. This lack of effects on docking might be due to insufficient miR‐335‐mediated knockdown (Fig. 2B–D) or the possibility that miR‐335 has other unidentified targets that normally inhibit granule docking. It is possible that other targets of miR‐335 contribute to the regulation of insulin secretion, resulting in a net cumulative negative effect of miR‐335 on insulin secretion. For instance, miR‐335 directly targets the transcription factors FOXA2 and SOX17 and miR‐335 thereby promotes mesendodermal lineage segregation (Yang et al. 2014). FOXA2 is not only involved in developmental β‐cell differentiation, but the protein also impact β‐cell function. Foxa2 deficient mice have increased insulin secretion and increased number of docked granules (Gao et al. 2007). Nevertheless, previous and current findings link miR‐335, SNAP‐25, STXBP1, and SYT11 to the exocytotic process where imbalances in any of them could contribute to impaired insulin exocytosis.
We provide data demonstrating that overexpression of miR‐335 in the human EndoC‐βH1 cells reduce insulin secretion and that miR‐335 expression was negatively correlated with insulin secretion in human islets from individuals with prediabetes. It is remarkable that miR‐335 expression levels negatively correlated with insulin secretion only in islets from high HbA1c donors. We can only speculate that as fine‐tuners of cellular processes (Bartel 2009), miRNAs may be mostly required during nonoptimal conditions, perhaps to be able to compensate for the detrimental effects brought about by pathophysiological conditions. In our previous study (Esguerra et al. 2011) we showed elevated miR‐335 expression in GK‐rat as compared to Wistar‐rat islets. This was true also when islets were incubated in hypoglycemic conditions (2.8 mmol/L glucose) for 24 h. However, under hyperglycemic conditions (16.7 mmol/L glucose) for the same period we found evidence of compensatory mechanisms bringing the levels of miR‐335 in the GK‐islets toward that of control. Interestingly, in short‐term incubation for 1 h miR‐335 expression in the GK‐islets was instead elevated after incubation in 16.7 mmol/L glucose as compared to 2.8 mmol/L glucose (or the Wistar control). We postulate therefore that under constant hyperglycemic environment in diabetic patients, miRNA‐mediated regulatory effects are more pronounced and hence the negative correlation between miR‐335 levels and HbA1c is more apparent in the diseased state.
We find our human data of specific interest since one of the signatures of T2D is reduced first‐phase insulin secretion prior to full‐blown diabetes (Gerich 2002). On a cellular level first‐phase insulin secretion has been suggested to be associated with exocytosis and the fusion of primed granules (Rorsman and Renstrom 2003, Eliasson et al., 2008). More so, our observation might be essential in a larger metabolic perspective. Expression of miR‐335 has been demonstrated to be increased in liver and white adipocyte tissue in ob/ob and db/db mice (Nakanishi et al. 2009; Oger et al. 2014), and in islets of diabetic GK‐rats (Esguerra et al. 2011). MiR‐335 is suggested to be involved in lipid metabolism, adipogenesis differentiation, and adipose inflammation (Nakanishi et al. 2009; Zhu et al. 2014), and miR‐335 expression is proposed to be regulated by adipokines (Zhu et al. 2014). It is well‐established that regulation of glucose‐transporters in adipose tissue, involves exocytosis proteins (Cheatham 2000). Hence, our observation that LNA335 and OE335 influence the exocytosis process might not only be valid for pancreatic β‐cells, but also for other metabolically relevant tissues. In this aspect it is noteworthy that miRNAs, as important players in gene regulation, have the potential to be innovative therapeutic drug targets against diabetes and associated complications (Eliasson and Esguerra 2014). To achieve this goal, a necessary first step is to understand the mechanistic principles by which miRNAs participate in cellular processes.
In conclusion, our data demonstrate the cellular mechanisms by which miR‐335 influence insulin secretion. Based on our observations we propose that miR‐335 overexpression negatively affects insulin secretion via the reduction in multiple exocytosis protein targets and impaired priming of insulin granules.
Conflict of Interests
The authors declare that there is no duality of interest associated with this manuscript.
Acknowledgment
We thank Anna‐Maria Veljanovska Ramsay and Britt‐Marie Nilsson at the Department of Clinical Sciences Malmö, Lund University for technical assistance.
Salunkhe V. A., Ofori J. K., Gandasi N. R., Salö S. A., Hansson S., Andersson M. E., Wendt A., Barg S., Esguerra J. L. S., Eliasson L.. MiR‐335 overexpression impairs insulin secretion through defective priming of insulin vesicles. Physiol Rep, 5 (21), 2017, e13493, https://doi.org/10.14814/phy2.13493
Funding Information
This project is financially supported by the Swedish Foundation for Strategic Research (IRC‐LUDC), the Swedish Research Council (Project grants to LE and SB; Linneus grant to LUDC; SFO‐EXODIAB), Region Skåne‐ALF, The Swedish Diabetes Foundation, Albert Påhlsson Foundation, Hjelt Diabetes Foundation, Novo Nordisk Foundation, and the Diabetes Wellness Network Sweden. NRG was supported by the EFSD/Lilly program.
Contributor Information
Jonathan L. S. Esguerra, Email: jonathan.esguerra@med.lu.se
Lena Eliasson, Email: lena.eliasson@med.lu.se.
References
- Ammala, C. , Eliasson L., Bokvist K., Larsson O., Ashcroft F. M., and Rorsman P.. 1993. Exocytosis elicited by action potentials and voltage‐clamp calcium currents in individual mouse pancreatic B‐cells. J. Physiol. 472:665–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, S. A. , Olsson A. H., Esguerra J. L., Heimann E., Ladenvall C., Edlund A., et al. 2012. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol. Cell. Endocrinol. 364:36–45. [DOI] [PubMed] [Google Scholar]
- Andersson, L. E. , Valtat B., Bagge A., Sharoyko V. V., Nicholls D. G., Ravassard P., et al. 2015. Characterization of stimulus‐secretion coupling in the human pancreatic EndoC‐betaH1 beta cell line. PLoS ONE 10:e0120879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango Duque, G. , Fukuda M., and Descoteaux A.. 2013. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J. Immunol. 190:1737–1745. [DOI] [PubMed] [Google Scholar]
- Barg, S. , Olofsson C. S., Schriever‐Abeln J., Wendt A., Gebre‐Medhin S., Renstrom E., et al. 2002. Delay between fusion pore opening and peptide release from large dense‐core vesicles in neuroendocrine cells. Neuron 33:287–299. [DOI] [PubMed] [Google Scholar]
- Bartel, D. P. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. [DOI] [PubMed] [Google Scholar]
- Bartel, D. P. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke, J. , Stark A., Russell R. B., and Cohen S. M.. 2005. Principles of microRNA‐target recognition. PLoS Biol. 3:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham, B. 2000. GLUT4 and company: SNAREing roles in insulin‐regulated glucose uptake. Trends Endocrinol. Metab. 11:356–361. [DOI] [PubMed] [Google Scholar]
- Dolai, S. , Xie L., Zhu D., Liang T., Qin T., Xie H., et al. 2016. Synaptotagmin‐7 functions to replenish insulin granules for exocytosis in human islet beta‐cells. Diabetes 65:1962–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson, L. , and Esguerra J. L.. 2014. Role of non‐coding RNAs in pancreatic beta‐cell development and physiology. Acta Physiol. (Oxf).211:273–284. [DOI] [PubMed] [Google Scholar]
- Eliasson, L. , Abdulkader F., Braun M., Galvanovskis J., Hoppa M. B., and Rorsman P.. 2008. Novel aspects of the molecular mechanisms controlling insulin secretion. J. Physiol. 586:3313–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esguerra, J. L. , Bolmeson C., Cilio C. M., and Eliasson L.. 2011. Differential glucose‐regulation of microRNAs in pancreatic islets of non‐obese type 2 diabetes model Goto‐Kakizaki rat. PLoS ONE 6:e18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadista, J. , Vikman P., Laakso E. O., Mollet I. G., Esguerra J. L., Taneera J., et al. 2014. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl Acad. Sci. USA 111:13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisano, H. Y. , Ostenson C. G., Sheu L., Wheeler M. B., and Efendic S.. 2002. Abnormal expression of pancreatic islet exocytotic soluble N‐ethylmaleimide‐sensitive factor attachment protein receptors in Goto‐Kakizaki rats is partially restored by phlorizin treatment and accentuated by high glucose treatment. Endocrinology 143:4218–4226. [DOI] [PubMed] [Google Scholar]
- Gandasi, N. R. , and Barg S.. 2014. Contact‐induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat. Commun. 5:399. [DOI] [PubMed] [Google Scholar]
- Gao, N. , White P., Doliba N., Golson M. L., Matschinsky F. M., and Kaestner K. H.. 2007. Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. 6:267–279. [DOI] [PubMed] [Google Scholar]
- Gauthier, B. R. , and Wollheim C. B.. 2008. Synaptotagmins bind calcium to release insulin. Am. J. Physiol. Endocrinol. Metab. 295:E1279–E1286. [DOI] [PubMed] [Google Scholar]
- Gerich, J. E. 2002. Is reduced first‐phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes? Diabetes 51(Suppl 1):S117–S121. [DOI] [PubMed] [Google Scholar]
- Gulyas‐Kovacs, A. , de Wit H., Milosevic I., Kochubey O., Toonen R., Klingauf J., et al. 2007. Munc18‐1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J. Neurosci. 27:8676–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, G. A. , Malintan N. T., Saw N. M., Li L., Han L., Meunier F. A., et al. 2011. Munc18‐1 domain‐1 controls vesicle docking and secretion by interacting with syntaxin‐1 and chaperoning it to the plasma membrane. Mol. Biol. Cell 22:4134–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmeier, H. E. , Mulder H., Chen G., Henkel‐Rieger R., Prentki M., and Newgard C. B.. 2000. Isolation of INS‐1‐derived cell lines with robust ATP‐sensitive K+ channel‐dependent and ‐independent glucose‐stimulated insulin secretion. Diabetes 49:424–430. [DOI] [PubMed] [Google Scholar]
- Kang, Y. , Huang X., Pasyk E. A., Ji J., Holz G. G., Wheeler M. B., et al. 2002. Syntaxin‐3 and syntaxin‐1A inhibit L‐type calcium channel activity, insulin biosynthesis and exocytosis in beta‐cell lines. Diabetologia 45:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovis, P. , Gattesco S., and Regazzi R.. 2008. Regulation of the expression of components of the exocytotic machinery of insulin‐secreting cells by microRNAs. Biol. Chem. 389:305–312. [DOI] [PubMed] [Google Scholar]
- Milochau, A. , Lagree V., Benassy M. N., Chaignepain S., Papin J., Garcia‐Arcos I., et al. 2014. Synaptotagmin 11 interacts with components of the RNA‐induced silencing complex RISC in clonal pancreatic beta‐cells. FEBS Lett. 588:2217–2222. [DOI] [PubMed] [Google Scholar]
- Nakanishi, N. , Nakagawa Y., Tokushige N., Aoki N., Matsuzaka T., Ishii K., et al. 2009. The up‐regulation of microRNA‐335 is associated with lipid metabolism in liver and white adipose tissue of genetically obese mice. Biochem. Biophys. Res. Commun. 385:492–496. [DOI] [PubMed] [Google Scholar]
- Oger, F. , Gheeraert C., Mogilenko D., Benomar Y., Molendi‐Coste O., Bouchaert E., et al. 2014. Cell‐specific dysregulation of microRNA expression in obese white adipose tissue. J. Clin. Endocrinol. Metab. 99:2821–2833. [DOI] [PubMed] [Google Scholar]
- Ohara‐Imaizumi, M. , Nishiwaki C., Kikuta T., Nagai S., Nakamichi Y., and Nagamatsu S.. 2004a. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic beta‐cells: different behaviour of granule motion between normal and Goto‐Kakizaki diabetic rat beta‐cells. Biochem. J. 381:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara‐Imaizumi, M. , Nishiwaki C., Nakamichi Y., Kikuta T., Nagai S., and Nagamatsu S.. 2004b. Correlation of syntaxin‐1 and SNAP‐25 clusters with docking and fusion of insulin granules analysed by total internal reflection fluorescence microscopy. Diabetologia 47:2200–2207. [DOI] [PubMed] [Google Scholar]
- Ohara‐Imaizumi, M. , Fujiwara T., Nakamichi Y., Okamura T., Akimoto Y., Kawai J., et al. 2007. Imaging analysis reveals mechanistic differences between first‐ and second‐phase insulin exocytosis. J. Cell Biol. 177:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostenson, C. G. , Gaisano H., Sheu L., Tibell A., and Bartfai T.. 2006. Impaired gene and protein expression of exocytotic soluble N‐ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 55:435–440. [DOI] [PubMed] [Google Scholar]
- Pang, Z. P. , and Sudhof T. C.. 2010. Cell biology of Ca2 + ‐triggered exocytosis. Curr. Opin. Cell Biol. 22:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portha, B. , Lacraz G., Kergoat M., Homo‐Delarche F., Giroix M. H., Bailbe D., et al. 2009. The GK rat beta‐cell: a prototype for the diseased human beta‐cell in type 2 diabetes? Mol. Cell. Endocrinol. 297:73–85. [DOI] [PubMed] [Google Scholar]
- del Prato, S. , and Tiengo A.. 2001. The importance of first‐phase insulin secretion: implications for the therapy of type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 17:164–174. [DOI] [PubMed] [Google Scholar]
- Ravassard, P. , Hazhouz Y., Pechberty S., Bricout‐Neveu E., Armanet M., Czernichow P., et al. 2011. A genetically engineered human pancreatic beta cell line exhibiting glucose‐inducible insulin secretion. J. Clin. Invest. 121:3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo, J. , and Sudhof T. C.. 2012. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu. Rev. Cell Dev. Biol. 28:279–308. [DOI] [PubMed] [Google Scholar]
- Rorsman, P. , and Renstrom E.. 2003. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46:1029–1045. [DOI] [PubMed] [Google Scholar]
- Salunkhe, V. A. , Esguerra J. L., Ofori J. K., Mollet I. G., Braun M., Stoffel M., et al. 2015. Modulation of microRNA‐375 expression alters voltage‐gated Na channel properties and exocytosis in insulin‐secreting cells. Acta Physiol. (Oxf).213:882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof, T. C. 2004. The synaptic vesicle cycle. Annu. Rev. Neurosci. 27:509–547. [DOI] [PubMed] [Google Scholar]
- Tomas, A. , Meda P., Regazzi R., Pessin J. E., and Halban P. A.. 2008. Munc 18‐1 and granuphilin collaborate during insulin granule exocytosis. Traffic 9:813–832. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , and Thurmond D. C.. 2009. Mechanisms of biphasic insulin‐granule exocytosis ‐ roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 122:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, H. , Walter A. M., Milosevic I., Gulyas‐Kovacs A., Riedel D., Sorensen J. B., et al. 2009. Synaptotagmin‐1 docks secretory vesicles to syntaxin‐1/SNAP‐25 acceptor complexes. Cell 138:935–946. [DOI] [PubMed] [Google Scholar]
- Yang, D. , Lutter D., Burtscher I., Uetzmann L., Theis F. J., and Lickert H.. 2014. miR‐335 promotes mesendodermal lineage segregation and shapes a transcription factor gradient in the endoderm. Development 141:514–525. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Efanov A., Yang S. N., Fried G., Kolare S., Brown H., et al. 2000. Munc‐18 associates with syntaxin and serves as a negative regulator of exocytosis in the pancreatic beta ‐cell. J. Biol. Chem. 275:41521–41527. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Khan A., Ostenson C. G., Berggren P. O., Efendic S., and Meister B.. 2002. Down‐regulated expression of exocytotic proteins in pancreatic islets of diabetic GK rats. Biochem. Biophys. Res. Commun. 291:1038–1044. [DOI] [PubMed] [Google Scholar]
- Zhu, L. , Chen L., Shi C. M., Xu G. F., Xu L. L., Zhu L. L., et al. 2014. MiR‐335, an adipogenesis‐related microRNA, is involved in adipose tissue inflammation. Cell Biochem. Biophys. 68:283–290. [DOI] [PubMed] [Google Scholar]