

Abstract
The regulation of glycerolipid biosynthesis is critical for homeostasis of cellular lipid stores and membranes. Here we review the role of lipin phosphatidic acid phosphatase enzymes in glycerolipid synthesis. Lipin proteins are unique among glycerolipid biosynthetic enzymes in their ability to transit among cellular membranes, rather than remain membrane tethered. We focus on the mechanisms that underlie lipin protein interactions with membranes and the versatile roles of lipins in several organelles, including the endoplasmic reticulum, mitochondria, endolysosomes, lipid droplets, and nucleus. We also review the corresponding physiological roles of lipins, which have been uncovered by the study of genetic lipin deficiencies. We propose that the growing body of knowledge concerning the biochemical and cellular activities of lipin proteins will be valuable for understanding the physiological functions of lipin proteins in health and disease.
Keywords: phosphatidic acid phosphatase, triacylglycerol, rhabdomyolysis, lipodystrophy
1. Triacylglycerol and phospholipid biosynthesis
Recent ‘omics’ studies (such as LIPID MAPS and other large-scale lipid analysis projects) have revealed a vast range of cellular lipids, with more than 1000 species detected in a typical eukaryotic cell [1,2]. It has been estimated that approximately 5% of genes have a role in lipid synthesis [3], reflecting the critical role that lipids play in cellular structure and metabolism. The glycerolipids are abundant cellular lipids with physiological roles in energy storage (primarily in the form of triacylglycerol) and membrane structure (phospholipids and other lipids, varying by organelle type) [3]. Enzymes that regulate glycerolipid levels also play a role in modulating lipid intermediates that serve as signaling nodes to influence a wide range of physiological processes [4–6]. Both in their roles in signaling pathways and in energy storage and homeostasis, the enzymes that modulate glycerolipid levels have been of interest as potential drug targets [4,7].
Here, we briefly review glycerolipid biosynthetic pathways and then focus on the function of lipin phosphatidate phosphatase enzymes at the molecular and physiological levels. Of the enzymes in the glycerol 3-phosphate pathway, lipin enzymes are unique in their ability to transit among cellular membranes, rather than remain membrane tethered as most other enzymes in the pathway. This property of lipins allows them to act at diverse cellular organelles, and to have a role in processes beyond lipid biosynthesis. Some of these processes have been uncovered through the study of genetic lipin deficiencies, which will also be reviewed here.
1.1. The glycerol 3-phosphate pathway
As elucidated by Eugene Kennedy decades ago, glycerolipids are synthesized through modification of a glycerol 3-phosphate backbone [8,9]. Through acylation and dephosphorylation reactions, glycerol 3-phosphate is converted to triacylglycerol for neutral lipid storage, or is converted through other modifications to zwitterionic or anionic phospholipids (Figure 1). The regulation of reactions in the glycerol 3-phosphate pathway is one mechanism for controlling glycerolipid levels in the cell. The key enzymes driving the addition of fatty acids onto the glycerol backbone are glycerol acyltransferases (GPATs), acylglycerol-phosphate acyltransferases (AGPATs), and diacylgylcerol acyltransferases (DGATs). In mammalian cells, each of these acyltransferase activities can be carried out by multiple proteins of the same class, including four GPAT enzymes and at least five AGPAT enzymes [10,11]. Some enzymes originally identified as AGPATs have subsequently been shown to have different enzyme activities.
Figure 1. Enzymes of the glycerol 3-phosphate pathway.
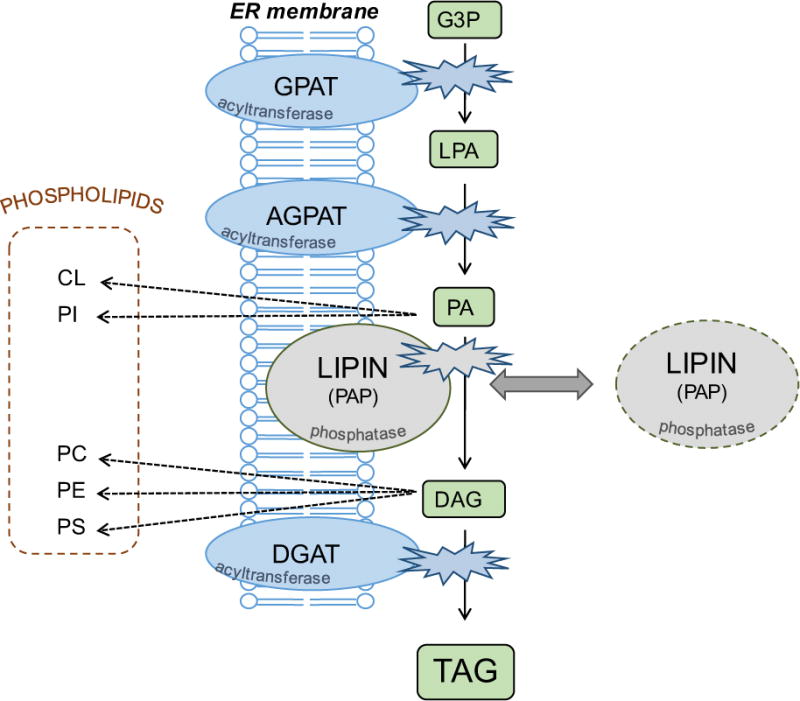
The primary route for triacylglycerol synthesis in most mammalian tissues involves the sequential transfer of acyl CoAs to a glycerol 3-phosphate backbone by the action of acyltransferases (GPAT, AGPAT, DGAT), and removal of the phosphate group by the action of lipin phosphatidic acid phosphatases (PAP). This pathway also provides building blocks for zwitterionic and anionic phospholipids. The acyltransferases are membrane-resident proteins, whereas lipins may reside in the cytosol and transiently interact with membranes containing the lipid substrate, PA. GPAT, glycerol 3-phosphate acyltransferase; AGPAT, acylglycerol 3-phosphate acyltransferase; DGAT, acyl CoA:diacylglycerol acyltransferase; LPA, lysophosphatidic acid; PA, phosphatidic acid; DAG, diacylglycerol; TAG, triacylglycerol; CL, cardiolipin; PI, phosphatidylinositol; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine.
In addition to the acyltransferase reactions, the de novo glyceropholipid synthesis pathway employs a phosphatidate phosphatase (PAP) enzyme (Figure 1). In mammals, three independent genes encode the lipin 1, lipin 2 and lipin 3 proteins that catalyze this reaction [12]. Lipin protein activity catalyzes the conversion of phosphatidic acid (PA) to diacylglycerol [13,14], and thus acts at a branch-point for the synthesis of triacylglycerol, zwitterionic phospholipids (phosphatidylcholine and phosphatidylethanolamine), or anionic phospholipids (phosphatidylglycerol, phosphatidylserine, or cardiolipin) [15] (Figure 1). Lipin regulation of local levels of PA and diacylglycerol may also influence activation of signaling cascades that are regulated by these lipid intermediates [16]. The pathologies of lipin deficiencies appear to be associated with the loss of normal glycerolipid synthetic capabilities as well as aberrant regulation of PA-mediated signaling cascades, as discussed throughout this review.
1.2 Alternative enzyme activities in triacylglycerol synthesis
It should be noted that while most mammalian cells primarily utilize the glycerol 3-phosphate pathway, intestinal enterocytes possess an alternate pathway for triacylglycerol synthesis from monoacylglycerol, which is generated at high concentrations from the hydrolysis of dietary lipids in the intestinal lumen [17,18]. The monoacylglycerol pathway uses acylCoA:monoacylglycerol acyltransferase (MGAT) enzymes to generate diacylglycerol from monoacylglycerol. The resulting diacylglycerol is acted upon by DGAT enzymes. Therefore, TAG synthesis through the monoacylglycerol pathway does not require GPAT, AGPAT or lipin activities. It has been estimated that the monoacylglycerol pathway is responsible for 70–80% of triacylglycerol synthesis for the production of chylomicrons in enterocytes [19,20]. However, these estimates are based on studies that were performed using chemical enzyme inhibitors prior to the isolation of genes for all of the enzymes shown in Figure 1. Recent studies in which MGAT2 or GPAT3 have been genetically ablated in the intestine suggest that the monoacylglycerol and glycerol phosphate pathways are non-redundant and each carries out a necessary role in intestinal lipid synthesis and metabolism [21,22]. Further studies are required to elucidate the relationship between the monoacylglycerol and glycerol phosphate pathways in intestinal lipid metabolism.
2. Activity and regulation of mammalian lipin proteins
The lipin genes were initially discovered by positional cloning in a spontaneous mutant mouse model known as fatty liver dystrophy (fld) [12]. The fld mice carry a null mutation in the gene now known as Lpin1, which prevents formation of any functional protein product. The Lpin2 and Lpin3 genes were identified based on their sequence similarity to Lpin1 [12]. Lpin1 mRNA undergoes alternative splicing to generate three isoforms: lipin 1α, lipin 1β and lipin-1γ [23,24]. Lipin 1α and lipin 1β exhibit different propensities for nuclear and cytoplasmic localization, although this may be cell-type dependent. Lipin 1α is localized predominantly in the nucleus, whereas lipin 1β is primarily present in the cytoplasm [23]. Lipin 1α and 1β isoforms are present in most of the tissues that express Lpin1, but the relative levels of the two isoforms varies with tissue type [23]. Lipin-1γ is present primarily in brain [24]. Lipin 1α and 1β isoforms may have distinct cellular functions in processes such as preadipocyte differentiation. Lipin 1α is associated with expression of adipogenic differentiation factors such as PPARγ and C/EBPα, and lipin 1β is associated with induction of lipogenic genes such as fatty acid synthase, stearoyl CoA desaturase, and DGAT [23]. Lipin 1β expression in adipose tissue of transgenic mice also leads to enhanced lipid accumulation in adipocytes, consistent with effects on lipogenesis [23,25]. It has been suggested that the ratio of α and β isoforms of human LPIN1 is regulated by the splicing factor SFRS10 (Splicing factor, Arginine/Serine-Rich 10), with decreased levels of SFRS10 in obese subjects leading to greater propensity for production of the lipogenic lipin 1β isoform [26]. However, the association between SFRS10 levels and LPIN1 isoform mRNA ratio was not replicated in a subsequent study [27].
Attesting to the fundamental role of lipin protein activity in biology, orthologous lipin proteins are present in humans and other mammals, as well as plants, invertebrates and yeast [12]. The identification of lipins as PAP enzymes resulted from ground-breaking work by Carman and colleagues, who purified and sequenced yeast phosphatidic acid phosphohydrolase (Pah1) activity, and revealed it to be the same sequence previously identified as the yeast lipin ortholog [13]. Additional studies of yeast Pah1 by the Carman and Siniossoglou groups have provided detailed mechanistic insights into Pah1 regulation and degradation [28–37]. Many excellent reviews on Pah1 and yeast glycerolipid synthesis have been presented [38–41]. Here we focus primarily on the function of mammalian lipin proteins and the current understanding of their roles in physiology and pathophysiology.
2.1 Lipin Phosphatidic Acid Phosphatase Activity
All three mammalian lipin proteins have PAP enzyme activity in vitro, with specific activity for lipin 1 somewhat higher than that for lipin 2 and lipin 3 [14]. Lipin PAP activity requires Mg2+, and is inhibited by N-ethylmaleimide [14,42]. Lipin enzymatic activity is specific for phosphatidate, with no detectable activity against a number of other lipid phosphates that have been tested [14].
The acyltransferase enzymes of the glycerol 3-phosphate pathway all possess a membrane-spanning domain and localize to the ER or mitochondrial membranes [11]. Unlike these proteins, lipin proteins do not possess a membrane spanning sequence, and lipin proteins may reside in the cytosol and associate with membranes in a transient manner. As discussed in subsequent sections, lipin proteins can also move to the nucleus and other cellular organelles. Prior to the molecular identification of lipins, PAP activity was known to re-locate from the cytosol to the ER/microsomal membranes in the presence of fatty acids [43,44]; this translocation has been confirmed for lipin proteins [45,46].
It should be noted that another group of lipid phosphate phosphatase/transferase enzymes (known as LPPs or LPTs), which are structurally unrelated to lipin proteins, can also dephosphorylate phosphatidate [16,47]. LPPs differ from lipin PAP enzymes in their lack of requirement for Mg2+ and failure to be inhibited by N-ethylmaleimide [48]. By contrast to lipin PAP enzymes, LPP activity is not specific for PA and can utilize a number of lipid phosphate substrates (e.g., lysophosphatidate, diacylglycerol pyrophosphate, sphingosine and ceramide phosphates) [49]. Furthermore, LPPs possess 6 transmembrane domains and reside at the outer surface of the plasma membrane, or the luminal surface of endomembrane compartments such as Golgi or ER membranes [50]. It is thought that a major portion of LPP activity may be carried out at the cell surface, where they hydrolyze extracellular lysophosphatidic acid or sphingosine-1-P and the resulting products activate intracellular signaling cascades in neighboring cells [16,50]. This ecto-enzymatic activity of LPPs may have a role in embryogenesis, tissue repair, development of atherosclerosis, and tumor growth and metastasis [51,52]. At intracellular membranes, LPPs have the potential to hydrolyze lipid substrates on the luminal side of ER or Golgi membranes, whereas lipin PAP activity acts at the cytosolic surface of these membranes. The intracellular lipid substrates for LPPs have not been clearly established. Consistent with distinct molecular roles of lipins and LPP enzymes, modulation of LPP or lipin levels in mouse models has distinct physiological effects [53].
2.2 Lipin transcriptional co-regulator activity
In addition to its role as a PAP enzyme in the glycerol 3-phosphate pathway, lipin proteins are implicated in the regulation of gene expression through interactions with nuclear receptors and other DNA-bound transcription factors [54]. The first demonstration of this was the requirement for lipin 1 in mouse hepatocytes for activation of genes regulated by peroxisome proliferator activated receptor α (PPARα) and it coactivator protein, PPARγ coactivator-1α (PCG-1α) by Finck and colleagues [55]. It is clear that lipin 1 also associates with additional transcription factors, including PPARγ, myocyte enhancer factor 2 (MEF2), and the nuclear factor of activated T-cells c4 (NFATc4) to co-activate, or in some cases, co-repress, gene expression [56–58]. Lipin 1 mediated co-repression of MEF2 may modulate inflammatory cytokine signaling in adipocytes [57]. Lipin 2 also has the ability to co-activate PPARα/PGC-1α [46], but it is unclear under what physiological context this is important. The ability of lipin proteins to regulate both triacylglycerol synthesis through PAP activity, and fatty acid oxidation through transcriptional co-activation, suggests a unique mechanism for coordination of fat synthesis and turnover [55,59].
3. Regulation of lipin membrane localization
Cytosolic lipin proteins redistribute to multiple organelle membranes in response to stimuli such as fatty acids, or in the presence of negative charges such as conferred by the lipin substrate, PA (Figure 2) [15,43]. The dichotomous role for lipin proteins in glycerolipid biosynthesis and gene expression may be modulated, in part, by protein subcellular localization. Translocation of lipin to membranes should favor activation of lipin PAP activity for lipid synthesis, whereas localization to the nucleus may be necessary for its role as a transcriptional co-regulator. The association of lipin proteins with ER and nuclear membranes is conserved from mammals to invertebrates such as C. elegans and Drosophila. Similar to mammalian lipins, the C. elegans lipin ortholog is enriched in the ER and nuclear envelope membrane in cells of the embryo [60,61]. In Drosophila, a proportion of lipin localizes to ER and nuclear envelope membranes in the fat body and ventral ganglion cells [62]. This has provided impetus for identifying mechanisms that regulate lipin protein localization to membranes. Below we discuss some of the known determinants of lipin membrane association.
Figure 2. Lipin localization to diverse organelles through association with membrane lipids.
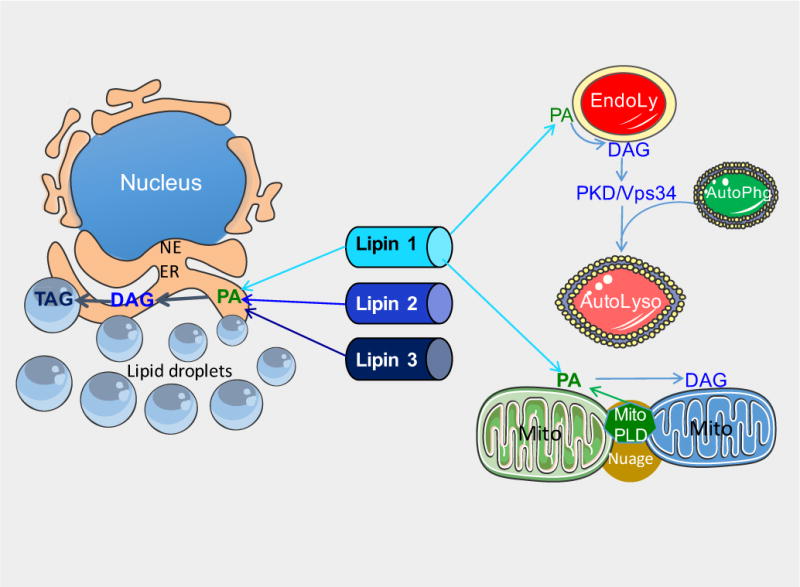
Lipin proteins associate with membranes of several organelles, including the ER, endolysosomes, mitochondria, and lipid droplets. See text for description of mechanisms. EndoLy: late endosome or lysosome; AutoLyso: autolysosome; AutoPhg: autophagososme; ER: endoplasmic reticulum; Mito: mitochondria; NE: nuclear envelope; Nuage: Nuage pi-body; PA: phosphatidic acid; DAG: diacylglycerol; TAG: triacylglycerol; PDK/Vps34, protein kinase D/phosphatidylinositol-3 kinase; PLD, phospholipase D; MitoPLD, a phospholipase D family member found on mitochondria.
3.1 PA-directed recruitment of lipin to membranes
Given that the ER is the primary site for synthesis of neutral lipids and membrane phospholipids, most studies of lipin membrane association have used ER membranes or artificial membranes. Collective evidence suggests that the biochemical basis of lipin protein– membrane association is the formation of a hydrogen bond between a polybasic domain in the lipin proteins and the PA phosphate group [63,64]. Lipin 1 and lipin 2 will physically associate with liposomes containing phosphatidylethanolamine and phosphatidylcholine in the absence of PA. However, the presence of PA greatly enhances the association efficiency [64]. It has been postulated that that cytosol-to-membrane redistribution of lipins may involve a two-step process. In the first step (known as the “bulk step”), soluble lipin enzyme associates with the membrane surface. This is followed by movement of the enzyme in the plane of the membrane to sites specifically containing PA substrate (known as the “surface step”) [64,65]. Little is known about how lipin proteins translocate to membrane surfaces in the “bulk step.” However, lipins (as well as other PA-binding proteins) may bind the PA substrate via hydrogen bonding between positively charged amino acids in lipins and the deprotonated form of PA [63,64,66]. Within the membrane, PA is an anionic lipid with a negatively charged phosphomonoester head group that can form hydrogen bonds with positive group donors, such as the primary amine group of phosphatidylethanolamine or other amphiphilic amines, in basic conditions. The formation of a hydrogen bond will deprotonate PA and create a di-anionic charged phosphate group that leads to increased electrostatic attraction.
It has been shown that a conserved polybasic motif in the N-terminal lipin domain is required for binding to PA-containing membranes [63]. In the three lipin paralogs, this motif is highly enriched in positively charged amino acids through a stretch of 9 lysine and arginine residues (amino acid residues 153–161 in mouse lipin 1). The hydrogen bond between lipin protein and PA serves both to enhance lipin–membrane association and to control PAP activity. Mutation of the polybasic motif in lipin 1 leads to reduced PA-mediated membrane association, consistent with lipin-membrane association occurring through an electrostatic interaction with negative charges in the membrane [63,64,67]. Conversely, the addition to liposomes of positive group donors such as PE or amphiphilic amines, and increasing the pH from 7 to 8, significantly enhances lipin 1 and lipin 2 membrane association and PAP activity [64,68]. Lipin 3 membrane interactions have not been experimentally tested, but conservation of the polybasic character of the corresponding region in lipin 3 suggests that similar interactions likely occur.
3.2 Fatty acid-induced lipin membrane translocation
As described above, fatty acids induce PAP enzyme translocation to the ER membrane. More specifically, unsaturated fatty acids, such as oleate, are potent inducers of lipin movement to the ER membrane, but saturated fatty acids are not [43–46,69,70]. It may be that unsaturated fatty acids increase membrane fluidity and PA movement, increasing the opportunity for interaction with the polybasic motif of lipin proteins [3].Fatty acid-induced translocation of lipin is nearly completely inhibited by insulin, which would be released in a post-prandial state [45]. This suggests that the regulation of lipin 1 PAP activity via translocation to the ER membrane is complex and requires integrated signals provided by nutrients and hormones. There appear to be differences in the regulation of the lipin family members, as lipin 1 and lipin 2 are both translocated to the ER in response to oleate, but lipin 2 is not sensitive to insulin [46]. This suggests that additional factors contribute to the regulation of PA flux into neutral lipid storage and membrane phospholipid synthesis to meet cellular demand under different environmental circumstances. Fatty acid type may contribute to the regulation, through effects on the lipin-1 kinase -mTOR, for example. The saturated fatty acid palmitate activates the mTOR complex, whereas unsaturated fatty acids (including oleate and eicosapentaenoic acid) block the activation, which might influence lipin-1 phosphorylation and membrane association [71,72].
3.3 Lipin phosphorylation and membrane association
As initially demonstrated by Harris and colleagues, lipin 1 protein association with membranes is influenced by reversible protein phosphorylation [45]. mTOR mediates insulin-induced lipin 1 phosphorylation at multiple sites leading to cytosolic localization and reduced enzyme activity [45,64,68,73]. Inhibition of mTOR favors lipin 1 membrane association and increases its PAP activity [45,64,68,74]. Although studies suggest that lipin 1 phosphorylation might influence PA binding via the polybasic motif, neither of the two well-established mTOR-mediated phosphorylation sites on lipin 1 (Ser106 and Ser472) is located within the polybasic motif, suggesting that phosphorylation alters the protein conformation or its interaction with other proteins [64,74]. Many kinases that phosphorylate yeast Pah1 have been identified, but the phosphorylation sites in yeast and mammalian lipin proteins are not directly comparable, such that the sites, the kinases, and the physiological consequences will require specific investigation in mammalian systems. Lipin 2 is also a highly phosphorylated protein, but unlike lipin 1, phosphorylation is not responsive to insulin or mTOR [68]. At present, little is known about the phosphorylation of lipin 3 and the effect of its subcellular translocation.
There is a complex relationship between mTOR, lipin 1 and PA: mTOR phosphorylates and regulates lipin 1, while the PA substrate of lipin 1 may itself regulate mTOR. Specifically, the PA generated from both the de novo synthesis pathway and hydrolysis by phospholipase D1 may bind to the FRB (FK506 binding protein-12-rapamycin binding) domain of mTOR protein to stabilize mTORC1 and mTORC2 complexes [75–77]. Interestingly, inhibition of de novo PA synthesis by AGPAT2 results in mTOR deactivation and lipin 1 subcellular redistribution in human pancreatic cancer cells [78]. On the other hand, exogenously supplied PA restores mTOR activation upon unsaturated fatty acid loading in two types of KRas-driven cancer cell lines [77]. These results suggest that PA dependence of mTOR activation might tune down lipin 1 PAP activity in rapidly proliferating cancer cells, which could promote PA flux into phospholipid synthesis for membrane biogenesis rather than neutral lipid storage. Thus, the interplay between mTOR and lipin 1 enzyme function appears to be more complex than simply kinase and phosphorylation substrate.
Lipin 1 protein phosphorylation is regulated by a phosphatase complex that was originally identified in yeast as Nem1 and Spo1 proteins, which were required to maintain formation of a spherical nucleus and for yeast sporulation [32]. The mammalian counterparts of these proteins are CTDNEP1 (formerly “dullard”) and NEP1-R1, which represent the catalytic and regulatory units, respectively, of the phosphatase complex [79,80]. The CTDNEP1/NEP1-R1 complex dephosphorylates lipin 1, promotes its association with the ER/nuclear envelope membrane, and stimulates its PAP activity in metazoan species from C. elegans to mammals [60,79,80]. A combination of three-dimensional structure analysis and in vitro steady-state kinetic analysis of CTDNEP1 showed that the phosphatase prefers to act at Ser106, one of the target sites for mTOR complex 1 phosphorylation [81]. However, to date there is no direct in vivo evidence that CTDNEP1/NEP1-R1 phosphatase activity counteracts the mTOR-mediated lipin 1 phosphorylation in mammalian cells. It remains unclear whether the dephosphorylation of lipin by CTDNEP1/NEP1-R1 affects the hydrogen bond formation between lipin and PA.
3.4 Lipin protein interactions
Lipin 1 can form homo-dimers, as well as heterodimers with lipin 2 or lipin 3 [82,83]. Despite the formation of oligomers, it appears that the individual lipin molecules present within oligomers act independently as phosphatases, rather than forming an active site through oligomerization [82]. The formation of lipin oligomers could influence lipin protein subcellular localization. This was demonstrated in a proof-of-principle experiment by fusing lipin 1 to a CAAX motif, which promotes membrane association through isoprenylation. This led to tethering of the tagged lipin 1 to membranes and the recruitment of other lipin molecules to the membrane site by interaction with the CAAX-tagged lipin 1 molecule [82]. These studies suggest that lipin proteins themselves may influence the localization of other lipin proteins within the cell.
Additional evidence for a role of lipin heterooligomers in protein localization comes from studies regarding the relationship between lipin 1 and lipin 3. Studies of mice with combined lipin 1 and lipin 3 deficiency revealed a cooperative role for lipin 1 and lipin 3 in adipose tissue development and triglyceride levels [83]. Investigation of the mechanism for lipin 1/lipin 3 cooperation in adipose tissue revealed that the combined presence of lipin 1 and lipin 3 influences their protein levels and subcellular localization of each. When expressed separately in HEK293 cells, lipin 1 and lipin 3 localized predominantly in the cytoplasm. When expressed in combination, a substantial amount of lipin 1 translocated from the cytoplasm to the peri-nuclear region, and lipin 3 appeared in the peri-nuclear region and in the nucleoplasm, in addition to the cytoplasm [83]. The co-expression of lipin 1 and lipin 3 from heterologous promoters that are not responsive to normal regulators of lipin gene expression led to increased lipin 1 protein and reduced lipin 3 protein levels [83]. This suggests that interaction of lipin 1 and lipin 3 influences their subcellular localization as well as protein concentration, the latter apparently occurring through a post-translational mechanism. Lipin 1 and lipin 2 also have been shown to reciprocally influence the levels of one another in cell systems and mouse tissues [84,85].
4. Lipin association with diverse organelles
In addition to the ER membrane, lipin proteins may associate with the nucleus, lipid droplets, endolysosomes, and mitochondrial membranes [45,46,68,80,86,87]. Lipin localization to these diverse membranes is most easily conceptualized when one considers that many of these organelles form a network throughout the cytoplasm with a continuous lumen. For example, subdomains of the ER include the peripheral ER, the interconnected tubular network, and the nuclear envelope [88]. Furthermore, the ER has numerous membrane contact sites at the Golgi, mitochondria, peroxisomes, endosomes, lipid droplets, and the plasma membrane [41,88]. An important goal in the study of lipin biology is to understand the regulation of lipin subcellular localization to these distinct membrane and organelle sites, and the cellular and physiological role of lipins at each site. Below we discuss some of the current information regarding lipin protein association with organelles beyond the ER membrane and nucleus (Figure 2).
4.1 Lipin 1 association with lipid droplets
As dynamic cytoplasmic organelles, lipid droplets (LDs) act as a reservoir for neutral lipids such as triacylglycerols and sterol esters, which serve as sources of energy and provide building blocks for membrane growth [89]. Lipid droplets are believed to initially form in specific regions of the ER where enzymes of the glycerol 3-phosphate pathway utilize fatty acids to synthesize triacylglycerol or sterol esters. LDs associate with specific proteins, such as members of the perilipin family. LDs may bud from the ER to form dispersed cytosolic LDs, or remain in contact with the ER to grow into large LDs through the association of glycerol 3-phosphate pathway enzymes such as GPAT4 and DGAT2, which migrate from the ER to LDs through ER-LD contact sites [90,91].
A comprehensive quantitative proteomics analysis in Drosophila S2 cells showed that at least one isoenzyme for each step of the triacylglycerol synthesis pathway (including GPAT4, AGPAT3, DGAT2, and the Drosophila lipin ortholog) is associated with the LD fraction following oleate treatment [90]. In human macrophages, lipin 1 was shown to associate with the surface of LDs upon stimulation with a variety of compounds, including oleic acid, calcium ionophore, phorbol acids, and lipopolysaccharide [92]. A brain-specific lipin 1 splice variant, lipin 1γ, translocates to LDs in response to oleate loading when expressed in COS-7 cells [24]. The source of PA on LDs that could recruit lipin enzymes to the LD surface is not known. One possibility is that lipin associates with PA that is generated through the action of LD-associated AGPAT3.
Some evidence points to a potential role of lipin 1 in LD biology through an interaction with seipin, an ER protein that has a role in LD biogenesis. Seipin has been implicated in the process of ER surveillance to identify nascent LDs and promote their maturation [91]. Deficiency of seipin (encoded by the BSCL2 gene) causes lipodystrophy in humans and mice [93–95], as does lipin 1 deficiency in the mouse [12,96,97]. Seipin knockdown leads to reduced lipin 1 binding to the ER membrane [98]. Physical interactions between seipin and lipin 1 proteins have been demonstrated in cell lines overexpressing these proteins, and this interaction was inducible by insulin [98]. Interestingly, seipin dodecamers were also shown to bind to AGPAT2 [99], which catalyzes the step preceding lipin action in the glycerol 3-phosphate pathway, and AGPAT2 deficiency also causes human lipodystrophy [100]. The lipin 1-seipin-AGPAT2 interactions are provocative and may provide a mechanism for channeling PA (the product of the AGPAT2 enzymatic reaction) to lipin 1 (which utilizes PA as a substrate). However, these findings require further investigation because the lipin 1-seipin-AGPAT2 interaction studies have all employed overexpression of epitope-tagged proteins in cell lines [98]. Verification of these interactions with endogenous proteins awaits the availability of specific antibodies against seipin and AGPAT2.
4.2 Lipin 1 association with mitochondria
As described above, the PA-directed recruitment mechanism plays a central role in lipin membrane association. This mechanism may also underlie lipin 1 translocation to mitochondrial and plasma membranes. The mitochondrial-resident phospholipase D (MitoPLD) hydrolyzes cardiolipin in the membrane of mitochondria to generate PA (Figure 2). The overexpression of MitoPLD serves to recruit lipin 1 (specifically the lipin 1β splice variant) to the outer mitochondrial membrane [101]. Unexpectedly, a central domain of lipin 1 (amino acid residues 430 to 570), which contains only a few dispersed basic amino acids, was required for recruitment of lipin 1β to the mitochondrial membrane in response to MitoPLD overexpression [101]. In addition, the brain-specific splice variant, lipin 1γ, was shown to translocate from mitochondria/mitochondria-associated membranes to LDs in response to oleate loading [24]. The role of lipin proteins at the mitochondrial membrane merits additional attention, both at the molecular level and in terms of corresponding physiological effects.
4.3 Lipin 1 association with endolysosomes/phagosomes
Lipin 1 activity at the endolysosomal membrane appears to play a role in autophagy by promoting the fusion of autophagosomes with lysosomes to form mature autolysosomes that are proficient for autophagic degradation [87]. As with the other membrane sites discussed here, the surface of late endosomes/lysosomes is known to contain PA, likely generated by phospholipase D1 activity (Figure 2) [102,103]. Recent studies in mouse muscle and fibroblasts revealed that lipin 1 translocates to the surface of endolysosomes upon mTOR inhibition [87]. The resulting PAP activity at the endolysosomal membrane serves to produce diacylglycerol and activate a protein kinase cascade that leads to the fusion of lysosome and autophagosome membranes. The lack of lipin 1 prevents the fusion step in the assembly of mature autolysosomes and leads to defective autophagy [87], which may contribute to lipin 1-related myopathy that occurs in lipin 1-deficient humans and mice (see section 5).
4.4 Lipin nuclear localization
Lipin localization to organelles such as lipid droplets, mitochondria, and endolysosomes appears to involve lipin interactions with PA on associated membranes. Additional factors influence lipin protein localization to the nucleus. As determined by studies of lipin 1, nuclear localization requires the same polybasic motif that is required for binding to PA-containing membranes [104]. Features in addition to the polybasic motif also appear to influence lipin 1 protein subcellular localization, since the lipin 1α and lipin 1β splice variants, which both contain the polybasic motif, exhibit different propensities for nuclear and cytoplasmic localization [23]. Post-translational modification of lipin 1 has also been shown to influence nuclear localization. Sumoylation of two independent lysine residues on lipin 1 leads to nuclear localization in cultured neuronal cells [58]. It is unknown whether sumoylation regulates lipin 1 nuclear localization in additional cell types. In neuronal cells, mutation of the sumoylation sites prevents lipin 1 nuclear localization, raising the possibility that this modification may regulate lipin 1 activity in transcriptional co-regulation versus lipid biosynthesis. The sumoylation sites in lipin 1 are not conserved in lipin 2 or lipin 3, and it does not appear that these become sumoylated [58].
5. Lipin gene mutations and disease
As described in the preceding sections, a great deal of work has been done to define the biochemical and cellular functions of the lipin proteins. Another key area is the identification of the physiological functions of each lipin family member. All three mammalian family members have the same enzymatic activity and substrate specificity, albeit with somewhat different specific activities [14]. Lipin 1 and lipin 2 also exhibit coactivator/corepressor activity in cellular assays (lipin 3 is likely to exhibit similar activity based on sequence conservation) [46,55]. This raises questions about whether the three lipin proteins each have specific physiological roles, and if so, what they are. Valuable clues have been provided by the study of humans and mouse strains that carry rare lipin mutations, as well as mouse models with engineered mutations in lipin genes.
As described earlier, a spontaneous mutation in the mouse Lpin1 gene produced a severe disease phenotype, indicating the physiological significance and non-redundancy of lipin 1 protein function. Rare mutations in human LPIN1 or LPIN2 genes have also been identified and found to cause severe, but substantially different, disease phenotypes. Individuals that carry bi-allelic pathogenic mutations in LPIN1 present with severe, potentially life-threatening rhabdomyolysis in childhood [105,106]. By contrast, individuals with bi-allelic pathogenic LPIN2 mutations develop an autoinflammatory syndrome (Majeed syndrome) characterized by recurrent fever, bone lesions, anemia and inflammatory skin lesions [107,108]. The disparate symptoms associated with lipin-1 or lipin-2 dysfunction/deficiency indicate that these lipin proteins each have unique roles in vivo. At present, it is unknown whether LPIN3 mutations cause disease. Large-scale exome sequencing in humans has identified numerous predicted pathogenic LPIN3 mutations within the human population [109]. However, from these sequence data it is unclear whether there are individuals with pathogenic mutations in both LPIN3 alleles, and if so, whether they are associated with disease.
Given the similar molecular activities of lipin 1 and lipin 2, what dictates the distinct disease pathologies? One contributor may be the tissue distribution of the two lipins. Recent high-throughput RNA sequence data for 50 human tissues from hundreds of individuals [110] allows the comparison of absolute expression levels of the three human lipin genes across tissues. As shown in Figure 3, mRNA counts reveal that LPIN1 is the only lipin gene with significant expression in human skeletal muscle, which corresponds with the severe muscle pathology that characterizes human lipin 1 deficiency. As with humans, mice have very high Lpin1 expression levels in muscle, as well as in adipose tissue and testes [5]. As in humans, Lpin1 deficiency in the mouse is associated with muscle disease [87], but also with adipose tissue abnormalities and reproductive phenotypes, consistent with prominent expression in adipose tissue and testes [12,96,97]. Mouse and human lipin 1 are both expressed prominently in nerve tissue, and lipin-1-deficient mice exhibit abnormalities in peripheral nerve [111,112], but neurological symptoms have not been reported in lipin 1-deficient humans.
Figure 3. Lipin mRNA expression levels in human tissues.
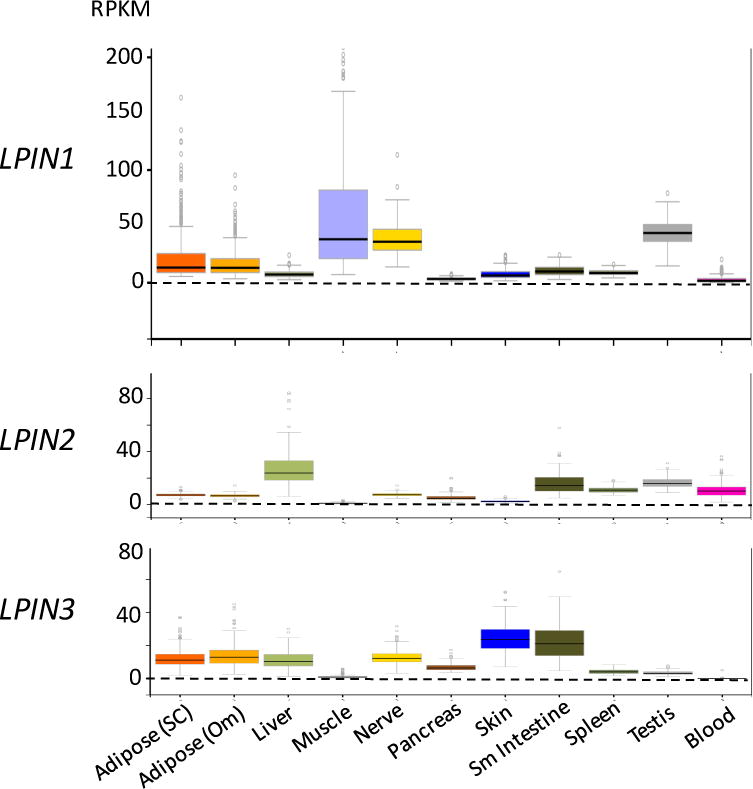
Data are from RNA-seq studies performed as part of the Genotype-Tissue expression Project [110]. RPKM, reads per kilobase of transcript per million mapped reads. Adipose (SC), subcutaneous adipose tissue; Adipose (om), omental adipose tissue; Sm intestine, small intestine; blood refers to white blood cells.
In contrast to LPIN1, the tissue expression pattern for LPIN2 does not provide clear associations with the disease phenotype. LPIN2 mRNA is most abundant in liver and intestine, tissues that are not known to be affected in Majeed syndrome. However, LPIN2 is also expressed in blood (white blood cells) in humans (Figure 3) and in mice [46], which likely relates to involvement of lymphocytes and/or monocytes in the autoinflammatory symptoms of Majeed syndrome [113,114]. The lack of striking liver and intestinal phenotypes in Majeed syndrome may relate to the expression of LPIN1 and/or LPIN3 in those tissues to compensate for loss of lipin 2. In the following sections, we discuss some of the key roles of lipin proteins in specific tissues as revealed through the study of human disease and mouse disease models.
5.1 Lipin 1 in muscle and myocytes
The identification of LPIN1 mutations in children with severe rhabdomyolysis was a revelatory discovery in lipin biology, which focused attention on a key role for lipin 1 in muscle [105,106,115]. Rhabdomyolysis is the destruction of skeletal muscle fibers with subsequent release of cellular contents, including high concentrations of myoglobin. In addition to the primary damage to muscle, the necessity to clear high levels of myoglobin from the circulation can lead to kidney failure. LPIN1-related rhabdomyolysis is a recessive condition, requiring pathogenic mutations in both alleles of the LPIN1 gene. Analysis of 21 individuals with childhood rhabdomyolysis revealed bi-alleleic LPIN1 mutations in 17 (59%), suggesting that mutations in the LPIN1 gene may account for a substantial proportion of childhood metabolic rhabdomyolysis cases [106]. In affected individuals, there is typically no evidence of the disease until a severe episode of muscle pain, often occurring at 2–5 years of age [105,116]. It is not fully understood how episodes of rhabdomyolysis are triggered, but they may be precipitated by fever, fasting, or anesthesia [105,116]. Sadly, up to one-third of affected individuals die during a childhood bout [106,115]. Fortunately, in recent years, the clinical awareness of LPIN1 mutation and disease course has grown, leading to better management of cases when they appear in hospital emergency rooms [117].
The majority of pathogenic LPIN1 mutations are null alleles that produce no protein product, resulting from nonsense, frame-shift, or deletion mutations [105,106,118,119]. Consistent with this, primary myoblasts from patients exhibit dramatically decreased PAP1 activity [120]. A few pathogenic missense LPIN1 alleles have been identified, and have been valuable to dissect the functional deficiency that underlies the observed muscle symptoms. For example, lipin 1 protein bearing the pathogenic missense mutation, Arg725His, retains co-activator function but lacks PAP activity, confirming that the pathology of lipin-1 rhabdomyolysis is a result of PAP deficiency [119]. Another missense mutation (Glu769Gly) was associated with severe myopathy in a heterozygous carrier when treated with statin drugs [105]. This raises the interesting possibility that LPIN1 mutation may be a risk factor for statin-related myopathy, which occurs in a proportion of individuals taking statins to combat hypercholesterolemia [121].
How does lipin 1 deficiency cause rhabdomyolysis? As mentioned earlier, lipin 1 is the only lipin family member with appreciable expression in skeletal muscle of humans and mice. This may explain why lipin 1 deficiency is most profoundly experienced in skeletal muscle, where other lipin family members are not available to compensate. Involvement of cardiac muscle has been noted in a minority of lipin 1–deficient patients, but this is not a typical finding [106,115]. Histological analysis of muscle biopsies isolated from lipin 1–deficient patients, as well as lipin 1–deficient mice, revealed an unexpected accumulation of neutral lipid droplets [87,120,122]. The neutral lipids accumulating in lipin 1–deficient muscle were determined in mice to be cholesteryl esters [87]. Cholesteryl ester accumulation likely occurs in response to impaired triacylglycerol synthesis in the absence of lipin 1, as a compensatory mechanism designed to minimize the accumulation of free fatty acids, which would normally be incorporated into triacylglycerols.
Further analysis of lipid content by quantitative mass spectrometry was performed in two human patients with LPIN1 mutations and in lipin 1-deficient mice [87,105]. The changes observed in phospholipid content is surprisingly mild, without significant changes in major membrane phospholipid classes, although some alterations in minor phospholipid species were observed. One of the human patients analyzed had elevated lyso-phospholipids, but the other did not [105]. In lipin 1-deficient mice, total phospholipids were not elevated, but the levels of ceramides and ether-phosphatidylcholine were increased, which my represent an adaptive mechanism for free fatty acid esterification in the absence of triacylglycerol formation [87]. Importantly, a common finding in mouse and human lipin 1-deficient muscle is an accumulation of PA [87,105], which may influence signaling pathways and processes such as autophagy (discussed below). Studies with primary myoblasts from lipin 1-deficient humans have revealed a possible interplay between altered lipid metabolism and inflammation as a contributing factor to lipin 1-associated rhabdomyolysis. Abnormalities in gene expression in human lipin 1-deficient myoblasts were observed when treated with pro-inflammatory cytokines (IL-1β and TNFα) that may influence fatty acid synthesis/oxidation [120].
The fld mouse model with lipin 1 deficiency provides a valuable model to elucidate the functions of lipin 1 in muscle in normo- and pathophysiology. The original studies of the fld mouse noted effects of the mutation in liver, nerve and adipose tissue [96,97,111]. No muscle defects were in evidence. Several years later, the identification of lipin 1–deficient humans provided an explanation: the clinical symptoms associated with lipin 1 deficiency (muscle pain, elevated creatine kinase levels due to muscle breakdown) are episodic, triggered by metabolic stress [105,116]. Furthermore, unlike humans, mice cannot easily communicate to their handlers that they are experiencing muscle pain, and so it was not evident that they are susceptible to muscle symptoms.
To make use of the fld model to study the basis for rhabdomyolysis in lipin 1-deficiency, it was necessary to identify conditions that could provoke ‘episodes’ of impaired muscle metabolism. Subjecting fld mice to a regimen of fasting for 16 hr followed by refeeding for 4 hr caused elevations in circulating creatine kinase levels, signifying muscle damage [87]. These conditions were used to characterize the effects of lipin 1 deficiency on muscle pathology, lipid profile, and mitochondrial function. It became clear that with the addition of fasting/refeeding as a metabolic stressor, the lipin 1-deficient mouse model recapitulates the changes in muscle lipid content observed in human muscle, including accumulation of PA and cholesteryl esters [87]. Muscle from fld mice also exhibited evidence of myocyte turnover, with > 10% of myocytes representing newly differentiated cells, presumably replacing dead myocytes. Lipin 1 deficient mice also have impaired muscle regeneration in response to injury [123], which could contribute to the severe muscle pathology in mice and humans lacking lipin 1.
In addition to muscle histopathlogical changes, lipin 1-deficient muscle has altered energy metabolism, including reduced mitochondrial respiratory reserve capacity [87]. Electron microscopy revealed an accumulation of aberrant mitochondria and presence of autophagosome structures. The latter are intermediates in the process of macroautophagy, and are typically short-lived. Their detection in the fld muscle, together with accumulation of potentially damaged mitochondria, suggested a defect in autophagic clearance. Interestingly, Drosophila melanogaster made deficient for the single lipin ortholog in the fruitfly fat body exhibit similar structural abnormalities of mitochondria and autophagososmes [124]. These observations led to the identification of a role for mammalian lipin 1 in autophagosome maturation. Lipin 1 conversion of PA to DAG on the surface of lysosomes activates a protein kinase cascade that leads to production of phosphatidylinositol 3-phosphate and fusion with autophagosomes to mature autolysosomes [87]. The impaired formation of autolysosomes in lipin 1-deficient muscle may contribute to dysfunction through impaired turnover of damaged organelles, particularly mitochondria, and subsequent changes in lipid and energy homeostasis. It is likely that lipin 1 deficiency also affects function of additional organelles in muscle, potentially through changes in membrane lipid composition, to bring about the severe consequences of lipin 1 deficiency. This merits further investigation.
5.2 Lipins in adipocytes
A hallmark feature of adipose tissue is the storage of triglyceride droplets, which can be lipolyzed during fasting and other conditions to supply fatty acids locally and to distant tissues. Consistent with an important role for lipin 1 and the glycerol 3-phosphate pathway in adipocyte triglyceride synthesis, the fld mouse exhibits generalized lipodystrophy, with 90% reduction in the mass of white adipose tissue depots (including epididymal/perioovarian fat, inguinal fat, retroperitoneal fat, and omental fat) [97]. Lipid profiles show a dramatic reduction in triacylglycerol levels, with an accumulation of PA [125]. The cells isolated from the primordial fat depots in fld mice exhibit dramatically reduced expression of PPARγ and other adipogenic transcription factors that are required for differentiation [126]. Similarly, stromal vascular preadipocytes and mouse embryonic fibroblasts isolated from fld mice fail to induce an adipogenic transcription program that occurs as the first steps in differentiation [125,126]. These observations point to a role for lipin 1 in the early stages of adipocyte differentiation that precede triglyceride synthesis and storage. One contributor appears to be the elevated levels of PA that accumulate in fld pre-adipocytes, which activate ERK signaling and inhibit adipocyte differentiation [125]. Differentiation of fld preadipocytes can be rescued by providing constitutively expressed PPARγ or by ERK inhibitor [125,126]. It can also be rescued by forced expression of lipin 1, lipin 2, or lipin 3, but not by introduction of lipin 1 protein that possesses coactivator function without PAP enzyme activity [125]. These results strongly suggest that lipin PAP activity is required for the early stages of adipocyte differentiation, potentially as a regulator of PA signaling events.
Beyond the fld mouse model, an engineered Lpin1 allele that leads to lipin 1 deficiency specifically in adipose tissue confirms the requirement for lipin 1 PAP activity for normal adipose tissue development in vivo. Finck and colleagues generated a “separation-of-function” mutant Lpin1 allele that encodes a lipin 1 protein lacking PAP activity but retaining coactivator function [127]. The resulting mice exhibit lipodystrophy similar to that in the fld mouse (including reduced glyceride content and increased PA levels in adipose tissue) indicating that this phenotype is associated with loss of lipin 1 PAP activity. Additionally, these studies revealed an unexpected role for lipin 1 in regulating adipose tissue triglyceride lipolysis. This occurs through lipin 1 modulation of PA levels to influence a phosphodiesterase 4-cAMP-protein kinase A signaling axis to control lipolysis [127].
In contrast to effects of lipin 1 deficiency, increased lipin 1 expression levels in the mouse via an adipocyte-directed transgene lead to obesity, with increased size and triglyceride content in adipocytes [23,25]. Interestingly, the obese adipose-specific lipin 1 transgenic mice have improved glucose and insulin homeostasis compared to their wild-type counterparts on both chow and high fat diets [23]. These findings suggest that increased capacity for triglyceride synthesis within adipocytes via lipin 1 is metabolically favorable, perhaps by partitioning fatty acids into adipose tissue and away from ectopic storage in other tissues. In humans, lipin 1 expression levels in adipose tissue are positively correlated with insulin sensitivity in both lean young adults and in obese subjects [128,129]. Lipin 1 expression levels increase both vivo and in vitro in response to thiazolidinedione drugs, which are PPARγ agonists [129,130]. In cultured adipocytes, lipin 1 was required for PPARγ/thiazolidinedione stimulated adipocyte differentiation [130]. The induction of lipin 1 by thiazolidinedione drugs may be a mechanism for increased adipocyte differentiation observed with activation of PPARγ [130], as well as a mediator of the improved insulin sensitivity that occurs on a background of increased adiposity with these drugs [131].
Lipin 2 and lipin 3 also influence lipid metabolism in mouse adipocytes. In the mouse 3T3-L1 adipocyte cell line, lipin 1 and lipin 2 are reciprocally regulated during adipocyte differentiation [85]. Lipin 2 protein is detectable in preadipocytes and diminishes in mature adipocytes, while lipin 1 protein is present primarily after initiation of differentiation and continuing to highest levels in mature adipocytes [85]. Knockdown of both lipin 1 and lipin 2 after the induction of adipocyte differentiation leads to increased PA levels, but does not alter total triglyceride content of the mature adipocytes. Instead, lack of lipin 1 and lipin 2 in mature 3T3-L1 adipocytes leads to fragmented lipid droplets of reduced size and increased number [85]. The fragmented lipid droplet phenotype can be rescued by introduction of lipin 1 that possesses PAP activity, but not by lipin 1 with coactivator function that lacks PAP activity. Interestingly, lipin 2-deficient mice do not exhibit lipodystrophy or an obvious adipose tissue phenotype [84], suggesting that in vivo compensation by lipin 1 is adequate for normal adipose tissue. Mouse lipin 3 is also induced during differentiation of 3T3-L1 cells, and has a role in adipose tissue PAP activity and lipid accumulation in combination with lipin 1 [83]. Stromal vascular cells isolated from lipin 3-deficient mice exhibit impaired differentiation in vitro, but lipin 3 deficiency in vivo does not cause lipodystrophy, likely due to compensation by lipin 1. Consistent with this, the loss of lipin 1 and lipin 3 in combination cause even more severe lipodystrophy than lipin 1 deficiency [83]. Thus, the three mammalian lipins each have a demonstrable effect on lipid metabolism in mouse adipocytes, but lipin 1 appears to be the dominant PAP enzyme for triglyceride accumulation in mouse adipose tissue in vivo.
As summarized above, a role for lipin 1 in mouse adipose tissue development and lipid accumulation is clear. It was therefore unexpected when human lipin 1-deficient subjects were found not to have lipodystrophy [105]. LPIN1, LPIN2 and LPIN3 are each expressed to some degree in human primary adipocytes [83] and in the human SGBS cell line (isolated from an individual with Simpson–Golabi–Behmel syndrome) [132]. LPIN1 knockdown in SGBS cells led to a 95% reduction in PAP activity, but TAG accumulated to the same level as wild-type cells [132]. LPIN2 or LPIN3 knockdown led to more modest (10–25%) reductions in PAP activity, with maintenance of normal (LPIN2) or slightly reduced (LPIN3) TAG levels. Surprisingly, combinatorial silencing of all three human lipins led to only a 40% reduction in cellular TAG accumulation, indicating that after dramatic reduction in cellular lipin content, alternative mechanisms act to promote TAG accumulation. However, its should be noted that the knockdown of the lipin proteins in SGBS cells was not complete; triple knockdown cells still retained lipin protein levels ranging from about 5% (for LPIN1) to 50% (for LPIN2) of levels in wild-type cells. Two additional mechanisms may compensate for reduced lipin levels as well. These are an approximately 50% reduction in lipolysis of stored TAG, as determined by glycerol release, and increased lipid phosphate phosphatase (LPP) activity in SGBS cells having triple lipin knockdown [132]. It will be interesting to determine whether similar compensatory mechanisms occur in adipose tissue of lipin 1-deficient humans to protect them from lipodystrophy, but cannot protect against other pathologies that occur in lipin 1-deficient muscle, such as altered energy metabolism and myocyte differentiation.
5.3 Lipins and inflammation
As described in an earlier section, lipin 2 deficiency in humans leads to an autoinflammatory disease known as Majeed syndrome, characterized by episodic sterile bone lesions, anemia, and skin inflammation in some patients. Majeed syndrome patients have elevated levels of proinflammatory cytokines. The LPIN2 mutations in Majeed syndrome include frame-shift mutations that lead to premature stop codons and a missense mutation, Ser734Leu [107,108,133]. Lipin 2 has both PAP activity and transcriptional coactivator activity [14,46], but it was unknown whether loss of one or both of these functions causes Majeed syndrome. Analysis of Ser734Leu lipin 2 revealed that this mutant protein has lost the ability to catalyze the PAP reaction, but retains transcriptional coactivator activity, indicating that lipin 2 PAP activity is the critical component in Majeed syndrome [46]. Interestingly, five other lipin 2 missense mutations are associated with psoriasis (Infevers database, https://fmf.igh.cnrs.fr/ISSAID/infevers).
A lipin 2-deficient mouse model was generated to characterize the physiological roles of lipin 2 [84]. These mice exhibited dyserythropoietic anemia similar to that reported in Majeed patients, as well as occasional skin lesions. This mouse model also revealed that loss of lipin 2 activity in liver is compensated by upregulation of lipin 1, and that lipin 2 is critical for maintenance of lipid homeostasis in the cerebellum as mice age. Lipin 2-deficint mice also had reduced trabecular bone density. However, analysis of tibiae via radiography and micro-computed tomography failed to reveal evidence of inflammatory bone lesions that are a hallmark of Majeed syndrome. It is possible that more sensitive techniques are required to observe bone lesions, such as isotope uptake techniques that are used in human diagnoses, or that the episodic nature of the appearance of Majeed symptoms prevented observation of the same effects in the mouse. Better understanding of triggers for severe episodes of bone inflammation may allow induction of similar episodes in the mouse model, which would be valuable for elucidating the molecular mechanisms.
Fortunately, recent studies have begun to elucidate the inflammatory dysregulation that occurs in Majeed syndrome. The treatment of Majeed syndrome patients with anti-inflammatory drugs provided an important clue to the inflammatory mechanism involved. Anti-inflammatory drugs that target tumor necrosis factor α had no effect on Majeed syndrome symptoms, but blockade of interleukin 1β (IL-1β) led to improvement, suggesting that elevated IL-1 β levels play a role in the disease pathogenesis [133]. Indeed, a recent study from Balboa and colleagues has shed light on the relationship between lipin 2 and IL-1β [114]. Lipin 2 knockdown in human macrophages, or lipin 2 deficiency in mouse bone marrow-derived macrophages, led to enhanced IL-1β production in response to activation of the NLRP3 inflammasome, a multiprotein complex that is normally activated by exposure to pathogens and other danger signals, and serves to initiate the innate immune response [134]. This led to the identification of several mechanisms by which lipin 2 influences inflammasome activation. The NLRP3 inflammasome requires two independent signals for activation. The first is a priming signal that occurs via activation of cellular receptors such as the toll like receptors, and leads to IL1β transcription. The second signal may consist of pathogens or cellular stress signals such as ATP, which signal through receptors such as the purinergic receptor, P2X7 [134]. Lipin 2 appears to influence multiple steps in inflammasome activation, including regulation of MAPK signaling to regulate IL-1β production during inflammasome priming, P2X7 sensitization, and processing of the caspase to its active form that ultimately leads to processing of IL-1β from its precursor to mature form [114]. IL-1β overproduction in response to lipin 2 deficiency could be prevented by MAPK inhibitors or by restoration of reduced cholesterol levels in lipin 2-deficient cells to wild-type levels. These studies reveal a novel nexus between lipin 2 and cholesterol metabolism and the innate immune response. The further understanding of this relationship may provide new insights into innate immunity, and provide novel treatment strategies for autoinflammatory diseases.
6. Summary and Future Perspectives
Studies of the multiple lipin family members in human disease and mouse models have led to unifying themes regarding the cellular function of lipin proteins, which in turn, impact their physiological roles. Themes that have arisen from the study of lipin biology include: (1) the ability of lipin proteins to traffic to multiple cellular locations, including membranes of different organelles; (2) the modulation by lipin PAP activity of the concentration of bioactive signaling lipids phosphatidic acid and diacylglycerol at specific membrane sites; (3) a need for lipin proteins in tissues that are not traditionally major sites of triacylglycerol synthesis and storage, such as skeletal muscle and macrophages; and (4) the three mammalian lipin family members each have unique physiological roles, but that they may cooperate with, and/or compensate for, one another. Future studies should take advantage of the growing body of knowledge concerning the biochemical and cellular activities of lipin proteins to inform investigations of lipin physiological function in health and disease.
Acknowledgments
We gratefully acknowledge support from Public Health Services PO1 HL090553 and PO1 HL028481. We thank Lauren Csaki for assistance with Figure 1.
References
- 1.Schmelzer K, Fahy E, Subramaniam S, Dennis EA. The Lipid Maps Initiative in Lipidomics. Methods Enzymol. 2007:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]
- 2.Han X. Lipidomics for studying metabolism. Nat Rev Endocrinol. 2016;12:668–679. doi: 10.1038/nrendo.2016.98. [DOI] [PubMed] [Google Scholar]
- 3.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott SA, Mathews TP, Ivanova PT, Lindsley CW, Brown HA. Chemical modulation of glycerolipid signaling and metabolic pathways. Biochim Biophys Acta - Mol Cell Biol Lipids. 2014;1841:1060–1084. doi: 10.1016/j.bbalip.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Csaki LS, Dwyer JR, Fong LG, Tontonoz P, Young SG, Reue K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog Lipid Res. 2013;52:305–316. doi: 10.1016/j.plipres.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prentki M, Madiraju SRM. Glycerolipid Metabolism and Signaling in Health and Disease. Endocr Rev. 2008;29:647–676. doi: 10.1210/er.2008-0007. [DOI] [PubMed] [Google Scholar]
- 7.Cao G, Konrad RJ, Li SD, Hammond C. Glycerolipid acyltransferases in triglyceride metabolism and energy homeostasis-potential as drug targets. Endocr Metab Immune Disord Drug Targets. 2012;12:197–206. doi: 10.2174/187153012800493459. http://www.ncbi.nlm.nih.gov/pubmed/22385114 (accessed December 29, 2016) [DOI] [PubMed] [Google Scholar]
- 8.KENNEDY EP, SMITH SW, WEISS SB. New synthesis of lecithin in an isolated enzyme system. Nature. 1956;178:594–5. doi: 10.1038/178594a0. http://www.ncbi.nlm.nih.gov/pubmed/13369471 (accessed December 29, 2016) [DOI] [PubMed] [Google Scholar]
- 9.KENNEDY EP. Biosynthesis of phospholipides. Fed Proc. 1957;16:847–53. http://www.ncbi.nlm.nih.gov/pubmed/13480372 (accessed December 29, 2016) [PubMed] [Google Scholar]
- 10.Coleman RA, Mashek DG. Mammalian Triacylglycerol Metabolism: Synthesis, Lipolysis, and Signaling. Chem Rev. 2011;111:6359–6386. doi: 10.1021/cr100404w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296:E1195–E1209. doi: 10.1152/ajpendo.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- 13.Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin Homolog Is a Mg2+-dependent Phosphatidate Phosphatase Enzyme. J Biol Chem. 2006;281:9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three Mammalian Lipins Act as Phosphatidate Phosphatases with Distinct Tissue Expression Patterns. J Biol Chem. 2006;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 15.Reue K, Brindley DN. Thematic Review Series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J Lipid Res. 2008;49:2493–2503. doi: 10.1194/jlr.R800019-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brindley DN, Pilquil C, Sariahmetoglu M, Reue K. Phosphatidate degradation: Phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim Biophys Acta - Mol Cell Biol Lipids. 2009;1791:956–961. doi: 10.1016/j.bbalip.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yen CLE, Nelson DW, Yen MI. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J Lipid Res. 2015;56:489–501. doi: 10.1194/jlr.R052902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Aquila T, Hung Y-H, Carreiro A, Buhman KK. Recent discoveries on absorption of dietary fat: Presence, synthesis, and metabolism of cytoplasmic lipid droplets within enterocytes. Biochim Biophys Acta - Mol Cell Biol Lipids. 2016;1861:730–747. doi: 10.1016/j.bbalip.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phan CT, Tso P. Intestinal lipid absorption and transport. Front Biosci. 2001;6:D299–319. doi: 10.2741/phan. http://www.ncbi.nlm.nih.gov/pubmed/11229876 (accessed December 29, 2016) [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol Endocrinol Metab. 2009;297:E10–8. doi: 10.1152/ajpendo.90949.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson DW, Gao Y, Yen MI, Yen CLE. Intestine-specific Deletion of Acyl-CoA:Monoacylglycerol Acyltransferase (MGAT) 2 Protects Mice from Diet-induced Obesity and Glucose Intolerance. J Biol Chem. 2014;289:17338–17349. doi: 10.1074/jbc.M114.555961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khatun I, Clark RW, Vera NB, Kou K, Erion DM, Coskran T, Bobrowski WF, Okerberg C, Goodwin B. Characterization of a Novel Intestinal Glycerol-3-phosphate Acyltransferase Pathway and Its Role in Lipid Homeostasis. J Biol Chem. 2016;291:2602–15. doi: 10.1074/jbc.M115.683359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Péterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem. 2005;280:32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Zhang J, Qiu W, Han GS, Carman GM, Adeli K. Lipin-1γ isoform is a novel lipid droplet-associated protein highly expressed in the brain. FEBS Lett. 2011;585:1979–84. doi: 10.1016/j.febslet.2011.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan J, Reue K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005;1:73–83. doi: 10.1016/j.cmet.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Pihlajamäki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, Dearie F, Crunkhorn S, Burak F, Jimenez-Chillaron JC, Kuulasmaa T, Miettinen P, Park PJ, Nasser I, Zhao Z, Zhang Z, Xu Y, Wurst W, Ren H, Morris AJ, Stamm S, Goldfine AB, Laakso M, Patti ME. Expression of the Splicing Factor Gene SFRS10 Is Reduced in Human Obesity and Contributes to Enhanced Lipogenesis. Cell Metab. 2011;14:208–218. doi: 10.1016/j.cmet.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brosch M, von Schönfels W, Ahrens M, Nothnagel M, Krawczak M, Laudes M, Sipos B, Becker T, Schreiber S, Röcken C, Schafmayer C, Hampe J. SFRS10—A Splicing Factor Gene Reduced in Human Obesity? Cell Metab. 2012;15:265–266. doi: 10.1016/j.cmet.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Barbosa AD, Sembongi H, Su WM, Abreu S, Reggiori F, Carman GM, Siniossoglou S. Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol Biol Cell. 2015;26:3641–3657. doi: 10.1091/mbc.E15-03-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karanasios E, Barbosa AD, Sembongi H, Mari M, Han GS, Reggiori F, Carman GM, Siniossoglou S. Regulation of lipid droplet and membrane biogenesis by the acidic tail of the phosphatidate phosphatase Pah1p. Mol Biol Cell. 2013;24:2124–2133. doi: 10.1091/mbc.E13-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Hara L, Han G-S, Peak-Chew S, Grimsey N, Carman GM, Siniossoglou S. Control of Phospholipid Synthesis by Phosphorylation of the Yeast Lipin Pah1p/Smp2p Mg2+ -dependent Phosphatidate Phosphatase. J Biol Chem. 2006;281:34537–34548. doi: 10.1074/jbc.M606654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos-Rosa H, Leung J, Grimsey N, Peak-Chew S, Siniossoglou S. The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J. 2005;24:1931–1941. doi: 10.1038/sj.emboj.7600672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siniossoglou S, Santos-Rosa H, Rappsilber J, Mann M, Hurt E. A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J. 1998;17:6449–6464. doi: 10.1093/emboj/17.22.6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pascual F, Hsieh LS, Soto-Cardalda A, Carman GM. Yeast Pah1p Phosphatidate Phosphatase Is Regulated by Proteasome-mediated Degradation. J Biol Chem. 2014;289:9811–9822. doi: 10.1074/jbc.M114.550103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pascual F, Soto-Cardalda A, Carman GM. PAH1-encoded Phosphatidate Phosphatase Plays a Role in the Growth Phase- and Inositol-mediated Regulation of Lipid Synthesis in Saccharomyces cerevisiae. J Biol Chem. 2013;288:35781–35792. doi: 10.1074/jbc.M113.525766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsieh LS, Su WM, Han GS, Carman GM. Phosphorylation Regulates the Ubiquitin-independent Degradation of Yeast Pah1 Phosphatidate Phosphatase by the 20S Proteasome. J Biol Chem. 2015;290:11467–11478. doi: 10.1074/jbc.M115.648659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh LS, Su WM, Han GS, Carman GM. Phosphorylation of Yeast Pah1 Phosphatidate Phosphatase by Casein Kinase II Regulates Its Function in Lipid Metabolism. J Biol Chem. 2016;291:9974–9990. doi: 10.1074/jbc.M116.726588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carman GM, Han GS. Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem Sci. 2006;31:694–699. doi: 10.1016/j.tibs.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siniossoglou S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. Biochim Biophys Acta - Mol Cell Biol Lipids. 2013;1831:575–581. doi: 10.1016/j.bbalip.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Pascual F, Carman GM. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim Biophys Acta - Mol Cell Biol Lipids. 2013;1831:514–522. doi: 10.1016/j.bbalip.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henry SA, Kohlwein SD, Carman GM. Metabolism and Regulation of Glycerolipids in the Yeast Saccharomyces cerevisiae. Genetics. 2012;190:317–349. doi: 10.1534/genetics.111.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Murray JP, McMaster CR. Lipid synthesis and membrane contact sites: a crossroads for cellular physiology. J Lipid Res. 2016;57:1789–1805. doi: 10.1194/jlr.R070920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butterwith SC, Hopewell R, Brindley DN. Partial purification and characterization of the soluble phosphatidate phosphohydrolase of rat liver. Biochem J. 1984;220:825–33. doi: 10.1042/bj2200825. http://www.ncbi.nlm.nih.gov/pubmed/6087797 (accessed January 28, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopewell R, Martin-Sanz P, Martin A, Saxton J, Brindley DN. Regulation of the translocation of phosphatidate phosphohydrolase between the cytosol and the endoplasmic reticulum of rat liver. Effects of unsaturated fatty acids, spermine, nucleotides, albumin and chlorpromazine. Biochem J. 1985;232:485–91. doi: 10.1042/bj2320485. http://www.ncbi.nlm.nih.gov/pubmed/3004406 (accessed January 28, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cascales C, Mangiapane EH, Brindley DN. Oleic acid promotes the activation and translocation of phosphatidate phosphohydrolase from the cytosol to particulate fractions of isolated rat hepatocytes. Biochem J. 1984;219:911–6. doi: 10.1042/bj2190911. http://www.ncbi.nlm.nih.gov/pubmed/6331400 (accessed January 28, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A, Lawrence JC. Insulin Controls Subcellular Localization and Multisite Phosphorylation of the Phosphatidic Acid Phosphatase, Lipin 1. J Biol Chem. 2007;282:277–286. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- 46.Donkor J, Zhang P, Wong S, O’Loughlin L, Dewald J, Kok BPC, Brindley DN, Reue K. A Conserved Serine Residue Is Required for the Phosphatidate Phosphatase Activity but Not the Transcriptional Coactivator Functions of Lipin-1 and Lipin-2. J Biol Chem. 2009;284:29968–29978. doi: 10.1074/jbc.M109.023663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.SIGAL YJ, McDERMOTT MI, MORRIS AJ. Integral membrane lipid phosphatases/phosphotransferases: common structure and diverse functions. Biochem J. 2005;387:281–293. doi: 10.1042/BJ20041771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jamal Z, Martin A, Gomez-Muñoz A, Brindley DN. Plasma membrane fractions from rat liver contain a phosphatidate phosphohydrolase distinct from that in the endoplasmic reticulum and cytosol. J Biol Chem. 1991;266:2988–96. http://www.ncbi.nlm.nih.gov/pubmed/1993672 (accessed January 28, 2017) [PubMed] [Google Scholar]
- 49.Waggoner DW, Gómez-Muñoz A, Dewald J, Brindley DN. Phosphatidate phosphohydrolase catalyzes the hydrolysis of ceramide 1-phosphate, lysophosphatidate, and sphingosine 1-phosphate. J Biol Chem. 1996;271:16506–9. doi: 10.1074/jbc.271.28.16506. http://www.ncbi.nlm.nih.gov/pubmed/8663293 (accessed January 28, 2017) [DOI] [PubMed] [Google Scholar]
- 50.Zhang QX, Pilquil CS, Dewald J, Berthiaume LG, Brindley DN. Identification of structurally important domains of lipid phosphate phosphatase-1: implications for its sites of action. Biochem J. 2000;345(Pt 2):181–4. http://www.ncbi.nlm.nih.gov/pubmed/10620492 (accessed January 28, 2017) [PMC free article] [PubMed] [Google Scholar]
- 51.Smyth SS, Mueller P, Yang F, Brandon JA, Morris AJ. Arguing the Case for the Autotaxin-Lysophosphatidic Acid-Lipid Phosphate Phosphatase 3-Signaling Nexus in the Development and Complications of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:479–486. doi: 10.1161/ATVBAHA.113.302737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benesch MGK, Tang X, Venkatraman G, Bekele RT, Brindley DN. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J Biomed Res. 2016;30:272–284. doi: 10.7555/JBR.30.20150058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang X, Benesch MGK, Brindley DN. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J Lipid Res. 2015;56:2048–2060. doi: 10.1194/jlr.R058362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris TE, Finck BN. Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol Metab. 2011;22:226–233. doi: 10.1016/j.tem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Koh YK, Lee MY, Kim JW, Kim M, Moon JS, Lee YJ, Ahn YH, Kim KS. Lipin1 is a key factor for the maturation and maintenance of adipocytes in the regulatory network with CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma 2. J Biol Chem. 2008;283:34896–906. doi: 10.1074/jbc.M804007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim HB, Kumar A, Wang L, Liu GH, Keller SR, Lawrence JC, Finck BN, Harris TE. Lipin 1 Represses NFATc4 Transcriptional Activity in Adipocytes To Inhibit Secretion of Inflammatory Factors. Mol Cell Biol. 2010;30:3126–3139. doi: 10.1128/MCB.01671-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu GH, Gerace L. Sumoylation Regulates Nuclear Localization of Lipin-1α in Neuronal Cells. PLoS One. 2009;4:e7031. doi: 10.1371/journal.pone.0007031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barroso E, Rodríguez-Calvo R, Serrano-Marco L, Astudillo AM, Balsinde J, Palomer X, Vázquez-Carrera M. The PPARβ/δ Activator GW501516 Prevents the Down-Regulation of AMPK Caused by a High-Fat Diet in Liver and Amplifies the PGC-1α-Lipin 1-PPARα Pathway Leading to Increased Fatty Acid Oxidation. Endocrinology. 2011;152:1848–1859. doi: 10.1210/en.2010-1468. [DOI] [PubMed] [Google Scholar]
- 60.Bahmanyar S, Biggs R, Schuh AL, Desai A, Müller-Reichert T, Audhya A, Dixon JE, Oegema K. Spatial control of phospholipid flux restricts endoplasmic reticulum sheet formation to allow nuclear envelope breakdown. Genes Dev. 2014;28:121–6. doi: 10.1101/gad.230599.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gorjanacz M, Mattaj IW. Lipin is required for efficient breakdown of the nuclear envelope in Caenorhabditis elegans. J Cell Sci. 2009;122:1963–1969. doi: 10.1242/jcs.044750. [DOI] [PubMed] [Google Scholar]
- 62.Valente V, Maia RM, Vianna MCB, Paçó-Larson ML. Drosophila melanogaster lipins are tissue-regulated and developmentally regulated and present specific subcellular distributions. FEBS J. 2010;277:4775–4788. doi: 10.1111/j.1742-4658.2010.07883.x. [DOI] [PubMed] [Google Scholar]
- 63.Ren H, Federico L, Huang H, Sunkara M, Drennan T, Frohman MA, Smyth SS, Morris AJ. A Phosphatidic Acid Binding/Nuclear Localization Motif Determines Lipin1 Function in Lipid Metabolism and Adipogenesis. Mol Biol Cell. 2010;21:3171–3181. doi: 10.1091/mbc.E10-01-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eaton JM, Mullins GR, Brindley DN, Harris TE. Phosphorylation of Lipin 1 and Charge on the Phosphatidic Acid Head Group Control Its Phosphatidic Acid Phosphatase Activity and Membrane Association. J Biol Chem. 2013;288:9933–9945. doi: 10.1074/jbc.M112.441493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin YP, Carman GM. Kinetic analysis of yeast phosphatidate phosphatase toward Triton X-100/phosphatidate mixed micelles. J Biol Chem. 1990;265:166–70. http://www.ncbi.nlm.nih.gov/pubmed/2152917 (accessed January 28, 2017) [PubMed] [Google Scholar]
- 66.Putta P, Rankenberg J, Korver RA, van Wijk R, Munnik T, Testerink C, Kooijman EE. Phosphatidic acid binding proteins display differential binding as a function of membrane curvature stress and chemical properties. Biochim Biophys Acta - Biomembr. 2016;1858:2709–2716. doi: 10.1016/j.bbamem.2016.07.014. [DOI] [PubMed] [Google Scholar]
- 67.Martin-Sanz P, Hopewell R, Brindley DN. Long-chain fatty acids and their acyl-CoA esters cause the translocation of phosphatidate phosphohydrolase from the cytosolic to the microsomal fraction of rat liver. FEBS Lett. 1984;175:284–8. doi: 10.1016/0014-5793(84)80752-8. http://www.ncbi.nlm.nih.gov/pubmed/6090213 (accessed January 28, 2017) [DOI] [PubMed] [Google Scholar]
- 68.Eaton JM, Takkellapati S, Lawrence RT, McQueeney KE, Boroda S, Mullins GR, Sherwood SG, Finck BN, Villen J, Harris TE. Lipin 2 Binds Phosphatidic Acid by the Electrostatic Hydrogen Bond Switch Mechanism Independent of Phosphorylation. J Biol Chem. 2014;289:18055–18066. doi: 10.1074/jbc.M114.547604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aridor-Piterman O, Lavie Y, Liscovitch M. Bimodal distribution of phosphatidic acid phosphohydrolase in NG108-15 cells. Modulation by the amphiphilic lipids oleic acid and sphingosine. Eur J Biochem. 1992;204:561–8. doi: 10.1111/j.1432-1033.1992.tb16668.x. http://www.ncbi.nlm.nih.gov/pubmed/1541271 (accessed January 28, 2017) [DOI] [PubMed] [Google Scholar]
- 70.Gomez-Muñoz A, Hamza EH, Brindley DN. Effects of sphingosine, albumin and unsaturated fatty acids on the activation and translocation of phosphatidate phosphohydrolases in rat hepatocytes. Biochim Biophys Acta. 1992;1127:49–56. doi: 10.1016/0005-2760(92)90200-f. http://www.ncbi.nlm.nih.gov/pubmed/1320939 (accessed January 28, 2017) [DOI] [PubMed] [Google Scholar]
- 71.Yasuda M, Tanaka Y, Kume S, Morita Y, Chin-Kanasaki M, Araki H, Isshiki K, Araki S, Koya D, Haneda M, Kashiwagi A, Maegawa H, Uzu T. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim Biophys Acta - Mol Basis Dis. 2014;1842:1097–1108. doi: 10.1016/j.bbadis.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 72.Kwon B, Querfurth HW. Palmitate activates mTOR/p70S6K through AMPK inhibition and hypophosphorylation of raptor in skeletal muscle cells: Reversal by oleate is similar to metformin. Biochimie. 2015;118:141–150. doi: 10.1016/j.biochi.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 73.Huffman TA, Mothe-Satney I, Lawrence JC. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc Natl Acad Sci. 2002;99:1047–1052. doi: 10.1073/pnas.022634399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic Acid-Mediated Mitogenic Activation of mTOR Signaling. Science (80-.) 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 76.Yoon MS, Rosenberger CL, Wu C, Truong N, Sweedler JV, Chen J. Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol Cell. 2015;58:549–56. doi: 10.1016/j.molcel.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menon D, Salloum D, Bernfeld E, Gorodetsky E, Akselrod A, Frias MA, Sudderth J, Chen PH, DeBerardinis R, Foster DA. Lipid Sensing by mTOR via de novo Synthesis of Phosphatidic Acid. J Biol Chem. 2017 doi: 10.1074/jbc.M116.772988. jbc.M116.772988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blaskovich MA, Yendluri V, Lawrence HR, Lawrence NJ, Sebti SM, Springett GM. Lysophosphatidic acid acyltransferase beta regulates mTOR signaling. PLoS One. 2013;8:e78632. doi: 10.1371/journal.pone.0078632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim Y, Gentry MS, Harris TE, Wiley SE, Lawrence JC, Dixon JE. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc Natl Acad Sci U S A. 2007;104:6596–601. doi: 10.1073/pnas.0702099104. http://www.ncbi.nlm.nih.gov/pubmed/17420445 (accessed January 28, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han S, Bahmanyar S, Zhang P, Grishin N, Oegema K, Crooke R, Graham M, Reue K, Dixon JE, Goodman JM. Nuclear envelope phosphatase 1-regulatory subunit 1 (formerly TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation pathway. J Biol Chem. 2012;287:3123–37. doi: 10.1074/jbc.M111.324350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu R, Garland M, Dunaway-Mariano D, Allen KN. Homo sapiens dullard protein phosphatase shows a preference for the insulin-dependent phosphorylation site of lipin1. Biochemistry. 2011;50:3045–7. doi: 10.1021/bi200336b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu GH, Qu J, Carmack AE, Kim HB, Chen C, Ren H, Morris AJ, Finck BN, Harris TE. Lipin proteins form homo- and hetero-oligomers. Biochem J. 2010;432:65–76. doi: 10.1042/BJ20100584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Csaki LS, Dwyer JR, Li X, Nguyen MHK, Dewald J, Brindley DN, Lusis AJ, Yoshinaga Y, de Jong P, Fong L, Young SG, Reue K. Lipin-1 and lipin-3 together determine adiposity in vivo. Mol Metab. 2014;3:145–154. doi: 10.1016/j.molmet.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dwyer JR, Donkor J, Zhang P, Csaki LS, Vergnes L, Lee JM, Dewald J, Brindley DN, Atti E, Tetradis S, Yoshinaga Y, De Jong PJ, Fong LG, Young SG, Reue K. Mouse lipin-1 and lipin-2 cooperate to maintain glycerolipid homeostasis in liver and aging cerebellum. Proc Natl Acad Sci. 2012;109:E2486–E2495. doi: 10.1073/pnas.1205221109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sembongi H, Miranda M, Han GS, Fakas S, Grimsey N, Vendrell J, Carman GM, Siniossoglou S. Distinct Roles of the Phosphatidate Phosphatases Lipin 1 and 2 during Adipogenesis and Lipid Droplet Biogenesis in 3T3-L1 Cells. J Biol Chem. 2013;288:34502–34513. doi: 10.1074/jbc.M113.488445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grimsey N, Han G-S, O’Hara L, Rochford JJ, Carman GM, Siniossoglou S. Temporal and Spatial Regulation of the Phosphatidate Phosphatases Lipin 1 and 2. J Biol Chem. 2008;283:29166–29174. doi: 10.1074/jbc.M804278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang P, Verity MA, Reue K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014;20:267–279. doi: 10.1016/j.cmet.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.English AR, Voeltz GK. Endoplasmic Reticulum Structure and Interconnections with Other Organelles. Cold Spring Harb Perspect Biol. 2013;5:a013227–a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walther TC, Farese RV. Lipid Droplets and Cellular Lipid Metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Fröhlich F, Liu X, Buhman KK, Coleman RA, Bewersdorf J, Farese RV, Walther TC. Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Dev Cell. 2013;24:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, Garg A, Olarte MJ, Lin Q, Fröhlich F, Hannibal-Bach HK, Upadhyayula S, Perrimon N, Kirchhausen T, Ejsing CS, Walther TC, Farese RV. Seipin is required for converting nascent to mature lipid droplets. Elife. 2016;5 doi: 10.7554/eLife.16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Valdearcos M, Esquinas E, Meana C, Gil-de-Gomez L, Guijas C, Balsinde J, Balboa MA. Subcellular Localization and Role of Lipin-1 in Human Macrophages. J Immunol. 2011;186:6004–6013. doi: 10.4049/jimmunol.1003279. [DOI] [PubMed] [Google Scholar]
- 93.Magré J, Delépine M, Khallouf E, Gedde-Dahl T, Van Maldergem L, Sobel E, Papp J, Meier M, Mégarbané A, Lathrop M, Capeau J, d’Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M, Lacombe D, Labrune P, Lanza M, Loret H, Matsuda F, Navarro J, Nivelon-Chevalier A, Polak M, Robert JJ, Tric P, Tubiana-Rufi N, Vigouroux C, Weissenbach J, Savasta S, Maassen JA, Trygstad O, Bogalho P, Freitas P, Medina JL, Bonnicci F, Joffe BI, Loyson G, Panz VR, Raal FJ, O’Rahilly S, Stephenson T, Kahn CR, Lathrop M, Capeau J, BSCL Working Group Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 94.Cui X, Wang Y, Tang Y, Liu Y, Zhao L, Deng J, Xu G, Peng X, Ju S, Liu G, Yang H. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20:3022–3030. doi: 10.1093/hmg/ddr205. [DOI] [PubMed] [Google Scholar]
- 95.Chen W, Chang B, Saha P, Hartig SM, Li L, Reddy VT, Yang Y, Yechoor V, Mancini MA, Chan L. Berardinelli-Seip Congenital Lipodystrophy 2/Seipin Is a Cell-Autonomous Regulator of Lipolysis Essential for Adipocyte Differentiation. Mol Cell Biol. 2012;32:1099–1111. doi: 10.1128/MCB.06465-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT, Gordon JI. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J Biol Chem. 1989;264:7994–8003. http://www.ncbi.nlm.nih.gov/pubmed/2722772 (accessed January 28, 2017) [PubMed] [Google Scholar]
- 97.Reue K, Xu P, Wang XP, Slavin BG. Adipose tissue deficiency, glucose intolerance, and increased atherosclerosis result from mutation in the mouse fatty liver dystrophy (fld) gene. J Lipid Res. 2000;41:1067–76. http://www.ncbi.nlm.nih.gov/pubmed/10884287 (accessed January 28, 2017) [PubMed] [Google Scholar]
- 98.Sim MFM, Dennis RJ, Aubry EM, Ramanathan N, Sembongi H, Saudek V, Ito D, O’Rahilly S, Siniossoglou S, Rochford JJ. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol Metab. 2013;2:38–46. doi: 10.1016/j.molmet.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Talukder MMU, Sim MFM, O’Rahilly S, Edwardson JM, Rochford JJ. Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol Metab. 2015;4:199–209. doi: 10.1016/j.molmet.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Agarwal AK, Arioglu E, de Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31:21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- 101.Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA. piRNA-Associated Germline Nuage Formation and Spermatogenesis Require MitoPLD Profusogenic Mitochondrial-Surface Lipid Signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Freyberg Z, Sweeney D, Siddhanta A, Bourgoin S, Frohman M, Shields D. Intracellular localization of phospholipase D1 in mammalian cells. Mol Biol Cell. 2001;12:943–55. doi: 10.1091/mbc.12.4.943. http://www.ncbi.nlm.nih.gov/pubmed/11294898 (accessed January 28, 2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, Duff K, Frohman MA, Wenk MR, Yamamoto A, Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nat Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Péterfy M, Harris TE, Fujita N, Reue K. Insulin-stimulated interaction with 14-3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J Biol Chem. 2010;285:3857–3864. doi: 10.1074/jbc.M109.072488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G, Hubert L, Saada A, de Keyzer Y, Eshel G, Vaz FM, Pines O, Elpeleg O. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. 2008;83:489–94. doi: 10.1016/j.ajhg.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Michot C, Hubert L, Brivet M, De Meirleir L, Valayannopoulos V, Müller-Felber W, Venkateswaran R, Ogier H, Desguerre I, Altuzarra C, Thompson E, Smitka M, Huebner A, Husson M, Horvath R, Chinnery P, Vaz FM, Munnich A, Elpeleg O, Delahodde A, de Keyzer Y, de Lonlay P. LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat. 2010;31:E1564–E1573. doi: 10.1002/humu.21282. [DOI] [PubMed] [Google Scholar]
- 107.Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, Munnich A, Lyonnet S, Majeed HA, El-Shanti H. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome) J Med Genet. 2005;42:551–557. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Al-Mosawi ZS, Al-Saad KK, Ijadi-Maghsoodi R, El-Shanti HI, Ferguson PJ. A splice site mutation confirms the role ofLPIN2 in Majeed syndrome. Arthritis Rheum. 2007;56:960–964. doi: 10.1002/art.22431. [DOI] [PubMed] [Google Scholar]
- 109.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffmann E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG. Exome Aggregation Consortium, Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segre AV, Djebali S, Niarchou A, T.G. Consortium. Wright FA, Lappalainen T, Calvo M, Getz G, Dermitzakis ET, Ardlie KG, Guigo R. The human transcriptome across tissues and individuals. Science (80-.) 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Langner CA, Birkenmeier EH, Roth KA, Bronson RT, Gordon JI. Characterization of the peripheral neuropathy in neonatal and adult mice that are homozygous for the fatty liver dystrophy (fld) mutation. J Biol Chem. 1991;266:11955–64. http://www.ncbi.nlm.nih.gov/pubmed/2050689 (accessed January 28, 2017) [PubMed] [Google Scholar]
- 112.Verheijen MHG, Chrast R, Burrola P, Lemke G. Local regulation of fat metabolism in peripheral nerves. Genes Dev. 2003;17:2450–2464. doi: 10.1101/gad.1116203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Valdearcos M, Esquinas E, Meana C, Pena L, Gil-de-Gomez L, Balsinde J, Balboa MA. Lipin-2 Reduces Proinflammatory Signaling Induced by Saturated Fatty Acids in Macrophages. J Biol Chem. 2012;287:10894–10904. doi: 10.1074/jbc.M112.342915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lordén G, Sanjuán-García I, de Pablo N, Meana C, Alvarez-Miguel I, Pérez-García MT, Pelegrín P, Balsinde J, Balboa MA. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med. 2016 doi: 10.1084/jem.20161452. jem.20161452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bergounioux J, Brassier A, Rambaud C, Bustarret O, Michot C, Hubert L, Arnoux JB, Laquerriere A, Bekri S, Galene-Gromez S, Bonnet D, Hubert P, de Lonlay P. Fatal Rhabdomyolysis in 2 Children with LPIN1 Mutations. J Pediatr. 2012;160:1052–1054. doi: 10.1016/j.jpeds.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 116.Hamel Y, Mamoune A, Mauvais FX, Habarou F, Lallement L, Romero NB, Ottolenghi C, de Lonlay P. Acute rhabdomyolysis and inflammation. J Inherit Metab Dis. 2015;38:621–628. doi: 10.1007/s10545-015-9827-7. [DOI] [PubMed] [Google Scholar]
- 117.Pichler K, Scholl-Buergi S, Birnbacher R, Freilinger M, Straub S, Brunner J, Zschocke J, Bittner RE, Karall D. A novel therapeutic approach for LPIN1 mutation-associated rhabdomyolysis-The Austrian experience. Muscle Nerve. 2015;52:437–439. doi: 10.1002/mus.24749. [DOI] [PubMed] [Google Scholar]
- 118.Jaradat SA, Amayreh W, Al-Qa’qa’ K, Krayyem J. Molecular analysis of LPIN1 in Jordanian patients with rhabdomyolysis. Meta Gene. 2016;7:90–94. doi: 10.1016/j.mgene.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schweitzer GG, Collier SL, Chen Z, Eaton JM, Connolly AM, Bucelli RC, Pestronk A, Harris TE, Finck BN. Rhabdomyolysis-Associated Mutations in Human LPIN1 Lead to Loss of Phosphatidic Acid Phosphohydrolase Activity, in. JIMD Rep. 2015:113–122. doi: 10.1007/8904_2015_440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Michot C, Mamoune A, Vamecq J, Viou MT, Hsieh LS, Testet E, Lainé J, Hubert L, Dessein AF, Fontaine M, Ottolenghi C, Fouillen L, Nadra K, Blanc E, Bastin J, Candon S, Pende M, Munnich A, Smahi A, Djouadi F, Carman GM, Romero N, de Keyzer Y, de Lonlay P. Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim Biophys Acta - Mol Basis Dis. 2013;1832:2103–2114. doi: 10.1016/j.bbadis.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thompson PD, Panza G, Zaleski A, Taylor B. Statin-Associated Side Effects. J Am Coll Cardiol. 2016;67:2395–2410. doi: 10.1016/j.jacc.2016.02.071. [DOI] [PubMed] [Google Scholar]
- 122.Michot C, Hubert L, Romero NB, Gouda A, Mamoune A, Mathew S, Kirk E, Viollet L, Rahman S, Bekri S, Peters H, McGill J, Glamuzina E, Farrar M, von der Hagen M, Alexander IE, Kirmse B, Barth M, Laforet P, Benlian P, Munnich A, JeanPierre M, Elpeleg O, Pines O, Delahodde A, de Keyzer Y, de Lonlay P. Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. J Inherit Metab Dis. 2012;35:1119–1128. doi: 10.1007/s10545-012-9461-6. [DOI] [PubMed] [Google Scholar]
- 123.Jiang W, Zhu J, Zhuang X, Zhang X, Luo T, Esser KA, Ren H. Lipin1 Regulates Skeletal Muscle Differentiation through Extracellular Signal-regulated Kinase (ERK) Activation and Cyclin D Complex-regulated Cell Cycle Withdrawal. J Biol Chem. 2015;290:23646–23655. doi: 10.1074/jbc.M115.686519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ugrankar R, Liu Y, Provaznik J, Schmitt S, Lehmann M. Lipin Is a Central Regulator of Adipose Tissue Development and Function in Drosophila melanogaster. Mol Cell Biol. 2011;31:1646–1656. doi: 10.1128/MCB.01335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang P, Takeuchi K, Csaki LS, Reue K. Lipin-1 Phosphatidic Phosphatase Activity Modulates Phosphatidate Levels to Promote Peroxisome Proliferator-activated Receptor (PPAR) Gene Expression during Adipogenesis. J Biol Chem. 2012;287:3485–3494. doi: 10.1074/jbc.M111.296681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Phan J, Péterfy M, Reue K. Lipin expression preceding peroxisome proliferator-activated receptor-γ is critical for adipogenesis in vivo and in vitro. J Biol Chem. 2004;279:29558–29564. doi: 10.1074/jbc.M403506200. [DOI] [PubMed] [Google Scholar]
- 127.Mitra MS, Chen Z, Ren H, Harris TE, Chambers KT, Hall AM, Nadra K, Klein S, Chrast R, Su X, Morris AJ, Finck BN. Mice with an adipocyte-specific lipin 1 separation-of-function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc Natl Acad Sci. 2013;110:642–647. doi: 10.1073/pnas.1213493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Donkor J, Sparks LM, Xie H, Smith SR, Reue K. Adipose Tissue Lipin-1 Expression Is Correlated with Peroxisome Proliferator-Activated Receptor α Gene Expression and Insulin Sensitivity in Healthy Young Men. J Clin Endocrinol Metab. 2008;93:233–239. doi: 10.1210/jc.2007-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yao-Borengasser A, Rasouli N, Varma V, Miles LM, Phanavanh B, Starks TN, Phan J, Spencer HJ, McGehee RE, Reue K, Kern PA. Lipin expression is attenuated in adipose tissue of insulin-resistant human subjects and increases with peroxisome proliferator-activated receptor gamma activation. Diabetes. 2006;55:2811–8. doi: 10.2337/db05-1688. [DOI] [PubMed] [Google Scholar]
- 130.Kim J, Lee YJ, Kim JM, Lee SY, Bae MA, Ahn JH, Han DC, Kwon BM. PPARγ agonists induce adipocyte differentiation by modulating the expression of Lipin-1, which acts as a PPARγ phosphatase. Int J Biochem Cell Biol. 2016;81:57–66. doi: 10.1016/j.biocel.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 131.Consoli A, Formoso G. Do thiazolidinediones still have a role in treatment of type 2 diabetes mellitus? Diabetes, Obes Metab. 2013;15:967–977. doi: 10.1111/dom.12101. [DOI] [PubMed] [Google Scholar]
- 132.Temprano A, Sembongi H, Han GS, Sebastián D, Capellades J, Moreno C, Guardiola J, Wabitsch M, Richart C, Yanes O, Zorzano A, Carman GM, Siniossoglou S, Miranda M. Redundant roles of the phosphatidate phosphatase family in triacylglycerol synthesis in human adipocytes. Diabetologia. 2016;59:1985–1994. doi: 10.1007/s00125-016-4018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, Ferguson PJ. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis. 2013;72:410–413. doi: 10.1136/annrheumdis-2012-201818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]