

Abstract
Postinjury fibrinolysis can manifest as three distinguishable phenotypes: 1) hyperfibrinolysis, 2) physiologic, and 3) hypofibrinolysis (shutdown). Hyperfibrinolysis is associated with uncontrolled bleeding due to clot dissolution; whereas, fibrinolysis shutdown is associated with organ dysfunction due to microvascular occlusion. The incidence of fibrinolysis phenotypes at hospital arrival in severely injured patients is: 1) hyperfibrinolysis 18%, physiologic 18%, and shutdown 64%. The mechanisms responsible for dysregulated fibrinolysis following injury remain uncertain. Animal work suggests hypoperfusion promotes fibrinolysis, while tissue injury inhibits fibrinolysis. Clinical experience is consistent with these observations. The predominant mediator of postinjury hyperfibrinolysis appears to be tissue plasminogen activator (tPA) released from ischemic endothelium. The effects of tPA are accentuated by impaired hepatic clearance. Fibrinolysis shutdown, on the other hand, may occur from inhibition of circulating tPA, enhanced clot strength impairing the binding of tPA and plasminogen to fibrin, or the inhibition of plasmin. Plasminogen activator inhibitor −1 (PAI-1) binding of circulating tPA appears to be a major mechanism for postinjury shutdown. The sources of PAI-1 include endothelium, platelets, and organ parenchyma. The laboratory identification of fibrinolysis phenotype, at this moment, is best determined with viscoelastic hemostatic assays (TEG, ROTEM). While D-dimer and plasmin antiplasmin (PAP) levels corroborate fibrinolysis, they do not provide real-time assessment of the circulating blood capacity. Our clinical studies indicate that fibrinolysis is a very dynamic process and our experimental work suggests plasma first resuscitation reverses hyperfibrinolysis. Collectively, we believe recent clinical and experimental work suggest antifibrinolytic therapy should be employed selectively in the acutely injured patient, and optimally guided by TEG or ROTEM.
The CRASH-2 trial1,2 provoked worldwide enthusiasm for routine delivery of tranexamic acid (TXA) to severely injured patients. Completion of this multinational, prospective, randomized study of 20,211 patients was a tremendous achievement. However, subsequently several investigators questioned the scientific validity of this trial and specifically its extrapolation to modern trauma centers in developed countries. 3,4 The objective of this counterpoint is not to critique the CRASH-2 trial, but to provide the rationale for the selective use of an antifibrinolytic in mature trauma systems. The fundamental basis is the potential physiologic benefit of systemic fibrinolysis under conditions produced by profound hemorrhagic shock and extensive tissue disruption.
HISTORICAL OBSERVATIONS
John Hunter recognized fibrinolysis in 1794, but the clinical significance was not appreciated for another 150 years. In 1948, MacFarlane and Briggs5 summarized prevailing thoughts that fibrinolysis was a process associated with death from rapid blood loss. In the ensuing decade, the plasminogen-plasmin system became well characterized, and the pathologic consequences of fibrinolysis were widely accepted.6 In the 1960s a number of investigators hypothesized that fibrinolysis could be a protective physiologic process. In 1964, Stafford7 proposed that “clotting is not episodic but a continuous process which is normally never allowed to program to a physiologic endpoint.” At the same time, Hardaway and Drake8 hypothesized that irreversible shock occurred when microvascular flow ceased due to fibrin accumulation and subsequently documented experimentally that induced fibrinolysis prevented irreversible hemorrhagic shock.9 Ultimately they conducted a Phase II trial showing tissue plasminogen activator (tPA) administration in the intensive care unit reduced acute lung injury in trauma patients.10 During this period, Starzl and colleagues11 documented hyperfibrinolysis during the anhepatic phase of liver transplant with thrombelastography (TEG), but found the intraoperative use of an antifibrinolytic resulted in a prohibitive risk of pulmonary emboli.12 In 1969, Chakrabarti and coworkers13 documented the extremes of fibrinolysis and proposed the term shutdown to describe impaired fibrinolysis.
MEASURING SYSTEMIC FIBRINOLYSIS
Fibrin clearance is an ongoing process to prevent low-flow occlusion of the microcirculation. The primary driver of intravascular fibrinolysis is tPA, derived from precapillary arterial and postvenular endothelial cells.14 The regional distribution of these endothelial cells underscores the physiologic importance of this process.15 After trauma, thrombin is generated from tissue injury resulting in extensive fibrin formation. The antifibrinolytic system localizes fibrin deposition to the site of injury. This critical regulation occurs via a number of counterbalancing mechanisms, including 1) binding tPA (e.g., plasminogen activator inhibitor 1 [PAI-1], C1 esterase inhibitor; 2) binding plasmin (e.g., α2-antiplasmin, α2-macroglobulin, α1-antitrypsin), and 3) limiting plasminogen access to the fibrin clot (e.g., factor [F]XIII cross-linking of fibrin, thrombin activatable fibrinolysis inhibitor, and platelet [PLT] aggregation/adherence).16,17 Consequently, after trauma as with elective surgery, there will be 1) elevated levels of D-dimer representing fibrin degradation following FXIII cross-linking, 2) increased levels of plasmin-antiplasmin, and 3) elevated tPA-PAI-1 complexes with secondary reduction in free PAI-1 levels. Thus, these circulating markers of fibrinolytic activity do not represent ongoing the systemic fibrinolytic status. For example, Raza and coworkers18 identified patients with high plasmin-antiplasmin levels but without thrombelastometry (ROTEM) evidence of lysis activity. The limitations of the “gold standard” plasma extraction euglobulin lysis test have been recognized since the time of its development.19 Perhaps less recognized is the important role PLTs play in modulating clot stability and, thus, susceptibility to plasminogen access or plasmin degradation.20 During primary hemostasis, PLTs aggregate and adhere to their surrounding environment including collagen and von Willebrand factor, creating a protected microenvironment for fibrin polymerization.21 This clot is further secured by the release of prothrombotic and antifibrinolytic products of PLT degranulation.22 Thus, whole blood assays are essential to measure systemic fibrinolytic activity. While a number of sophisticated assays have been employed experimentally, at this moment, whole blood viscoelastic hemostasis assays (e.g., TEG, ROTEM) are the best clinical tests of systemic fibrinolysis.23,24
FIBRINOLYSIS PHENOTYPES: HYPERFIBRINOLYSIS, PHYSIOLOGIC FIBRINOLYSIS, AND FIBRINOLYSIS SHUTDOWN
Our interest in systemic fibrinolysis was piqued by our early experience with TEG, indicating an 18% incidence of hyperfibrinolysis in patients requiring a massive transfusion.25 This observation appeared compatible with the proposed concept of activated protein C as the dominant mechanism of trauma-induced coagulopathy with degradation of activated FV and PAI-1.26 However, subsequent principal component analyses by the San Francisco group27 and our team28 suggested that the mechanisms responsible for impaired thrombin generation and hyperfibrinolysis are distinct. Because systemic fibrinolysis is difficult to replicate in animal models, we developed a TEG tPA challenge test to unmask the fibrinolysis phenotype.29 We defined fibrinolysis shutdown as a relative resistance to tPA. In both our animal models, shock (ischemia-reperfusion) consistently evoked systemic hyperfibrinolysis, whereas tissue injuring (thoracotomy, laparotomy, bowel crush, and femur fracture) provoked fibrinolysis shutdown.22 We then interrogated our prospectively collected TEG database and limited the analysis to those tested within 12 hours of injury.30 Patients were stratified into three groups: hyperfibrinolysis (LY30 > 3%), physiologic fibrinolysis (LY30 0.81%–2.9%), and fibrinolysis shutdown (LY30 < 0.81%). Our study population consisted of 180 critically injured patients (median Injury Severity Score was 29 and median initial base deficit was nine) with a mortality of 20%. Somewhat unexpectedly, fibrinolysis shutdown was the dominant phenotype (64%) with physiologic fibrinolysis and hyperfibrinolysis accounting for 18%, respectively (Fig. 1). Mortality among the phenotypes had a U-shaped distribution (Fig. 2), with the lowest in the physiologic group (3%) compared with hyperfibrinolysis (44%) and fibrinolysis shutdown (17%). Of note, acute blood loss was the predominant attributable cause of death in the hyperfibrinolysis phenotype while multiple organ failure was the etiology in 40% of the shutdown phenotype (Fig. 3).
Fig. 1.
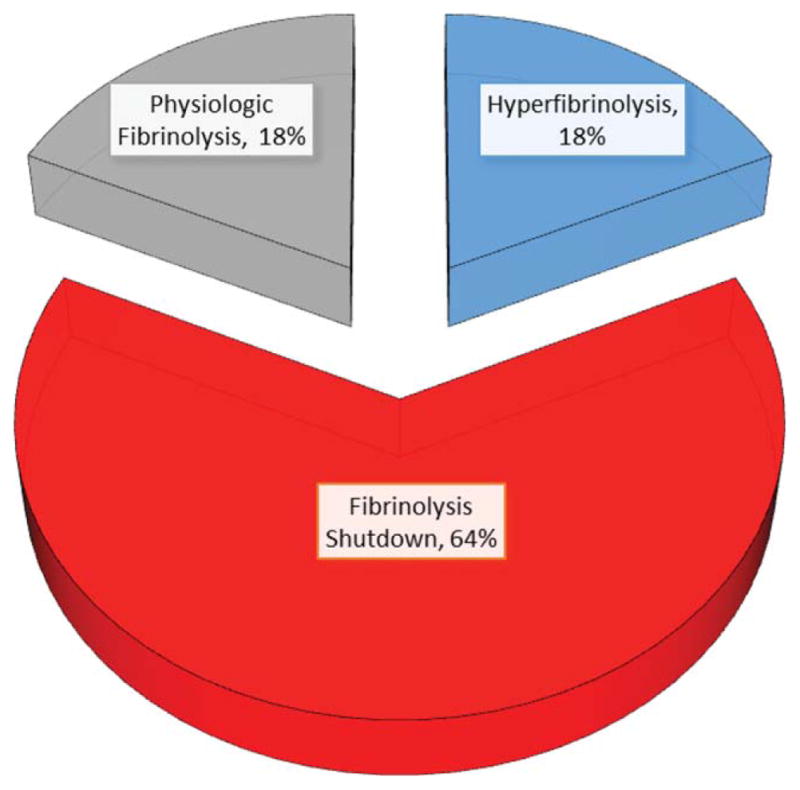
The distribution of fibrinolytic phenotype in seriously injured patients (median ISS=29, mortality=20%), based on LY30 % in TEG assays.
Fig. 2.
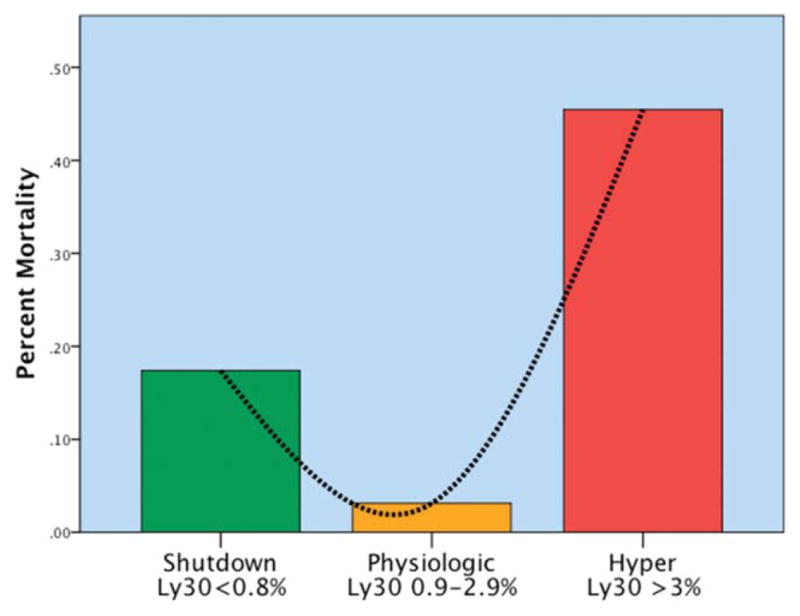
U-shaped distribution of mortality related to fibrinolysis phenotype. The y axis represents the percentage of mortality per phenotype. There is a U-shaped distribution of mortality, with a nadir in mortality identified in the physiologic group (Ly30 between 0.9% and 2.8%). Percentage of Ly30 higher and lower than this range had statistical increases in mortality. Hyper, hyperfibrinolysis; Ly30, percentage of fibrinolysis 30 minutes after reaching maximum amplitude measured by thombelastography; Physiologic, physiologic fibrinolysis; Shutdown, fibrinolysis shutdown. Copied with permission from the Journal of Trauma and Acute Care Surgery.
Fig. 3.
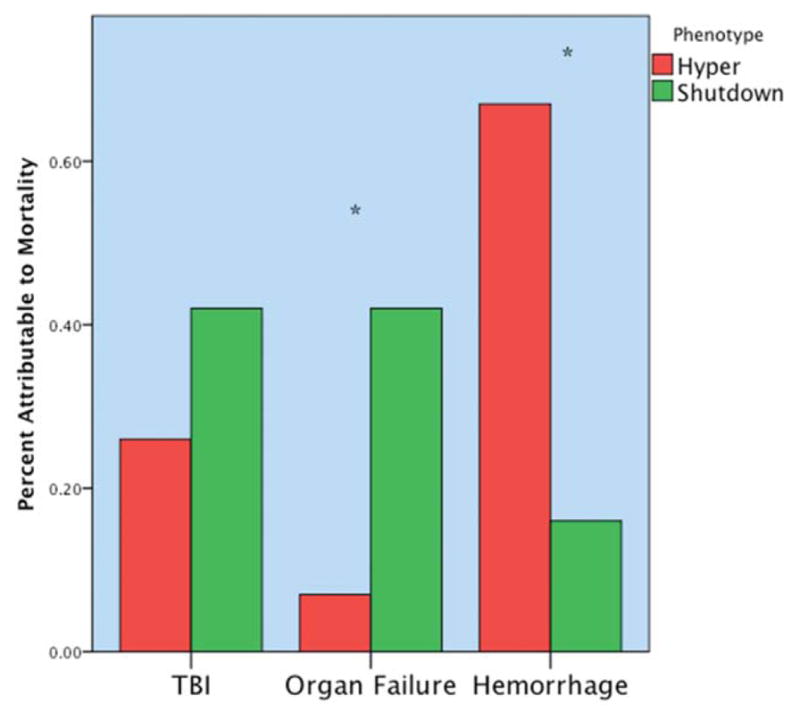
Distribution of mortality according to fibrinolytic phenotype. The y axis represents the percentage of total mortality per phenotype. The hyperfibrinolytic phenotype had a high frequency of mortality associated with hemorrhage. The shutdown phenotype has a high frequency of organ failureYrelated death. TBI did not reach statistical difference between phenotypes but was more common in the shutdown cohort. *p < 0.05. Hyper, hyperfibrinolysis; Shutdown, fibrinolysis shutdown; TBI, traumatic brain injury. Copied with permission from the Journal of Trauma and Acute Care Surgery.
While fibrinolysis shutdown was the most common phenotype in our seriously injured patients, the responsible mechanisms remain to be established. As discussed previously, normal plasma contains proteins that directly inhibit tPA or the effector of tPA, plasmin. In addition, PLT-α granules store α2-antiplasmin, α2-macroglobulin, and thrombin-activatable fibrinolysis inhibitor, and dense granules harbor FXIII. PLT-rich plasma is reported to have a 19-fold higher plasminogen activator inhibitor 1 antigen than PLT-poor plasma.31 Our clinical work further implicates metabolic byproducts in the regulation of systemic fibrinolysis.32
On the other hand, the most consistent factor associated with hyperfibrinolysis is systemic hypoperfusion. Schöchl and colleagues33 found a high rate of hyperfibrinolysis in patients with out-of-hospital cardiac arrest, and our observation is that injured patients with near exsanguination virtually always manifest advanced fibrinolysis. Others have similarly found that prolonged shock is a dominant feature of injured patients who are hyperfibrinolytic, and the CRASH-2 trial2 indicated TXA had the most benefit in those with a systolic blood pressure < 75 mmHg. Furthermore, the effects of circulating tPA are exaggerated when tPA clearance by the liver is impaired, as well documented with liver transplantation.11
THE UNCERTAIN MECHANISMS OF TXA
Terminal amines such as lysine, aminocaproic acid, and TXA are believed to inhibit plasminogen activation by binding to kringle domains, thus preventing rearrangement to the disulfide-linked, two-chain, active plasmin form. Plasminogen contains five kringle domains. To date, only aprotinin is recognized as a catalytic site inhibitor and problems with its clinical use are well documented.34 However, the coagulation system in mammals abounds with proteins bearing plasminogen-like kringle domains (e.g., t-PA prothrombin, FXII, and apolipoprotein-A). Since the binding of lysine analogs such as TXA is expected to alter the conformation of plasminogen-like kringle domains, actual activation or inactivation of an enzyme is unpredictable without knowing what role such kringles play in any multidomain protein.35 Since t-PA is itself a serine protease (with plasminogen as its best known substrate), the systemic consequences of its activation are concerning. Similarly, we have noted a small activation of clot formation with TXA in whole blood.36 Furthermore, recent experimental work demonstrates that TXA promotes urinary plasminogen activator activation of plasminogen to plasmin.37 Both angiostatin (contains the first four kringles of plasminogen) and apolipoprotein-A (contains > 10 such kringles) impact fibrinolysis, but the ramifications of TXA on these interactions remain to be clarified. On the other hand, recognizing that many cells of the immunoinflammatory response to stress contain plasminogen receptors on their surface, it is conceivable that TXA has beneficial effects in the injured patient independent of the regulation of fibrinolysis.
TREATMENT STRATEGIES TO ATTENUATE PATHOLOGIC FIBRINOLYSIS
High-volume crystalloid resuscitation has been associated with hyperfibrinolysis,38 and in vitro studies corroborate the adverse effects of hemodilution on tPA-mediated fibrinolysis.39 Colloids have been shown to impair fibrin polymerization40 and, thus, likely promote hyperfibrinolysis. The enthusiasm for permissive hypotensive resuscitation must be tempered by the fact that prolonged hypoperfusion stimulates tPA release.41 Our recent animal work suggests plasma resuscitation may be the optimal fluid strategy,42 and our ongoing clinical experience with plasma-first resuscitation is consistent with our experimental findings.43
In conclusion, we believe that the empiric administration of TXA warrants careful evaluation. We continue to use TXA early (within 2 hr of injury) in severely injured patients with TEG-documented TXA-reversible hyperfibrinolysis. On the other hand, we do not attempt to inhibit fibrinolysis in patients with isolated extremity vascular injuries because we believe this a tissue preserving physiologic process. Furthermore, we believe that enhancing fibrinolysis shutdown with untimely TXA administration in trauma patients may be critical in the pathogenesis of postinjury organ failure and venous thromboembolism.44 We eagerly await the results of ongoing prospective, randomized trials currently under way in mature trauma systems.45
ABBREVIATIONS
- PAI-1
plasminogen activator inhibitor 1
- TEG
thrombelastography
- tPA
tissue plasminogen activator
- TXA
tranexamic acid
References
- 1.Roberts I, Shakur H, Afolabi A, et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377:1096–101. doi: 10.1016/S0140-6736(11)60278-X. [DOI] [PubMed] [Google Scholar]
- 2.Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376:23–32. doi: 10.1016/S0140-6736(10)60835-5. [DOI] [PubMed] [Google Scholar]
- 3.Napolitano LM, Cohen MJ, Cotton BA, et al. Tranexamic acid in trauma: how should we use it? J Trauma Acute Care Surg. 2013;74:1575–86. doi: 10.1097/TA.0b013e318292cc54. [DOI] [PubMed] [Google Scholar]
- 4.Pusateri AE, Weiskopf RB, Bebarta V, et al. Tranexamic acid and trauma: current status and knowledge gaps with recommended research priorities. Shock. 2013;39:121–6. doi: 10.1097/SHK.0b013e318280409a. [DOI] [PubMed] [Google Scholar]
- 5.Macfarlane RG, Biggs R. Fibrinolysis: its mechanism and significance. Blood. 1948;3:1167–87. [PubMed] [Google Scholar]
- 6.Sherry S, Fletcher AP, Alkjaersig N. Fibrinolysis and fibrinolytic activity in man. Physiol Rev. 1959;39:343–82. doi: 10.1152/physrev.1959.39.2.343. [DOI] [PubMed] [Google Scholar]
- 7.Stafford JL. The fibrinolytic mechanism in haemostasis: a review. J Clin Pathol. 1964;17:520–30. doi: 10.1136/jcp.17.5.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardaway RM, Drake DC. Prevention of irreversible hemorrhagic shock with fibrinolysin. Ann Surg. 1963;157:39–47. doi: 10.1097/00000658-196301000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardaway RM, James PM, Anderson RW, et al. Intensive study and treatment of shock in man. JAMA. 1967;199:779–90. [PubMed] [Google Scholar]
- 10.Hardaway RM, Harke H, Tyroch AH, et al. Treatment of severe acute respiratory distress syndrome: a final report on a phase I study. Am Surg. 2001;67:377–82. [PubMed] [Google Scholar]
- 11.Starzl TE, Marchioro TL, Von Kaulla KN, et al. Homotrans-plantation of the liver in humans. Surg Gynecol Obstet. 1963;117:659–76. [PMC free article] [PubMed] [Google Scholar]
- 12.Groth CG, Pechet L, Starzl TE. Coagulation during and after orthotopic transplantation of the human liver. Arch Surg. 1969;98:31–4. doi: 10.1001/archsurg.1969.01340070049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakrabarti R, Hocking ED, Fearnley GR. Reaction pattern to three stresses–electroplexy, surgery, and myocardial infarction–of fibrinolysis and plasma fibrinogen. J Clin Pathol. 1969;22:659–62. doi: 10.1136/jcp.22.6.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin EG, Santell L, Osborn KG. The expression of endothelial tissue plasminogen activator in vivo: a function defined by vessel size and anatomic location. J Cell Sci. 1997;110:139–48. doi: 10.1242/jcs.110.2.139. [DOI] [PubMed] [Google Scholar]
- 15.Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost. 2005;7:1392–406. doi: 10.1111/j.1538-7836.2005.01328.x. [DOI] [PubMed] [Google Scholar]
- 16.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 17.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5:116–24. doi: 10.1111/j.1538-7836.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 18.Raza I, Davenport R, Rourke C, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11:307–14. doi: 10.1111/jth.12078. [DOI] [PubMed] [Google Scholar]
- 19.Chakrabarti R, Fearnley GR. The ‘fibrinolytic potential’ as a simple measure of spontaneous fibrinolysis. J Clin Pathol. 1962;15:228–30. doi: 10.1136/jcp.15.3.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore HB, Moore EE, Chapman MP, et al. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J Thromb Haemost. 2015;13:1878–87. doi: 10.1111/jth.13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115:3385–92. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore HB, Moore EE, Gonzalez E, et al. Hemolysis exacerbates hyperfibrinolysis, whereas platelolysis shuts down fibrinolysis: evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock. 2015;43:39–46. doi: 10.1097/SHK.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez E, Moore EE, Moore HB, Chapman MP, Chin TL, Ghasabyan A, Wohlauer MV, Barnett CC, Bensard DD, Biffl WL, Burlew CC, Johnson JL, Pieracci FM, Jurkovich GJ, Banerjee A, Silliman CC, Sauaia A. Goal-directed Hemostatic Resuscitation of Trauma-induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann Surg. 2015 Dec 28; doi: 10.1097/SLA.0000000000001608. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Görlinger KL, Dirkmann D, Solomon C, et al. Fast interpretation of thromboelastometry in non-cardiac surgery: reliability in patients with hypo-, normo-, and hypercoagulability. Br J Anaesth. 2013;110:222–30. doi: 10.1093/bja/aes374. [DOI] [PubMed] [Google Scholar]
- 25.Kashuk JL, Moore EE, Sawyer M, et al. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252:434–42. doi: 10.1097/SLA.0b013e3181f09191. [DOI] [PubMed] [Google Scholar]
- 26.Brohi K, Cohen MJ, Ganter MT, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64:1211–7. doi: 10.1097/TA.0b013e318169cd3c. discussion 1217. [DOI] [PubMed] [Google Scholar]
- 27.Kutcher ME, Ferguson AR, Cohen MJ. A principal component analysis of coagulation after trauma. J Trauma Acute Care Surg. 2013;74:1223–9. doi: 10.1097/TA.0b013e31828b7fa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin TL, Moore EE, Moore HB, et al. A principal component analysis of postinjury viscoelastic assays: clotting factor depletion versus fibrinolysis. Surgery. 2014;156:570–7. doi: 10.1016/j.surg.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore HB, Moore EE, Lawson P, et al. Fibrinolysis shutdown phenotype masks changes in rodent coagulation in tissue injury versus hemorrhagic shock. Surgery. 2015;158:386–92. doi: 10.1016/j.surg.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore HB, Moore EE, Gonzalez E, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77:811–7. doi: 10.1097/TA.0000000000000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Booth NA, Simpson AJ, Croll A, et al. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. 1988;70:327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x. [DOI] [PubMed] [Google Scholar]
- 32.Wiener G, Moore HB, Moore EE, et al. Shock releases bile acid inducing platelet inhibition and fibrinolysis. J Surg Res. 2015;195:390–5. doi: 10.1016/j.jss.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schöchl H, Cadamuro J, Seidl S, et al. Hyperfibrinolysis is common in out-of-hospital cardiac arrest: results from a prospective observational thromboelastometry study. Resuscitation. 2013;84:454–9. doi: 10.1016/j.resuscitation.2012.08.318. [DOI] [PubMed] [Google Scholar]
- 34.Fergusson DA, Hébert PC. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. 2008;358:2319–31. doi: 10.1056/NEJMoa0802395. [DOI] [PubMed] [Google Scholar]
- 35.Ho-Tin-Noé B, Rojas G, Vranckx R, et al. Functional hierarchy of plasminogen kringles 1 and 4 in fibrinolysis and plasmin-induced cell detachment and apoptosis. FEBS J. 2005;272:3387–400. doi: 10.1111/j.1742-4658.2005.04754.x. [DOI] [PubMed] [Google Scholar]
- 36.Ramos CR, Moore EE, Silliman CC, et al. Tranexamic acid accelerates the enzymatic phase of coagulation in trauma patients. J Am Coll Surg. 2013;S217:136. [Google Scholar]
- 37.Silva MM, Thelwell C, Williams SC, et al. Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue-type plasminogen activator. J Thromb Haemost. 2012;10:2354–60. doi: 10.1111/j.1538-7836.2012.04925.x. [DOI] [PubMed] [Google Scholar]
- 38.Cotton BA, Harvin JA, Kostousouv V, et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73:365–70. doi: 10.1097/TA.0b013e31825c1234. [DOI] [PubMed] [Google Scholar]
- 39.Kostousov V, Wang YW, Cotton BA, et al. Influence of resuscitation fluids, fresh frozen plasma and antifibrinolytics on fibrinolysis in a thrombelastography-based, in-vitro, whole-blood model. Blood Coagul Fibrinolysis. 2013;24:489–97. doi: 10.1097/MBC.0b013e32835e4246. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen VG. Colloids decrease clot propagation and strength: role of factor XIII-fibrin polymer and thrombin-fibrinogen interactions. Acta Anaesthesiol Scand. 2005;49:1163–71. doi: 10.1111/j.1399-6576.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- 41.Brown JB, Cohen MJ, Minei JP, et al. Goal-directed resuscitation in the prehospital setting: a propensity-adjusted analysis. J Trauma Acute Care Surg. 2013;74:1207–12. doi: 10.1097/TA.0b013e31828c44fd. discussion 1212–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore HB, Moore EE, Morton AP, et al. Shock induced systemic hyperfibrinolysis in attenuated by plasma first-resuscitation. J Trauma Acute Care Surg. 2015;79:897–904. doi: 10.1097/TA.0000000000000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore EE, Chin TL, Chapman MP, et al. Plasma first in the fields for postinjury hemorrhagic shock. Shock. 2014;41(Suppl 1):35–8. doi: 10.1097/SHK.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez E, Moore EE, Moore HB, et al. Is fibrinolysis the missing link leading to post-injury hypercoagulability. J Am Coll Surg. 2014;219:S47–8. [Google Scholar]
- 45.Binz S, McCollester J, Thomas S, et al. CRASH-2 study of tranexamic acid to treat bleeding in trauma patients: a controversy fueled by science and social media. J Blood Transfus. 2015;2015:874920. doi: 10.1155/2015/874920. [DOI] [PMC free article] [PubMed] [Google Scholar]