

Abstract
Pharmacological perturbation is a powerful tool for understanding mRNA synthesis, but identification of the specific steps of this multi-step process that are targeted by small molecules remains challenging. Here we applied strand-specific total RNA sequencing (RNA-seq) to identify and distinguish specific pharmacological effects on transcription and pre-mRNA processing in human cells. We found unexpectedly that the natural product isoginkgetin, previously described as a splicing inhibitor, inhibits transcription elongation. Compared to well-characterized elongation inhibitors that target CDK9, isoginkgetin caused RNA polymerase accumulation within a broader promoter-proximal band, indicating that elongation inhibition by isoginkgetin occurs after release from promoter-proximal pause. RNA-seq distinguished isoginkgetin and CDK9 inhibitors from topoisomerase I inhibition, which alters elongation across gene bodies. We were able to detect these and other specific defects in mRNA synthesis at low sequencing depth using simple metagene-based metrics. These metrics now enable total-RNA-seq-based screening for high-throughput identification of pharmacological effects on individual stages of mRNA synthesis.
Pharmacological manipulation is an important tool for understanding the regulation of transcription1. For example, the identification of DRB (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole) as a transcription elongation inhibitor2 facilitated the identification of the protein factors pTEFb, DSIF, and NELF that regulate transcription elongation3–5. The importance of these factors in regulating elongation was subsequently reinforced by the discovery that DRB shares a common target with a second elongation inhibitor, flavopiridol (FP)6,7. Both DRB and FP inhibit the kinase activity of the pTEFb subunit CDK9, blocking phosphorylation of serine 2 in the RNA polymerase C-terminal domain (CTD)8–11. Regulation of transcription elongation via pTEFb is now recognized as an important mode of gene regulation12,13, although pTEFb is not the only CTD kinase14,15. The usefulness of DRB and FP in understanding transcription elongation suggests that the identification of additional compounds that regulate specific stages of mRNA synthesis will reveal new regulatory mechanisms.
Current approaches for screening small molecules that affect mRNA synthesis have substantial limitations. In both screens and subsequent characterization, specific steps of mRNA synthesis (e.g., transcription initiation, elongation, and splicing) are typically tested separately despite their interdependence16. Consequently, the predominant regulatory effects of a small molecule may be missed. One potential example is the biflavonoid isoginkgetin (IsoG), first isolated from the extract of Ginkgo biloba leaves. IsoG is one of the few reported chemical inhibitors of splicing and the first that does not target the SF3b subunit of the U2 small nuclear ribonucleoprotein particle (snRNP)17,18. Splicing inhibition of IsoG was first reported in an in vivo screen in which splicing inhibition was linked to an increase in the activity of a luciferase reporter gene19. IsoG causes an accumulation of unspliced endogenous pre-mRNAs19–21 as well as an enlargement of splicing factor compartments22. In vitro, IsoG blocks splicing and stalls spliceosome assembly in the A complex before assembly of a catalytically active spliceosome19. In addition to its reported inhibition of splicing, IsoG was also recently reported to cause accumulation of transcription start site (TSS)-proximal antisense RNAs (PROMPTs) at the human telomerase RNA gene, suggesting that IsoG may inhibit nuclear RNA exosome-mediated degradation21. Unfortunately, both in vivo splicing and in vivo exosome functions are typically assessed under the implicit assumption that transcription is unaffected. However, it is reasonable to suspect that compounds such as IsoG that influence splicing may also influence transcription; splicing factors are required for transcription elongation23–27, and transcription influences splicing as well16,28. The specific targets of IsoG are not known, and it remains possible that the primary effect of IsoG is on transcription rather than splicing or exosome function.
We previously demonstrated that strand-specific total RNA sequencing (RNA-seq), with appropriate analysis, can be used to separately probe transcription initiation, elongation, and splicing20,29. Here we demonstrate the power of strand-specific total RNA-seq for screening and classifying small molecules that regulate mRNA synthesis. Surprisingly, we found that IsoG inhibits promoter-proximal transcription elongation. The effects of IsoG treatment on elongation superficially resemble those of CDK9 inhibitors. Both reduce pre-mRNA expression levels and decrease phosphoserine 2 in the RNA polymerase II CTD. However, IsoG does not inhibit CDK9, and it causes RNA polymerase to accumulate within a broader promoter-proximal band than is seen with CDK9 inhibitors. Our data reveal at least three major locations at which small-molecule-mediated transcription–elongation inhibition occurs: at the promoter (CDK9 inhibition), near the promoter (IsoG), and across the remainder of gene bodies (topoisomerase I inhibition). Strand-specific total RNA-seq can distinguish these effects on transcription elongation from deficits in exosomal processing, splicing, and transcription termination. Our results highlight the power of total RNA-seq as a tool for screening and target identification.
RESULTS
Total RNA-seq reveals that IsoG inhibits elongation
To identify specific stages of mRNA synthesis influenced by IsoG, we performed strand-specific total RNA-seq on IsoG-treated HeLa cells (see Supplementary Table 1 for a summary of all RNA-seq data). We had previously performed a similar experiment in which we confirmed a modest splicing defect in IsoG-treated cells20. Here we sought to develop simple total RNA-seq analysis metrics that would allow us to comprehensively assess any effects on mRNA synthesis. We first asked whether IsoG causes alterations in the abundances of promoter-proximal RNA transcripts, since changes in their levels can reflect defects in multiple gene-expression processes. We examined the abundance of promoter-proximal RNA transcripts using TSS metaplots that bioinformatically exclude mature mRNA transcripts by omitting sequencing reads that align to annotated exons. We reasoned that the exclusion of mature mRNA would improve sensitivity for detecting defects in transcription and pre-mRNA processing, similar to performing RNA-seq on chromatin-associated RNAs30. Upon IsoG treatment we observed a global increase in the abundance of promoter-proximal RNA transcripts, including increases in the abundance of both sense and antisense transcripts (Fig. 1a). In addition to increasing promoter-proximal transcripts, IsoG also decreases promoter-distal pre-mRNA transcripts, beginning ~5 kb downstream of the TSS. The promoter-proximal transcripts induced by IsoG are largely eliminated by poly(A) purification (Fig. 1b), highlighting the advantage of performing total RNA-seq when characterizing the gene regulatory effects of small molecules (Fig. 1).
Figure 1. Strand-specific total RNA-seq reveals global accumulation of nonpolyadenylated promoter-proximal RNA transcripts upon isoginkgetin (IsoG) treatment.

(a,b) Metaplots showing mean nonexonic read density across annotated promoter regions, aligned at transcription start sites (TSSs) and normalized to ERCC spike-in controls. (a) Strand-specific total RNA-seq in which ribosomal RNAs were depleted from total RNA by hybridization. (b) mRNA-seq in which poly(A)+ RNAs were purified from total RNA by oligo-dT hybridization. The traces represent an average of n = 2 replicates of 30 μM IsoG treatment of HeLa cells for 6 h. In these and subsequent TSS metaplots, only genes longer than the maximum x-axis value (here 10 kb) are included. S, sense; AS, antisense.
We reasoned that strand-specific total RNA-seq should be able to distinguish between two classes of explanations for the increase in promoter-proximal RNA transcripts in IsoG-treated cells: a defect in the exosomal degradation of promoter-proximal transcripts and a defect in transcription elongation. We first asked whether the increase in promoter-proximal transcripts upon IsoG treatment could, as previously suggested21, be due to inhibition of the nuclear exosome, a complex that degrades promoter-proximal RNAs31. In an analysis of published total RNA-seq data, we found that IsoG and knockdown of exosome subunits RRP40 and RRP6 all lead to a global increase of sense and antisense promoter-proximal RNAs relative to mature mRNAs (Fig. 2a; Supplementary Results, Supplementary Fig. 1a). However, the effects of exosome knockdown and IsoG treatment are distinct: compared to IsoG treatment, exosome knockdown leads to a greater increase in antisense compared to sense promoter-proximal transcripts (Fig. 2a), consistent with the exosome having a specific function in the degradation of antisense transcripts31. This distinction is captured by an ‘antisense TSS/sense TSS metric’ based on the ratio of promoter-proximal antisense to sense RNA-seq signal, which is significantly elevated upon exosome knockdown but not IsoG treatment (Fig. 2b; see Supplementary Fig. 2a–e and Online Methods for schematization and detailed description of this and subsequent metrics). Additionally, unlike IsoG treatment, exosome knockdown does not decrease promoter-distal pre-mRNA transcript abundance (Supplementary Fig. 1a). These results suggest that the increase in promoter-proximal RNAs in IsoG-treated cells is not due to a defect in exosomal degradation.
Figure 2. RNA-seq metagene-based metrics classify IsoG as a transcription elongation inhibitor.

(a,c,e) TSS-centered or cleavage and polyadenylation site (PAS)-centered metaplots showing average nonexonic RNA expression, normalized to the maximum density of sense reads downstream of the TSS or PAS, respectively. (b,d,f,g) Metagene-based metrics for each treatment, normalized to the corresponding ratios from the appropriate control samples. Quoted names (y-axis) indicate the process that metrics are designed to query and are followed by metric descriptions. (a) Comparison of IsoG with knockdown of the exosomal subunit RRP40 from analysis of previously collected data21. (c) Comparison of IsoG with the transcriptional elongation inhibitor 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB). (e) Comparison of IsoG with the splicing inhibitor meayamycin (MY). In a–g, conditions are treatment with: IsoG, elongation inhibitors (DRB and flavopiridol (FP)), siRNAs targeting exosomal subunits (siRRP40 and siRRP6, published data21), or splicing inhibitors (SSA, published data21; MY). Controls (Cntl) are side-by-side vehicle- or control-siRNA-treated (siCntl) conditions. In all experiments except MY treatment, IsoG treatment was also performed side-by-side. See Supplementary Figure 2a–e and Online Methods for a full description of how each metric is computed. Traces in a,e are means from n = 2 replicates. Traces in c are means from n = 3 replicates. Stars in b,d,f,g indicate significant increases (P < 0.05) based on a one-tailed t-test with Bonferroni correction.
We used strand-specific total RNA-seq to investigate the alternative hypothesis that IsoG inhibits transcription elongation by comparing IsoG to the well-characterized transcription elongation inhibitors DRB and FP in side-by-side total RNA-seq experiments. We found that all three compounds decrease promoter-distal pre-mRNA transcript abundance (compared to External RNA Control Consortium (ERCC) spike-ins; Supplementary Fig. 2f). In addition, all three compounds increase the ratio of promoter-proximal to promoter-distal transcript abundance, quantified in a ‘TSS/gene-body metric’ (P < 0.05; Bonferroni-adjusted one-tailed t-test; Fig. 2c,d). However, only IsoG causes a large increase in promoter-proximal transcript abundance (Supplementary Fig. 1b). Our inability to detect increases in promoter-proximal transcripts that were previously observed with global run-on sequencing (GRO–seq) upon FP treatment32 may be due to the fact that short (<60 bp) transcripts are not detectable by our total RNA-seq method. Another distinction between IsoG and DRB (or FP) is that RNA-seq read density remains elevated at greater distances from the TSS upon IsoG than upon DRB or FP treatment (Fig. 2c; Supplementary Fig. 1b). The similarity between the FP, DRB, and IsoG profiles is consistent with the idea that IsoG causes slowing, stalling, or termination of RNA polymerase near promoters, although further from the TSS than that caused by CDK9 inhibition.
Splicing inhibition does not generally block elongation
We investigated whether the apparent defect in transcription elongation in IsoG-treated cells is a general consequence of IsoG’s inhibition of splicing. This hypothesis was appealing because transcription and splicing are mechanistically coupled16,23–25. We therefore performed strand-specific total RNA-seq on cells treated with the splicing factor 3b (SF3b) complex inhibitor meayamycin (MY), which inhibits U2 binding to the branchpoint of pre-mRNA (Fig. 2e)33,34. We found that, unlike IsoG, meayamycin does not affect promoter-proximal transcript abundance or increase the TSS/gene-body metric (Fig. 2d; Supplementary Fig. 1c). Nor does spliceostatin A (SSA), a related splicing inhibitor derived from the same natural product (Fig. 2d; Supplementary Fig. 1d). However, as expected, these inhibitors do inhibit splicing. Meayamycin treatment leads to accumulation of unspliced IkB pre-mRNA (Supplementary Fig. 3a), and both meayamycin and SSA increase an ‘intron/exon metric’, defined as the ratio of intronic to exonic RNA-seq read densities (Fig. 2f). This metric is ill-suited for assessing splicing defects of compounds that also affect transcription elongation, as these compounds will tend to reduce pre-mRNA more than mature mRNA levels (e.g., DRB, FP, and IsoG; Fig. 2f). However, for compounds that do not affect elongation, the intron/exon metric is a useful proxy for splicing because of its simplicity as well as the minimal read depth that it requires compared to more accurate total-RNA-seq-based metrics20. The observation that meayamycin and SSA inhibit splicing without inhibiting promoter-proximal elongation indicates that inhibition of splicing per se does not interfere with transcription elongation; hence, the effect of IsoG on transcription elongation is not a simple consequence of its inhibition of splicing.
Our total RNA-seq analysis suggests that IsoG treatment also causes an apparent increase in transcriptional read through, wherein RNA polymerase transcription fails to terminate downstream of annotated cleavage and polyadenylation sites (PASs) (Fig. 2a; Supplementary Fig. 1e). This apparent defect in transcription termination can be captured by a ‘PAS-downstream/PAS metric’, based on the decrease in RNA-seq signal over the 10 kb downstream of the PAS (Fig. 2g). Based on this metric, we observe no defect in termination upon treatment with DRB or FP (P < 0.05; Bonferroni-adjusted one tailed t-test), suggesting that the termination defect observed upon IsoG treatment is not a general consequence of interfering with promoter-proximal transcription elongation. Exosome knockdown increases the abundance of PAS-proximal transcripts, consistent with its function in RNA degradation (Supplementary Fig. 1a). However, unlike IsoG, it does not increase the PAS-downstream/PAS metric (Fig. 2g). By contrast, SSA and meayamycin treatment do cause an apparent defect in transcription termination (Fig. 2e,g; Supplementary Fig. 1d), similarly to IsoG treatment. It is possible that IsoG, SSA, and meayamycin influence termination indirectly via their inhibition of splicing, since splicing facilitates recruitment of cleavage and polyadenylation machinery35. These results suggest that pharmacological effects on exosomal degradation and transcription termination can be distinguished via their distinctive PAS-proximal total RNA-seq signatures.
Metagene metrics classify inhibitors by affected process
Having developed a set of metagene-based metrics that enable quantification of small-molecule effects on individual stages of mRNA synthesis, we also assessed the ability of these metrics to classify compounds based on their specific effects on gene expression, via unbiased clustering. Clustering based on our metrics ‘correctly’ classifies three transcription elongation inhibitors (DRB, FP, and IsoG), two splicing inhibitors (MY and SSA), and two exosomal siRNAs (RRP6 and RRP40) into groups reflecting their specific effects on these processes (Fig. 3a). We compared this approach to traditional gene-expression analysis, in which samples are clustered on the basis of their effects on the mRNA expression levels of many genes. Clustering based on mRNA expression levels did not correctly classify inhibitors according to the specific stage of mRNA synthesis they affect (Fig. 3b). Instead, mRNA expression was more likely to cluster samples by experimental batch, laboratory, or other criteria. Thus, simple ratiometric analysis of meta-gene profiles can be more effective than traditional gene-expression analysis for classifying compounds based on their influences on specific stages of gene expression.
Figure 3. Clustering by metrics outperforms clustering by gene expression.

(a) Euclidean-distance-based clustering of samples using the metrics shown in Figure 2b,d,f,g. Rn refers to replicate number n and En to experiment number n. (b) Clustering of samples based on mRNA expression profiles from the same data, using the same clustering method as in a.
IsoG does not function analogously to CDK9 inhibition
Given that IsoG appears to have a defect in transcription elongation, we asked whether it might function by inhibiting CDK9, similar to other transcriptional elongation inhibitors1. The CDK9 inhibitors DRB and FP prevent phosphorylation of the CTD of RNA polymerase II (Pol II), specifically at the Ser2 residue8–10. We found that IsoG also reduces the levels of pSer2 Pol II (Fig. 4a; Supplementary Fig. 3b). The reduction in pSer2 Pol II by IsoG is dose responsive and reversible, as is with DRB or FP treatment (Supplementary Fig. 3c). Moreover, the pSer2 reduction occurs without substantial alteration in the levels of pSer5 Pol II (Fig. 4a; Supplementary Fig. 3b), consistent with the idea that the transcriptional disruption caused by IsoG occurs after transcription initiation. We therefore tested the ability of IsoG to inhibit CDK9 itself. We found that IsoG has no effect on CDK9 activity as assessed using an in vitro kinase assay (Fig. 4b). In control experiments, FP and DRB have the expected inhibitory effect on CDK9 activity (Fig. 4b). These results, together with the distinct shape of the promoter-proximal RNA signal in IsoG-treated cells (Fig. 2c; Supplementary Fig. 1b), raise the possibility that IsoG inhibits promoter-proximal transcription elongation via a different mechanism than CDK9 inhibitors.
Figure 4. IsoG’s effect on promoter-proximal RNA polymerase accumulation is distinct from that of CDK9 inhibition.
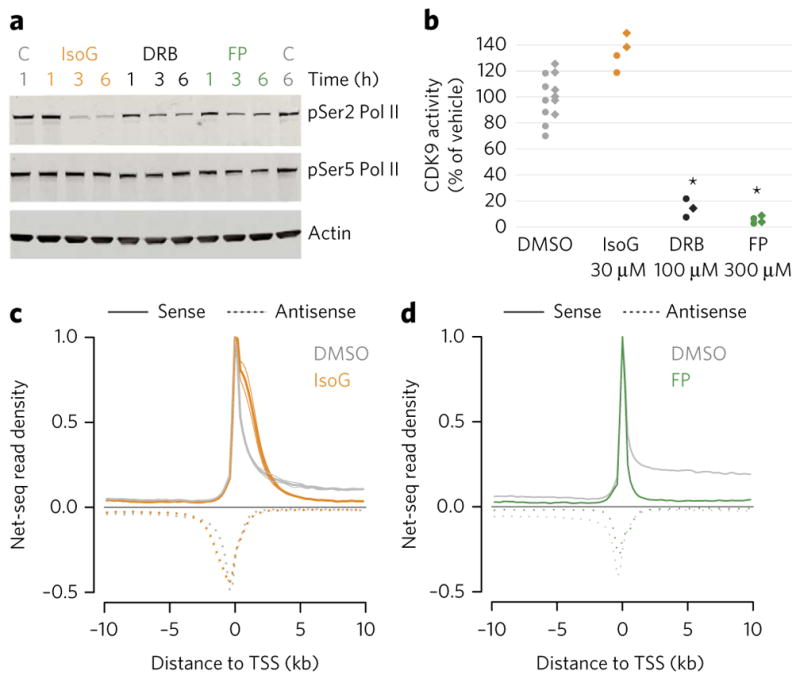
(a) A representative western blot showing RNA polymerase II (Pol II) CTD serine 2 and serine 5 phosphorylation (pSer2 and pSer5, respectively) after treatment with 30 μM IsoG, 100 μM DRB, or 300 nM FP for the indicated times. C, vehicle control (DMSO). For the full gel image, see Supplementary Figure 3b. (b) In vitro kinase activity of CDK9. Kinase activity was determined via quantification of ADP production at ten different concentrations of each compound with n = 2 for each concentration. For the dose shown, each point is an independent technical measurement. Biological replicate 1, circles; biological replicate 2, diamonds. Stars denote drug treatments that deviate significantly from the control samples (P < 0.05; one-tailed t-test with Bonferroni correction). (c) Metagene analysis of NET-seq data from HeLa S3 cells treated with 30 μM IsoG for 6 h. The solid lines are the average of two replicates, and the lighter lines are the individual replicates. The mean read density is normalized to its maximum. (d) Metagene analysis of NET-seq data from HeLa S3 cells treated with FP for 1 h as previously published36 and re-analyzed as in c.
CDK9 inhibitors inhibit elongation most dramatically within the first several dozen nucleotides downstream of TSSs. To determine where elongation is inhibited in IsoG-treated cells, we used native elongating transcript sequencing (NET-seq), which maps Pol II with single nucleotide resolution36. The NET-seq read density at a genomic position is proportional to its Pol II occupancy. Upon IsoG treatment, NET-seq reveals a dramatic increase in promoter-proximal relative to gene-body occupancy of RNA polymerase (Fig. 4c). Because we did not use spike-in standards with NET-seq, these data do not indicate whether there is an absolute, as opposed to a relative, increase in promoter-proximal RNA polymerase. Nonetheless, they strongly support the IsoG-mediated promoter-proximal elongation defect suggested by strand-specific total RNA-seq (Fig. 2; Supplementary Fig. 1). Additionally, NET-seq revealed that IsoG, but not FP, treatment leads to an apparent transcription termination defect (Supplementary Fig. 4; compare to IsoG vs. DRB in Fig. 2c), further confirming that strand-specific total RNA-seq can detect defects in specific transcriptional processes. Finally, the high resolution of NET-seq reveals that the promoter-proximal polymerase accumulation seen with IsoG treatment occurs in an extended, multi-kilobase region downstream of TSSs (Fig. 4c). This extended region accumulation is in contrast to the more tightly promoter-proximal accumulation observed with NET-seq upon FP treatment (Fig. 4d)36. The distinct promoter-proximal signatures of IsoG and FP further support the hypothesis that IsoG may inhibit a previously uncharacterized regulatory step in transcription elongation, specifically one that occurs subsequent to the CDK9-dependent RNA polymerase release from pausing.
Three distinct effects on elongation by IsoG, DRb/FP, CPT
Having used strand-specific total RNA-seq to identify specific pre-mRNA synthesis defects caused by IsoG, we sought to further test the ability of RNA-seq to disentangle complex effects on pre-mRNA synthesis by testing a second small molecule, the topoisomerase I (Topo I) inhibitor camptothecin (CPT). Like IsoG, CPT has complex effects on pre-mRNA synthesis37. It has been reported to stimulate initiation of transcription38 but also to inhibit transcription elongation38 and slow transcription39,40. Upon treatment with 1.0 μM CPT for 18 h, we observe decreasing pre-mRNA expression from 5′ to 3′ along the lengths of genes over tens to hundreds of kilobases (Fig. 5a,b). This profile is consistent with inhibition of gene-body elongation that is independent of the polymerase’s distance to its TSS, and it would be expected based on CPT-mediated introduction of covalent Topo I-DNA complexes at random locations throughout transcribed regions. To quantify this elongation defect, we created a ‘density change across long gene bodies metric’, consisting of the ratio of pre-mRNA transcripts at 100–150 kb from the TSS compared to 200–250 kb from the TSS (Fig. 5c). No other compounds that we tested had a gene-body elongation defect, with the notable exception of the splicing inhibitor SSA (Fig. 5c). We speculate that splicing inhibition may affect gene-body elongation in a general but subtle manner that is detectable only when examining long genes. We also asked whether CPT affected other aspects of pre-mRNA synthesis. We observed a modest increase in sense and antisense promoter-proximal transcripts upon CPT treatment (Fig. 5b, inset), possibly due to previously reported increases in transcription initiation or defects in promoter-proximal elongation. Using our metrics, we did not detect defects in splicing or in transcription termination upon CPT treatment (Fig. 5d), as was reported for higher CPT concentrations41. The ability of strand-specific total RNA-seq not only to detect an elongation defect caused by CPT but also to clearly distinguish it from the elongation defects caused by IsoG and DRB further establishes total RNA-seq as having the ability to identify and isolate pharmacological effects on distinct stages of mRNA synthesis.
Figure 5. The topoisomerase inhibitor camptothecin (CPT) decreases pre-mRNA expression along the bodies of long genes.

(a) CPT treatment of HeLa cells (1 μM for 18 h) leads to a decrease in intron expression at TSS-distal locations detectable by strand-specific total RNA-seq at the human Gpc5 locus. (b) A TSS metaplot reveals decreased intronic expression at TSS-distal locations upon CPT treatment (quantified and statistically assessed in c), normalized to the number of mRNA (exon)-aligned reads at least 10 kb from the TSS. Only the 850 genes longer than 250 kb are included. The inset shows the metaplot for −5 to 10 kb relative to the TSS, as in Figure 2. (c) Density change across long gene bodies metric based on the metaplot in b: the ratio of the sense RNA-seq signal +100 to +150 kb to +200 to +250 kb relative to the TSS, divided by the same ratio in controls. Stars indicate significant increases (P < 0.05) based on a one-tailed t-test with Bonferroni correction. (d) Antisense TSS/sense TSS, TSS/gene body, intron/exon, and PAS-downstream/PAS metrics calculated for CPT treatment as in Figure 2b,d,f,g; these values are repeated here for reference. None of the CPT effects are statistically significant. CPT data in b–d are means from n = 2 replicates.
DISCUSSION
We show here that metagene analysis of strand-specific total RNA-seq can be used to detect and distinguish pharmacological effects on transcription elongation, splicing, exosome function, and transcription termination. In the case of transcription elongation, our method can distinguish at least three major locations at which small-molecule-mediated transcription elongation inhibition occurs: at the promoter (CDK9 inhibition), near the promoter (IsoG), and across the remainder of gene bodies (topoisomerase I inhibition). In a demonstration of the utility of our approach, we have recast the natural product IsoG, previously identified as an inhibitor of splicing19, as a blocker of transcription elongation. In light of IsoG’s effect on transcription elongation, the effects of IsoG on splicing in vivo become difficult to assess. Transcription elongation in IsoG-treated cells is so profoundly altered that traditional metrics of splicing, including the ratios of introns to exons, are not easily interpretable. While IsoG does appear to inhibit splicing in vitro19 and in vivo20 after controlling for its effects on transcription, the specific in vivo effects on splicing are relatively small (~2-fold)20 compared to those on transcription elongation (~10-fold; Fig. 2d). More generally, our results suggest that the current paucity of splicing inhibitors may be in part due to the difficulty of designing in vivo screening assays42,43 that exclude the kinds of effects on transcription seen with IsoG.
Our application of total-RNA-seq-based metrics for small-molecule characterization opens the door to identification of many more compounds that block specific stages of pre-mRNA synthesis. At the same time, it will be important to follow up strand-specific total RNA-seq analysis with more directed assays, for several reasons. First, while powerfully simplifying, our metrics are not unambiguously interpretable on a stand-alone basis. In one example, the apparently stronger effect of IsoG compared to DRB and FP on promoter-proximal elongation (Fig. 2d) could be caused by a bias in our RNA-seq method disfavoring detection of the short (<60 bp) transcripts that are produced upon FP or DRB treatment32. Second, our metrics rely on built-in assumptions that may need to be altered to interpret the effects of new compounds. IsoG’s elevation of promoter-proximal transcript abundance is observed <5 kb from the TSS, so 9–10 kb here served as a useful gene-body reference point for the TSS/gene-body ratio metric (Supplementary Fig. 2a–e). For other compounds, or potentially even alternative doses of the same compound, it may be necessary to choose a different gene-body reference point. It should be possible to develop more refined and more general metrics as RNA-seq profiles for additional compounds are analyzed. Finally, our metrics would benefit from more extensive validation. For example, it will be important to confirm that the PAS-downstream/PAS metric is able to capture known termination defects caused by changes in the rates of polymerization or Xrn2-mediated exonuclease activity44,45.
Crucial to strand-specific total RNA-seq-based screening efforts will be two practical elements that make our approach highly scalable. First is the availability of multiplex RNA-seq library preparation methods46, with sample pooling at an early stage to reduce the cost of reagents and labor. This kind of highly scalable multiplexing has not yet been achieved for related methods such as NET-seq36, GRO-seq47, or metabolic labeling48,49, perhaps in part because each of these methods is substantially more complex. Consequently, these other methods have not yet been applied to simultaneously assess the effects of many small molecules on multiple steps of RNA processing. Equally importantly, we focus our method here on metagene-based metrics, which enables us to achieve high sensitivity with just one million sequencing reads (Supplementary Fig. 5) and makes it possible to analyze hundreds of libraries in a single sequencing run. We anticipate that this strategy could rapidly enable the identification of hundreds of small molecules that influence gene expression and could facilitate secondary screening to classify compounds based on their effects on specific stages of gene expression.
ONLINE METHODS
Cell culture and pharmacological treatments
HeLa cells were obtained from the ATCC and were not authenticated for this study. HeLa cells were maintained in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin, 1% non-essential amino acids. Cells were plated to about 70% confluence the day before any drug treatments on 10-cm plates. Cells were treated with inhibitors at the following doses unless otherwise noted: 30 uM isoginketin (IsoG, Millipore), 100 μM 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB, Sigma), 300 nM flavopiridol (FP, Sigma), or 80 nM meayamycin (gift from K. Koide) for 6 or 18 h as indicated. For time-course data, cells were treated at different times, such that all cells were lysed at the same time. For reversibility washout experiments, cells were treated with drug for 3 h then washed two times with warm PBS before adding fresh warmed media.
RNA extraction
At the time of harvest for RNA, cells were washed 1× with PBS and then lysed with RLT buffer (Qiagen). Samples were immediately processed with QIAshredder and RNeasy kits (Qiagen) and frozen until further use. 9 μg of RNA was DNase treated and cleaned up with RNeasy MinElute kit (Qiagen). RNA quality was assessed by Bioanalyzer (Agilent) and all correct samples had RNA integrity numbers (RINs) of 9.0 or higher.
RNA-seq experiment summary
We performed four RNA-seq experiments and one NET-seq experiment, submitted to GEO (accession GSE86857). The first RNA-seq experiment tested IsoG and DRB in HeLa cells, and compared strand-specific total RNA to strand-specific poly(A)+ RNA. The second compared IsoG, DRB, and FP treatments in HeLa cells. The third tested meayamycin (MY) in HeLa cells. Each of the first three experiments used the dUTP (2′-deoxyuridine, 5-triphosphate) method. We also used previously published NET-seq data for DMSO vs. FP (GSM1505442 and GSM1505443)36.
Illumina library construction and sequencing
dUTP method
RNA-seq libraries were constructed using the strand-specific dUTP method50, with minor modifications. Input amounts of RNA varied between 3–5 μg with the proportional amount of ERCC spike in RNA (Ambion). For each batch experiment total amounts of input RNA were consistent across the samples. For poly(A)+ RNA, DNAse-treated RNA was enriched for poly(A)+ RNA using Dynabeads mRNA purification (Ambion). For strand-specific total RNA libraries, DNAse-treated RNA was depleted of rRNA using Ribo-Zero (Epicentre), then samples were cleaned up using the RiboMinus concentration module (Invitrogen) and fragmented at 90 °C for 3 min using NEB fragmentation buffer. First-strand synthesis was followed by cleanup with RNAClean XP SPRI beads (Agencourt). Second-strand synthesis incorporated dUTP, which was followed by sample clean up with MinElute PCR purification kit (Qiagen). Fragment ends were repaired, adenylated, and then ligated to True-seq barcoded adaptors and cleaned up with AMPure XP SPRI beads (Agencourt). The libraries were then amplified by PCR for 9–12 cycles and cleaned up with AMPure XP SPRI beads. Illumina sequencing (1× 50 bp read length) was performed on a HiSeq 2000.
NET-seq
HeLa S3 cells (ATCC, CCL-2.2) were maintained in DMEM media containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin to 75% confluency. For drug treatment, media was replaced with DMEM containing either 30 μM isoginkgetin (IsoG, Millipore) or 0.6% DMSO for 6 h. The NET-seq protocol was performed as described in ref. 51 with modifications described below. In brief, chromatin-associated RNA is enriched by cellular fractionation52. A DNA linker is ligated specifically to the 3′ hydroxyl group on nascent RNA, representing the active site of Pol II53. The ligation step was performed overnight at 16 °C using a DNA linker with a random hexamer sequence to improve ligation efficiency and computationally remove library preparation biases. All RNA purification steps were performed on a column (RNA Clean & Concentrator-5 kit, Zymo Research). DNA sequencing libraries were prepared from ligated nascent transcripts and sequenced on an Illumina NextSeq 500 instrument with a read length of 75 base pairs. Sequencing reads were processed and aligned exactly as described in ref. 36 using STAR (v2.4.2a)54. Aligned reads were then processed and plotted exactly as with total RNA-seq (see below). For our NET-seq experiment, there were no spike-ins, so we normalized each sample to the maximum (sense) bin in each metaplot.
Protein extraction and western blotting
At the time of protein harvest, cells were washed 2× with cold PBS then lysed with protein lysis buffer: PBS, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 10 mM sodium pyrophosphate, 50 mM B-glycerol phosphate, and EDTA-free cOmplete Protease Inhibitor Cocktail (Roche). Lysates were immediately frozen in liquid nitrogen until being lysed by two freeze–thaw cycles and vortexed. Equal amounts of protein per sample were separated by SDS–PAGE and transferred to a nitro-cellulose membrane by iBlot electroblotting (Life Technologies). After the membranes had been incubated with primary (1:1,000 dilutions) and secondary antibodies (1:2,000 dilutions), chemiluminescent detection was performed with the Western-Lighting ELC (PerkinElmer) and developed using CL-XPosure Film (Thermo). Antibodies used for western blogging were: Actin (A2066; Sigma), RNA pol II CTD phospho Ser2 (3E10; Active Motif), and the RNA pol II CTD phospho Ser5 (3E8; Active Motif).
RNA-seq analysis, normalization, and metaplot construction
The total RNA-seq analysis framework and pipeline used here is distinct from our previously published ones20. We trimmed reads with fastx v0.0.13, aligned them to hg37 using STAR v2.2.1, and mapped to RefSeq exons and introns using bedtools v2.25.0. We summed ERCC alignment counts using FeatureCounts v1.5.3. To make TSS and PAS metaplots that show average expression across many genes aligned at their TSS or PAS, we used the metaseq v0.5.6 module in Python to export text files that we analyzed in R. For these metaplots, we excluded any reads aligning to RefSeq annotated exons, as our interest was in pre-mRNA and other nascent transcripts. TSS- and PAS-centered metaplots were made by summing all nonexonic reads of genes longer than the maximum x-axis value in the metaplot (typically 10 kb). To create metaplots showing absolute levels of expression, we normalized to the total number of aligned ERCC spike-in control reads. For each of these ERCC spike-in-normalized metaplots, we subtracted genomic background transcription by subtracting the mean sense read density in the [−10 kb to −5 kb] region (upstream of the TSS). We used this background subtraction because some compounds (i.e., IsoG) appear to increase genic and extragenic background transcription (Supplementary Fig. 1e). For our CPT experiment, we did not include ERCC spike-ins, so we instead normalized to the number of mRNA (exon)-aligned reads at least 10 kb from TSSs. The latter criterion filters out potentially confounding effects of changes in promoter-proximal transcript abundance between samples. To create metaplots to compare promoter-proximal versus pre-mRNA (promoter distal) expression, we normalized to the maximum average expression without upstream genomic background subtraction. In all metaplots, we removed genes above the 99.9th percentile of expression in the region plotted, as a method of spike filtering.
RNA-seq metrics
Supplementary Figure 2a–e schematizes all metrics. Our TSS/gene-body metric assesses the change in the (nonexonic) RNA transcript abundance in the region downstream of the TSS, defined as the ratio of RNA-seq read density [0.4–1 kb] divided by [9 kb–10 kb] downstream of annotated RefSeq TSSs of genes longer than 10 kb. The absolute gene-body metric assesses nonexonic read density from [9–10 kb] downstream of TSSs for genes longer than 10 kb, normalized to ERCC spike-ins. The antisense TSS/sense TSS metric assesses exosome function via the antisense to sense ratio at the TSS, specifically the ratio of antisense reads [−0.4 to −1 kb] to sense reads [0.4 to 1 kb] relative to annotated TSSs of genes longer than 10 kb. The PAS-downstream/PAS metric assesses the change in transcript abundance across the 10 kb region downstream of cleavage and polyadenylation sites via the ratio of the sense RNA-seq signal [8.2–10 kb] to [0.2–2 kb] downstream of annotated PASs. The intron/exon metric assesses splicing by comparing the overall ratio of intronic to exonic read densities, based on intronic and exonic read densities [9–10 kb] relative to annotated TSSs. The choice of [9–10 kb] for the splicing defect metric minimizes the contribution of promoter-proximal transcripts that would dominate this metric if we included more promoter-proximal regions. The density change across long gene bodies metric assesses the ratio of reads [100–150 kb] to [200–250 kb] relative to the TSS for genes that are at least 250 kb in length. This metric also uses an additional spike filter. Read densities were calculated for bins of 100 bp. If any bin differed from its neighboring bins by over ten-fold in density (ignoring bins with zero reads) the value of the bin was replaced with the average of its neighbors. This filter modifies a small fraction of the total bins; the elevated counts are likely due to features that were missed in the RefSeq genome annotation. All metrics use background-subtracted [−10 to −5 kb] average read densities, with the exception of the long gene-body elongation metric. In the case of the density change across long gene bodies metric, the small number of long (>250 kb) genes leads to significant noise in the upstream background, so we did not perform this correction. To allow for comparison between experiments, all small-molecule or knockdown metrics are normalized to their matching control samples.
Data analysis and statistics
RNA-seq of drug treatments were typically performed in duplicate or triplicate. This is not enough measurement to directly test whether each treatment has the same variance. Instead we confirmed that across all measurements the RNA-seq technique replicates follow a normal distribution and used this confirmation of normality as a prior when assessing the significance of all other measurements. In conformance with journal policy, given the number of measurements, we display the data points instead of mean and variance. Because we did not have a predetermined hypothesis, we Bonferroni corrected our statistical tests by the number of metrics we examined. Clustering was performed using MATLAB. For clustering, metrics and mRNA expression values were ratioed to within-experiment controls before clustering, and the TSS/gene-body ratio and absolute gene-body metrics were inverted to avoid introduction of noise due to division by small numbers.
Code availability
Custom MATLAB code was written to calculate the metrics. Code available upon request.
Data availability
Four RNA-seq experiments and one NET-seq experiment were submitted to GEO (accession code GSE86857). Previously published data sets used in this study can also be found on GEO. NET-seq data on HeLa S3 cells treated with FP is GSM1505443 (ref. 36). Strand-specific total RNA-seq data from exosome knockdown and SSA-treated HeLa cells with matched IsoG treatment is from GSE73776 (ref. 21).
Supplementary Material
Acknowledgments
We thank M. Hemberg and D. Harmin for advice on bioinformatic analyses. This work was funded by R01 MH101528-01. M.S. is also supported by National Science Foundation grant 1349248. S.A.B. is supported by the Harvard Medical School Center of Excellence in Systems Pharmacology NIH grant P50 GM107618 and the Giovanni Armenise-Harvard Foundation. H.M.L. and L.S.C’s contributions were funded by NHGRI: R01 HG007173. We thank K. Koide (University of Pittsburgh) for sharing meayamycin.
Footnotes
Author contributions
M.S. and J.M.G. conceived the study. S.A.B. performed all of the experiments except those mentioned below. A.S. performed the western blot in Figure 4a and data analysis of meayamycin experiments. H.M.L. performed the NET-seq protocol. H.M.L. and L.S.C. helped analyze the NET-Seq data. J.M.G., S.A.B., and M.S. performed bioinformatics analysis. J.M.G., S.A.B., A.S., and M.S. interpreted the results. J.M.G., S.A.B., and M.S. wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
References
- 1.Bensaude O. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J Biol Chem. 1989;264:2250–2257. [PubMed] [Google Scholar]
- 3.Marshall NF, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;12:2078–2090. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi Y, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97:41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 6.Chao SH, et al. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem. 2000;275:28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- 7.Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 8.Pirngruber J, et al. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3′-end processing. EMBO Rep. 2009;10:894–900. doi: 10.1038/embor.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laitem C, et al. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol. 2015;22:396–403. doi: 10.1038/nsmb.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwak H, Lis JT. Control of transcriptional elongation. Annu Rev Genet. 2013;47:483–508. doi: 10.1146/annurev-genet-110711-155440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bösken CA, et al. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun. 2014;5:3505. doi: 10.1038/ncomms4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev. 2013;113:8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 16.Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Pawellek A, et al. Identification of small-molecule inhibitors of pre-mRNA splicing. J Biol Chem. 2014;289:34683–34698. doi: 10.1074/jbc.M114.590976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Disney MD. Short-circuiting RNA splicing. Nat Chem Biol. 2008;4:723–724. doi: 10.1038/nchembio1208-723. [DOI] [PubMed] [Google Scholar]
- 19.O’Brien K, Matlin AJ, Lowell AM, Moore MJ. The biflavonoid isoginkgetin is a general inhibitor of pre-mRNA splicing. J Biol Chem. 2008;283:33147–33154. doi: 10.1074/jbc.M805556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray JM, et al. SnapShot-seq: a method for extracting genome-wide, in vivo mRNA dynamics from a single total RNA sample. PLoS One. 2014;9:e89673. doi: 10.1371/journal.pone.0089673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tseng CKK, et al. Human telomerase RNA processing and quality control. Cell Rep. 2015;13:2232–2243. doi: 10.1016/j.celrep.2015.10.075. [DOI] [PubMed] [Google Scholar]
- 22.Huranová M, et al. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J Cell Biol. 2010;191:75–86. doi: 10.1083/jcb.201004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ntini E, et al. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 26.Ji X, et al. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD. The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol. 2008;15:819–826. doi: 10.1038/nsmb.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misteli T, Spector DL. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol Cell. 1999;3:697–705. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 29.Gaidatzis D, Burger L, Florescu M, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015;33:722–729. doi: 10.1038/nbt.3269. [DOI] [PubMed] [Google Scholar]
- 30.Khodor YL, et al. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011;25:2502–2512. doi: 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 32.Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albert BJ, et al. Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol Cancer Ther. 2009;8:2308–2318. doi: 10.1158/1535-7163.MCT-09-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, Vogt A, Forsyth CJ, Koide K. Comparison of splicing factor 3b inhibitors in human cells. ChemBioChem. 2013;14:49–52. doi: 10.1002/cbic.201200558. [DOI] [PubMed] [Google Scholar]
- 35.Lutz CS, et al. Interaction between the U1 snRNP-A protein and the 160-kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev. 1996;10:325–337. doi: 10.1101/gad.10.3.325. [DOI] [PubMed] [Google Scholar]
- 36.Mayer A, et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;161:541–554. doi: 10.1016/j.cell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capranico G, et al. The effects of camptothecin on RNA polymerase II transcription: roles of DNA topoisomerase I. Biochimie. 2007;89:482–489. doi: 10.1016/j.biochi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 38.Ljungman M, Hanawalt PC. The anti-cancer drug camptothecin inhibits elongation but stimulates initiation of RNA polymerase II transcription. Carcinogenesis. 1996;17:31–35. doi: 10.1093/carcin/17.1.31. [DOI] [PubMed] [Google Scholar]
- 39.Darzacq X, et al. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veloso A, et al. Genome-wide transcriptional effects of the anti-cancer agent camptothecin. PLoS One. 2013;8:e78190. doi: 10.1371/journal.pone.0078190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci USA. 2008;105:11218–11223. doi: 10.1073/pnas.0801661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Younis I, et al. Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol Cell Biol. 2010;30:1718–1728. doi: 10.1128/MCB.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fong N, et al. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol Cell. 2015;60:256–267. doi: 10.1016/j.molcel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sansó M, et al. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 2016;30:117–131. doi: 10.1101/gad.269589.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shishkin AA, et al. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat Methods. 2015;12:323–325. doi: 10.1038/nmeth.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabani M, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwalb B, et al. TT-seq maps the human transient transcriptome. Science. 2016;352:1225–1228. doi: 10.1126/science.aad9841. [DOI] [PubMed] [Google Scholar]
- 50.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayer A, Churchman LS. Genome-wide profiling of RNA polymerase transcription at nucleotide resolution in human cells with native elongating transcript sequencing. Nat Protoc. 2016;11:813–833. doi: 10.1038/nprot.2016.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wuarin J, Schibler U. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol. 1994;14:7219–7225. doi: 10.1128/mcb.14.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Four RNA-seq experiments and one NET-seq experiment were submitted to GEO (accession code GSE86857). Previously published data sets used in this study can also be found on GEO. NET-seq data on HeLa S3 cells treated with FP is GSM1505443 (ref. 36). Strand-specific total RNA-seq data from exosome knockdown and SSA-treated HeLa cells with matched IsoG treatment is from GSE73776 (ref. 21).