

Abstract
The once nascent field of immunoengineering has recently blossomed to include approaches to deliver and present biomolecules to program diverse populations of lymphocytes to fight disease. Building upon improved understanding of the molecular and physical mechanics of lymphocyte activation, varied strategies for engineering surfaces to activate and deactivate T-Cells, B-Cells and natural killer cells are in preclinical and clinical development. Surfaces have been engineered at the molecular level in terms of the presence of specific biological factors, their arrangement on a surface, and their diffusivity to elicit specific lymphocyte fates. In addition, the physical and mechanical characteristics of the surface including shape, anisotropy, and rigidity of particles for lymphocyte activation have been fine-tuned. Utilizing these strategies, acellular systems have been engineered for the expansion of T-Cells and natural killer cells to clinically relevant levels for cancer therapies as well as engineered to program B-Cells to better combat infectious diseases.
Keywords: lymphocyte engineering, nanoparticle, microparticle, artificial antigen presenting cell
Graphical abstract
1. Introduction
The field of drug delivery has in many ways focused on the controlled delivery of soluble biomolecules to tissue types of interest and increasingly to targeted cell types. While this mode of delivery covers many categories of therapeutics, including both small molecule drugs and biologics such as peptides, proteins, and nucleic acids, certain types of biologics require presentation from a surface, rather than soluble presentation, for their desired cellular function. Biomimetic materials, in particular, that aim to mimic the physical, chemical, and biological aspects of natural biological materials for cellular engineering, must take into account this feature of surface presentation.
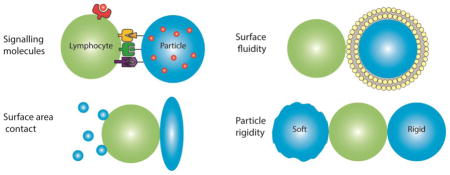
One of the varied areas of biology where the proper balance of physical, chemical, and biological interactions appears most critical is in the signaling of the immune system. Thus, for the engineering of lymphocytes, surface engineering of biomolecules is key to deliver the proper signals for lymphocyte programming. This review highlights lymphocyte immunoengineering approaches including the components of the necessary chemical and biological signals that engender cellular responses, the required features of the surfaces that best present these signals such as surface fluidity, and the geometric and physical properties of the supporting substrate that also modulate lymphocyte behavior (Figure 1)(Table 1).
Figure 1.

Lymphocyte interaction with engineered surfaces is affected by particle (A) signaling molecules, (B) surface fluidity, (C) shape and size and (D) particle rigidity.
Table 1.
Summary of various surface engineering features for lymphocyte programming.
| Lymphocyte Target | Surface Feature1 | Material2 | Signal Protein3 | Result | Ref |
|---|---|---|---|---|---|
| T-Cells | MHC Dimer for antigen specific activation | Dynal® microbead | S1: MHC Dimer + S2: Anti-CD28 | Micro-aAPC gave 106 expansion ex vivo over two months of tumor specific T-Cells | [14] |
| Paracrine delivery of IL-2 | PLGA | S1: Anti- CD3 + S2: Anti-CD28 | Enhanced CD8+ expansion with IL-2 delivery | [36] | |
| Ellipsoidal shape – micron scale | PLGA | S1: MHC Dimer + S2: Anti-CD28 | 20-fold increase in T-Cell proliferation in ellipsoidal aAPC compared to spherical | [22] | |
| Ellipsoidal shape – nano scale | PLGA | S1: MHC Dimer + S2: Anti-CD28 | 3-fold stronger in vivo induction of T-Cells by ellipsoidal aAPC compared to spherical aAPC | [23] | |
| MHC Dimer with FasL | Dynal® microbead | S1: MHC Dimer + S2: FasL | Killer aAPC induced T- Cell apoptosis with both high efficacy and specificity | [40] | |
| SLB with variable ligand mobility | DMPC and DPPC | S1: Anti-CD3 | T-Cell activation and IS formation preferentially induced by more fluid membranes | [65] | |
| Paramagnetic nanoparticle | Iron-dextran | S1: MHC Dimer + S2: Anti-CD28 | 5.5-fold increase in T cell expansion with magnetic clustering of nano aAPC compared to non-clustered nano aAPC | [76] | |
| Planar substrate with variable rigidity | PDMS | S1: Anti- CD3 + S2: Anti-CD28 | 4-fold increase in T cell expansion on softest substrate compared to stiffest substrate. | [135] | |
| B-Cells | Patterned antigen with surface fluidity | Liposome | Trimeric HIV-1 spikes | B-Cell activation against HIV-1 antigen ex vivo | [50] |
| Combine B-Cell and CD4+ T- Cell stimulation | Copolymer | OVA antigen + B- Cell epitope | Enhanced activation of B- Cells in combination with CD4+ cells | [51] | |
| Planar substrate with variable rigidity | Polyacrylamide | Anti-IgM | Enhanced B-Cell activation on stiffer substrates | [137] | |
| NK Cells | Membrane bound IL-15 | K562 cells | sIL-15 + 4- 1BBL | Ex vivo expansion of NK cells | [53] |
| Membrane bound IL-15 and IL-21 | K562 cells | sIL-15 + sIL-21 | Enhanced NK cell stimulation with both IL- 15 and IL-21 | [54] |
MHC = major histocompatibility complex, SLB = supported lipid bilayer.
PLGA = poly(lactic-co-glycolic acid), DMPC = 1,2 dimyristoyl-sn-glycero-3-phosphocholine, DPPC = dipalmitoylphosphatidylcholine, PDMS = polydimethylsiloxane.
S1 = Signal 1 protein, S2 = Signal 2 protein.
Lymphocytes include T-Cells, B-Cells and natural killer (NK) cells, all of which arise from the common lymphoid progenitor [1]. T-Cells and B-Cells are primarily responsible for the effector functions of the adaptive immune system, while NK cells serve as innate effector cytotoxic lymphocytes. Lymphocyte development begins in the bone marrow during hematopoiesis with lymphocytes migrating to peripheral lymphoid tissue following maturation. In the body, lymphocytes interact with a variety of cell types and signaling molecules that provide the cues necessary to initiate expansion, activation, anergy or cell death. T-Cell interaction with professional antigen presenting cells (pAPC), as well as stromal cells, largely shapes the adaptive immune response to pathogens and plays a role in auto-immunity [1]. pAPCs include dendritic cells, macrophages and to a lesser extent B-Cells that all express the major histocompatibility (MHC) class II molecule to allow presentation of exogenous antigens. B-Cells, in contrast, are capable of interacting with soluble antigen directly, allowing for a varied approach to targeting their response. After the immune system is activated by a specific pathogen and that pathogen is subsequently cleared, some of the pathogen-specific T-Cells and B-Cells become memory cells, ready to respond quickly if the same pathogen is ever seen again in the future. While lymphocytes are the chief cells involved in adaptive immunity and the long-term immunological memory necessary for effective vaccination, this review focuses on lymphocytes in the context of direct programming to elicit primary functions. Much work has been done in the area of vaccine design, although it has often focused on soluble antigen, small molecule adjuvants and release formulations for optimal temporal stimulation instead of surface engineering, as well as focusing on delivery to dendritic cells rather than to lymphocytes [2, 3]. In particular, Purcell et al. provides a good review of the interplay between lymphocytes in response to peptide antigens and their involvement in long term immunity [4] and Irvine et al. provides a good review of nanoparticles for use in vaccines [5].
Modulating the immune system through cellular based systems, particularly for anti-cancer immunotherapies has seen great success in recent trials with therapies targeting the anti-tumor response both through the direct modulation of lymphocytes and through ex vivo expansion of dendritic cells. In particular, chimeric antigen receptor (CAR) T-cell therapies for a subset of otherwise non-responsive cancers have seen high levels of efficacy and are in various stages of clinical trials in the USA [6, 7]. Most CAR-T-cell therapies rely on adoptive transfer strategies that have certain risks associated with the genetic modulation of T-cells for the purpose and have had adverse advents resulting from antigen recognition leading to cytokine storms [8]. In contrast to CAR-T-cell expansion for adoptive transfer, Sipuleucel-T therapy was recently approved for refractory prostate cancer with high efficacy in a subset of patients but carries its own costs and associated risks [9]. For Sipuleucel-T therapy, patient specific ex vivo expansion of dendritic cell populations in the presence of immunostimulatory molecules followed by reinfusion had an initial cost-per-patient of $93,000 in 2010 that has since risen [9]. Many of the challenges associated with these therapies in terms of cost and regulatory hurdles could be overcome with sufficiently effective acellular strategies currently in pre-clinical stages as discussed in this review.
As understanding of these natural systems has advanced, investigators have sought to design artificial systems capable of mimicking and controlling these interactions to shape the lymphocyte response. Moving towards this goal, engineered particle and surface based systems have been designed that can activate a variety of lymphocyte sub-types in vitro and in vivo for purposes of anti-cancer therapies. Across multiple stages of translation to the clinic, activation of lymphocytes ex vivo and in vivo have been studied. As the majority of lymphocyte engineering strategies in the past two decades have focused on cancer therapies, engineering of cytotoxic T lymphocytes (CTLs) has arguably advanced the furthest and strategies for genetically engineering T-Cells have already reached the clinic in the form of CAR-T-Cells [10]. Similar in some contexts, cellular based artificial antigen presentation systems have likewise seen significant development [11], but face challenges related to the manufacturing and amplification of dendritic cells or other professional APCs ex vivo [12].
Unlike strategies to modify lymphocytes directly such as with CAR T-Cell engineering [13], artificial antigen presenting cells (aAPCs) [14] and surface engineering for lymphocyte modulation function within the domain of activating lymphocytes through their existing molecular machinery. This approach of lymphocyte activation has the benefit of being potentially safer in terms of lower risk of run-away activation, induced tumorigenesis, or mutagenesis due to the lack of viral modification of the lymphocytes. Particle and surface based acellular aAPC engineering have fewer of the translational challenges associated with the use of live cellular aAPC systems in terms of cost and regulatory hurdles for translation [15].
The approach of lymphocyte engineering through outside-in signaling mediated by intact cellular machinery maintains similarities to traditional drug delivery approaches in that it requires determination of the biological factors, dosages and manner of presentation necessary to result in particular cellular responses. Similar to the identification of small drug molecules that mediate specific biological responses, engineered surfaces must possess the specific three-dimensional structures needed to bind to key biomolecule targets, including proteins, often along with targeting, such as to immunological tissues. Similar to the formulation of release systems for small molecule drug delivery, engineered systems for the controlled presentation of specific biological factors from a surface may need to also consider spatial and temporal factors relating to local dosages and the avoidance of undesirable clearance. Development of these technologies applies knowledge of chemical engineering, immunology, and materials science. A challenge with this approach is that the requirements for modulating lymphocyte responses have been determined primarily through empirical means and are not fully explored for all cell types or intended cellular responses and can also be different in varying model systems. Despite this limitation, lymphocyte engineering through the rational design of acellular materials has the potential to enable unique safe and specific in vivo cellular therapies without the challenges, risks, and expense of ex vivo cellular engineering. The presentation of chemical and biological factors from defined biomaterial surfaces could significantly impact many areas of medicine.
2. Chemical and Biological Factors (Signal Proteins)
This area of lymphocyte engineering best matches the traditional approach of the drug delivery field as it involves the selection of bioactive molecules as well as their dosages to elicit particular cellular responses. Lymphocytes primarily interact with their environment through intramembrane protein complexes that allow for outside-in signaling to occur when they bind other protein complexes or biomacromolecules. It is important to design and select both the specific biological factors to be included in each system, as well as the ratios between the signals involved.
Particle and surface based strategies for engineering lymphocyte responses have been primarily focused on engineering T-Cells for expansion to cytotoxic T lymphocyte populations as cancer therapies [10]. For therapeutic cancer applications, activation of T-Cells with MHC class I is of particular interest for its ability to induce expansion of cytotoxic T lymphocytes that can infiltrate the tumor and trigger apoptosis of cells presenting the same signal. Surface engineering for B-Cell programming has progressed more slowly due to additional challenges including recapitulation of all the signals present in the spleen and peripheral lymphoid tissues, but progress has been made in recapitulation of specific signaling molecules using acellular systems. Natural killer cell surface engineering using specific biological factors has likewise presented challenges as it has advanced.
2.1 T-Cells
Biochemical activation or deactivation of T-Cells specifically requires two signals, which have been recapitulated with a variety of approaches to guide activation or deactivation [16]. T-Cell activation in vivo occurs primarily in lymph nodes, spleen or Peyer’s patches, where T-Cells interact with professional APCs. pAPCs interact with T-Cells primarily through two protein complexes termed Signal 1 and Signal 2 for simplicity. Signal 1 provides antigen specificity for T-Cell expansion via interaction of the T-Cell receptor (TCR) with MHC class I or II with bound cognate antigen for activation of CD8+ or CD4+ T-Cells, respectively. Signal 2, in contrast, provides the context for Signal 1 to allow for a variety of T-Cell responses, among them expansion, anergy and apoptosis. In addition to these two primary signals, soluble Signal 3 and adhesion molecules assist in providing guiding cues to lymphocytes as shown in Figure 2. Signals 1 and 2 have historically been used in a 1:1 mole ratio, but this factor has not specifically been studied.
Figure 2.

Surface engineering for T-Cell immunomodulation primarily aims to recapitulate the signaling molecules and context provided by professional APCs in vivo. Presentation of Signal 1 for antigen presentation and Signal 2 for costimulation can be achieved with MHC class II and anti-CD28 respectively. Signal 3 has been shown to enhance T-Cell activation the form of soluble secretory molecules such as IL-2 and additional adhesion molecules such as ICAM-1 and LFA-3.
2.1.1 Signal 1: Cognate Antigen Presentation
Recapitulation of Signal 1 in acellular T-Cell engineering approaches have traditionally been achieved in vitro using a constitutively active, agonist anti-CD3 antibody to trigger T-Cell expansion [17–20], but this approach is not suitable for in vivo activation due to the lack of specificity for TCRs of expanded T-Cells [16, 21]. Furthermore, expansion of T-cells with agonist anti-CD3 antibodies has been shown to lead to dwindling CD8+ T-cell expansion in successive generations of cells, as cells become exhausted [14]. As an alternative, MHC class I or II have been bound directly to particles [14, 22–26] or surfaces for both antigen specific as well as CD4+ or CD8+ TCR activation; for this reason, recombinant forms of MHC class I or class II are much more promising for antigen specific CTL expansion either in vivo or ex vivo for clinical applications. To further increase avidity of the MHC molecule for specific TCRs, dimers [25] and tetramers [27] of MHC have been engineered at the molecular level that have been shown to be highly potent for antigen specific T-Cell expansion. In particular, the use of a dimerized MHC class I peptide complex on microbeads was shown to give a 106 expansion of CTLs ex vivo with the estimated possibility of generating 1011 antigen specific CTLs from a 500 mL leakuopack in under two months [14]. This level of CTL expansion is sufficient to be clinically relevant for human cancer cases [28, 29], but requires substantial investment for ex vivo expansion. In contrast, activation in vivo with an acellular material could be the gold-standard for therapy, as it would not necessitate the manufacturing and regulatory hurdles associated with cellular therapies.
Approaching the goal of fully acellular systems that can stimulate robust T-cell expansion in vivo following administration, degradable particle systems using dimerized MHC IgG pre-loaded with selected antigen and co-injected with T-cells intravenously in an adoptive transfer murine model have demonstrated improved survival in aggressive B16-F10 melanoma model [26]. These particles were shown to be capable of stimulating antigen specific T-Cells to proliferate up to 30-fold over a span of seven days, however, the cells showed high levels of PD-1 expression [26]; treatment of the expanded T-Cells with anti-PD-1, however, protected the T-Cells and allowed them to remain effective in vivo.
2.1.2 Signal 2: Costimulation
Whereas Signal 1 provides antigen specificity for TCR selection, costimulatory Signal 2 provides context to trigger a specific fate for T-Cells interacting with the engineered system. Most aAPCs are engineered to interact with T-Cells through at least two costimulatory signals, targeting the CD28 receptor on T-Cells with either B7.1/B7.2 (CD80/86) or anti-CD28 to provide Signal 2, while simultaneously providing activation of the TCR with MHC or anti-CD3 [30]. The use of anti-CD28 presents benefits over B7.1/7.2 due to the fact that it can be engineered to specifically bind only to CD28 and not the inhibitory CTLA-4, thus avoiding T-Cell deactivation [30]. This is particularly important in cases of tumor models that have upregulated deactivating signals, as the binding affinity of B7.1/7.2 to CTLA-4 is higher than to CD28, making it a less suitable costimulatory molecule than alternatives such as anti-CD28 [31]. As an additional alternative Signal 2, 4-1BBL with affinity for T-Cell expressed 4-1BB (CD137) has been identified as a potent costimulatory molecule that works synergistically when combined with anti-CD28 for CTL expansion [27, 32–35].
2.1.3 Signal 3: Soluble Factors
Tertiary signals for T-Cell engineering includes soluble signals such as IL-2, which has been included in biodegradable particle formations for controlled release with strong evidence of improved activation. Incorporation of IL-2 for local paracrine delivery to T-Cells from biodegradable microparticles improved activation and expansion of CD8+ T-Cells compared to exogenous IL-2 delivery in the bulk solution, though the local particle based release of IL-2 was noted to result in CD4+ cell apoptosis [36]. In this specific study, particle degradation and release rates were shown to be important for IL-2 release, with sustained release over multiple days being necessary for benefits in expansion [36]. In addition to IL-2, IL-21 has been demonstrated to result in expansion of highly potent CD8+ CTLs when using a cellular aAPC system [37]. For more specific lymphocyte engineering, other soluble signals including TGF-β1, TNF-α, IL-12, IL-1 can be utilized to induce specific T-Cell subtype responses including Treg, TH17, TH1 and TH2 CD4+ T-Cells [38]. Incorporation of these soluble protein molecules in degradable polymeric microparticles or nanoparticles for acellular aAPCs can be achieved using water-in-oil-in-water double emulsion methods, similarly to published approaches for IL-2 release [36].
2.1.4 Adhesion Molecules
Interaction between professional APCs and T-Cells in vivo is facilitated by binding of T-Cell expressed LFA-2 (CD2) and LFA-1 with APC expressed ICAM-1 and LFA-3. Utilization of ICAM-1 and LFA-3 adhesion molecules has been shown to greatly assist in the artificial recapitulation of immunological synapse formation on micropatterned surfaces as well as liposome and particle based systems [17, 19, 20, 39]. Utilization of adhesion molecules on microparticles and nanoparticles for activation of T-Cells in vivo may improve the ability of these particles to stay adhered to T-Cells encountered in the blood stream long enough for activation to be initiated or for the conjugates to be trafficked to a lymph node.
2.1.5 Killer aAPCs: T-Cell Deactivation
In contrast to most T-Cell engineering approaches, another strategy for lymphocyte engineering includes “killer aAPCs” designed to eliminate T-Cells possessing a highly specific TCR for a certain antigen [40]. Killer aAPCs have been designed as microparticles coated with a combination of MHC dimer for TCR specificity and FasL (CD95L) as the Signal 2 molecule to deliver an apoptotic command to T-Cells in an antigen specific manner. These particles were shown to mediate 80% antigen specific killing of cognate CD8+ T-Cells only whereas the non-cognate T-Cells were not affected [40]. This strategy of selective T-Cell elimination may prove to be highly useful in cases of autoimmunity where T-Cells are erroneously attacking a patients’ own cells or in cases of host versus graft disease in allograft organ rejection [24, 40–42]. Later generations of these killer aAPCs have been designed with anti-Fas antibodies as an alternative to FasL [42].
2.2 B-Cells
Although not as frequently investigated as T-Cells, B-Cells are another class of lymphocytes that have been engineered with the intent to elicit humoral immunity as opposed to cellular immunity. While B-Cells are capable of responding to soluble antigen, the manner of antigen presentation to the B-Cells can alter the subsequent response.
2.2.1 Requirements of B-Cell/Antigen Interaction
B-Cell activation has classically been viewed as the result of crosslinking of surface bound immunoglobulin (sIg) receptors by repeated antigenic patterns on a target. Upon crosslinking of these sIg molecules, a phosphate kinase cascade is initialized which results in maturation of the B-Cell into a form that can produce soluble antibodies [43]. Further interactions with cognate helper T-Cells results in further maturation and differentiation into plasma cells for rapid synthesis and secretion of high affinity antibodies [44].
More recent studies suggest that the primary means of activation of B-Cells in vivo is not by soluble antigen, but rather by APCs presenting membrane bound antigen [45]. This membrane bound antigen is not presented in the context of an MHC protein as would be required for T-Cell activation. The primary means of B-Cell signaling in this context is the microcluster [46] which contains 50–100 sIg receptors clustered together [45]. It was recently discovered by dSTORM imaging that these receptor microclusters initially exist as smaller clusters ranging from 1–10 receptors but upon activation by simulated surface bound antigen, they would aggregate into larger microclusters accompanied by and increased contact area with the antigen presenting surface [47]. Microcluster formation was recently mechanistically linked to a decrease in the lateral mobility of the BCR upon antigenic activation, thus restricting the normally diffusive single receptor to a relatively rigid microstructure [48]. Interestingly, upon initial BCR engagement, the B-Cell will undergo a morphological change to flatten out along the antigen presenting surface, similar to the related T-Cell [46]. The resulting prolonged contact allows for the formation of an immunological synapse similar to the T-Cell/APC interaction [45].
2.2.2 Particulate Systems for B-Cell Activation
Numerous particulate systems have been created to activate B-Cells. A repeated antigenic pattern on the surface of calcium phosphate nanoparticles resulted in a significantly stronger B-Cell response to soluble cognate antigens. This activation was determined to be 100-fold stronger than the soluble antigen case [49]. Similarly, a lipid nanoparticle system was developed with HIV antigens loaded onto the surface with a regular spacing of 12–14 nm [50]. This regular spacing of B-Cell antigens was linked to a 12-fold increase in B-Cell stimulation as measured by cytokine secretion compared to a soluble control [50].
For B-Cells, it has also been shown that a solid particle surface is not necessary for efficient stimulation as polymerized antigen was found to be capable of eliciting an effective B-Cell response. This reaction was found to be correlated to the number of repeat units in the construct with an approximately 10-fold increase in B-Cell mediated T-Cell activation for a 6-fold increase in the number of repeating units [51].
2.3 Natural Killer Cells
Natural killer (NK) cells are a lymphocyte population with crucial involvement in anti-cancer activity [52]. NKs are difficult to expand in vivo, but have been expanded ex vivo using a variety of strategies [53]. In particular, NK cell expansion ex vivo has primarily utilized cellular based aAPC systems with membrane bound IL-15 in combination with 4-1BBL to generate clinically relevant numbers of NK cells for anti-cancer applications [53]. This strategy, while thus far has only been implemented using K562 cellular aAPC systems, could be readily adapted to acellular particle aAPC or surface based systems in the future to alleviate concerns over the use of another human cell line. In addition to the use of membrane bound IL-15, membrane bound IL-21 has been shown to be very potent for NK cell expansion ex vivo using the same K562 cellular aAPC system [54]. Over the course of 42 days it was shown that proliferation of natural killer cells was 10,000 times higher in the presence of membrane bound IL-21 compared to comparable aAPC without membrane bound IL-21
3. Surface patterning and fluidity
Interactions between lymphocytes and APCs or soluble antigens are highly complex in spatial arrangement. An immunological synapse (IS) is formed at the interface between lymphocytes and APCs. The IS is characterized by dynamic rearrangements of proteins into specific clusters. Randomly distributed, immobilized proteins cannot recapitulate this organization and clustering [55]. As a result, patterned and fluid surfaces have been engineered to replicate the IS and enhance lymphocyte modulation.
3.1 T-Cells
When a biological APC engages with a T-Cell, peptide antigen-MHC (pMHC) on the APC is recognized by the TCR, leading to the formation of the immunological synapse. The IS is composed of concentric rings containing clusters of specific proteins (Fig. 3). In order for the IS to form, relevant molecules must migrate towards the interaction site and form clusters. The IS can be recapitulated and studied by patterning relevant proteins in a certain spatial arrangement [20, 56, 57] or by engineering surface fluidity so that the proteins have lateral mobility to form the IS when engaged with a T-Cell [17, 58, 59]. The IS has been studied in depth using a variety of techniques including supported lipid bilayers, microfabricated surfaces and super-resolution optical microscopy to ascertain the identity of the signaling molecules involved and their precise spatial arrangement for maximal activation [60].
Figure 3.

Immunological synapse formation between T cells and a supported lipid bilayer. Images over time of MHC-peptide (green) and ICAM-1 (red) at area of contact show eventual formation of immunological synapse with outer pSMAC and inner cSMAC. Reproduced with permission from Science [63].
3.1.1 Spatial arrangement of immunological synapse
The small size of the immunological synapse of approximately 20 nm in diameter for individual receptor clusters [60, 61] and the Abbe diffraction limit of approximately 250 nm for light microscopy necessitates the use of single molecule localization super-resolution microscopy techniques including PALM or STORM to study individual immunological synapses [60]. Single molecule localization microscopy techniques have allowed individual supramolecular activation complexes (SMAC) of the immunological synapse to be probed in live cells, showing evidence of a bullseye-like structure of a central SMAC (cSMAC) with distinct composition compared to the surrounding peripheral SMAC (pSMAC) and exterior distal SMAC (dSMAC) of the immunological synapse (Figure 3) [60, 62, 63]. The cSMAC, where initial activation occurs, contains the TCR/CD3 complex, as well as signaling molecules LAT and LCK. The pSMAC, in an annulus around the cSMAC, is composed almost entirely of LFA-1 [60, 62]. Interestingly, TCR signaling occurs in the dSMAC following initial activation at the cSMAC, which has implications for the design of aAPCs as individual focal spots of anti-CD3 or pre-clustered MHC are unlikely to be optimal [60].
3.1.2 Surface Patterning
Microfabrication allows for deposition of proteins in a site-specific manner on surfaces at resolutions down to approximately 1 μm to allow for effective pre-clustering of signal molecules to study lymphocyte activation [56]. Using microfabrication with biotinylated photoresist to pattern anti-CD3 onto a glass substrate, solid focal spots were shown to be more effective for T-Cell activation than annulus shaped protein deposition, demonstrating the importance of receptor clustering for T-Cell activation [20]. This study further concluded that differences in T-Cell activation were due specifically to the shape of the protein deposition spots and not due to density of ligand or surface area of the receptor, which has implications for particle systems that are capable of only displaying a certain surface area to T-Cells based on their geometry [20, 22].
In contrast to microfabricated surfaces that allow for receptor localization at the micron level, nanoarrays of gold nanoparticles to specifically localize ligands at defined intervals between 35–150 nm have been used to study the importance of inter-receptor spacing for T-Cell activation [57]. Using anti-CD3 conjugated to gold nanoparticles, a spacing of 60 nm was shown to be optimal for maximal threshold stimulation of T-Cells [57], which has implications for the design of particle-based aAPC systems with surface conjugated ligands where ligand localization is more challenging to define.
3.1.3 Surface fluidity
The fluidity of proteins on a synthetic surface of a particle is an important component of mimicking the membrane fluidity of a cell. Surface fluidity allows particles to behave in a more biomimetic manner by enabling receptor clustering, which is an important aspect of the physiological interaction between T cells and APCs.
Supported lipid bilayers (SLB) on planar substrates have long been utilized to study receptor interactions between cells, in particular as they enable membrane fluidity and facilitate receptor clustering. Artificial APC surfaces have been engineered to mimic the lateral diffusivity of the IS by coating a support substrate with an SLB containing Signal 1, Signal 2, and adhesion molecules. SLBs with immobilized ligands separated by chromium strips for precise localization have been used to demonstrate the importance of receptor clustering for T-Cell activation [18]. This study was one of the first to demonstrate the necessity of ligand positioning and fluidity for the formation an immunological synapse required for robust T-Cell activation [18]. SLBs have also been used to study the requirements for TCR triggering, which were found to include surface-anchoring of pMHC, T-Cell surface adhesion, and subsequent ability of the T-Cell to move, suggesting that a fluid aAPC/T-Cell contact area is optimal [64]. Ligand mobility, specifically, has been shown to be an important parameter in modulating T-Cell response [65]. SLBs of different lipid compositions were engineered to vary ligand mobility, and those with greater ligand mobility had increased CD3 accumulation at the IS and increased phosphotyrosine (pY) signaling at the TCR microclusters (Figure 4).
Figure 4.

Receptor clustering is critical for T cell activation. (A) Supported lipid bilayers conjugated with biotinylated anti-CD3 mAb via NTA (neutravidin) were synthesized with different compositions of DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) to produce ligand mobility. SLBs with a higher diffusion coefficient (D), which corresponds to greater ligand mobility, induced higher levels of TCR signaling [44]. (B) Paramagnetic nano-aAPC induce clustering of the TCR/CD3 complex on T cells under the influence of a magnetic field. Reproduced and modified with permission from ACS Nano [76].
Knowledge of the importance of membrane fluidity for immunological synapse formation has implications for lymphocyte surface engineering that have been investigated through the use of liposomes with conjugated signal molecules that allow for free movement and clustering of the biomolecules [17, 59]. As planar SLBs are only relevant for ex vivo T cell expansion, 3D aAPCs with membrane fluidity have been engineered by incorporating Signal 1, Signal 2, and adhesion molecules into liposomes [17, 58, 59]. TCR molecules on T-Cells engaging with liposome-based aAPCs formed clusters over time that co-localized with the aAPCs, suggesting the formation of an IS [58]. In the first generation of liposome-based aAPCs, the pMHC molecules were randomly distributed throughout the membrane [58]. However, pre-clustering MHC class II on lipid rafts for highly concentrated spots of signaling molecules has been shown to increase activation of T-Cells [66]. As a result, liposome-based aAPCs were enhanced by pre-clustering the T-Cell ligands in microdomains on the aAPC surface, which significantly increased antigen-specific T cell stimulation [59]. Microdomains composed of anti-CD3, anti-CD28, and anti-LFA-1 were anchored to GM-1-enriched liposomes by neutravidin bound cholera toxin. This liposome-based aAPC generated almost a 1.5-fold increase in the expansion of T-Cells compared to bead-based aAPCs with immobilized ligands, with an overall expansion of over 150-fold over the course of 14 days. Liposome-based aAPCs allow receptor clustering but are limited by shape, size, and the number of signaling molecules that can interact with the T-Cell. SLBs atop a particle core would allow for control over these parameters, in addition to surface fluidity. Particles coated with lipid bilayers have been successfully engineered for targeted drug delivery applications [67, 68], and are an interesting approach for next generation aAPCs.
Coating particles with cell membranes is an additional technique for generating surface fluidity that provides a biomimetic surface in terms of both fluidity and protein content and enables particles to evade clearance. Particles have been coated with membranes of many cell types, such as red blood cells [69], platelets [70], and macrophages [71], and cell membrane coated nanoparticles have been shown to induce receptor clustering [72]. Nanoporous silicon (NPS) particles coated with leukocyte membranes bound to endothelial cells and led to clustering of ICAM-1 [72], suggesting that this technique could be applied to aAPCs to drive receptor clustering on T-Cells.
3.1.4 Alternative methods to enable receptor clustering
Alternative engineering methods allow for receptor clustering on T-Cells without patterning the particle surface or using a fluid lipid bilayer. One way to accomplish this is through the use of a semi-flexible polymer based on poly(isocyano peptides) conjugated with anti-CD3 and anti-CD28 to allow for receptor clustering, as well as efficient multivalent binding [68, 73]. The polymers were able to activate T-Cells at significantly lower concentrations in vitro than rigid spherical poly(lactic-co-glycolic acid) (PLGA) counterparts, but their efficacy and biodistribution in vivo has yet to be studied.
With the knowledge of receptor clustering necessary for TCR activation, nanoparticle systems capable of self-clustering in response to paramagnetic activation have been designed that allow for larger focal spot formation, with a length scale similar to that of micron-sized particles [74, 75]. Perica et al. engineered paramagnetic iron-dextran nano-aAPC to drive receptor clustering [74, 76]. Under the influence of an external magnetic field, nanoparticles bound to T-Cells aggregate, leading to receptor clustering on the T-Cell and an increase in TCR cluster size (Figure 4). T-Cells activated by the nano-aAPC in a magnetic field mediated tumor rejection in a melanoma adoptive immunotherapy model and were more effective in activating naïve T-Cells than nano-aAPC without magnetic field counterparts. In addition, magnetic preclustering of these nano aAPC resulted in 80% long term survival of mice compared to unclustered controls [74, 76].
3.2 B-Cells
Similar to T-Cells, B-Cells form an immunological synapse when engaged with APCs, and receptor clustering is an important component of B-cell activation [77, 78]. Antigen binding to the BCR triggers BCR crosslinking and the formation of microclusters, which initiate downstream signaling that leads to B-Cell activation [77, 79]. BCR/antigen clusters in the cSMAC, while LFA-1/ICAM-1 migrate to the pSMAC [78, 80]. However, unlike T-Cells, adhesion molecules are not required for mature synapse formation if the affinity of BCR-antigen binding is high enough [78]. SLBs and liposomes have been used to recapitulate this spatial organization for B-Cell modulation.
3.2.1 Surface Fluidity
Supported lipid bilayers have been used to study B-Cell activation by membrane-anchored antigen in vitro [46, 48, 78–84]. Typically, SLBs contain biotinylated anti-IgM, anti-IgD, or anti-IgG as surrogate antigen tethered to biotinylated lipids with a strepdavidin linker [85]. Anti-IgG SLBs with lateral mobility result in enhanced BCR microcluster formation and signaling compared to immobilized IgG, which form small and unstable microclusters that lead to inefficient signaling [79]. Although adhesion molecules are not required to induce BCR clustering and synapse formation, incorporating ICAM-1 into SLBs can enhance contact formation for lower-avidity antigens [78, 82].
Although biotinylation is widely used to conjugate IgG to SLBs, it is difficult to control the location of biotin binding on the antibody, which can lead to inaccessibility of the antigen to BCRs, and multiple IgG molecules can bind to one streptavidin molecule [85]. Zhang et al. addressed these issues by using a modified D domain from staphylococcal protein A molecule fused with a polyhistidine tag, which binds to nickel-containing SLBs [85]. Protein A binds IgG with high affinity only in the constant region. This technique enhanced lateral mobility compared to tethering IgG with strepdavidin. However, this approach cannot be used to target B-Cells expressing IgG BCRs since the linker protein will bind the BCRs. Others have similarly utilized nickel-containing SLBs to conjugate histidine-tagged antigen and adhesion molecules [86–89], as well as the inhibitory receptor FcγRIIB, which blocks BCR microcluster formation and prevents downstream signaling [90].
Results using planar lipid bilayers suggest that a liposome-based design for B-Cell therapies may be more effective than particles with immobilized antigen [91]. Researchers have used liposomes presenting peptide antigen to stimulate a B-Cell IgG response by including a costimulatory TLR ligand [91] and to induce tolerance by incorporating the ligand for CD22, an inhibitory co-receptor [92]. Studies have focused to a greater extent on delivering free antigen encapsulated in liposomes to B-Cells [93]. However, given that membrane-anchored antigen stimulates B-Cells more strongly, surface presentation on fluid particles is a promising new direction [80].
4. Surface Area Contact
The surface topography and area of contact with substrates has been shown to dramatically influence cellular behavior and aspects of cellular phenotypes [94]. Lymphocytes are one cell type where the physical surface topography has been shown to be highly influential on cellular programming. Topographical designs have been engineered to replicate the primary agents with which lymphocytes, specifically T-Cells and B-Cells, interact for increased influence on cellular activity. The interactions of T-Cells with particulate systems have frequently been investigated and have been demonstrated more effective at T-Cell stimulation than soluble or flat surface bound immune signal proteins [95].
4.1 T-Cells
The primary mimetic target for topographical design in the context of T-Cells is the APC. Taking cues from the natural biology of a T-Cell/APC interaction, several particulate and high surface area structures have been designed for T-Cell activation.
4.1.1 Requirements of APC/T-Cell Surface Area Interactions
The APC can be thought of as a critical bridge between the innate and adaptive immune responses. The dendritic cell is the cell subtype in the body that has been designated as the most effective APC to direct T-Cell activity [96]. Thus, the dendritic cell/T-Cell interaction is likely the most important phenomenon to consider when designing a surface topography for T-Cell programming.
Immature dendritic cells in the periphery undergo rapid micropinocytosis and act as sentinels for the immune system [97]. Upon encountering a danger signal such as a pathogen associated molecular pattern or an inflammatory cytokine, they will detach from the periphery and migrate to the lymph nodes. The dendritic cell will then undergo a maturation process to halt rapid micropinocytosis, and upregulate expression of T-Cell stimulatory proteins on the surface. Following lymph node infiltration, resident T-Cells will begin to rapidly scan the antigen presenting cell surface. If a cognate interaction is formed between a TCR and the associated MHC complex, a conjugate between the T-Cell and APC will be formed. Typically the area of this contact is on the order of microns [98].
Following the initial interaction of T-Cell and APC, dramatic morphological changes have been observed in both cell types during the formation of the immunological synapse. During this interaction, the dendritic cell has been shown to increase the area of contact for the T-Cell to engage by flattening the cell surface through cytoskeletal remodeling. Interruption of this morphological change is associated with significant reduction in T-Cell stimulatory capacity [99]. During the formation of a mature immunological synapse, the T-Cell will also undergo morphological change by flattening its surface to allow for maximal cellular contact. Within 2–15 minutes of a T-Cell contacting a cognate dendritic cell, it has been observed that T-Cells will flatten out over the surface of the APC (Figure 5) [100, 101]. This morphological change observed in T-Cells has been attributed to intracellular calcium signaling which subsequently controls several aspects of T-Cell polarization and migration [102].
Figure 5.

Diagram of the changes in cellular morphology during the formation of the immune synapse. The microcluster forms the initial point of contact and then leads to the initial spreading of the T-Cell membrane on the surface of the APC. As more microclusters are engaged, the T-Cell spreads out along the flat surface of the aAPC. Reproduced and modified with permission from Immunology [101].
4.1.2 Particle Size as a Parameter to Control Surface Area Interaction
The simplest way to control the available area for an artificial T-Cell/particle interaction is through the use of isotropic particles and the modulation of particle size. For a sphere, the surface area increases proportional to the square of the radius. It has been generally accepted that the more area available for a T-Cell to interact with on a particle, the better the subsequent T-Cell stimulation will be [55]. This has been exhibited using biodegradable polymeric particles. In one study, particles that were approximately 8 μm in size (similar order of magnitude of T-Cell size) and 130 nm in size were compared for T-Cell stimulation capacity [103]. It was found the microparticles could elicit a three-fold better immune response than the nanoparticles as evidenced by IL-2 secretion [103]. In addition to particle size, Janus particles have been appropriated to illustrate this point as well. By patterning the surface of polymeric microparticles using particle lithography, Chen et. al. demonstrated that particles with higher surface areas coated with stimulatory molecules resulted in approximately 1.5-fold increased T-Cell proliferation [104].
Despite the drawback exhibited in vitro by nanoparticles as T-Cell stimulatory agents, other modifications have been made in particle synthesis that enable them to be more potent potentiates of T-Cell action. Steenblock et al. demonstrated that biodegradable nanoparticles encapsulating IL-2 could result in 3–4 fold higher T-Cell activation than equivalent amounts of soluble IL-2, suggesting a high local concentration at T-Cell mediated by the proximity of the nanoparticles when engaged with cognate T-Cells [36]. A similar effect was exploited in the local release of TGF-β from biodegradable nanoparticles for the induction of T-Reg cells from naïve T-Cells [105].
Interestingly, despite the poor performance of nanoparticles in vitro for T-Cell activation, in vivo effects of stimulatory nanoparticles on T-Cells can be more pronounced. For example, Fifis et. al. examined antigen loaded particles of sizes ranging from 20 nm to 2 μm in terms of in vivo T-Cell stimulatory capacity [106]. It was found that 40 nm was the optimal size for targeting the particles to the lymph nodes. It was found that this advantage enabled superior T-Cell stimulation in vivo as evidenced by a 10-fold increase in IFN-γ secretion by T-Cells in response to antigen stimulation for the 40 nm particles compared to the 1 and 2 μm particles [106].
4.1.3 Particle Shape as a Parameter to Control Surface Area Interaction
Another way to modulate the available area for particle/lymphocyte interaction is by alteration of particle shape. There have been several protocols developed for synthesis of non-spherical particles [107], however a popular and widely accepted method is the thin film stretching method. Pioneered by Ho et. al., this protocol involves casting the particles into a thin plastic film, heating the particles above their glass transition temperature, stretching the film, cooling the film, and releasing the particles from the film [108]. Although this method was originally developed for the synthesis of 1D-stretched prolate ellipsoidal particles, it has been extended to the synthesis of particles of a wide variety of shapes [109]. Furthermore, this method has recently been automated to allow for scaled up production of anisotropic particles [110].
The impact of shape on the particle’s interaction with cells has been extensively studied in the past decade [107]. With respect to anisotropic particles as a drug delivery vector, they have been determined to have two key advantages compared to equivalent spherical particles. The first is reduced non-specific cellular uptake by phagocytosis. This effect has been shown in anisotropic particles of various shapes [111] and sizes [112] resulting in reduced phagocytic uptake. Through a series of experimental and modeling studies, this effect has been linked to the orientation of the particle as it approaches the cell membrane [113]. Due to anisotropy, an ellipsoidal particle was shown to prefer to bind non-specifically to a cell along its longer axis. However, the cell prefers to engulf the particle along a shorter axis. Combined, these two phenomenon result in the reduction in non-specific phagocytosis. The second and more crucial feature of the anisotropic particle for lymphocyte programming is the increased frequency of targeted binding. Antibody bound anisotropic ellipsoidal particles have been shown to bind to target cells two-fold more compared to spherical particles [114]. This has been attributed to the larger radius of curvature and the flatter surface afforded by particle anisotropy. This effect was extended in vivo where rod-shaped particles were shown to have 7.5 times more accumulation to brain endothelium when targeted to that location with transferrin [115].
Due to these advantageous properties of anisotropic particles, they have recently been investigated as platforms for T-Cell stimulation. Sunshine et. al. constructed aAPCs from spherical and prolate ellipsoidal microparticles of approximately 4 μm in size [22]. Despite similar levels of protein and protein density for the two shapes, the ellipsoidal particles elicited a much stronger 20-fold higher T-Cell proliferation response compared to spherical particles. Furthermore, it was observed under confocal microscopy that the T-Cell preferentially translocated to the long axis of the ellipsoid with the flatter surface (Figure 6) [22]. In addition, the increased T-Cell stimulatory capacity of ellipsoidal particles was observed to mediate a significantly better anti-tumor T-Cell response in vivo compared to spherical particles, with 20% long term survivors compared to 0% for spherical [22]. Particle shape has also been determined to be an important design parameter in the construction of nanodimensional aAPC [23]. Comparing prolate ellipsoidal and spherical nanoparticles of size 200 nm, it was shown that the non-spherical particles could trigger a 15-fold stronger T-Cell proliferative response compared to their spherical equivalents. This effect was also observed in vivo with non-spherical particles resulting in three-fold higher concentrations of antigen specific T-Cells in the periphery compared to spherical particles [23].
Figure 6.

Non-spherical ellipsoidal particles allow for more surface area of contact between the T-Cell and the aAPC. (A) Image of the T-Cell (red) and the aAPC (green) interacting in a conjugate. Cognate (B) spherical and (C) non-spherical aAPC interacting with T-Cells. Non-cognate (D) spherical and (E) non-spherical aAPC show minimal interaction with T-Cells. (F) Ellipsoidal shape leads to a higher percent of aAPC/T-Cell conjugates. (G) The measured length of contact between T-Cells and aAPC is higher with ellipsoidal aAPC. (H) A time lapse image of a non-spherical aAPC interacting with the T-Cell demonstrates preference for the flat surface of the aAPC over time. Reproduced with permission from Biomaterials [12].
4.1.4: Nanotopography as a Parameter to Control Surface Area Interaction
Nanotopographical features have also been shown to influence the efficiency of T-Cell programming by artificial constructs. This has recently been investigated with the use of single walled carbon nanotubes (SWNTs). Fadel et. al. was able to synthesize these constructs with varying degrees of surface roughness as mediated by treatment with different acids [116]. Increased surface roughness at the nanodimensional level was shown to increase available surface area for protein adsorption. Subsequently this increased surface roughness was correlated to an increase of 2–6 fold IL-2 secretion by anti CD-3 based stimulation of T-Cells [116]. The impact of surface roughness was partly attributed to the presence of protein clusters that allowed for rapid rebinding of the T-Cell following dissociation of the T-Cell [117]. This technology was extended to antigen specific stimulation of T-Cells through the immobilization of MHC Class I proteins for CD8+ T-Cell stimulation [118].
SWNTs with high surface roughness and antigen specific T-Cell stimulatory capacity were synthesized with immobilized PLGA particles encapsulating IL-2 and hydrophobic magnetite [119]. It was found that encapsulation of the IL-2 at the SWNT/T-Cell binding site resulted in comparable levels of T-Cell stimulation compared to 1000-fold greater soluble IL-2. Furthermore, the stimulation was noted to be stronger than spherical Dynabeads with soluble IL-2. T-Cells activated and produced using this method were then used therapeutically in a melanoma treatment model where it was found that the T-Cells stimulated by the SWNTs with encapsulated IL-2 had near identical tumor treatment capabilities to Dynabeads with 1000-fold greater concentration of soluble IL-2 [119].
4.2 B-Cells
B-Cells have the ability to respond to soluble antigens with repeated patterns. In this context, designing particle based systems capable of reaching B-Cells in vivo following administration is the primary criterion for designing the size and shape of systems designed to stimulate B-Cells [120, 121]. Nanoparticles less than 200 nm in diameter are known to reach lymph nodes following intravenous injection [122] and the use of ellipsoidal, rod-shaped [107] or biomimetic nanoparticles [123] that demonstrate enhanced circulation time may further enhance the fraction of nanoparticles that reach lymphatic tissue as opposed to accumulating in the liver and spleen, although further research is needed [124].
More recent work has demonstrated that the B-Cell is most likely in vivo to encounter antigen sequestered on the surface of an antigen presenting cell and engage the APC in a manner similar to the immunological synapse of the T-Cell [125]. It has been shown that the B-Cell will initially spread out along a flat surface of the APC to engage as many of the cognate antigens presented on the surface as possible. Following this phase, the B-Cell/APC membrane contact will contract resulting in a mature synapse between the two cells [80]. During this time, the B-Cell can continue to collect antigen from the surface and move along the surface of the APC [78]. Existing particle based platforms for B-Cell activation are designed for the delivery of soluble antigen to B-Cells. In order to accurately mimic this interaction, future systems for B-Cell stimulation could consider antigen presentation from a surface that is biomimetic. For example, such a surface could contain a radius of curvature and a particle size of comparable character to a dendritic cell and also follow T-Cell aAPC technology.
5. Surface rigidity and mechanical properties
The effect of substrate stiffness on lymphocyte modulation has been relatively unexplored but has been shown to have an important role in the differentiation and activation of other cell types, such as mesenchymal stem cells (MSCs) [126–129] and endothelial cells [130–132]. For example, Engler et al. showed that substrate stiffness alone can drive MSC differentiation fate [127]. A limited number of studies have shown that lymphocyte activation and differentiation may be affected by planar substrate rigidity [133–138]. This is an interesting area for further investigation of particle-based systems, as particle stiffness is an important component of cellular biomimicry.
5.1 T-Cells
It has been shown that mechanical forces can mediate T-Cell signaling, through both the TCR/CD3 complex and the costimulatory receptor, CD28 [139, 140]. T-Cells cultured on elastomer pillar arrays presenting CD3 and CD28 activating antibodies generated traction forces on the pillars [133]. Traction forces were also generated through the TCR on pillars presenting antigen-loaded MHC. Additionally, inhibition of actin polymerization [18, 64, 141] and myosin [142] significantly inhibits TCR signaling. This evidence suggests that cytoskeletal and cellular forces play a role in T-Cell activation at the immunological synapse. Optimizing particle rigidity may allow T-Cells to perform mechanosensing during the formation of the immune synapse as they do with biological APCs.
While particle stiffness has not been explored, it has been shown that the ridigity of planar surfaces plays a role in T-Cell activation, proliferation, and differentiation. Kam and colleagues have investigated the effect of substrate stiffness on ex vivo T-Cell activation and expansion [135, 136]. Human T-Cells were cultured on PDMS substrates of varying elastic modulus coated with anti-CD3 and anti-CD28, and softer substrates (E < 100 kPa) exhibited enhanced polyclonal expansion compared to stiffer substrates (E > 2 MPa) [135]. Specifically, softer substrates yielded a higher level of CD4+ and CD8+ T-Cell stimulation, as evidenced by increased proliferation and IL-2 production. Additionally, softer substrates generated a greater proportion of IFN-γ producing Th1-differentiated cells. In a different study, mouse T-Cells expanded on polyacrylamide gels of elastic modulus varying from 10 to 200 kPa showed increased IL-2 secretion on stiffer substrates [136]. The seemingly opposite trends in the two studies are likely due to the different ranges of elastic moduli tested—the mouse T-Cells were cultured on polyacrylamide gels ranging in elastic modulus from 10 to 200 kPa [136], while the human T-Cell study tested PDMS substrates ranging from 50–100 kPa to >2 MPa [135]. Both papers show that the optimal elastic modulus for T-Cell stimulation in the ranges tested is approximately 100 kPa. A different group looked at a larger range of elastic moduli—Jurkat T-Cells were cultured on polyacrylamide gels of elastic modulus (E) ranging from 200 Pa to ~6 kPa [134]. T-Cell signaling persisted for a longer period of time on softer substrates, but the efficiency of T-Cell activation and expansion on the different substrates needs to be further investigated.
Additional research needs to be performed to determine the effect of surface rigidity of particles, in addition to planar substrates, on T-Cell activation and differentiation. In engineering particle-based aAPCs, mimicking the low compressive modulus of biological APCs may optimize the T-Cell-aAPC interaction. Dendritic cells have been measured to have an elastic modulus of ~800 Pa when activated [143], so T-Cell activation may be optimal on surfaces with lower elastic moduli. The effect of particle stiffness on T-Cells needs to be explored to better understand the mechanics of lymphocyte signaling and to optimize particle therapies designed to modulate lymphocytes.
5.2 B-Cells
There has been a limited amount of research on the effect of substrate stiffness on B-Cell stimulation. Unlike T-Cells, B-Cells encounter antigen in vivo on substrates of varying stiffness—for example, B-Cells sense antigen on the surface of stiff viral capsids, on the membranes of infected host cells, and in soluble form. Liu and colleagues found that B-Cells cultured on antigen-coated polyacrylamide gels of elastic moduli varying from 2.6 to 22.1 kPa were more strongly activated and were better able to perform antigen affinity discrimination on the stiffest substrate [137]. B-Cells were even more efficiently activated on a PDMS surface with an elastic modulus of 1100 kPa compared to a 20 kPa substrate, as measured by formation of the immune synapse [138]. However, the softer substrate showed enhanced B-Cell expansion and antibody response. Further evaluation with a larger number of substrate elastic moduli is necessary. Nonetheless, these studies suggest that B-Cells perform mechanosensing and that optimizing particle rigidity based on the desired response could be beneficial for therapies designed to modulate B-Cells, such as particle-based vaccines and B-Cell lymphoma treatments.
6. Conclusion
Recent advances in the field of immunoengineering have enabled the development of technologies designed to activate and deactivate lymphocytes, including T-Cells, B-Cells, and NK cells. Particle-based systems that program lymphocytes aim to mimic biological cells in terms of physical, chemical, and biological properties in order to improve the particle-lymphocyte interaction. Surfaces have been engineered to present various chemical and biological factors designed to induce a specific lymphocyte response, and properties, such as shape, size, surface area, and rigidity, have been optimized. Researchers have attempted to recapitulate the complex spatial arrangement of proteins and membrane reorganization that occurs during the formation of the immunological synapse by engineering surfaces with patterned proteins and surface fluidity. While these features have all been individually implemented, in the future, enhanced biological factors, surface diffusivity, surface area, and mechanical properties will likely be combined to create highly effective advanced biomimetic materials for immunotherapy.
Acknowledgments
The authors wish to thank the NIH (R01EB022148 and R01CA195503), the Johns Hopkins University Discovery Award, and the Johns Hopkins Bloomberg~Kimmel Institute for Cancer Immunotherapy for their support. RAM thanks the National Cancer Institute at the National Institute of Health (NIH F31CA214147), the Institute for Nanobiotechnology, and the Achievement Rewards for College Scientists program for fellowship support. DRW thanks the NSF Graduate Research Fellowship for support (DGE-0707427).
Abbreviations
- TCR
T-Cell receptor
- APC
antigen presenting cell
- aAPC
artificial antigen presenting cell
- CTL
cytotoxic T lymphocytes
- MHC
major histocompatibility complex
- BCR
B-Cell receptor
- NK
natural killer
- IS
immunological synapse
- Ig
immunoglobulin
- IL
interleukin
- SMAC
supramolecular activation cluster
- SLB
supported lipid bilayer
- PLGA
poly(lactic-co-glycolic acid)
- NPS
nanoporous silicon
- SWNTs
single walled carbon nanotubes
- MSCs
mesenchymal stem cells
- PDMS
polydimethylsiloxane
- E
Young’s elastic modulus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parkin J, Cohen B. An overview of the immune system. The Lancet. 2001;357:1777–1789. doi: 10.1016/S0140-6736(00)04904-7. [DOI] [PubMed] [Google Scholar]
- 2.Koup RA, Douek DC. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harbor perspectives in medicine. 2011;1:a007252. doi: 10.1101/cshperspect.a007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tzeng SY, Guarecuco R, McHugh KJ, Rose S, Rosenberg EM, Zeng Y, Langer R, Jaklenec A. Thermostabilization of inactivated polio vaccine in PLGA-based microspheres for pulsatile release. Journal of Controlled Release. 2016;233:101–113. doi: 10.1016/j.jconrel.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature reviews Drug discovery. 2007;6:404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 5.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem Rev. 2015;115:11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine. 2014;6:224ra225–224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anassi E, Ndefo UA. Sipuleucel-T (provenge) injection: the first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharmacy and Therapeutics. 2011;36:197. [PMC free article] [PubMed] [Google Scholar]
- 10.Levine BL. T lymphocyte engineering ex vivo for cancer and infectious disease. Expert opinion on biological therapy. 2008;8:475–489. doi: 10.1517/14712598.8.4.475. [DOI] [PubMed] [Google Scholar]
- 11.Hasan AN, Selvakumar A, O’Reilly RJ. Artificial Antigen Presenting Cells: An Off the Shelf Approach for Generation of Desirable T-Cell Populations for Broad Application of Adoptive Immunotherapy. Advancements in Genetic Engineering. 2015;2015 [PMC free article] [PubMed] [Google Scholar]
- 12.Steenblock ER, Wrzesinski SH, Flavell RA, Fahmy TM. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy. Expert opinion on biological therapy. 2009;9:451–464. doi: 10.1517/14712590902849216. [DOI] [PubMed] [Google Scholar]
- 13.June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nature Reviews Immunology. 2009;9:704–716. doi: 10.1038/nri2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA–Ig coated artificial antigen-presenting cells. Nature medicine. 2003;9:619–625. doi: 10.1038/nm869. Oelke et al. demonstrates the use of a form of dimerized MHC with cognate antigen for TCR specific T-Cell expanion ex vivo. [DOI] [PubMed] [Google Scholar]
- 15.Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends in Biotechnology. 2014;32:456–465. doi: 10.1016/j.tibtech.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer RA, Green JJ. Biomaterials in Regenerative Medicine and the Immune System. Springer; 2015. Artificial Antigen-Presenting Cells: Biomimetic Strategies for Directing the Immune Response; pp. 257–277. [Google Scholar]
- 17.Zappasodi R, Di Nicola M, Carlo-Stella C, Mortarini R, Molla A, Vegetti C, Albani S, Anichini A, Gianni AM. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. haematologica. 2008;93:1523–1534. doi: 10.3324/haematol.12521. [DOI] [PubMed] [Google Scholar]
- 18.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 19.Shen K, Thomas VK, Dustin ML, Kam LC. Micropatterning of costimulatory ligands enhances CD4+ T cell function. Proceedings of the National Academy of Sciences. 2008;105:7791–7796. doi: 10.1073/pnas.0710295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doh J, Irvine DJ. Immunological synapse arrays: patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proceedings of the National Academy of Sciences. 2006;103:5700–5705. doi: 10.1073/pnas.0509404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine. 2013;8:1173–1189. doi: 10.2217/nnm.13.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sunshine JC, Perica K, Schneck JP, Green JJ. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials. 2014;35:269–277. doi: 10.1016/j.biomaterials.2013.09.050. Sunshine et. al. provides evidence that non-spherical aAPC activate T-Cells much more effectively than spherical aAPC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ. Biodegradable Nanoellipsoidal Artificial Antigen Presenting Cells for Antigen Specific T-Cell Activation. Small. 2014 doi: 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oelke M, Schneck JP. Overview of a HLA-Ig based “Lego-like system” for T cell monitoring, modulation and expansion. Immunologic research. 2010;47:248–256. doi: 10.1007/s12026-009-8156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Herrin SM, Slansky JE, Tang Q, Markiewicz MA, Gajewski TF, Pardoll DM, Schneck JP, Bluestone JA. Antigen-specific blockade of T cells in vivo using dimeric MHC peptide. The Journal of Immunology. 2001;167:2555–2560. doi: 10.4049/jimmunol.167.5.2555. [DOI] [PubMed] [Google Scholar]
- 26.Kosmides AK, Meyer RA, Hickey JW, Aje K, Cheung KN, Green JJ, Schneck JP. Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma. Biomaterials. 2016 doi: 10.1016/j.biomaterials.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu X, Jiang X, Liu R, Zhao H, Liang Z. Adoptive transfer of pTRP2-specific CTLs expanding by bead-based artificial antigen-presenting cells mediates anti-melanoma response. Cancer letters. 2008;271:129–139. doi: 10.1016/j.canlet.2008.05.049. [DOI] [PubMed] [Google Scholar]
- 28.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler MO, Lee J-S, Ansén S, Neuberg D, Hodi FS, Murray AP, Drury L, Berezovskaya A, Mulligan RC, Nadler LM, Hirano N. Long-Lived Antitumor CD8+ Lymphocytes for Adoptive Therapy Generated Using an Artificial Antigen-Presenting Cell. Clinical Cancer Research. 2007;13:1857–1867. doi: 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 30.Kim JV, Latouche J-B, Rivière I, Sadelain M. The ABCs of artificial antigen presentation. Nature biotechnology. 2004;22:403–410. doi: 10.1038/nbt955. [DOI] [PubMed] [Google Scholar]
- 31.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunological Reviews. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, Bühring H-J, Jung G, Kwon BS, Rammensee H-G. Potent costimulation of human CD8 T cells by anti-4–1BB, anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunology, Immunotherapy. 2008;57:175–183. doi: 10.1007/s00262-007-0360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, Weber J, Hwu P, Radvanyi L. Co-stimulation through 4–1BB/CD137 improves the expansion and function of CD8+ melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PloS one. 2013;8:e60031. doi: 10.1371/journal.pone.0060031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chacon J, Wang Y, Wu R, Hwu P, Radvanyi L. Costimulation through 4–1BB/CD137 improves the expansion and function of CD8+ melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy (66.21) The Journal of Immunology. 2011;186:66–21. [Google Scholar]
- 35.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, Buhring HJ, Jung G, Kwon BS, Rammensee HG, Stevanovic S. Potent costimulation of human CD8 T cells by anti-4–1BB, anti-CD28 on synthetic artificial antigen presenting cells. Cancer immunology, immunotherapy: CII. 2008;57:175–183. doi: 10.1007/s00262-007-0360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. Journal of Biological Chemistry. 2011;286:34883–34892. doi: 10.1074/jbc.M111.276329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santegoets SJ, Turksma AW, Suhoski MM, Stam AG, Albelda SM, Hooijberg E, Scheper RJ, van den Eertwegh AJ, Gerritsen WR, Powell DJ, Jr, June CH, de Gruijl TD. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J Transl Med. 2013;11:37. doi: 10.1186/1479-5876-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. Hubbell et al. provides an excellent review of the use of various materials for immunoengineering. [DOI] [PubMed] [Google Scholar]
- 39.Delcassian D, Depoil D, Rudnicka D, Liu M, Davis DM, Dustin ML, Dunlop IE. Nanoscale ligand spacing influences receptor triggering in T cells and NK cells. Nano letters. 2013;13:5608–5614. doi: 10.1021/nl403252x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schütz C, Fleck M, Mackensen A, Zoso A, Halbritter D, Schneck JP, Oelke M. Killer artificial antigen-presenting cells: a novel strategy to delete specific T cells. Blood. 2008;111:3546–3552. doi: 10.1182/blood-2007-09-113522. Schütz et al. engineers aAPC particles for TCR specific T-Cell elimination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schütz C, Fleck M, Schneck JP, Oelke M. Killer artificial antigen presenting cells (KaAPC) for efficient in vitro depletion of human antigen-specific T cells. Journal of visualized experiments: JoVE. 2014:e51859. doi: 10.3791/51859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schütz C, Oelke M, Schneck JP, Mackensen A, Fleck M. Killer artificial antigen-presenting cells: the synthetic embodiment of a ‘guided missile’. 2010 doi: 10.2217/imt.10.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desiderio SV. B-cell activation. Current opinion in immunology. 1992;4:252–256. doi: 10.1016/0952-7915(92)90073-n. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez MI, Catalan-Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine. 2015;74:318–326. doi: 10.1016/j.cyto.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harwood NE, Batista FD. Early events in B cell activation. Annual review of immunology. 2009;28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- 46.Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VLJ, Batista FD. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol. 2008;9:63–72. doi: 10.1038/ni1547. [DOI] [PubMed] [Google Scholar]
- 47.Lee J, Sengupta P, Brzostowski J, Lippincott-Schwartz J, Pierce SK. The nanoscale spatial organization of B cell receptors on IgM-and IgG-expressing human B cells. Molecular Biology of the Cell. 2016 doi: 10.1091/mbc.E16-06-0452. mbc. E16-06-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Tang S, Wan Z, Gao Y, Cao Y, Yi J, Si Y, Zhang H, Liu L, Liu W. Utilization of a photoactivatable antigen system to examine B-cell probing termination and the B-cell receptor sorting mechanisms during B-cell activation. Proceedings of the National Academy of Sciences. 2016;113:E558–E567. doi: 10.1073/pnas.1517612113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Temchura VV, Kozlova D, Sokolova V, Überla K, Epple M. Targeting and activation of antigen-specific B-cells by calcium phosphate nanoparticles loaded with protein antigen. Biomaterials. 2014;35:6098–6105. doi: 10.1016/j.biomaterials.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 50.Ingale J, Stano A, Guenaga J, Sharma SK, Nemazee D, Zwick MB, Wyatt RT. High-Density Array of Well-Ordered HIV-1 Spikes on Synthetic Liposomal Nanoparticles Efficiently Activate B Cells. Cell reports. 2016;15:1986–1999. doi: 10.1016/j.celrep.2016.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett NR, Zwick DB, Courtney AH, Kiessling LL. Multivalent Antigens for Promoting B and T Cell Activation. ACS chemical biology. 2015;10:1817–1824. doi: 10.1021/acschembio.5b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nature Medicine. 2016 doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shook DR, Campana D. Natural killer cell engineering for cellular therapy of cancer. Tissue antigens. 2011;78:409–415. doi: 10.1111/j.1399-0039.2011.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS one. 2012;7:e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perica K, Kosmides AK, Schneck JP. Linking form to function: biophysical aspects of artificial antigen presenting cell design. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2015;1853:781–790. doi: 10.1016/j.bbamcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Falconnet D, Csucs G, Grandin HM, Textor M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials. 2006;27:3044–3063. doi: 10.1016/j.biomaterials.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 57.Matic J, Deeg J, Scheffold A, Goldstein I, Spatz JP. Fine tuning and efficient T cell activation with stimulatory aCD3 nanoarrays. Nano letters. 2013;13:5090–5097. doi: 10.1021/nl4022623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prakken B, Wauben M, Genini D, Samodal R, Barnett J, Mendivil A, Leoni L, Albani S. Artificial antigen-presenting cells as a tool to exploit the immunesynapse’. Nature medicine. 2000;6:1406–1410. doi: 10.1038/82231. Prakken et al. develops liposome-based aAPC, which induced T-Cell receptor clustering. [DOI] [PubMed] [Google Scholar]
- 59.Giannoni F, Barnett J, Bi K, Samodal R, Lanza P, Marchese P, Billetta R, Vita R, Klein MR, Prakken B, Kwok WW, Sercarz E, Altman A, Albani S. Clustering of T Cell Ligands on Artificial APC Membranes Influences T Cell Activation and Protein Kinase C θ Translocation to the T Cell Plasma Membrane. The Journal of Immunology. 2005;174:3204–3211. doi: 10.4049/jimmunol.174.6.3204. [DOI] [PubMed] [Google Scholar]
- 60.Shannon MJ, Burn G, Cope A, Cornish G, Owen DM. Protein clustering and spatial organization in T-cells. Biochemical Society Transactions. 2015;43:315–321. doi: 10.1042/BST20140316. [DOI] [PubMed] [Google Scholar]
- 61.Owen DM, Rentero C, Rossy J, Magenau A, Williamson D, Rodriguez M, Gaus K. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. Journal of biophotonics. 2010;3:446–454. doi: 10.1002/jbio.200900089. [DOI] [PubMed] [Google Scholar]
- 62.Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harbor perspectives in biology. 2010;2:a002311. doi: 10.1101/cshperspect.a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. Grakoui et al. demonstrates the geometrical configuration of the immulogical synapse using supported lipid bilayers, establishing the importance for T-Cell activation. [DOI] [PubMed] [Google Scholar]
- 64.Chen HH, Ho Y-P, Jiang X, Mao H-Q, Wang T-H, Leong KW. Quantitative comparison of intracellular unpacking kinetics of polyplexes by a model constructed from quantum dot-FRET. Molecular therapy: the journal of the American Society of Gene Therapy. 2008;16:324–332. doi: 10.1038/sj.mt.6300392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hsu C-J, Hsieh W-T, Waldman A, Clarke F, Huseby ES, Burkhardt JK, Baumgart T. Ligand Mobility Modulates Immunological Synapse Formation and T Cell Activation. PLoS ONE. 2012;7:e32398. doi: 10.1371/journal.pone.0032398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson HA, Hiltbold EM, Roche PA. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nature immunology. 2000;1:156–162. doi: 10.1038/77842. [DOI] [PubMed] [Google Scholar]
- 67.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandal B, Bhattacharjee H, Mittal N, Sah H, Balabathula P, Thoma LA, Wood GC. Core–shell-type lipid–polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine: Nanotechnology, Biology and Medicine. 2013;9:474–491. doi: 10.1016/j.nano.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 69.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xuan M, Shao J, Dai L, Li J, He Q. Macrophage Cell Membrane Camouflaged Au Nanoshells for in Vivo Prolonged Circulation Life and Enhanced Cancer Photothermal Therapy. ACS Applied Materials & Interfaces. 2016;8:9610–9618. doi: 10.1021/acsami.6b00853. [DOI] [PubMed] [Google Scholar]
- 72.Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E. Biomimetic functionalization with leukocyte membranes imparts cell like functions to synthetic particles. Nature nanotechnology. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mandal S, Hammink R, Tel J, Eksteen-Akeroyd ZH, Rowan AE, Blank K, Figdor CG. Polymer-Based Synthetic Dendritic Cells for Tailoring Robust and Multifunctional T Cell Responses. ACS Chemical Biology. 2015;10:485–492. doi: 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- 74.Perica K, Medero ADL, Durai M, Chiu YL, Bieler JG, Sibener L, Niemöller M, Assenmacher M, Richter A, Edidin M. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomedicine: Nanotechnology, Biology and Medicine. 2014;10:119–129. doi: 10.1016/j.nano.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perica K, Bieler JG, Schütz C, Varela JC, Douglass J, Skora A, Chiu YL, Oelke M, Kinzler K, Zhou S. Enrichment and expansion with nanoscale artificial antigen presenting cells for adoptive immunotherapy. ACS nano. 2015;9:6861–6871. doi: 10.1021/acsnano.5b02829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perica K, Tu A, Richter A, Bieler JG, Edidin M, Schneck JP. Magnetic Field-Induced T Cell Receptor Clustering by Nanoparticles Enhances T Cell Activation and Stimulates Antitumor Activity. ACS Nano. 2014;8:2252–2260. doi: 10.1021/nn405520d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Batista FD, Iber D, Neuberger MS. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411:489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- 78.Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 Interaction Lowers the Threshold of B Cell Activation by Facilitating B Cell Adhesion and Synapse Formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- 79.Ketchum C, Miller H, Song W, Upadhyaya A. Ligand Mobility Regulates B Cell Receptor Clustering and Signaling Activation. Biophysical Journal. 2014;106:26–36. doi: 10.1016/j.bpj.2013.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B Cell Ligand Discrimination Through a Spreading and Contraction Response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- 81.Arana E, Vehlow A, Harwood NE, Vigorito E, Henderson R, Turner M, Tybulewicz Victor LJ, Batista FD. Activation of the Small GTPase Rac2 via the B Cell Receptor Regulates B Cell Adhesion and Immunological-Synapse Formation. Immunity. 2008;28:88–99. doi: 10.1016/j.immuni.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 82.Lin KBL, Freeman SA, Zabetian S, Brugger H, Weber M, Lei V, Dang-Lawson M, Tse KWK, Santamaria R, Batista FD, Gold MR. The Rap GTPases Regulate B Cell Morphology, Immune-Synapse Formation and Signaling by Particulate B Cell Receptor Ligands. Immunity. 2008;28:75–87. doi: 10.1016/j.immuni.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 83.Decuzzi P, Godin B, Tanaka T, Lee S-Y, Chiappini C, Liu X, Ferrari M. Size and shape effects in the biodistribution of intravascularly injected particles. Journal of Controlled Release. 2010;141:320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 84.Xu L, Auzins A, Sun X, Xu Y, Harnischfeger F, Lu Y, Li Z, Chen Y-H, Zheng W, Liu W. The synaptic recruitment of lipid rafts is dependent on CD19-PI3K module and cytoskeleton remodeling molecules. Journal of Leukocyte Biology. 2015;98:223–234. doi: 10.1189/jlb.2A0614-287RR. [DOI] [PubMed] [Google Scholar]
- 85.Zhang S, Xu L, Zhao X, Chen X, Fan Y, Wan Z, Xu Y, Liu W. A New and Robust Method of Tethering IgG Surrogate Antigens on Lipid Bilayer Membranes to Facilitate the TIRFM Based Live Cell and Single Molecule Imaging Experiments. PloS one. 2013;8:e63735. doi: 10.1371/journal.pone.0063735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tolar P, Hanna J, Krueger PD, Pierce SK. The Constant Region of the Membrane Immunoglobulin Mediates B Cell-Receptor Clustering and Signaling in Response to Membrane Antigens. Immunity. 2009;30:44–55. doi: 10.1016/j.immuni.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davey A, Liu W, Sohn HW, Brzostowski J, Pierce SK. Understanding the initiation of B cell signaling through live cell imaging. Methods in enzymology. 2012;506:265–290. doi: 10.1016/B978-0-12-391856-7.00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang C, Cen L, Yin S, Liu Q, Liu W, Cao Y, Cui L. A small diameter elastic blood vessel wall prepared under pulsatile conditions from polyglycolic acid mesh and smooth muscle cells differentiated from adipose-derived stem cells. Biomaterials. 2010;31:621–630. doi: 10.1016/j.biomaterials.2009.09.086. [DOI] [PubMed] [Google Scholar]
- 89.Wan Z, Liu W. The growth of B cell receptor microcluster is a universal response of B cells encountering antigens with different motion features. Protein & Cell. 2012;3:545–558. doi: 10.1007/s13238-012-2054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu W, Won Sohn H, Tolar P, Meckel T, Pierce SK. Antigen-Induced Oligomerization of the B Cell Receptor Is an Early Target of FcγRIIB Inhibition. The Journal of Immunology. 2010;184:1977–1989. doi: 10.4049/jimmunol.0902334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pihlgren M, Silva AB, Madani R, Giriens V, Waeckerle-Men Y, Fettelschoss A, Hickman DT, López-Deber MP, Ndao DM, Vukicevic M, Buccarello AL, Gafner V, Chuard N, Reis P, Piorkowska K, Pfeifer A, Kündig TM, Muhs A, Johansen P. TLR4- and TRIF-dependent stimulation of B lymphocytes by peptide liposomes enables T cell–independent isotype switch in mice. Blood. 2013;121:85–94. doi: 10.1182/blood-2012-02-413831. [DOI] [PubMed] [Google Scholar]
- 92.Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. The Journal of Clinical Investigation. 123:3074–3083. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang C, Liu P, Zhuang Y, Li P, Jiang B, Pan H, Liu L, Cai L, Ma Y. Lymphatic-targeted cationic liposomes: A robust vaccine adjuvant for promoting long-term immunological memory. Vaccine. 2014;32:5475–5483. doi: 10.1016/j.vaccine.2014.07.081. [DOI] [PubMed] [Google Scholar]
- 94.Flemming R, Murphy CJ, Abrams G, Goodman S, Nealey P. Effects of synthetic micro-and nano-structured surfaces on cell behavior. Biomaterials. 1999;20:573–588. doi: 10.1016/s0142-9612(98)00209-9. [DOI] [PubMed] [Google Scholar]
- 95.Shalaby WS, Yeh H, Woo E, Corbett JT, Gray H, June CH, Shalaby SW. Absorbable microparticulate cation exchanger for immunotherapeutic delivery. J Biomed Mater Res B Appl Biomater. 2004;69:173–182. doi: 10.1002/jbm.b.20040. [DOI] [PubMed] [Google Scholar]
- 96.Rossi M, Young JW. Human Dendritic Cells: Potent Antigen-Presenting Cells at the Crossroads of Innate and Adaptive Immunity. The Journal of Immunology. 2005;175:1373–1381. doi: 10.4049/jimmunol.175.3.1373. [DOI] [PubMed] [Google Scholar]
- 97.Knight SC, Stagg AJ. Antigen-presenting cell types. Current opinion in immunology. 1993;5:374–382. doi: 10.1016/0952-7915(93)90056-x. [DOI] [PubMed] [Google Scholar]
- 98.Valitutti S, Coombs D, Dupre L. The space and time frames of T cell activation at the immunological synapse. FEBS letters. 2010;584:4851–4857. doi: 10.1016/j.febslet.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 99.Al-Alwan MM, Rowden G, Lee TD, West KA. Cutting edge: the dendritic cell cytoskeleton is critical for the formation of the immunological synapse. The Journal of Immunology. 2001;166:1452–1456. doi: 10.4049/jimmunol.166.3.1452. [DOI] [PubMed] [Google Scholar]
- 100.Donnadieu E, Bismuth G, Trautmann A. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Current Biology. 1994;4:584–595. doi: 10.1016/s0960-9822(00)00130-5. [DOI] [PubMed] [Google Scholar]
- 101.Alarcon B, Mestre D, Martinez-Martin N. The immunological synapse: a cause or consequence of T-cell receptor triggering? Immunology. 2011;133:420–425. doi: 10.1111/j.1365-2567.2011.03458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility and sensitivity to antigen. Immunity. 1996;4:421–430. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- 103.Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Molecular Therapy. 2008;16:765–772. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 104.Chen B, Jia Y, Gao Y, Sanchez L, Anthony SM, Yu Y. Janus particles as artificial antigen-presenting cells for T cell activation. ACS applied materials & interfaces. 2014;6:18435–18439. doi: 10.1021/am505510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Paracrine co-delivery of TGF-β and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015;59:172–181. doi: 10.1016/j.biomaterials.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fifis T, Gamvrellis A, Crimeen-Irwin B, Pietersz GA, Li J, Mottram PL, McKenzie IF, Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. The Journal of Immunology. 2004;173:3148–3154. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 107.Meyer RA, Green JJ. Shaping the future of nanomedicine: anisotropy in polymeric nanoparticle design. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2015 doi: 10.1002/wnan.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ho C, Keller A, Odell J, Ottewill R. Preparation of monodisperse ellipsoidal polystyrene particles. Colloid and Polymer Science. 1993;271:469–479. [Google Scholar]
- 109.Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proc Natl Acad Sci U S A. 2007;104:11901–11904. doi: 10.1073/pnas.0705326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meyer RA, Meyer RS, Green JJ. An automated multidimensional thin film stretching device for the generation of anisotropic polymeric micro-and nanoparticles. Journal of Biomedical Materials Research Part A. 2015 doi: 10.1002/jbm.a.35399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma G, Valenta DT, Altman Y, Harvey S, Xie H, Mitragotri S, Smith JW. Polymer particle shape independently influences binding and internalization by macrophages. J Control Release. 2010;147:408–412. doi: 10.1016/j.jconrel.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Toy R, Peiris PM, Ghaghada KB, Karathanasis E. Shaping cancer nanomedicine: the effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine. 2013;9:121–134. doi: 10.2217/nnm.13.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barua S, Yoo J-W, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S. Particle shape enhances specificity of antibody-displaying nanoparticles. Proceedings of the National Academy of Sciences. 2013;110:3270–3275. doi: 10.1073/pnas.1216893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proc Natl Acad Sci U S A. 2013;110:10753–10758. doi: 10.1073/pnas.1308345110. Kolhar et. al. provides the evidence for non-spherical particles resulting in enhanced targeted tissue accumulation compared to spherical equivalents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fadel TR, Steenblock ER, Stern E, Li N, Wang X, Haller GL, Pfefferle LD, Fahmy TM. Enhanced cellular activation with single walled carbon nanotube bundles presenting antibody stimuli. Nano letters. 2008;8:2070–2076. doi: 10.1021/nl080332i. [DOI] [PubMed] [Google Scholar]
- 117.Fadel TR, Look M, Staffier PA, Haller GL, Pfefferle LD, Fahmy TM. Clustering of stimuli on single-walled carbon nanotube bundles enhances cellular activation. Langmuir. 2010;26:5645–5654. doi: 10.1021/la902068z. [DOI] [PubMed] [Google Scholar]
- 118.Fadel TR, Li N, Shah S, Look M, Pfefferle LD, Haller GL, Justesen S, Wilson CJ, Fahmy TM. Adsorption of Multimeric T Cell Antigens on Carbon Nanotubes: Effect on Protein Structure and Antigen-Specific T Cell Stimulation. Small. 2013;9:666–672. doi: 10.1002/smll.201201684. [DOI] [PubMed] [Google Scholar]
- 119.Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, Hong E, Li N, Haller GL, Pfefferle LD. A carbon nanotube–polymer composite for T-cell therapy. Nature nanotechnology. 2014;9:639–647. doi: 10.1038/nnano.2014.154. Fadel et. al. demonstrates the use of high surface area structures in conjunction with IL-2 release results in rapid expansion of CD8+ T-Cells for melanoma immunotherapy. [DOI] [PubMed] [Google Scholar]
- 120.Thomas SN, Schudel A. Overcoming transport barriers for interstitial-lymphatic- and lymph node-targeted drug delivery. Current opinion in chemical engineering. 2015;7:65–74. doi: 10.1016/j.coche.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Swartz MA, Hirosue S, Hubbell JA. Engineering Approaches to Immunotherapy. Science Translational Medicine. 2012;4:148rv149–148rv149. doi: 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- 122.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry kinetics and molecular patterns. Nature Reviews Immunology. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 123.Meyer RA, Sunshine JC, Green JJ. Biomimetic particles as therapeutics. Trends in biotechnology. 2015;33:514–524. doi: 10.1016/j.tibtech.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoo J-W, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Current pharmaceutical design. 2010;16:2298–2307. doi: 10.2174/138161210791920496. [DOI] [PubMed] [Google Scholar]
- 125.Kuokkanen E, Šuštar V, Mattila PK. Molecular control of B cell activation and immunological synapse formation. Traffic. 2015;16:311–326. doi: 10.1111/tra.12257. [DOI] [PubMed] [Google Scholar]
- 126.Yang C, DelRio FW, Ma H, Killaars AR, Basta LP, Kyburz KA, Anseth KS. Spatially patterned matrix elasticity directs stem cell fate. Proceedings of the National Academy of Sciences. 2016;113:E4439–E4445. doi: 10.1073/pnas.1609731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 128.Li Z, Gong Y, Sun S, Du Y, Lü D, Liu X, Long M. Differential regulation of stiffness, topography and dimension of substrates in rat mesenchymal stem cells. Biomaterials. 2013;34:7616–7625. doi: 10.1016/j.biomaterials.2013.06.059. [DOI] [PubMed] [Google Scholar]
- 129.Wong ST, Teo S-K, Park S, Chiam K-H, Yim EKF. Anisotropic rigidity sensing on grating topography directs human mesenchymal stem cell elongation. Biomechanics and Modeling in Mechanobiology. 2014;13:27–39. doi: 10.1007/s10237-013-0483-2. [DOI] [PubMed] [Google Scholar]
- 130.Onken MD, Mooren OL, Mukherjee S, Shahan ST, Li J, Cooper JA. Endothelial monolayers and transendothelial migration depend on mechanical properties of the substrate. Cytoskeleton. 2014;71:695–706. doi: 10.1002/cm.21203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chang H, Zhang H, Hu M, Chen J-y, Li B-c, Ren K-f, Martins MCL, Barbosa MA, Ji J. Stiffness of polyelectrolyte multilayer film influences endothelial function of endothelial cell monolayer. Colloids and Surfaces B: Biointerfaces. 2017;149:379–387. doi: 10.1016/j.colsurfb.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 132.Chang H, Liu X-q, Hu M, Zhang H, Li B-c, Ren K-f, Boudou T, Albiges-Rizo C, Picart C, Ji J. Substrate Stiffness Combined with Hepatocyte Growth Factor Modulates Endothelial Cell Behavior. Biomacromolecules. 2016;17:2767–2776. doi: 10.1021/acs.biomac.6b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, Hone JC, Kam LC. CD28 and CD3 have complementary roles in T-cell traction forces. Proceedings of the National Academy of Sciences. 2014;111:2241–2246. doi: 10.1073/pnas.1315606111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hui KL, Balagopalan L, Samelson LE, Upadhyaya A. Cytoskeletal forces during signaling activation in Jurkat T-cells. Molecular Biology of the Cell. 2015;26:685–695. doi: 10.1091/mbc.E14-03-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, Milone MC. Substrate rigidity regulates human T cell activation and proliferation. The Journal of Immunology. 2012;189:1330–1339. doi: 10.4049/jimmunol.1102757. O’Connor et al. shows that substrate rigidity affects T cell proliferation, differentiation, and activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Judokusumo E, Tabdanov E, Kumari S, Dustin Michael L, Kam Lance C. Mechanosensing in T Lymphocyte Activation. Biophysical Journal. 2012;102:L5–L7. doi: 10.1016/j.bpj.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wan Z, Zhang S, Fan Y, Liu K, Du F, Davey AM, Zhang H, Han W, Xiong C, Liu W. B Cell Activation Is Regulated by the Stiffness Properties of the Substrate Presenting the Antigens. The Journal of Immunology. 2013;190:4661–4675. doi: 10.4049/jimmunol.1202976. [DOI] [PubMed] [Google Scholar]
- 138.Zeng Y, Yi J, Wan Z, Liu K, Song P, Chau A, Wang F, Chang Z, Han W, Zheng W, Chen Y-H, Xiong C, Liu W. Substrate stiffness regulates B-cell activation, proliferation, class switch and T-cell-independent antibody responses in vivo. European Journal of Immunology. 2015;45:1621–1634. doi: 10.1002/eji.201444777. [DOI] [PubMed] [Google Scholar]
- 139.Li Y-C, Chen B-M, Wu P-C, Cheng T-L, Kao L-S, Tao M-H, Lieber A, Roffler SR. Cutting Edge: Mechanical Forces Acting on T Cells Immobilized via the TCR Complex Can Trigger TCR Signaling. The Journal of Immunology. 2010;184:5959–5963. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- 140.Kim ST, Takeuchi K, Sun Z-YJ, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, Reinherz EL. The αβ T Cell Receptor Is an Anisotropic Mechanosensor. Journal of Biological Chemistry. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rivas FV, O’Keefe JP, Alegre M-L, Gajewski TF. Actin Cytoskeleton Regulates Calcium Dynamics and NFAT Nuclear Duration. Molecular and Cellular Biology. 2004;24:1628–1639. doi: 10.1128/MCB.24.4.1628-1639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bufi N, Saitakis M, Dogniaux S, Buschinger O, Bohineust A, Richert A, Maurin M, Hivroz C, Asnacios A. Human Primary Immune Cells Exhibit Distinct Mechanical Properties that Are Modified by Inflammation. Biophysical Journal. 2015;108:2181–2190. doi: 10.1016/j.bpj.2015.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]