

Abstract
Molecular recognition is central to biological processes, function, and specificity. Proteins associate with ligands with a wide range of association rate constants, with maximal values matching the theoretical limit set by the rate of diffusional collision. As less is known about RNA association, we compiled association rate constants for all RNA/ligand complexes that we could find in the literature. Like proteins, RNAs exhibit a wide range of association rate constants. However, the fastest RNA association rates are considerably slower than those of the fastest protein associations and fall well below the diffusional limit. The apparently general observation of slow association with RNAs has implications for evolution and for modern-day biology. Our compilation highlights a quantitative molecular property that can contribute to biological understanding and underscores our need to develop a deeper physical understanding of molecular recognition events.
Keywords: molecular recognition, binding kinetics, RNA structure
INTRODUCTION
Molecular recognition underlies all of life. The individual molecules that determine the accuracy of what complexes form and what reactions occur are responsible for the fidelity of biological responses and ultimately for the cellular and organismal order that is emblematic of life. Nevertheless, the most accurate process is of no value to an organism if it cannot be carried out rapidly, to overcome stray chemical and physical processes and to outcompete other organisms.
The rate of association between molecules sets a speed limit for metabolic functions and regulatory responses, with a theoretical maximum set by the rate of diffusional collision (see Eigen and Hammes 1963; Berg and von Hippel 1985; Fersht 1999; Schreiber et al. 2009 and below). Underscoring the importance of speed, nature has evolved clever mechanisms to overcome the physical limit of three-dimensional diffusion, for example, allowing proteins to slide in one dimension along DNA or hop between DNAs and harnessing electrostatic interactions to speed associations (von Hippel and Berg 1989; Halford and Marko 2004; Schreiber et al. 2009). On the other end of the spectrum, association rates can fall many orders of magnitude below diffusion control, and in some cases slow association can contribute to regulation and specificity (e.g., Wu et al. 2002; James and Tawfik 2005).
Given the importance of molecular recognition and its speed, and the generally adopted model of an early RNA world that was later overcome by a protein-dominant world (Gilbert 1986; Cech 2009), we wondered whether there might be differences in molecular recognition between RNA and proteins at the level of association rates. Such differences could have contributed to the course of evolution and may impact current-day biology. On the molecular level, differences in association rates may reflect atomic-level properties critical in molecular recognition and may provide a unique window into understanding these processes.
RESULTS
To broadly assess the kinetics of association, we gathered all of the RNA/ligand association rate constants we could find in the literature (n = 64) (Supplemental Tables S1, S2). For comparison, we gathered a large number of protein/ligand association rate constants (n = 207) (Supplemental Table S3). We also examined a third category of association events, those between proteins and RNA molecules (n = 22) (Supplemental Table S4). While we expect that our RNA/ligand list is nearly complete, there are too many protein/ligand measurements to fully assemble (see Materials and Methods). Nevertheless, we expect our list to be reasonably representative and suitable for revealing trends and stimulating ideas and discussion about their physical origins and biological implications.
RNA and protein ligand association rate constants
Figure 1 shows rate constants for ligands binding to structured RNAs and to proteins (leftmost and middle columns, respectively). We expect a generic upper limit corresponding to diffusional collision at ∼109 M−1 sec−1 (Eigen and Hammes 1963; Berg and von Hippel 1985; Fersht 1999; Schreiber et al. 2009). For protein ligands, the values span a wide range of over 108-fold and approach and even exceed the diffusional limit, as electrostatic attraction between the protein and ligand can increase their rate of association (e.g., Lambeth et al. 1980; Wallis et al. 1995; Schreiber and Fersht 1996; Radic et al. 1997; Polticelli et al. 1998; and see Schreiber et al. 2009 and references therein). There is also a large range observed for RNA/ligand association rate constants. However, the highest values are distinctly lower than those for proteins, falling well below the diffusional limit (Fig. 1, compare left and middle columns).
FIGURE 1.
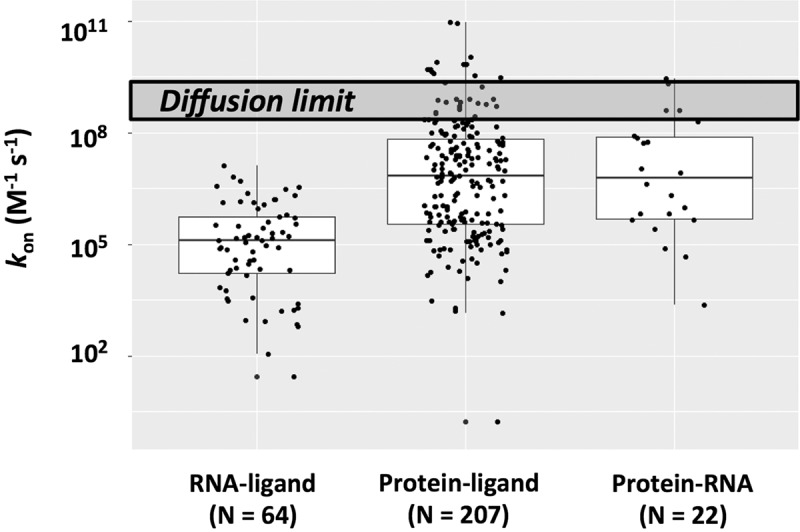
Rate constants for ligands binding to structured RNAs (left column) and to proteins (middle column) and for RNA•protein association (right column). Values are presented in Supplemental Tables S1–S3, and “N” is the number of examples in each column. The upper dark gray box represents a generic diffusion limit centered around 109 M−1 sec−1. In the box plot representations, the line inside the rectangle shows the median, the box spans the first quartile to the third quartile (representing the span from 0.25 to 0.75 of the ranked values), and the whiskers are shown as guides. The median values are as follows: RNA–ligand: 1.0 × 105 M−1 sec−1; Protein–ligand: 6.6 × 106 M−1 sec−1; and Protein–RNA: 6.3 × 106 M−1 sec−1.
As a crude test of whether there is some type of intrinsic limitation to the rate of associations involving RNA, we separately plotted rate constants for the association between RNA and proteins (Fig. 1, rightmost column). The values exceeded those for RNA with non-protein binding partners (Fig. 1, compare left and right columns) and reach the upper range observed for other protein/ligand associations (Fig. 1, compare middle and right columns), indicating that there is no inherent limitation to association involving RNA.
To further probe association rates involving RNA we parsed its association rates (Fig. 2), first separating out RNA/RNA associations (“RNA ligands”) from those with non-RNA ligands (“Small molecule ligands”), and, for the RNA/RNA group, further demarcating subgroups that involve formation of base pairs only (“BP”), formation of base pairs and additional tertiary interactions (“BP + 3°”), and formation of non-Watson–Crick tertiary interactions only (“3°”).4 RNA binding to structured RNAs that involve base-pair formation is significantly faster than binding that does not (Fig. 2, compare “BP” and “3°”), although the number and diversity of RNAs binding via only tertiary contacts is small. Interestingly, the binding of structured RNAs via base-pairing occurs at rates similar to those for simple duplex formation between short RNA oligomers (Fig. 2, compare “BP” and “RNA duplex”; “RNA duplex” values are from Turner et al. 1990 and are listed in Supplemental Table S5), suggesting that there are no general constraints or accelerants to base-pairing in structured RNAs. Non-RNA ligands bind to structured RNAs with rate constants extending up to those for recognition via base-pair formation (Fig. 2, compare “Small molecule ligands” and “BP”), although on average the values are somewhat lower and a wide range of values is observed. Interestingly, the measured values for naturally occurring and in vitro–selected RNAs have similar means and span similar ranges (Fig. 2, compare “Natural” and “In vitro” columns above “Small molecule ligands”).
FIGURE 2.

Rate constants for association involving RNA, separated into RNA/RNA associations (left), RNA/small molecule associations (middle), and RNA duplex formation (right). Rate constants between structured RNAs and RNA ligands are shown in the first three columns, and are further subdivided into interactions that involve formation of base pairs only (BP), formation of base pairs and additional tertiary interactions (BP + 3°), and formation of non-Watson–Crick, tertiary interactions only (3°). For the binding of non-RNA ligands to structured RNAs, association rate constants are broken down into naturally occurring and in vitro–selected RNAs. Values are presented in Supplemental Tables S1–S3 and S5, and “N” is the number of examples in each column. The median values are as follows: RNA-ligand: BP: 3.3 × 105 M−1 sec−1; (BP + 3°): 5.7 × 106 M−1 sec−1; 3°: 2.0 × 103 M−1 sec−1. The median values for RNA-small molecule ligands are 5.5 × 104 and 8.1 × 104 M−1 sec−1 for natural and in vitro RNAs, respectively. The median value for RNA duplex is 1.3 × 106 M−1 sec−1.
Implications and mechanisms of slow RNA association
We consider the assembled RNA association data from biological and then physical perspectives. Our goal is to stimulate discussion about potential implications and origins of the observed trends and, ultimately, experiments that reveal the physical bases for the observed behavior and the ensuing biological manifestations.
Implications of slow RNA recognition in early evolution
We previously proposed that nonspecific RNA chaperones provided the first foothold into the RNA world for peptides and then proteins (Tsuchihashi et al. 1993; Herschlag 1995), and it is possible that association rates played a role in these transitions.
According to this model, short peptide sequences that nonspecifically bind RNA acted as chaperones; short polymers and nonspecific interactions would be more likely to arise evolutionarily than long polymers and specific interactions, and RNA's tendency to fall into kinetic traps (Gartland and Sueoka 1966; Lindahl et al. 1966; Herschlag 1995; Uhlenbeck 1995; Russell 2008) would allow a selective advantage from nonspecific chaperones that could aid RNAs’ escape from these traps and thereby yield a higher fraction of RNA molecules in functional conformations (Fig. 3). Considering association rates, it is also possible that early peptides provided selective advantages by increasing association rates, as has been seen in model RNA/peptide and protein systems (e.g., Kumar and Wilson 1990; Munroe and Dong 1992; Herschlag et al. 1994; Portman and Dreyfuss 1994).
FIGURE 3.

Schematic depiction of the progression from an RNA world to the modern biological world. We use the RNA and protein subunits of RNase P as well as tRNA synthetase to illustrate this progression. RNA's tendency to form long-lived misfolded states is depicted on the far left. (i) Short peptide sequences that nonspecifically bind RNA can act as chaperones and provide a selective advantage for an organism that can escape these traps to yield a higher fraction of RNA molecules in functional conformations. (ii) Once peptides gained a foothold there would be a selective advantage for longer and more accurate peptide production that could lead to specific binding and to structured proteins. (iii) As protein catalytic function emerged and competed with ribozymes, RNA association rates may have limited catalytic efficiency and contributed to the current widespread use of proteins for catalysis. The blue circle corresponds to the amino acid covalently attached to tRNA by tRNA synthetase.
The next evolutionary step, according to this model, was the transition to longer peptides and specific functions. Once peptides gained a foothold there would be advantages and successive selection for longer and more accurate peptide production, presumably leading to specific binding and structured proteins (Fig. 3, “RNA + protein”) (Herschlag 1995; Cech 2009). But why would proteins have supplanted RNAs in functional roles so resoundingly? Common and reasonable ideas include an ability of proteins to create more specific folds and binding pockets (Herschlag 1995; Russell 2008) and to carry out more efficient and a broader range of chemical catalysis with their >20 amino acid side chains than the four canonical RNA side chains (Herschlag 1995; Narlikar and Herschlag 1997). Instead or additionally, the diversity of protein side chains instead of RNA side chains may have arisen because of the constraint on RNA to also serve as genetic material, because of more facile routes to synthesize and incorporate additional amino acid side chains (e.g., Huber et al. 2003; Leman et al. 2004) and/or because of limits imposed on RNA folding, molecular recognition, and catalytic function by its uniformly charged backbone (Narlikar and Herschlag 1997).
The observations herein suggest that an additional driving force could have been operative: faster molecular recognition by proteins than by RNA (Figs. 1, 3). Especially if the metabolic milieu in the RNA world was rather complex, with efficient ribozymes limited by their recognition speed, increasing association rates and thus enzyme efficiency beyond that possible with ribozymes could have provided a selective advantage for proteins. Indeed, the potential for faster recognition by proteins may also have provided some of the selective advantage for additional amino acid side chains with different “shapes” that can better preorganize protein structures and thereby speed ligand binding (see below).
Implications of slow RNA recognition in modern-day biology
The simplest model for the presence of complex structured RNAs that carry out functions in modern biology is that these are vestiges from an earlier RNA world (Gilbert 1986; Cech 2009). Indeed, the RNA core of the ribosome provides the strongest single argument for functional RNAs predating proteins (Cech 2000; Nissen et al. 2000). While it is possible that all modern-day RNAs are evolutionary vestiges that would be eliminated in a perfectly engineered—rather than evolved—organism, it is also possible that certain processes benefit from utilization of RNA, and that some of this utilization is connected to how RNA recognizes ligands.
Base-pairing is a highly effective and highly specific means to recognize and discriminate between RNAs, and is highly modular. While there are cases of modular proteins that bind individual bases, each protein module involves multiple amino acids and thus requires a longer and less probable an evolutionary search to switch recognition specificity to another base than the single base swap needed for RNA. For this reason, recognition by RNA may be more evolvable (Houle 1992; Wagner and Altenberg 1996; Kirschner and Gerhart 1998; Wagner 2005; Bloom et al. 2006). It may also be harder to achieve near-uniform affinity for any base accompanied by high specificity for that base with protein modules than with individual RNA bases. Finally, base pairs in RNA duplexes are themselves accessible and can therefore be recognized by additional RNA elements (or proteins) to increase (or decrease) base-pair affinity and specificity (e.g., Leontis et al. 2002; Carlson et al. 2003; Arya 2011; Masliah et al. 2013 and Hougland et al. 2006 and references therein).
When else might RNA prove advantageous? Folding studies have demonstrated that RNA readily adopts stable alternative structures—and we and others have argued and shown that these alternative structures can be inactive and deleterious (Gartland and Sueoka 1966; Lindahl et al. 1966; Herschlag 1995; Uhlenbeck 1995; Russell 2008); however, riboswitches are biological RNAs that use such metastable states for regulation (Haller et al. 2011; Serganov and Patel 2012; Furtig et al. 2015), and ribosome and spliceosome function rely on multiple conformational states (Guthrie and Patterson 1988; Madhani and Guthrie 1994; Voorhees and Ramakrishnan 2013; Chen and Moore 2014; Warnasooriya and Rueda 2014; Rodnina et al. 2017). Thus, kinetically trapped metastable states could have been co-opted through evolution to provide a series of functional conformational steps, with these steps controlled by proteins such as ATPases and GTPases in pre-mRNA splicing and translation (Staley and Guthrie 1998; Chang et al. 2013; Jarmoskaite and Russell 2014; Bourgeois et al. 2016).
Finally, the slow observed ligand recognition by RNA provides an opportunity for kinetic control of specificity when binding is rate limiting (Karbstein and Herschlag 2003; Gleitsman and Herschlag 2014). In this mechanism, referred to as “kon specificity,” added neighboring interactions can increase the lifetime of an initial encounter complex and thereby provide an attached ligand multiple chances at productive binding prior to diffusing away and thus a higher kon. For RNA, adding neighboring interactions can be as simple as adding residues that can base pair with the structured RNA, and indeed there is evidence for this mechanism with the Tetrahymena group I intron (Karbstein and Herschlag 2003). There, residues neighboring the guanosine at the 3′-intron–exon junction form base pairs with intron sequences near the guanosine binding site and make association of that guanosine faster and more probable than other G residues. Similarly, positively charged functional groups on ligands can provide an “anchor” that increases residence times and thus association rate constants. There is evidence for such an effect for a positively charged guanosine analog binding to a group I intron (RN Sengupta and D Herschlag, unpubl.) and analogous effects may be operative in aminoglycoside antibiotics binding to specific ribosome states (Vicens and Westhof 2003; Trylska and Kulik 2016) and other structured RNAs (Stage et al. 1995; Walter et al. 1999). These interactions can allow a ligand to “wait” for the formation of an inhibitable state and need not even be maintained in the final bound state, and it may be possible to co-opt this strategy for new therapeutics that target RNA.
Physical origins of slow RNA recognition
Molecular binding events are typically represented as simple association processes (Fig. 4A). The widespread awareness that the predominant conformational states of free and bound RNAs often differ has led to considerable discussion about induced fit or conformational capture models in which conformational changes occur after the initial encounter or prior to the encounter, respectively (Fig. 4B) (e.g., Leulliot and Varani 2001; Rose and Weeks 2001; Webb et al. 2001; Boehr et al. 2009; Daniels et al. 2014). Most kinetic experiments cannot distinguish these pathways (Chakraborty and Di Cera 2017), but even more critically, these alternatives represent limiting models for a wide range of possible pathways (Fig. 4C); labeling a process by one of these two designations does not reveal the myriad of molecular rearrangements and atomic-level forces that make up the actual pathway and determine the efficiency of association.
FIGURE 4.
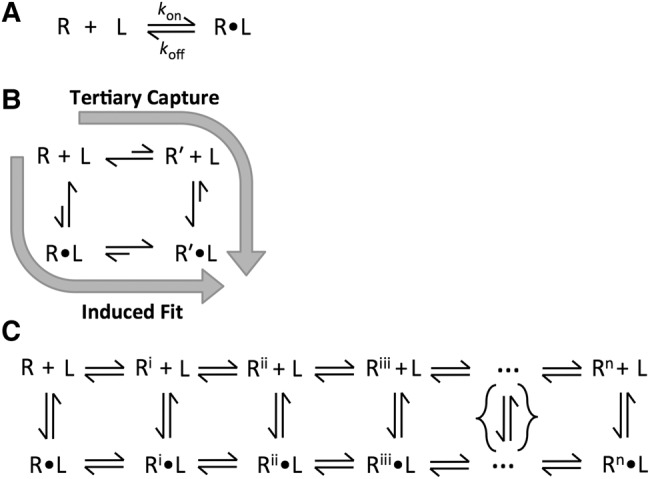
Descriptions of molecular binding events. (A) Simple model of ligand binding. Binding event between receptor (R) and ligand (L) to form receptor-ligand complex (R•L) represented as simple association processes governed by a single set of association and dissociation rates. When association is slower than diffusion-limited collision, additional steps are needed to describe binding. (B) Induced fit and conformational capture models of ligand binding. These models account for slower-than-diffusional binding. The tertiary capture model invokes conformational changes prior to the encounter between R and L, while the induced fit model represents the opposite extreme in which conformational changes occur after the initial encounter. (C) Complex model of ligand binding considering an ensemble of conformational states and series of conformational changes to the receptor during the association process. Even the complex representation in the figure is a vast simplification, as it depicts a linear order of conformation changes (and a limited number of conformational steps) rather than a larger and more realistic multi-dimensional energy landscape that can account for multiple orders for the local conformational changes and multiple possible binding pathways.
The nucleic acid association process that we know the most about is duplex formation (Fig. 5; see Bloomfield et al. 2000 and references therein). Early work on simple, short DNA oligos showed that duplex formation was much slower than diffusion and revealed a large temperature dependence (Ross and Sturtevant 1960; Wetmur and Davidson 1968), something not observed for diffusional processes (Berg and von Hippel 1985), and similar rates and temperature dependences are observed for RNA duplexes (Pörschke and Eigen 1971; Pörschke et al. 1973; Nelson and Tinoco 1982). The observed enthalpies of activation (ΔH‡) derived from these temperature dependences match enthalpies for the formation of 2–3 base pairs, leading to a zipper model involving nucleation via the formation of 2–3 bp, after which subsequent base-pair formation was favored over the dissociation of the 2–3 bp nucleation complex (Fig. 5; Pörschke 1977). More recent optical trap experiments with DNA hairpins provided strong support for this model (Woodside et al. 2006).
FIGURE 5.
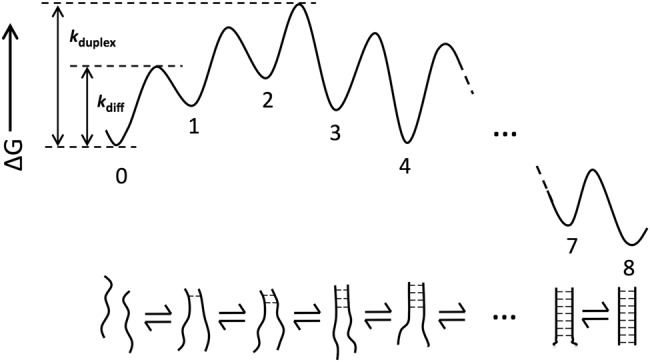
Free energy diagram for simple duplex formation zipper model. In the zipper model, nucleation involves the formation of 2–3 bp, after which subsequent base-pair formation is favored over the dissociation of the nucleation complex. The bimolecular association rate between two ssRNA molecules is limited by this nucleation rate (kduplex). Because the formation of these 2–3 bp is uphill in energy from an initial encounter between the two strands, the association rate for duplex formation (kduplex) is slower than diffusional encounter (kdiff).
While the above results provide a compelling model, they do not provide the detailed timing and extent of the individual conformational transitions that together constitute the binding process and, correspondingly, they do not explain “why” the process has its maximal barrier subsequent to the formation of two base pairs. Indeed, even duplex formation is extraordinarily complex, involving many bond rotations, changes in solvation, and the breaking and making of multiple hydrogen bonds, van der Waals, and stacking interactions. As the steps involved in association represent basic RNA conformational transitions, learning more about association processes has the potential to deepen our fundamental understanding of RNA. More generally, the observation of slower association with structured RNAs than structured proteins provides a perspective for considering the question of the molecular features involved in RNA conformational dynamics and ligand association. Below we briefly consider molecular properties that differ between proteins and RNA and may contribute to their different association rates.
Limited conformational preorganization: The limited number and diversity of RNA side chains and the greater number of backbone rotatable bonds, relative to protein side chains and its backbone, have been suggested to limit RNA packing and preorganization, and electrostatic repulsion from the uniformly charged RNA backbone may provide additional limitations (Narlikar and Herschlag 1997). Limited preorganization in the conformational substates matching that for the bound complex introduce the need for rearrangements prior to and/or accompanying binding and thus additional free energy barriers to binding. Indeed, it has been suggested that protein•RNA complexes recognize RNA or DNA by base-pairing faster than naked RNA because of preorganization of the bound RNA strand, although electrostatic attraction introduced with the protein partner could also or instead be responsible (Gorski et al. 2017 and references therein; see also below). Finally, we note that intrinsically disordered proteins (IDPs) can bind ligands faster than observed for structured RNAs (Shammas et al. 2016), indicating that an absence of structure alone does not compel slow binding, and we discuss this further below.
Binding pockets: The concave geometry of many protein binding sites may facilitate association, by increasing the probability of encountering multiple tethering interactions, once the ligand enters the pocket. The shape of the binding pocket may also facilitate desolvation, by not allowing the water–water interactions that are most favorable in bulk water. Nevertheless, concavity may also decrease the number of collisions, underscoring the multiplicity of features that influence complex association events.
Steric accessibility: Limited steric accessibility can slow association, but even proteins that surround their bound ligands can bind those ligands rapidly, presumably because the needed loop opening and closings and domain motions associated with binding are sufficiently rapid (e.g., Rozovsky and McDermott 2001; Henzler-Wildman et al. 2007). We know much less about the steric accessibility of binding sites for structured RNAs in solution and their opening and closing or breathing rates.
Backbone degrees of freedom: Limited conformational preorganization within binding sites, as noted above, necessitates structural rearrangements to access configurations that allow or favor binding. In the course of these rearrangements, RNA may in general explore more conformational states than proteins that require rearrangement, due to RNA's more flexible backbone (Narlikar and Herschlag 1997). A broader search landscape would be expected to lengthen searches and lower the probability of finding the “right” subset of conformational states—those that allow productive binding to proceed.
RNA base and sugar motions: When RNA rearrangements are needed for ligand binding, the bulkiness and rigidity of their base and sugar components might slow the needed motions, as they would require a larger vacated space to move into than a more flexible linear protein side chain, which might be likened to the ability of octopus to fit through narrow crevices (see https://www.youtube.com/watch?v=949eYdEz3Es). Indeed, as noted above, even intrinsically disordered proteins (IDPs) can bind ligands faster than observed for structured RNAs (Shammas et al. 2016). This faster binding is consistent with significant kinetic barriers for RNA rearrangements to binding-competent conformations that are absent for the IDPs. Nevertheless, electrostatic and/or hydrophobic IDP•ligand interactions could also or instead increase residence times and thus association rates, as described above for kon specificity, underscoring the complexity of molecular association events and the need for careful and systematic investigation.
Hydrophobic interactions: Hydrophobic interactions, which occur in many protein–ligand binding processes (Fersht 1999), are less directional than hydrogen bonds and less constrained than stacking interactions and thus may allow facile exploration in early association complexes to “find” the additional binding interactions needed to favor formation of the final bound state. Hydrophobic groups may also shed solvating water more readily than polar and charged groups to allow native contacts to form. Nevertheless, protein ligands range in their hydrophobicity, polarity, and charge, so this strategy is unlikely to account for all fast protein associations; different association events are likely to differ substantially.
Hydrogen bonds: The ability of RNA bases to form two or three hydrogen bonds with interaction partners would seem to be an advantage for association by increasing the residence time of precursor complex states. Perhaps these hydrogen bonds are weaker and short-lived, relative to at least some protein–ligand hydrogen bonds. Alternatively, hydrogen bonds (and other interactions) stabilizing the alternative state of the RNA may also be more stable or more numerous and thus slower to resolve, thereby leading to more dissociation of encounter complexes prior to formation of a stably bound state.
Solvation and electrostatics: Macromolecular motions require or are coupled to solvent motions, and the charged nature of RNA may result in greater coupling and more restricted motion. It is also possible that reorganization of the ion atmosphere that surrounds RNA as a polyelectrolyte (Lipfert et al. 2014) must also readjust to facilitate motions and engenders an additional impediment to rearrangement.
The myriad interactions and possibilities outlined above highlight that we have a long way to go to understand association involving both RNA and proteins. We hope that comparing and contrasting these different biomolecules has helped illuminate underlying factors; nevertheless, the factors are too numerous and varied to expect mechanistic clarity from these rudimentary comparisons alone. Understanding RNA and protein associations at the atomic level will require an integration of information-rich experiments that provide detailed information at high time resolution and, given the large number of interactions and degrees of freedom in these systems, tight feedback between these experiments and predictions from atomic-level molecular dynamics models. Finally, we have not proven that RNA cannot bind ligands faster; it will be fascinating to explore this question through SELEX-type experiments that increase the selective pressure for fast and not just strong binding.
Closing remarks
Nature is powered by but also limited by the physical and chemical properties of its constituent materials and by the laws of physics. The slow association observed for ligands binding to structured RNAs may reflect basic physical properties of RNA and may have impacted the course of evolution and the molecules and molecular behaviors seen in modern-day biology. We hope that the observations and initial discussion presented herein will stimulate discussions, models, and new experiments that deepen our understanding across the interface of physics and biology.
MATERIALS AND METHODS
Compiling kinetic data from the literature
Association rate constants were assembled from peer-reviewed literature through an extensive internet search using both Google Scholar and the PubMed database. Search terms included “association rate constant,” “on rate,” “kon,” “binding rate,” “ligand binding kinetics,” “association kinetics,” “fast association rate,” and “slow association rate.” For nucleic acids, the terms “duplex formation,” “duplex association,” and “base-pair association” were also used. Papers identified through this search were manually scanned for relevant information including the association rate constant and the identity of the binding partners and their classification (RNA, DNA, protein, small molecule). For RNA binding processes, we also compiled the method used for determining the association rate constant (e.g., surface plasmon resonance, NMR, etc.), experimental conditions (buffer, temperature, etc.), and, when available, the dissociation rate constant (koff) and equilibrium dissociation constant (Kd). This information was used to create Supplemental Tables S1–S5.
Our aim was to create a database that was representative of the repertoire of macromolecular association rate constants. To avoid overrepresentation, for papers that contained multiple mutants of a single protein, typically only the wild-type association rate constant was entered. A notable exception comes from papers where the mutations were specifically aimed at altering binding kinetics; in these cases typically the extrema and wild-type values were included (e.g., work on barnase/barstar [Schreiber and Fersht 1996]). Similarly, for binding experiments where a large number of binding conditions were explored, association rate constants for every condition may not be included in the table; however, the conditions for the listed association rate constant are indicated.
Potential sources of bias
Whereas the protein data set contains different types of proteins from various organisms and biochemical pathways (Supplemental Table S3), the RNA data set (Supplemental Tables S1, S2) is less diverse, having a few predominant RNA types. Further, many of these RNAs have been artificially truncated to facilitate handling and biochemical analyses and these alterations have the potential to affect activity (e.g., Uhlenbeck 2003) and kon (e.g., Soulière et al. 2013).
Second, while in principle molecular constants are independent of the technique used to obtain them, there are on occasion technique-specific effects. For example, surface plasmon resonance (SPR) involves association at a surface, so surface effects and accessibility can affect the rate of binding (e.g., Myszka 1997). Also, care is needed in applying this and other techniques and in analyzing results to prevent errors in measurement and interpretation (e.g., Fried 1989; Hellman and Fried 2007; Suddala and Walter 2014; Vander Meulen et al. 2016). For the RNA data presented, there were no systematic differences in association rate constants for SPR versus the other methods (not shown).
Finally, binding interactions with a strong electrostatic component will be highly sensitive to salt concentration. For example, the kon value for the theophylline RNA aptamer increases by two orders of magnitude from 0 to 10 mM MgCl2 (Latham et al. 2009). Similarly, the Barnase–Barnstar association rate constant increases by four orders of magnitude from 5 mM to 2 M NaCl (Schreiber and Fersht 1996). These studies underscore the importance of varying reaction conditions to evaluate the sensitivity of kon toward salt concentration, and the ability to learn about this component of binding through such studies.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This paper is dedicated to Olke Uhlenbeck on the occasion of his retirement in honor of his contributions to RNA biochemistry and in celebration of his ceaseless and contagious enthusiasm for science. This work was supported by a grant from the National Institutes of Health to D.H. (GM049243 and GM066275) and a National Institutes of Health training grant to R.N.S. (5 T32 GM007276). K.R.G. gratefully acknowledges a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund. We thank members of the Herschlag laboratory for helpful comments and discussion.
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062026.117.
For RNAs that form non-Watson–Crick tertiary interactions only (3°), kon values were obtained for tertiary contact partners physically separated in two molecules (see Supplemental Table S2 and references therein).
REFERENCES
- Arya DP. 2011. New approaches toward recognition of nucleic acid triple helices. Accounts Chem Res 44: 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg OG, von Hippel PH. 1985. Diffusion-controlled macromolecular interactions. Annu Rev Biophys Biophys Chem 14: 131–160. [DOI] [PubMed] [Google Scholar]
- Bloom JD, Labthavikul ST, Otey CR, Arnold FH. 2006. Protein stability promotes evolvability. Proc Natl Acad Sci 103: 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield VA, Crothers DM, Tinoco I. 2000. Nucleic acids: structure, properties, and functions. University Science Books, Sausalito, CA. [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. 2009. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 5: 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois CF, Mortreux F, Auboeuf D. 2016. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol 17: 426–438. [DOI] [PubMed] [Google Scholar]
- Carlson CB, Stephens OM, Beal PA. 2003. Recognition of double-stranded RNA by proteins and small molecules. Biopolymers 70: 86–102. [DOI] [PubMed] [Google Scholar]
- Cech TR. 2000. Structural biology. The ribosome is a ribozyme. Science 289: 878–879. [DOI] [PubMed] [Google Scholar]
- Cech TR. 2009. Crawling out of the RNA world. Cell 136: 599–602. [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Di Cera E. 2017. Induced fit is a special case of conformational selection. Biochemistry 56: 2853–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Tung L, Yeh FL, Chen JH, Chang SL. 2013. Functions of the DExD/H-box proteins in nuclear pre-mRNA splicing. Biochim Biophys Acta 1829: 764–774. [DOI] [PubMed] [Google Scholar]
- Chen WJ, Moore MJ. 2014. The spliceosome: disorder and dynamics defined. Curr Opin Struct Biol 24: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels KG, Tonthat NK, McClure DR, Chang YC, Liu X, Schumacher MA, Fierke CA, Schmidler SC, Oas TG. 2014. Ligand concentration regulates the pathways of coupled protein folding and binding. J Am Chem Soc 136: 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M, Hammes GG. 1963. Elementary steps in enzyme reactions (as studied by relaxation spectrometry). Adv Enzymol Relat Subj Biochem 25: 1–38. [DOI] [PubMed] [Google Scholar]
- Fersht A. 1999. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W.H. Freeman, New York. [Google Scholar]
- Fried MG. 1989. Measurement of protein-DNA interaction parameters by electrophoresis mobility shift assay. Electrophoresis 10: 366–376. [DOI] [PubMed] [Google Scholar]
- Furtig B, Nozinovic S, Reining A, Schwalbe H. 2015. Multiple conformational states of riboswitches fine-tune gene regulation. Curr Opin Struct Biol 30: 112–124. [DOI] [PubMed] [Google Scholar]
- Gartland WJ, Sueoka N. 1966. Two interconvertible forms of tryptophanyl sRNA in E. coli. Proc Natl Acad Sci 55: 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W. 1986. Origin of life: the RNA world. Nature 319: 618. [Google Scholar]
- Gleitsman KR, Herschlag DH. 2014. A kinetic and thermodynamic framework for the Azoarcus group I ribozyme reaction. RNA 20: 1732–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski SA, Vogel J, Doudna JA. 2017. RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol 18: 215–228. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Patterson B. 1988. Spliceosomal snRNAs. Annu Rev Genet 22: 387–419. [DOI] [PubMed] [Google Scholar]
- Halford SE, Marko JF. 2004. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res 32: 3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller A, Souliere MF, Micura R. 2011. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc Chem Res 44: 1339–1348. [DOI] [PubMed] [Google Scholar]
- Hellman LM, Fried MG. 2007. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc 2: 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, et al. 2007. Intrinsic motions along an enzymatic reaction trajectory. Nature 450: 838–844. [DOI] [PubMed] [Google Scholar]
- Herschlag D. 1995. RNA chaperones and the RNA folding problem. J Biol Chem 270: 20871–20874. [DOI] [PubMed] [Google Scholar]
- Herschlag D, Khosla M, Tsuchihashi Z, Karpel RL. 1994. An RNA chaperone activity of non-specific RNA binding proteins in hammerhead ribozyme catalysis. EMBO J 13: 2913–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougland JL, Piccirilli JA, Forconi M, Lee J, Herschlag D. 2006. How the group I intron works: a case study of RNA structure and function. In RNA world, 3rd ed, Vol. 43, pp. 133–205. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Houle D. 1992. Comparing evolvability and variability of quantitative traits. Genetics 130: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber C, Eisenreich W, Hecht S, Wachtershausher G. 2003. A possible primordial peptide cycle. Science 301: 938–940. [DOI] [PubMed] [Google Scholar]
- James LC, Tawfik DS. 2005. Structure and kinetics of a transient antibody binding intermediate reveal a kinetic discrimination mechanism in antigen recognition. Proc Natl Acad Sci 102: 12730–12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I, Russell R. 2014. RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem 83: 697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbstein K, Herschlag D. 2003. Extraordinarily slow binding of guanosine to the Tetrahymena group I ribozyme: implications for RNA preorganization and function. Proc Natl Acad Sci 100: 2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M, Gerhart J. 1998. Evolvability. Proc Natl Acad Sci 95: 8420–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Wilson SH. 1990. Studies of the strand-annealing activity of mammalian hnRNP complex protein A1. Biochemistry 29: 10717–10722. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Seybert DW, Kamin H. 1980. Adrenodoxin reductase. Adrenodoxin complex. Rapid formation and breakdown of the complex and a slow conformational change in the flavoprotein. J Biol Chem 255: 4667–4672. [PubMed] [Google Scholar]
- Latham MP, Zimmermann GR, Pardi A. 2009. NMR chemical exchange as a probe for ligand-binding kinetics in a theophylline-binding RNA aptamer. J Am Chem Soc 131: 5052–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman L, Orgel L, Ghadiri MR. 2004. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 306: 283–286. [DOI] [PubMed] [Google Scholar]
- Leontis NB, Stombaugh J, Westhof E. 2002. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res 30: 3497–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N, Varani G. 2001. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry 40: 7947–7956. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Adams A, Fresco JR. 1966. Renaturation of transfer ribonucleic acids through site binding of magnesium. Proc Natl Acad Sci 55: 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Doniach S, Das R, Herschlag D. 2014. Understanding nucleic acid-ion interactions. Annu Rev Biochem 83: 813–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994. Dynamic RNA–RNA interactions in the spliceosome. Annu Rev Genet 28: 1–26. [DOI] [PubMed] [Google Scholar]
- Masliah G, Barraud P, Allain FHT. 2013. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci 70: 1875–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe SH, Dong XF. 1992. Heterogeneous nuclear ribonucleoprotein A1 catalyzes RNA.RNA annealing. Proc Natl Acad Sci 89: 895–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myszka DG. 1997. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotechnol 8: 50–57. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Herschlag D. 1997. Mechanistic aspects of enzymatic catalysis: lessons from comparison of RNA and protein enzymes. Annu Rev Biochem 66: 19–59. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Tinoco I. 1982. Comparison of the kinetics of ribooligonucleotide, deoxyribooligonucleotide, and hybrid oligonucleotide double-strand formation by temperature-jump kinetics. Biochemistry 21: 5289–5295. [DOI] [PubMed] [Google Scholar]
- Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289: 920–930. [DOI] [PubMed] [Google Scholar]
- Polticelli F, Battistoni A, O'Neill P, Rotilio G, Desideri A. 1998. Role of the electrostatic loop charged residues in Cu, Zn superoxide dismutase. Protein Sci 7: 2354–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pörschke D. 1977. Elementary steps of base recognition and helix-coil transitions in nucleic acids. Mol Biol Biochem Biophys 24: 191–218. [DOI] [PubMed] [Google Scholar]
- Pörschke D, Eigen M. 1971. Co-operative non-enzymic base recognition. III. Kinetics of the helix–coil transition of the oligoribouridylic · oligoriboadenylic acid system and of oligoriboadenylic acid alone at acidic pH. J Mol Biol 62: 361–381. [DOI] [PubMed] [Google Scholar]
- Pörschke D, Uhlenbeck OC, Martin FH. 1973. Thermodynamics and kinetics of the helix-coil transition of oligomers containing GC base pairs. Biopolymers 12: 1313–1335. [Google Scholar]
- Portman DS, Dreyfuss G. 1994. RNA annealing activities in HeLa nuclei. EMBO J 13: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic Z, Kirchhoff PD, Quinn DM, McCammon JA, Taylor P. 1997. Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J Biol Chem 272: 23265–23277. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Fischer N, Maracci C, Stark H. 2017. Ribosome dynamics during decoding. Philos Trans R Soc B 372: 20160182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MA, Weeks KM. 2001. Visualizing induced fit in early assembly of the human signal recognition particle. Nat Struct Biol 8: 515–520. [DOI] [PubMed] [Google Scholar]
- Ross PD, Sturtevant JM. 1960. The kinetics of double helix formation from polyriboadenylic acid and polyribouridylic acid. Proc Natl Acad Sci 46: 1360–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovsky S, McDermott AE. 2001. The time scale of the catalytic loop motion in triosephosphate isomerase. J Mol Biol 310: 259–270. [DOI] [PubMed] [Google Scholar]
- Russell R. 2008. RNA misfolding and the action of chaperones. Front Biosci 13: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. 1996. Rapid, electrostatically assisted association of proteins. Nat Structl Biol 3: 427–431. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Haran G, Zhou HX. 2009. Fundamental aspects of protein-protein association kinetics. Chem Rev 109: 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, Patel DJ. 2012. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu Rev Biophys 41: 343–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas SL, Crabtree MD, Dahal L, Wicky BI, Clarke J. 2016. Insights into coupled folding and binding mechanisms from kinetic studies. J Biol Chem 291: 6689–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulière MF, Altman RB, Schwarz V, Haller A, Blanchard SC, Micura R. 2013. Tuning a riboswitch response through structural extension of a pseudoknot. Proc Natl Acad Sci 110: E3256–E3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage TK, Hertel KJ, Uhlenbeck OC. 1995. Inhibition of the hammerhead ribozyme by neomycin. RNA 1: 95–101. [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Suddala KC, Walter NG. 2014. Riboswitch structure and dynamics by smFRET microscopy. Methods Enzymol 549: 343–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trylska J, Kulik M. 2016. Interactions of aminoglycoside antibiotics with rRNA. Biochem Soc Trans 44: 987–993. [DOI] [PubMed] [Google Scholar]
- Tsuchihashi Z, Khosla M, Herschlag D. 1993. Protein enhancement of hammerhead ribozyme catalysis. Science 262: 99–102. [DOI] [PubMed] [Google Scholar]
- Turner DH, Sugimoto N, Freier SM. 1990. Thermodynamics and kinetics of base-pairing and of DNA and RNA self-assembly and helix coli transition: nucleic acids. In Landolt-Börnstein, Vol. 1: Nucleic acids. Group 7: Biophysics, pp. 201–227. Springer-Verlag, Berlin. [Google Scholar]
- Uhlenbeck OC. 1995. Keeping RNA happy. RNA 1: 4–6. [PMC free article] [PubMed] [Google Scholar]
- Uhlenbeck OC. 2003. Less isn't always more. RNA 9: 1415–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Meulen KA, Horowitz S, Trievel RC, Butcher SE. 2016. Measuring the kinetics of molecular association by isothermal titration calorimetry. Methods Enzymol 567: 181–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicens Q, Westhof E. 2003. RNA as a drug target: the case of aminoglycosides. Chembiochem 4: 1018–1023. [DOI] [PubMed] [Google Scholar]
- von Hippel PH, Berg OG. 1989. Facilitated target location in biological systems. J Biol Chem 264: 675–678. [PubMed] [Google Scholar]
- Voorhees RM, Ramakrishnan V. 2013. Structural basis of the translational elongation cycle. Annu Rev Biochem 82: 203–236. [DOI] [PubMed] [Google Scholar]
- Wagner A. 2005. Robustness and evolvability in living systems. Princeton University Press, Princeton, NJ. [Google Scholar]
- Wagner GP, Altenberg L. 1996. Perspective: complex adaptations and the evolution of evolvability. Evolution 50: 967–976. [DOI] [PubMed] [Google Scholar]
- Wallis R, Moore GR, James R, Kleanthous C. 1995. Protein-protein interactions in colicin E9 DNase-immunity protein complexes. 1. Diffusion-controlled association and femtomolar binding for the cognate complex. Biochemistry 34: 13743–13750. [DOI] [PubMed] [Google Scholar]
- Walter F, Vicens Q, Westhof E. 1999. Aminoglycoside–RNA interactions. Curr Opin Chem Biol 3: 694–704. [DOI] [PubMed] [Google Scholar]
- Warnasooriya C, Rueda D. 2014. Single-molecule fluorescence-based studies on the dynamics, assembly and catalytic mechanism of the spliceosome. Biochem Soc Trans 42: 1211–1218. [DOI] [PubMed] [Google Scholar]
- Webb AE, Rose MA, Westhof E, Weeks KM. 2001. Protein-dependent transition states for ribonucleoprotein assembly. J Mol Biol 309: 1087–1100. [DOI] [PubMed] [Google Scholar]
- Wetmur JG, Davidson N. 1968. Kinetics of renaturation of DNA. J Mol Biol 31: 349–370. [DOI] [PubMed] [Google Scholar]
- Woodside MT, Behnke-Parks WM, Larizadeh K, Travers K, Herschlag D, Block SM. 2006. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc Natl Acad Sci 103: 6190–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. 2002. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature 418: 552–556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.