

Abstract
Protein-only ribonuclease P (PRORP) is an enzyme responsible for catalyzing the 5′ end maturation of precursor transfer ribonucleic acids (pre-tRNAs) encoded by various cellular compartments in many eukaryotes. PRORPs from plants act as single-subunit enzymes and have been used as a model system for analyzing the function of the metazoan PRORP nuclease subunit, which requires two additional proteins for efficient catalysis. There are currently few molecular details known about the PRORP–pre-tRNA complex. Here, we characterize the determinants of substrate recognition by the single subunit Arabidopsis thaliana PRORP1 and PRORP2 using kinetic and thermodynamic experiments. The salt dependence of binding affinity suggests 4–5 contacts with backbone phosphodiester bonds on substrates, including a single phosphodiester contact with the pre-tRNA 5′ leader, consistent with prior reports of short leader requirements. PRORPs contain an N-terminal pentatricopeptide repeat (PPR) domain, truncation of which results in a >30-fold decrease in substrate affinity. While most PPR-containing proteins have been implicated in single-stranded sequence-specific RNA recognition, we find that the PPR motifs of PRORPs recognize pre-tRNA substrates differently. Notably, the PPR domain residues most important for substrate binding in PRORPs do not correspond to positions involved in base recognition in other PPR proteins. Several of these residues are highly conserved in PRORPs from algae, plants, and metazoans, suggesting a conserved strategy for substrate recognition by the PRORP PPR domain. Furthermore, there is no evidence for sequence-specific interactions. This work clarifies molecular determinants of PRORP–substrate recognition and provides a new predictive model for the PRORP–substrate complex.
Keywords: RNase P, PRORP, PPR, pre-tRNA, molecular recognition
INTRODUCTION
Ribonuclease P (RNase P) enzymes are essential endonucleases with diverse macromolecular composition that are responsible for catalyzing the maturation of the 5′ end of pre-tRNA (Howard et al. 2013). In many biological settings, RNase P is a ribonucleoprotein complex containing a large catalytic RNA capable of processing pre-tRNAs in vitro (Guerrier-Takada et al. 1983). Additionally, one or more associated protein components are required for function in vivo (Walker and Engelke 2006; Marvin and Engelke 2009). In bacterial RNase P, the protein subunits increase substrate affinity and the ability of divalent metal ions to bind at specific sites (Crary et al. 1998; Kurz et al. 1998; Niranjanakumari et al. 1998; Kurz and Fierke 2002).
In many eukaryotic species, including protists, algae, land plants, and metazoans, protein-only RNase Ps (PRORPs) have been identified (Holzmann et al. 2008; Gobert et al. 2010; Lai et al. 2011; Taschner et al. 2012). Human mitochondrial RNase P (mtRNase P) was the first PRORP described and it requires two additional protein subunits for activity (Holzmann et al. 2008). These subunits are an m1G/A9 tRNA-methyltransferase (TRMT10C, also MRPP1) and a hydroxysteroid dehydrogenase/reductase (HSD17B10, also MRPP2), which form a subcomplex (Holzmann et al. 2008; Vilardo et al. 2012). MRPP1 and MRPP2 are proposed to contribute primarily to substrate recognition.
In contrast to the metazoan PRORP, the PRORPs from algae, protists, and plants do not require additional subunits for efficient catalysis in vitro (Gobert et al. 2010; Lai et al. 2011; Gutmann et al. 2012; Taschner et al. 2012; Sugita et al. 2014; Howard et al. 2015; Bonnard et al. 2016), suggesting differences in substrate recognition. The three PRORPs from Arabidopsis thaliana are designated PRORP1–3. AtPRORP1 localizes to the mitochondria and chloroplasts where it is responsible for catalyzing pre-tRNA maturation (Gobert et al. 2010), while AtPRORP2 and AtPRORP3 colocalize to the nucleus and are not fully redundant in nuclear pre-tRNA processing (Gutmann et al. 2012). AtPRORP1 utilizes a metal ion-dependent mechanism similar to the mechanism of the ribozyme, relying on ionization of metal-bound waters for nucleophile activation in catalysis (Chen et al. 1997; Howard et al. 2015). Given the additional mechanistic information and the relative simplicity of the AtPRORPs, they have been used as a model system to study PRORP–substrate molecular recognition.
PRORPs contain a unique three domain architecture (Fig. 1A). An N-terminal pentatricopeptide repeat (PPR) domain significantly enhances the affinity for substrate, and truncation of the first 3–4 repeats abolishes catalytic activity (Howard et al. 2012; Imai et al. 2014; Karasik et al. 2016). Thus, the PPR domain is proposed to both bind and orient substrate with respect to the metallonuclease domain (Howard et al. 2012). In addition, the nuclease domain is a member of the Nedd4-BP1, YacP nuclease (NYN) family (Anantharaman and Aravind 2006). Lastly, a bipartite CC/HC Zn2+-binding domain flanks the NYN domain (Howard et al. 2012). Our current understanding of how each domain, in particular the PPR domain (Fig. 1B; Supplemental Fig. S1), contributes to PRORP substrate recognition is limited.
FIGURE 1.
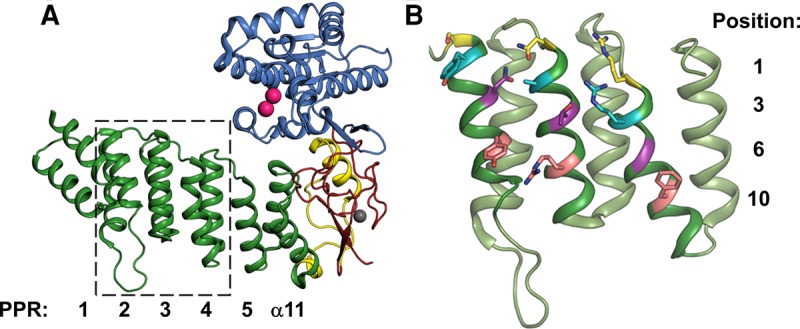
Structure of Arabidopsis thaliana PRORP1 (PDB 4g24), generated in PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4, Schrödinger, LLC). (A) Overall architecture of AtPRORP1. PPR motifs are in green and numbered from the N-terminal end, a plant-specific helical insertion in yellow (α12–13), central domain in red, and NYN domain in blue. Mn2+ ions displayed as pink spheres and Zn2+ ion displayed as a gray sphere. The region marked by the dashed box is expanded in panel B. (B) AtPRORP1 PPR domain motifs 2–4, generated in PyMOL. For each PPR motif, position 1 is colored yellow, position 3 is colored cyan, position 6 is colored purple, and position 10 is colored pink.
Previous results suggest differences in substrate recognition between the bacterial RNA-dependent RNase P and PRORP. Unlike the ribozyme, the PRORP active site metal ions apparently do not contact the pro-RP oxygen (Pavlova et al. 2012), but rather contact the pro-SP oxygen of the scissile phosphodiester bond (Walczyk et al. 2016). Furthermore, while the 3′-CCA is specifically recognized by bacterial RNase P RNA, it is either inhibitory or immaterial to AtPRORP activity (Gobert et al. 2013; Brillante et al. 2016; Mao et al. 2016). Additionally, AtPRORP1 and AtPRORP3 do not significantly contact either the 5′ leader sequence beyond N-2 or the 3′ trailer (Fig. 2A); these regions do not alter substrate affinity or catalytic activity (Brillante et al. 2016; Howard et al. 2016). The minimal 5′ and 3′ end interactions indicate that PRORP substrate recognition lies primarily within the tRNA body.
FIGURE 2.
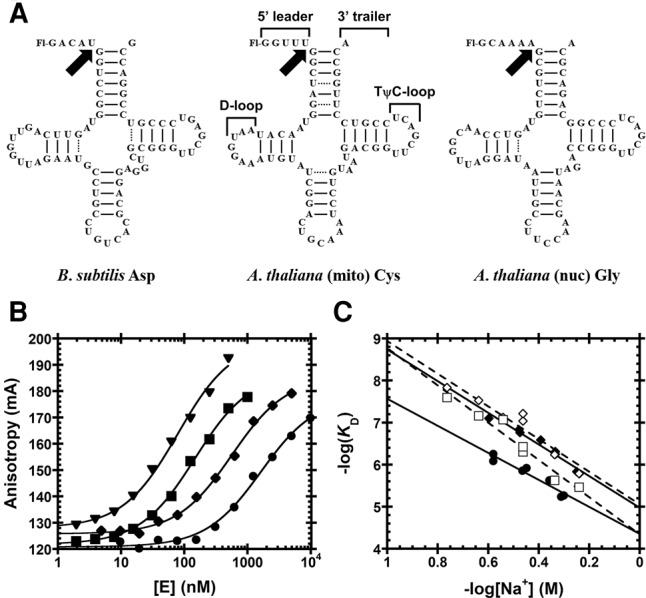
Substrates used for functional assays. (A) Substrates containing a 5′-fluorescein label include pre-tRNAAsp from Bacillus subtilis and Arabidopsis thaliana pre-tRNACys and pre-tRNAGly from the mitochondrial and nuclear genomes, respectively. Structural features of pre-tRNA are detailed on pre-tRNACys, including the 5′ leader, 3′ trailer, and the D- and TψC-loops. Black arrows indicate canonical RNase P cleavage site. (B) Fluorescence anisotropy binding curves for AtPRORP1 binding to B. subtilis pre-tRNAAsp (log scale). A hyperbola (Equation 1, Materials and Methods) was fit to the data. Data were measured in 30 mM MOPS pH 7.8, 1 mM TCEP, and 20 mM CaCl2 with 250 mM (▾), 330 mM (▪), 450 mM (♦), and 550 mM NaCl (•). (C) Na+ dependence of AtPRORP affinity. Equation 4 (Materials and Methods) was fit to the data. The slope of the line (Z) reports on the apparent number of ionic interactions with substrate phosphodiester bonds, while the intercept [log(K0)] reports on the nonionic contributions to affinity. Data include AtPRORP1 binding to pre-tRNAAsp (♦) and pre-tRNACys (•) in 20 mM Ca2+, as well as AtPRORP2 in 6 mM Ca2+ binding to pre-tRNAAsp (⋄) and pre-tRNAGly (□).
Previously, a nuclease footprinting assay demonstrated that there was significant protection of bases in the D- and TψC-loops (Fig. 2A) by AtPRORP1 (Gobert et al. 2013). Given these data and the likelihood that the NYN domain binds at the scissile phosphodiester bond, it was proposed that the PPRs recognized the pre-tRNA elbow, the structure formed by interaction between the D- and TψC-loops (Gobert et al. 2013). However, this proposal remains to be tested. Recent attempts to alter base specificity of the PPR domain in AtPRORP3 were unsuccessful (Brillante et al. 2016). Furthermore, while the TψC-arm is sufficient for recognition and catalysis by plant PRORPs, the presence of a D-arm increases the affinity significantly (Brillante et al. 2016; Howard et al. 2016). These data provide a basis for examining the features of PRORP that contribute to recognition of pre-tRNA.
A model of substrate-bound AtPRORP1 was previously generated using molecular dynamics and includes the PPR domain docked to the TψC-loop (Imai et al. 2014). The authors assumed that PRORPs use the recognition strategy used by several single-stranded RNA binding PPR proteins. The ssRNA-binding PPR proteins recognize nucleobases utilizing residues in two tandem repeats at positions 6 and 1′ (Fig. 1B; Supplemental Fig. S1), as well as hydrophobic amino acids at position 3 that contribute to binding affinity by van der Waals or stacking interactions (Barkan et al. 2012; Yagi et al. 2013). Cleavage assays catalyzed by AtPRORP1 indicated that mutations to position 6 of PPR motifs 2, 3, and 4 reduce activity modestly (≤70% reduction) (Imai et al. 2014). However, the full suite of PPR residues important for PRORP substrate recognition remains to be identified.
Here, we characterize the mode of substrate binding and recognition by the highly conserved AtPRORP1 and 2 using a variety of biochemical techniques. The salt dependence of pre-tRNA affinity indicates that AtPRORP1 and AtPRORP2 make at least four direct contacts to substrate backbone phosphodiester bonds, including a single phosphodiester bond contact with the pre-tRNA leader. Importantly, these interactions with the backbone are not sequence specific. However, the salt dependence of affinity for mature tRNA also demonstrates that a significant portion of the affinity for substrate stems from interactions with the sugars and/or bases in the body of the substrate, in contrast to the bacterial ribozyme, which makes more contacts with the 5′ leader and 3′ CCA. To test whether AtPRORP1 uses canonical PPR–nucleobase interactions, we mutated residues in both the PRORP PPR domain and nucleotides in a pre-tRNA substrate and assessed how the mutations impact the PRORP–pre-tRNA affinity. In contrast to other known PPR proteins, PRORP does not exhibit demonstrable sequence selectivity for substrate affinity, suggesting that substrate recognition relies instead on the three-dimensional structure of pre-tRNA. These experiments provide a biochemical framework for understanding molecular recognition of complex RNA structures by the noncanonical PPRs of plant PRORPs.
RESULTS
AtPRORP–substrate recognition mode
To begin characterizing how AtPRORPs recognize their cognate substrates, we set out to determine the general mode of substrate binding. We first measured the dependence of the protein–nucleic acid interaction on the concentration and identity of ions in solution. These data parse the dependence of affinity on ionic interactions with backbone phosphodiester bonds, compared to that of nonionic interactions. Monovalent and divalent cations directly interact with backbone phosphodiester bonds on nucleic acids. These ions must be released for a protein to directly contact those sites, thus affinity depends on the cation concentration (Record et al. 1978; Barkley et al. 1981). Cations associate with nucleic acids through an ionic atmosphere also containing anions, which inhibits protein–nucleic acid interactions through a related screening mechanism (Record et al. 1978).
We measured the dependence of the substrate binding affinity of AtPRORP1 and AtPRORP2 on ions in solution to estimate the number of backbone phosphodiester bond contacts. We determined dissociation constants (KD) for AtPRORP1 and 2 by fluorescence anisotropy (FA) assays using three substrates: a B. subtilis pre-tRNAAsp (AtPRORP1 and 2), an A. thaliana mitochondrial pre-tRNACys (AtPRORP1), each with 5-nt leaders, and an A. thaliana nuclear pre-tRNAGly (AtPRORP2) with a 6-nt leader; all substrates have a fluorescein label at the 5′ end (Fig. 2A). The pre-tRNAAsp substrate has been used extensively with the bacterial ribozyme, allowing us to make direct comparisons to PRORPs, while the pre-tRNACys is a cognate substrate for AtPRORP1 and the pre-tRNAGly is a cognate substrate for AtPRORP2. We obtained thermodynamic affinities (KD) by fitting a hyperbola (Equation 1, Materials and Methods) to the data (Fig. 2B).
The Na+ dependence of KD shows a linear dependence in a log–log plot (Fig. 2C), as described by Equation S1 (Supplemental Methods), which was adapted from Equation 18 of deHaseth et al. (1977). Divalent cations are required to fold the pre-tRNA, so CaCl2, which does not activate AtPRORP1 or AtPRORP2 (Howard et al. 2012; Karasik et al. 2016), was supplied at a constant value for each measurement. The primary effect of Ca2+ in our assays is to increase the anisotropy of free pre-tRNA (see the next section), while the affinity we measure at different CaCl2 concentrations varied ≤65% at a given NaCl concentration. Thus, we selected CaCl2 concentrations that allowed us to best measure the affinity under high concentrations of NaCl (20 or 6 mM CaCl2 for AtPRORP1 or AtPRORP2, respectively). We observe minimal competition between Ca2+ and Na+ for the RNA substrate under the concentrations used for the binding assays for AtPRORP1, as evidenced by the relatively linear Na+ dependence in the log–log plot (Fig. 2C). We maintained constant pH during the experiments and anion effects are precluded based on the CaCl2 alone and Na2SO4 data described below. In the absence of these effects, Equation S1 can be reduced to Equation 4 (Materials and Methods), which was fit to the data.
The slope of a −log(KD) versus −log[Na+] plot is given by Zϕ (Equation 4), represented as Zφ for divalent cations, where ϕNa is the fraction of Na+ associated thermodynamically with each backbone phosphodiester bond and Z is the number of cations (M+) that are released from the nucleic acid upon binding to the protein, which approximates the number of protein–phosphodiester bond contacts. Previous data suggest that the value of ϕNa for dsRNA and structured RNAs, such as pre-tRNA, are comparable to the ϕNa for dsDNA (Latt and Sober 1967; Day-Storms et al. 2004). Thus, we used the value for dsDNA, ϕNa = 0.88, in fitting Equation 4 to the data (Table 1). The Z-values for AtPRORP1 suggest the formation of four protein–phosphodiester bond contacts upon binding pre-tRNA. The values for AtPRORP2 are higher, possibly suggesting contacts with five phosphodiester groups. Furthermore, these Z-values do not have a high dependence on the estimated value of ϕNa. Specifically, the Z-value for AtPRORP2 binding to pre-tRNAGly increases to 6 if ϕNa < 0.85. However, Z remains ≤5 for the other PRORP/pre-tRNA pairs until ϕNa < 0.75 and is ≤6 until ϕNa < 0.6, which is likely to be well below the actual value.
TABLE 1.
Na+ dependence of binding affinity
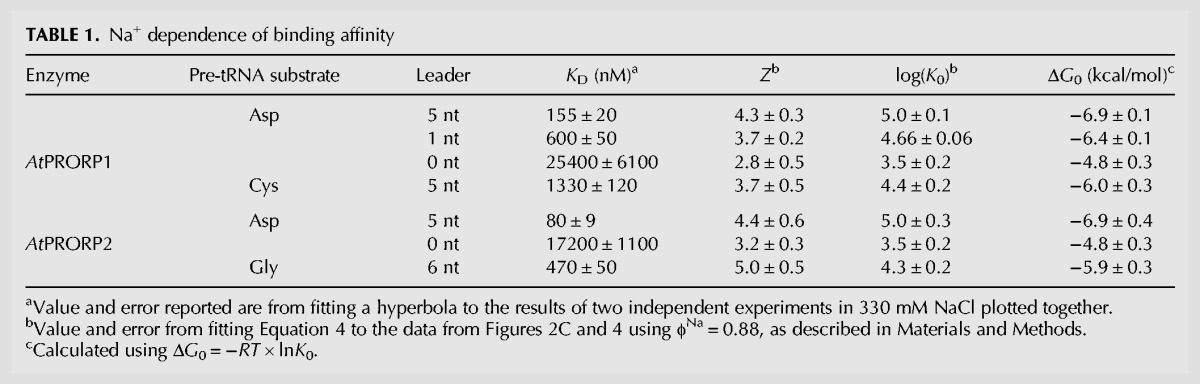
The extrapolated substrate affinity at 1 M NaCl has been used to estimate the contribution of nonionic interactions to affinity in model systems, using normal Gibbs free energy definitions (Equation 5) (Record et al. 1976). For AtPRORP1 at 27°C, the −log(KD) at 1 M NaCl indicates values of −6.9 ± 0.1 and −6.0 ± 0.3 kcal/mol for pre-tRNAAsp and pre-tRNACys, respectively (Fig. 2C). For AtPRORP2, the values are −6.9 ± 0.2 and −5.9 ± 0.4 kcal/mol for pre-tRNAAsp and pre-tRNAGly, respectively (Fig. 2C). The reduced affinity for pre-tRNACys or pre-tRNAGly compared to pre-tRNAAsp represents only a minor loss of nonionic interactions.
AtPRORP1 does not have specific anion binding sites that compete with substrate binding
Specific anion binding sites on proteins can decrease the binding affinity of nucleic acids, in an ion-dependent manner similar to the effect of cations binding to nucleic acids. We screened the AtPRORP1 affinity for pre-tRNAAsp in several Na+ salts at a single Na+ concentration; the data largely follow the lyotropic series (Supplemental Methods; Supplemental Table S1). This trend suggests that the primary effect of the anions is to alter protein stability, as opposed to directly binding to the pre-tRNA binding sites on AtPRORP1 to inhibit pre-tRNA binding. Anion sites on proteins have also been probed by comparing the dependence of binding affinity on the concentrations of monovalent (M+) and divalent (M2+) cations for a given anion (deHaseth et al. 1977; Barkley et al. 1981).
For a protein binding to dsDNA in the absence of specific anion binding sites, the theoretical φMg/ϕNa is 0.53, which corresponds to the difference in the cations’ occupancy on the phosphodiester bonds in the backbone (deHaseth et al. 1977). To further test whether anion binding to AtPRORP1 contributes to the salt dependence of binding affinity, we measured dissociation constants in the presence of varying concentrations of CaCl2 (alone) or Na2SO4 (Fig. 3B). Fitting Equation 4 to the CaCl2 data with Z = 4, we obtained φCa = 0.51 ± 0.04. This value is in relatively good agreement with φMg = 0.47 for dsDNA (deHaseth et al. 1977). For the PRORP/pre-tRNA complex, the ratio of the slopes (Ca2+ /Na+) for the dependence of log KD on log concentration is 0.54, similar to the Mg2+/Na+ ratio of 0.53 for protein/DNA (deHaseth et al. 1977). Thus, the salt dependence of PRORP binding affinity can be explained using only the occupancies of the cations on backbone phosphodiester bonds and excluding anions.
FIGURE 3.

Cation (Mn+) dependence of dissociation constants for AtPRORP1 binding to B. subtilis fluorescein-labeled pre-tRNAAsp. (A) Anisotropy of Bacillus subtilis pre-tRNAAsp in the absence of PRORP is dependent on CaCl2 concentration. Data reported as the mean and standard deviation of four independent experiments. A hyperbola (Equation 1, Materials and Methods) was fit to the data (KD,app = 11±3 mM). (B) Equation 4 (Materials and Methods) was fit to the data with the dsDNA ϕNa = 0.88 or φMg = 0.47. Data include the dependence of AtPRORP1 affinity on NaCl [♦, Z = 4.3 ± 0.3, log(K0) = 5.0 ± 0.1], Na2SO4 [◯, Z = 3.6 ± 0.1, log(K0) = 5.62 ± 0.03], and CaCl2 [▪, Z = 4.3 ± 0.3, log(K0) = 5.1 ± 0.2]. The slope of the line (Zϕ or Zφ) reports on the apparent number of ionic interactions made to substrate phosphodiester bonds, while the intercept [log(K0)] reports on the nonionic contributions to affinity. (C) Ionic strength (I) dependence of AtPRORP1 binding to pre-tRNAAsp, plotted as the −log(KD) versus −log[I]. Equation 4 (Materials and Methods) was fit to the data with ϕNa = 0.88 or φCa = 0.47. Data include AtPRORP1 binding in NaCl [♦; log(K0) = 6.11 ± 0.04], Na2SO4 [◯; log(K0) = 6.18 ± 0.02], and CaCl2 [▪; log(K0) = 6.08 ± 0.09].
The slope of −log(KD) as a function of log[Na2SO4] is smaller than the slope of the NaCl data, resulting in tighter binding at higher Na+ concentrations in Na2SO4 (Fig. 3B). However, when −log(KD) is plotted against ionic strength (−log[I]), the NaCl and Na2SO4 data nearly overlay, while the CaCl2 data remain distinct (Fig. 3C). Thus, the modest differences in affinity observed in NaCl compared to Na2SO4 are most likely due mainly to nonspecific screening by the ionic atmosphere, to which Na2SO4 contributes less due to the lower concentration of SO42− at a given concentration of Na+. Given these data, we exclude the term for specific anion binding sites from our fits. The 1 M NaCl, 1 M Na2SO4, and 1 M CaCl2 ionic strength intercepts are within error (Fig. 3C), confirming that the nonionic contributions to binding are ion-independent.
AtPRORP1 makes fewer contacts to substrate leader than the bacterial ribozyme
Varying the leader length of pre-tRNA substrates beyond 1-nt (Howard et al. 2016) or 2-nt (Brillante et al. 2016) was previously shown to have little effect on the single-turnover activity and binding affinity with AtPRORPs. From these data, it is apparent that AtPRORPs can process a substrate with short 1- and 2-nt leaders, and that AtPRORP1 discriminates against binding the tRNA product (>30-fold lower affinity for tRNA than pre-tRNA). In contrast, the B. subtilis RNA-based RNase P relies on extensive contacts with the leader and trailer sequences for substrate recognition and displays a significant dependence on leader length beyond 2 nt (Crary et al. 1998; Rueda et al. 2005).
We determined the Na+ dependence of affinity for the fluorescein-labeled 1-nt pre-tRNAAsp and tRNAAsp product to evaluate the nature of the AtPRORP1 interactions with the leader (Table 1; Fig. 4). For the 1-nt substrate, the main effect is a value for K0 that is increased twofold compared to the 5-nt substrate, suggesting a 0.5 kcal/mol reduction in nonionic interactions with the shorter leader. The value of Z is also reduced modestly, although not enough to indicate the full loss of a phosphodiester contact. In contrast, the Z-value for tRNAAsp is reduced to 2.8 ± 0.5, consistent with the loss of one full phosphodiester bond contact. Interaction between PRORP and a phosphodiester bond in the 5′ leader is a feature of recognition in common with bacterial RNase P, which contacts the N-3/N-2 phosphodiester bond (Hansen et al. 2001). The value of K0 for tRNAAsp also increases significantly compared with pre-tRNAAsp containing either a 5 nt or 1 nt leader, equivalent to a loss of 2.1 and 1.6 kcal/mol, respectively, indicative of nonionic interactions with the leader. Apart from the interactions in the leader, the data also demonstrate that nonionic interactions with the tRNA body are important determinants of binding affinity, contributing 4.8 kcal/mol, or ∼70% of the nonionic binding energy (Table 1).
FIGURE 4.
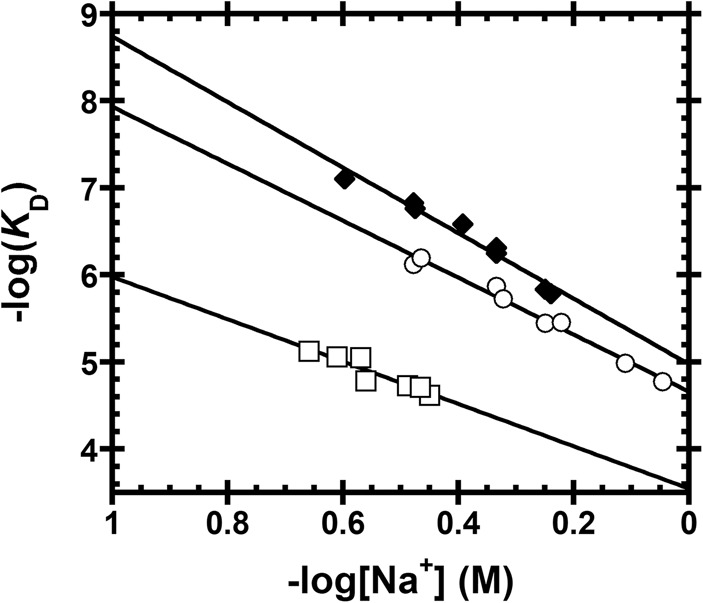
Sodium dependence of AtPRORP1 affinity for substrates with varied leader lengths. Data include AtPRORP1 binding to 5′ fluorescein-labeled substrates, including 5-nt pre-tRNAAsp [♦, Z = 4.3 ± 0.3, log(K0) = 5.0 ± 0.1], 1-nt pre-tRNAAsp [◯, Z = 3.7 ± 0.2, log(K0) = 4.7 ± 0.1], and tRNAAsp [□, Z = 2.8 ± 0.5, log(K0) = 3.5 ± 0.2]. Equation 4 (Materials and Methods) was fit to the data. The slope of the line (Z) reports on the apparent number of ionic interactions made to substrate phosphodiester bonds, while the intercept [log(K0)] reports on the nonionic contributions to affinity.
The PRORP PPR domain recognizes tRNAs using noncanonical positions
Previous work demonstrated that mutations of N136T, T180N, and G215N, each at position 6 of an AtPRORP1 PPR motif, resulted in minor pre-tRNA processing defects (Imai et al. 2014). Likewise, T113S and T113N mutations in AtPRORP2, equivalent to T180 in AtPRORP1, resulted in little or no processing defects (Brillante et al. 2016). To further characterize substrate recognition by the PRORP PPR domain, we measured the pre-tRNA binding affinity and salt dependence for variants of seven residues in AtPRORP1. The targeted side chains (i) are highly or fully conserved among plant PRORPs, as judged from alignment of PRORPs from 16 species; (ii) are located on the PPR surface facing the NYN domain; and (iii) have the potential to make hydrogen bonding, ionic, or base-stacking interactions (Fig. 5). In addition to the residues at the 1, 3, and 6 positions that have been shown to be involved in base selection in other PPR domains (Barkan et al. 2012; Yagi et al. 2013), we also targeted residues at position 10, which was not identified in the canonical base-selection motifs (Kobayashi et al. 2012). While residues at position 3 in ssRNA binding PPR proteins are typically hydrophobic (i.e., Leu, Phe), in PRORPs the residues at this position are mostly small or hydrophilic. Figure 5A shows the position of the residues that we targeted: Y133 (position 3; PPR2), N136 (position 6; PPR2), Y140 (position 10; PPR2), N175 (position 1; PPR3), T180 (position 6; PPR3), R184 (position 10; PPR3), and R212 (position 3; PPR4). We examined the effects of alanine mutations at each position, as well as more conservative mutations including Y133F, T180S, and R184K.
FIGURE 5.
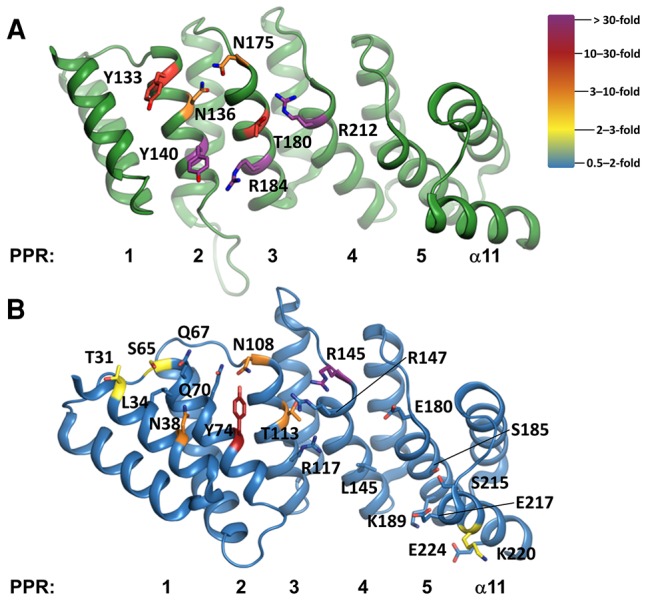
Residues selected for mutation in the PPRs of AtPRORPs, generated in PyMOL (The PyMOL Molecular Graphics System, Version 1.5, Schrödinger, LLC). (A) AtPRORP1 (PDB 4g24) PPRs are numbered left (PPR1) to right (helix α11). Residues that were targeted for mutation are numbered. Carbon atoms are color-coded by the largest effect on binding as indicated by the color ramp. (B) AtPRORP2 (PDB 5diz) PPRs are numbered left (PPR1) to right (helix α11). Residues that were targeted for mutation are numbered. Carbon atoms are color-coded as in A. Alanine mutants were not soluble for Q70, R117, and R147, so no data were collected for these variants.
We measured the binding affinities of the AtPRORP1 mutants for the B. subtilis 5-nt pre-tRNAAsp substrate using the FA assay at 330 mM NaCl; these data are summarized in Table 2. We observed the largest reductions in binding affinity for the Y140A and R184A variants with decreases of >190- and 67-fold, respectively, as well as for the R212A mutant (see below). Representative binding data for Y140A and R184A at 330 mM NaCl are shown in Supplemental Figure S2. T180 was the only residue in a canonical PPR base-selection position that we tested with a strong effect on binding; T180A reduces the binding affinity by approximately 20-fold compared to WT AtPRORP1. The other canonical base-selecting residues that we mutated led to modest decreases in binding affinity: N136A had a 6.3-fold effect and N175A had a ninefold effect. Finally, R212A eliminated binding as measured with the anisotropy assay (KD > 30 µM) (Supplemental Fig. S3B). Additionally, the enzymatic activity of this mutant in an STO assay remained lower than the wild-type value even with >35 µM enzyme and high Mg2+ concentrations and several significant miscleavage bands were observed (Supplemental Fig. S3A).
TABLE 2.
Na+ dependence of AtPRORP1 variants affinity for B. subtilis pre-tRNAAsp
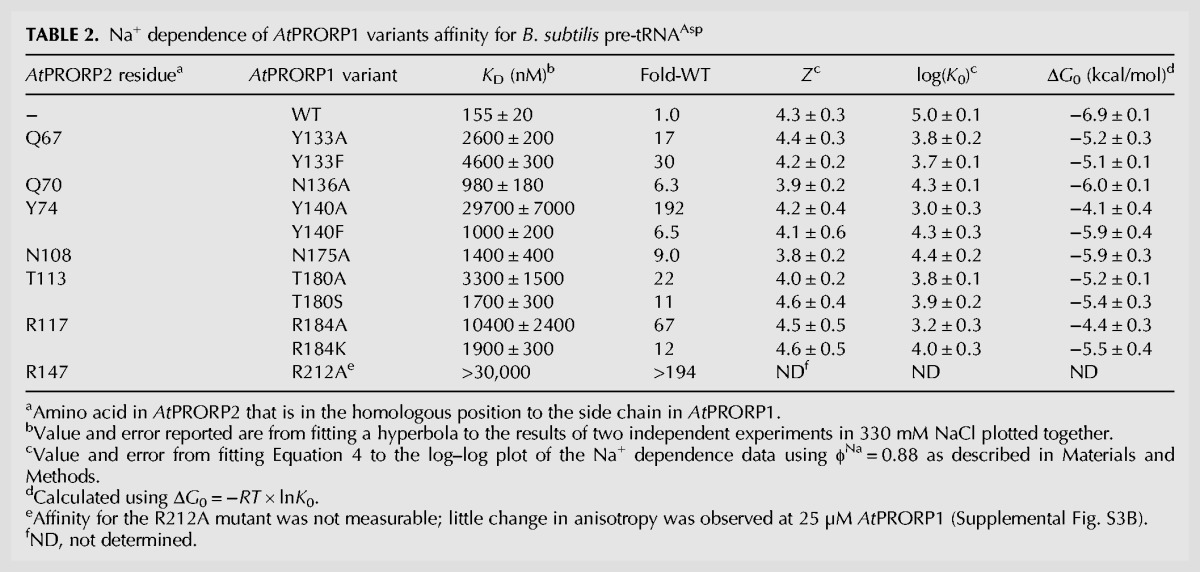
We parsed the determinants of substrate binding by AtPRORP1 in more detail by analyzing the salt dependence of the mutants. In general, the mutations had little effect on the Z-value for the Na+ dependence of binding affinity, but they affected the intercept value, K0 (Table 2). These results indicate that the mutated side chains do not form ionic interactions with the phosphodiester backbone of pre-tRNA, rather mediating nonionic interactions with the substrate. The largest measurable reduction in affinity (>190-fold) was observed for the Y140A variant, corresponding to a loss of 2.8 kcal/mol of nonionic binding energy. However, the Y140F mutation only increased K0 by 6.5-fold, corresponding to a loss of ∼1 kcal/mol in nonionic interactions compared to WT AtPRORP1. These results are consistent with PRORP interacting with pre-tRNA with both the tyrosine hydroxyl and the phenyl ring (Guckian et al. 2000). For the R184A mutant, the 67-fold reduced affinity corresponds to a reduction of 2.5 kcal/mol compared to WT, while the R184K mutation increased the K0 by 10-fold, corresponding to a loss of 1.4 kcal/mol of binding energy compared to WT.
The Y133 variants reveal a relationship different from that of the Y140 variants. The Y133F and Y133A variants reduce pre-tRNA affinity by comparable values, 1.8 kcal/mol and 1.6 kcal/mol, respectively. These data suggest that the hydroxyl group, but not the phenyl ring of Y133, contributes to substrate affinity. The N136A and N175A mutations reduce the value of the intercept (K0) corresponding to approximately the loss of 1 kcal/mol in nonionic binding energy apiece. We estimated comparable free energy losses for T180A and T180S variants, 1.7 and 1.5 kcal/mol, respectively, compared to that of WT.
The AtPRORP2 PPR domain recognizes tRNAs using a similar binding surface
To determine whether this binding surface is shared among Arabidopsis PRORPs and whether additional PRORP PPR side chains are important for substrate binding, we screened the pre-tRNA binding affinity of 22 alanine variants in AtPRORP2 (Fig. 5B). Previously, we and others proposed that the first, third, sixth, and tenth residues (numbered as in Barkan et al. 2012) in each PPR motif could periodically contribute to substrate binding in AtPRORPs (Supplemental Fig. S1; Barkan et al. 2012; Kobayashi et al. 2012; Yin et al. 2013; Karasik et al. 2016). Therefore, we systematically targeted residues in these positions for all five PPR motifs and the PPR C-terminal helix (α11) for alanine mutagenesis. This analysis necessarily excludes three alanine residues (A110, A150, and A182).
We examined the binding affinity of the alanine mutants with A. thaliana nuclear 6-nt pre-tRNAGly and B. subtilis 5-nt pre-tRNAAsp substrates. The residues that we identified with alanine scanning mutagenesis of AtPRORP2 are consistent with the binding surface identified in AtPRORP1, although the effects on binding affinity are smaller. The largest decreases in binding affinity compared to wild-type AtPRORP2 were observed for Q67A (position 3; PPR2), N108A (position 1; PPR3), T113A (position 6; PPR3), and R145A (position 1; PPR4), which increased the KD values for pre-tRNA by at least 1.5-fold (in the highest fold-increase that we observed, Supplemental Table S2). Importantly, these residues (excluding R145) correspond to three of the seven positions we identified in the AtPRORP1 PPR domain. This analysis also identified additional residues that decrease pre-tRNA binding affinity beyond those evaluated in AtPRORP1, all of which fall primarily within the nearby PPR surface. These include N38A (position 10; PPR1), S65A (position 1; PPR2), T31A (position 3; PPR1), and K220A (position 6; α11), each of which increased the KD by at least twofold compared to wild-type AtPRORP2 (in the highest fold-increase that we observed, Supplemental Table S2). Four AtPRORP2 alanine mutants—Q70A (position 6; PPR2), Y74A (position 10; PPR2), R117A (position 10; PPR3), R147A (position 3; PPR4)—did not express as soluble proteins, suggesting that mutation of these residues may affect the stability of AtPRORP2. These residues are all located on the proposed substrate binding surface, and include four of the seven residues that alter substrate affinity in AtPRORP1.
Given the importance of Y140 in AtPRORP1 for pre-tRNA affinity, we further investigated the interaction between this amino acid and pre-tRNA by analyzing the Y74S and Y74F mutants in AtPRORP2. We found that these mutations significantly decrease substrate affinity (Supplemental Table S2), as observed for AtPRORP1. These results further demonstrate that Y140 interacts with substrate using both the phenyl ring and the hydroxyl group. Taken together, the above results suggest that the surface we identified in AtPRORP1, primarily in PPR2 and PPR3, generalizes as the major surface involved in PRORP substrate binding.
PRORP PPR domain appears not to recognize tRNAs with base-specificity
To investigate whether AtPRORPs recognize pre-tRNA in a base-specific manner, we mutated tRNA residues that the previously established PPR recognition codes suggest should recognize PRORP1 PPR base recognition sites (Barkan et al. 2012; Yagi et al. 2013). AtPRORP1 contains amino acids located between PPR motifs 2 and 3 (Y133/N136/N175) that should recognize pyrimidines (Barkan et al. 2012; Yagi et al. 2013). For this analysis, we examined pyrimidines in pre-tRNA that would likely interact with PRORPs. The D- and TψC-loops (tRNA elbow) in pre-tRNA have been proposed to interact with the PPR domain (Gobert et al. 2013). We assumed that the pyrimidines should not be in secondary/tertiary contacts or otherwise buried and inaccessible in the unbound tRNA structure, limiting the proposed interaction to uridines at positions 16, 17, 20, and 21 in pre-tRNAAsp (Fig. 2A). Mutation to adenosine at each of these positions altered the affinity by at most twofold (Supplemental Table S3), demonstrating a lack of sequence-specific interaction at these sites. In combination with the previous data regarding the effects of tRNA mutations on PRORP binding/catalysis (Imai et al. 2014; Brillante et al. 2016), these data reinforce the hypothesis that PRORPs utilize a mode of substrate recognition different from the previously described PPR base-selection determined in ssRNA binding proteins. However, the possibility remains that there are other sites in pre-tRNA that interact with PRORP PPRs in a sequence-specific manner.
Na+ screening inhibits AtPRORP1 single-turnover activity
While it is possible that the ionic strength affects only the binding affinity, it might also affect other aspects of PRORP catalysis. To determine whether the NaCl concentration affects cleavage catalyzed by PRORP, we performed single-turnover (STO) activity assays with 5 µM AtPRORP1, which is saturating under low NaCl conditions, and limiting (30 nM) B. subtilis pre-tRNAAsp. We used concentrations of MgCl2 that we previously determined to be either saturating (20 mM) or subsaturating (1.25 mM) for catalysis (Howard et al. 2015). The observed rate constant (kobs) is independent of the NaCl concentration between 25 and 200 mM, but is reduced at higher NaCl concentrations (Fig. 6). The concentration dependence of NaCl inhibition above 400 mM is similar for saturating and subsaturating MgCl2. Fitting a general inhibition model (Equation 3, weighted fit, Materials and Methods) with a variable Hill coefficient (nNa) to the data yields similar IC50 (310 ± 70 mM for 20 mM MgCl2 and 360 ± 70 mM for 1.25 mM MgCl2) and nNa values (4.5 ± 1.3 for 20 mM MgCl2 and 5.0 ± 1.3 for 1.25 mM MgCl2) for both MgCl2 conditions (Fig. 6). The Hill coefficients for STO inhibition (4.5–5) by sodium are in good agreement with the cooperativity we observe for inhibition of pre-tRNA binding.
FIGURE 6.
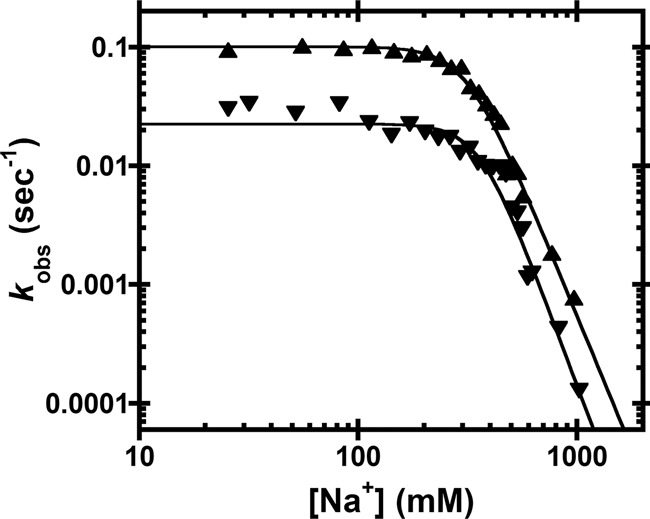
Na+ dependence of AtPRORP1 cleavage activity. The dependence of the AtPRORP1-catalyzed cleavage of fluorescein-labeled pre-tRNAAsp on the [NaCl] was measured under single-turnover conditions (kobs). Data include AtPRORP1-catalyzed cleavage in 20 mM (▴) or 1.25 mM (▾) MgCl2. Equation 3 (Materials and Methods, weighted fit) was fit to the data. For 20 mM MgCl2, IC50 = 310 ± 70, nNa = 4.5 ± 1.3. For 1.25 mM MgCl2, IC50 = 360 ± 70, nNa = 5.0 ± 1.3.
If the observed inhibition is due to decreased affinity of AtPRORP1, the activity should begin to decrease at NaCl ∼530 mM (the point at which KD reaches >1 µM; thus, [E] becomes <5 times the KD, as measured in Ca2+), assuming that the binding affinity is equivalent in Ca2+ and Mg2+. However, the activity is inhibited at lower NaCl concentrations (∼300 mM). To test whether this observation is only due to an effect of NaCl on binding affinity, as opposed to NaCl affecting other steps in catalysis, we measured the STO kobs at 20 mM MgCl2 and 350 mM NaCl with varying enzyme concentration (data not shown). The STO kobs is strongly dependent on enzyme concentration under these conditions, indicating that 5 µM PRORP1 is subsaturating, thus requiring that the STO K1/2 in MgCl2 is >30-fold higher than the thermodynamic KD (155 ± 20 nM) in CaCl2. These data also indicate that PRORP–substrate affinity is reduced in MgCl2 as compared to CaCl2. This cation-dependent decrease in affinity is consistent with an EcoRV catalytic mutant that binds its cognate DNA ≈ 40-fold weaker in Mg2+ than in Ca2+ (Martin et al. 1999). In summary, the STO data are consistent with NaCl disrupting PRORP–substrate binding, but not other kinetic steps.
DISCUSSION
The goal of this work was to characterize the molecular interactions between AtPRORPs and pre-tRNA using the ion dependence of binding, catalysis, and mutations in the PPR domain. The NaCl dependence of pre-tRNA affinity of AtPRORPs revealed 4–5 interactions with phosphodiester bonds in pre-tRNA and ∼6–7 kcal/mol of nonionic binding energy. These narrow ranges support previous observations that pre-tRNA recognition by AtPRORPs is similar across paralogs and largely independent of sequence. Depending on the context, biological hydrogen bonds can supply 0.5–3 kcal/mol of free energy (Fersht et al. 1985), while van der Waals interactions such as base stacking can supply 0.5–2 kcal/mol (Guckian et al. 2000). AtPRORPs likely form a combination of hydrogen bonds and van der Waals interactions with pre-tRNA.
Comparison of the Na+ dependence and mutation of residues in the substrate-binding domain in both AtPRORP1 and 2 indicate a similar binding surface with modest differences in substrate recognition, as previously evaluated (Karasik et al. 2016). In general, alanine mutations affect the substrate affinity of AtPRORP1 to a greater extent than AtPRORP2. This might be due to the lower CaCl2 conditions used for affinity measurements with AtPRORP2, which also reduced the differences between wild-type and mutant AtPRORP1 binding affinity (Supplemental Table S4). The variety of ion-dependent effects on PRORP binding affinity that we observe can be synthesized as follows.
Cations such as Na+ and Ca2+ inhibit PRORP/pre-tRNA binding by interacting with and competing for phosphodiester bonds on pre-tRNA. We observe a log-linear decrease in PRORP/pre-tRNA binding affinity with respect to [Na+] above 180 mM NaCl (Figs. 2C, 3B,C, 4). The PPR mutations we generated (described in further detail below) do not affect the slope of the Na+ dependence of binding at high [Na+] (Table 2). However, we observe lesser effects from these mutations on binding affinity below 180 mM NaCl (Supplemental Tables S2, S4). Overall, these data are most informative with respect to the differences we observe between WT and variant PRORPs and pre-tRNAs, which allow us to parse the interactions between various aspects of each.
PRORP PPR domain
Our extensive mutagenesis data in the PPR domain allow us to contrast PRORP-RNA binding with previously described PPR proteins. PPR-containing proteins are a large family with the structurally conserved ≈35 residue helix–turn–helix motif found in tandem repeats that have been implicated in RNA metabolism (Small and Peeters 2000; Schmitz-Linneweber and Small 2008). PPR proteins are found broadly in eukaryotes, with land plants having the largest set of PPR proteins (O'Toole et al. 2008). Some PPR proteins bind target RNAs in a sequence-specific manner, with recognition of a nucleobase achieved primarily by residues 6 and 1′ (also numbered 4 and ii by Yagi and coworkers, or 5 and 35 by Yin and coworkers) on the A helices of two tandem PPR motifs (Fig. 1B; Supplemental Fig. S1; A helices colored dark green; Barkan et al. 2012; Yagi et al. 2013; Yin et al. 2013). In some cases, the binding sites have been identified in the UTRs of mRNAs, where the PPR proteins are proposed to regulate splicing, translation, and/or stability of the mature transcript (Schmitz-Linneweber and Small 2008; Chen et al. 2016). In contrast, our mutagenesis experiments indicate that PRORP PPRs use a different mode of RNA recognition. The PPR domains of AtPRORP1 and AtPRORP2 do not rely on interactions with a limited number of residues from every repeat, but rather a more extensive surface primarily located in PPR2 and PPR3, which is consistent with previous data indicating impaired activity for ΔPPR2 and ΔPPR3 AtPRORP1 variants (Imai et al. 2014).
We initially hypothesized that PRORP PPRs would not recognize RNA using base selection as observed with ssRNA-binding PPR proteins because the base-specifying residues in the five tandem PPR repeats in PRORPs are frequently noncanonical (e.g., 6 and 1′ are not often asparagine, aspartate, or threonine, Supplemental Fig. S1). Several substrate binding residues that we identified, such as Y140 and R184, are conserved in metazoan PRORPs (Y183 and R218 in humans). Thus, even though the metazoan PRORPs require additional subunits for catalysis, pre-tRNA recognition by the human PRORP PPR domain could use the same interaction surface as plant PRORP PPRs.
The alanine mutations most detrimental to B. subtilis pre-tRNAAsp affinity were Y140A in AtPRORP1 PPR2 (>190-fold), R184A in AtPRORP1 PPR3 (67-fold), and R212A in AtPRORP1 PPR4; the equivalent mutations in AtPRORP2 (Y74A, R117A and R147A) rendered the protein insoluble. Despite these insoluble variants, we find no evidence that the soluble AtPRORP1 mutants are less stable than the WT enzyme. The melting temperatures (Tm) (Pantoliano et al. 2001) for Y140A, R184A, and R212 AtPRORP1 are not significantly different from WT (Supplemental Fig. S2C; data not shown), and the CD spectrum for Y74S AtPRORP2 does not reveal a significantly different secondary structure from WT (Supplemental Fig. S2D). Furthermore, R184A catalyzes STO cleavage at high concentrations (Supplemental Fig. S2E) with no apparent miscleavage (Supplemental Fig. S2F). These data support the conclusion that the PPR mutations primarily reduce the affinity for pre-tRNA.
Y140A and R184A AtPRORP1 mutations similarly bind the A. thaliana pre-tRNACys substrate weaker than WT in 20 mM CaCl2 and 330 mM NaCl conditions (>93-fold and >3.8-fold, respectively). Unexpectedly, the effects of the AtPRORP1 mutations are greater than the 34-fold decrease in binding affinity reported for a Δ245 AtPRORP1, which fully lacks the first four PPR motifs (Howard et al. 2012). However, this measurement was carried out at 1 mM CaCl2 and 100 mM NaCl with the A. thaliana mitochondrial pre-tRNACys substrate. Under these conditions, the individual mutations have little effect on binding affinity (Supplemental Table S4). This effect could be explained by two different possibilities, either formation of additional electrostatic contacts at lower ionic strength, or the increased proportion of the PRORP–substrate affinity due to ionic interactions under lower ionic strength, such that individual nonionic interactions we identify contribute less significantly to the overall affinity.
The loss of 1 kcal/mol that we observe for the Y140F is consistent with Y140 interacting through a hydrogen bond to substrate with the tyrosine hydroxyl. The additional 1.8 kcal/mol loss observed with the alanine substitution is consistent with the energy supplied by stacking a phenyl ring with a nucleic acid base (Guckian et al. 2000). For the R184A mutant, the 67-fold reduced affinity corresponds to a loss of 2.5 kcal/mol in nonionic binding energy compared to WT, which could indicate loss of 1–3 hydrogen bonds with the guanidinium group and/or hydrophobic interactions with the arginine methylene groups. By comparison, the R184K mutation increased the K0 by 10-fold, corresponding to a loss of 1.4 kcal/mol compared to WT, consistent with the loss of a hydrogen bond. In AtPRORP1, Y140 and R184 are located at position 10 of neighboring helices forming a nonsequential, structural pair. This YR pair is widely conserved in PRORP PPRs, including metazoan PRORPs (Supplemental Fig. S1).
The C-terminal La domain of the telomerase protein p65 also contains a conserved YR structural pair (Y407 and R465), situated on neighboring β-strands, that are important for recognition of the conserved GA bulge in stem IV of the telomerase RNA (Singh et al. 2012). Given the significant contribution of the phenyl ring and the guanidinium group revealed by the Y140A/F and R184A/K mutations in AtPRORP1 and Y74S/F in AtPRORP2, we propose that these residues make similar interactions with the tRNA elbow, the conserved structural feature that results from the interaction of the D- and TψC-loops, which were previously proposed to interact with PRORPs (Gobert et al. 2013). These interactions do not need to be sequence specific, but like the p65 YR pair (Singh et al. 2012), they could favor purines such as the conserved G18G19 in the D loop. Consistent with this, mutation of these resides to adenine in a canonical pre-tRNAGly leads to an ≈4.5-fold (G18) or ≈1.5-fold (G19) increase in the STO K1/2 (Brillante et al. 2016).
In contrast to our Y140 data, the data for Y133 indicate that the hydroxyl group, but not the phenyl ring, contributes to substrate affinity. Mutation of N136 and N175 in PRORP1 results in the loss of ∼1 kcal/mol apiece, consistent with a hydrogen bond from the amide side chain of each. The T180A and T180S mutations in PRORP1 lead to a 1.5–1.7 kcal/mol loss while the homologous T113A in PRORP2 had at most a 3.5-fold effect on binding affinity. However, substitutions of T113S and T113N in PRORP2 were not sufficient for significant processing defects (Brillante et al. 2016). These data suggest that the threonine methyl group is required to sterically position the hydroxyl for substrate interaction or to make a hydrophobic contact to substrate.
While AtPRORP1 T180 (T113 in AtPRORP2) is at a base-selecting position 6, its corresponding 1′ partner in the putative base-selection position would be R210 (R145 in AtPRORP2). Arginine residues have not previously been identified as base-selecting residues for PPRs (Barkan et al. 2012; Yagi et al. 2013). Furthermore, the potential nucleobase binding cleft is occluded by an interaction between T180 (T113) and R212 (R147), as visualized in the AtPRORP1 and AtPRORP2 crystal structures (PDB 4g24 and 5diz) (Howard et al. 2012; Karasik et al. 2016). Taken together, the mutagenesis data for AtPRORP1 and AtPRORP2 indicate that the PRORP PPR domain does not interact with pre-tRNA in the same manner as ssRNA-binding PPR proteins.
An alternative recognition mode could include structural recognition, like the tRNA elbow recognition used by the specificity (S) domain of the P RNA subunit in the bacterial ribozyme. The S-domain makes stacking interactions with the conserved G19–C56 tertiary interaction between the D- and TψC-loops, and sugar face interactions with the conserved G53–C61 pair at the end of the TψC arm (Reiter et al. 2010). These interactions recognize conserved pre-tRNA structural elements and allow P RNA to recognize the entire set of pre-tRNA transcripts without specificity for the tRNA body sequence. Likewise, we propose that the PPRs of PRORPs recognize tRNA using structure-specific interactions. A similar mode of recognition has been proposed previously (Gobert et al. 2013). However, our data do not completely rule out base-recognition strategies such as those used by canonical PPR proteins.
PRORP–substrate recognition model
Combining our data with published data (Gobert et al. 2013; Imai et al. 2014), we propose a new, predictive model to describe how AtPRORP1 and AtPRORP2 bind their substrates. A previous model of the complex utilized AtPRORP1 bound to tRNA with base-specifying interactions between a TψC-loop cytosine and PPR motifs 3 and 4 (Imai et al. 2014). The tRNA coordinates were derived from the bacterial RNase P holoenzyme in complex with a tRNA product (Reiter et al. 2010) and tRNA bound to a pseudouridine synthetase (Hoang and Férre-D'Amaré 2002), which has significant alterations to the TψC-loop bases and backbone. While this paper was under review, a similar model was published that largely supports our model (Pinker et al. 2017).
For constructing our model, we used the crystal structures of AtPRORP1 (PDB 4g24) and AtPRORP2 (PDB 5diz) (Howard et al. 2012; Karasik et al. 2016). The two structures are highly conserved, yet there is a significant difference in the angle between specific PPR motifs and between the central domain and the metallonuclease domain; as a consequence, AtPRORP2 is in a more “open” conformation. We propose that the two distinct structural snapshots may represent two different conformations that potentially play a role in substrate binding (Karasik et al. 2016). The more “open” AtPRORP2 conformation can more readily accommodate tRNA, since the distance between the active site and the proposed substrate binding region in the PPR domain is ∼50 Å. Therefore, we used the AtPRORP2 structure to generate a tRNA interaction model and used this model as a template to generate a model for AtPRORP1–tRNA recognition. There are no crystal structures available for any of the tRNAs used in this study. However, since the 3D structure of tRNAs is highly conserved, we used a canonical eukaryotic tRNA, yeast tRNAPhe, with high resolution crystal structure (PDB 1ehz) (Shi and Moore 2000) with only slight modifications to bases predicted to interact with the protein.
The tRNA substrate can be accommodated well between the metallonuclease and PPR domains in our model (Fig. 7), with one exception. There is a short PRORP helix (α21) and part of the loop that precedes it near the active site that sterically clashes with the 3′ strand of the tRNA acceptor stem (Fig. 7B). We posit that this NYN helix will adopt a different conformation upon pre-tRNA binding and may be directly involved in recognition. This region could serve as a hinge that allows or blocks substrate binding to the metallonuclease domain. Interestingly, the invariant and solvent exposed R496 (R443 in PRORP2) and H498 (H445 in PRORP2) residues in this region are ideally placed for interaction with the phosphodiester backbone in the acceptor stem. Mutational analysis of the residues H498 (H498A/H498Q, AtPRORP1) and H445 (H445A, AtPRORP2) demonstrate that these mutations reduce the STO kobs without significantly affecting the KD (Howard et al. 2015; Karasik et al. 2016). We proposed that these residues are involved in positioning the substrate, and our model provides potential contacts with substrate for testing this hypothesis.
FIGURE 7.

Model of the PRORP–substrate complex. (A) Overall view of the modeled complex. AtPRORP2 (PDB 5diz, blue) is bound to tRNA (PDB 1ehz, orange backbone with green and blue rings). The AtPRORP1 (PDB 4g24, yellow) NYN and PPR domains are aligned to the corresponding residue domains in AtPRORP2. Purple spheres are the Mn2+ ions bound to the AtPRORP1 active site. Close-up view of the PPR domain highlights the positions of residues for which mutation affected binding affinity (greater than threefold), including D105/N38, N136/Q70, Y140/Y74, N175/N108, T180/T113, R210/R145, and R212/R147 in AtPRORP1/AtPRORP2, respectively. (B) Close-up of the AtPRORP2-tRNA complex with a potential steric clash between NYN helix α21 (red) and the 3′ side of the tRNA acceptor stem. (C,D) Close-up of the AtPRORP2– (C) or AtPRORP1–substrate complex (D) showing the NYN active site. The loop for which AtPRORP2–3 have a four-residue insertion is highlighted in red (C). In both structures, the 5′ leader of pre-tRNA would extend forward from the panel, while the 3′ trailer would extend behind the NYN domain.
Our model suggests an exit groove for the 5′ leader that would place the N-3/N-2 phosphodiester bond and N-3 nucleoside outside the bounds of the NYN domain (Fig. 7C,D). This is consistent with data suggesting few contacts with the leader beyond the N-2 nucleoside. Moreover, our model places the N-2/N-1 phosphodiester bond near the invariant H438 (H386 in PRORP2) and R441 (R389 in PRORP2), implicating these residues for interactions with the negatively charged backbone. This aspect of our model is congruent with our data indicating that PRORPs form one non-base-specific phosphodiester backbone contact with the tRNA leader. Furthermore, the α16–α17 loop is positioned to separate the 5′ leader and 3′ trailer. Interestingly, AtPRORP1 (Fig. 7D) has four fewer amino acids in the loop than AtPRORP2/3 (Fig. 7C, red). These loop differences might explain the variations in 5′ end discrimination observed previously, in which AtPRORP2/3 had a stronger propensity to miscleave at the N-1 position when an N-1/N73 base pair was possible (Brillante et al. 2016; Howard et al. 2016).
The elbow region of tRNAs are highly structured and numerous tRNA binding enzymes recognize this part of the tRNA using a variety of interaction motifs (Zhang and Férre-D'Amaré 2016). Importantly, our model predicts that (i) R210 (R145 in AtPRORP2) and R212 (R147 in PRORP2) contact TψC stem phosphodiester bonds; (ii) Asp 105 (N38 in PRORP2), N136 (Q70 in PRO RP2), and N175 (N108 in PRORP2) are positioned to hydrogen bond with bases of the D loop (first residue) or the TψC (last two nucleotides) loop, respectively; and (iii) Y140 (Y74 in PRORP2) is capable of both base stacking and hydrogen bonding with bases of the TψC loop. Although our proposed model needs to be further tested, it provides insight into the details of precursor tRNA binding of PRORPs in the absence of crystal structures of PRORP–tRNA complexes and a rough framework for the design of future experiments.
Conclusions
The data we present herein support a novel model for PRORP–pre-tRNA recognition that shares similarities with the mode of substrate recognition by the RNase P ribozyme. The salt dependence of PRORP–substrate binding parses the ionic and nonionic contributions to PRORP–substrate binding affinity. The data reveal that AtPRORPs make at least four contacts with pre-tRNA phosphodiester bonds. Only one of these is contained in the leader sequence, most likely at the N-2/N-1 phosphodiester bond. Additionally, we identified an extended surface on the PPR domains of AtPRORP1 and AtPRORP2 that interacts with substrate. Mutations on this surface suggested a mode of binding different from that of sequence-specific ssRNA-binding PPR proteins. The biochemical and modeling data we have presented will facilitate the development of additional hypotheses for single-subunit PRORP substrate recognition. Given that metazoan PRORPs require two additional proteins for catalysis, there are likely differences that will need to be determined.
MATERIALS AND METHODS
Reagents
A full list of reagents used in this manuscript is detailed in the Supplemental Information.
Enzyme preparation
Variants of Δ76 AtPRORP1 and full-length AtPRORP2 were generated by site-directed mutagenesis (Hutchison et al. 1978). Sequences were verified at the University of Michigan DNA Sequencing Core facility. Variants were expressed in Rosetta, Rosetta 2 or BL21(DE3) E. coli (Novagen/EMD Millipore) from the T7 promoter on a pETM-11 (encoding His6-TEV-AtPRORP1) or pMCSG7 (His6-TEV-AtPRORP2) vector in LB media with 50 µg/mL kanamycin and 33 µg/mL chloramphenicol for selection of pETM-11 and pRARE (a plasmid encoding rare-codon tRNAs in the Rosetta cell lines) or 100 µg/mL ampicillin for selection of pMCSG7. Wild-type Δ76 AtPRORP1 and full-length AtPRORP2 and variants of these enzymes were purified as described previously (Howard et al. 2012, 2015; Karasik et al. 2016).
Substrate preparation
Substrates were prepared as described previously (Howard et al. 2012, 2015). Briefly, substrates were synthesized by run-off transcription from restriction-digested plasmid encoding pre-tRNA, a PCR-amplified template DNA, or a commercially synthetized ultramer oligo(IDT) (Milligan and Uhlenbeck 1989). In vitro transcription was carried out in the presence of 5′-O-monophosphorothioate guanosine (GMPS) in 5:1 excess of GTP. The pre-tRNA containing a 5′-GMPS was reacted with 5-iodoacetamidofluorescein (5-IAF) to generate a 5′-fluorescein label. The pre-tRNA product was gel purified using 12% urea–PAGE and substrate was eluted from the gel using the crush-soak method (Milligan and Uhlenbeck 1989). The purified pre-tRNAs were washed and concentrated using 10-kDa MWCO Amicon Ultra Centrifugal Filters, then ethanol precipitated. Substrate stocks were resuspended in 10 mM tris(hydroxymethyl)aminomethane (Tris) pH 8.0 with 1 mM EDTA, quantified by absorbance and stored at −20 or −80°C. The extinction coefficients at 260 nm for total RNA concentration are: 685,000 M−1 cm−1 for Bacillus subtilis pre-tRNAAsp (experimentally determined by alkaline hydrolysis), 674,390 M−1 cm−1 for A. thaliana mitochondrial pre-tRNACys (experimentally determined by alkaline hydrolysis), and 870,700 M−1 cm−1 for A. thaliana nuclear pre-tRNAGly (calculated). The fluorescein concentration was measured at 492 nm (extinction coefficient = 78,000 M−1 cm−1). Variants of pre-tRNAAsp were generated by site-directed mutagenesis (Hutchison et al. 1978). Immediately before initiating an assay, substrates were thawed, diluted with H2O, and heated at 95°C for 60–90 sec. Substrates were refolded by cooling to 25°C for ≥10 min, then incubating with buffer (as specified for each assay) for ≥10 min.
Anisotropy binding assays
Thermodynamic binding assays were performed in a 96-well plate format as previously described (Howard et al. 2012, 2015). Briefly, WT AtPRORP1 was serially diluted and mixed 1:1 with a concentration of prefolded pre-tRNA containing a 5-fluorescein. In all experiments, the maximum enzyme concentration ([P]) was at least three times greater than the KD and the pre-tRNA concentration at least five times lower than the KD. In all cases, the data were well described by a hyperbolic binding curve (Equation 1). Reactions were incubated at 28 ± 1°C in 30 mM MOPS pH 7.8, and 1 mM TCEP (AtPRORP1) or 1 mM DTT (AtPRORP2). The NaCl concentration was varied between 0.025 and 1.0 M. Unless otherwise specified, assays contained 20 or 6 mM CaCl2 for AtPRORP1 and AtPRORP2, respectively. The CaCl2 concentrations were chosen to maintain the KD values in the measurable range. For AtPRORP1, decreasing the CaCl2 concentration to 6 mM increased the KD values (65%) but altered the slope of the Na+ dependence <10% (data not shown). When varying the Na+ salt, we maintained constant Na+ at 330 mM (see Supplemental Methods). Changes in anisotropy of the 5′-fluorescein–pre-tRNA were measured with a Tecan Ultra plate reader with polarizing filters using excitation and emission wavelengths of 485 and 535 nm, respectively. Readings were taken 3–5 times over the course of 15–20 min to ensure that the reading was stable.
| (1) |
Single-turnover assays
Single-turnover kinetic assays for AtPRORP1 were performed in a stopped-format as previously described (Howard et al. 2012, 2015). Briefly, enzyme was mixed with prefolded B. subtilis pre-tRNAAsp with a 5-nt leader and a 5′-fluorescein to final concentrations of 5 µM and 30 nM, respectively. For R184A assays, 30 nM substrate was incubated with 1–50 µM enzyme. Reactions were incubated at 25°C in 30 mM MOPS pH 7.8, 1 mM TCEP, with MgCl2 and NaCl varied as indicated for a given assay. Aliquots were removed at various times and mixed 1:1 with a 2× quench dye (6 M urea [MP Biochemicals], 100 mM EDTA [Acros Organics], 0.1% bromophenol blue [BPB; Fisher Scientific], 0.1% xylene cyanol [XC; United States Biochemical Corporation], and 2 µg/µL bulk yeast tRNA [Fisher Scientific]). Products were resolved from substrate by fractionation on a ≥20% urea–PAGE, and the gels were scanned using a Typhoon 9410 (GE Life Sciences) in fluorescence mode with a 532 nm green laser and fluorescein emission filter. Assays for AtPRORP2 were carried out using the same conditions, but changes in polarization upon cleavage were detected by ClarioStar (BMG Labtech) in 96-well plate format. The observed rate constants (kobs) were determined by quantifying the fraction product using ImageQuant 5.2 software and fitting a single exponential (Equation 2, where A is the endpoint, B is the amplitude, and t is the time) to the data using KaleidaGraph 4.0 software. At low concentrations of NaCl (below ≈90 mM), the 5′ leader product degraded after it appeared and did not accumulate to 100%. A double exponential was fit to these data and the kobs from the phase with increasing product is reported. The IC50 for inhibition of AtPRORP1 by NaCl was determined by fitting Equation 3 to the dependence of the STO kobs on the NaCl concentration (as described in the Results section).
| (2) |
| (3) |
Sodium dependence
Equation 4 is an approximation of Equation S1 (shown in the Supplemental Methods) that describes the dependence of the affinity on cations when effects from pH, anions, and divalent ions are negligible or can otherwise be precluded by maintaining constant pH and divalent ions and observing the log-linear region of the decreasing affinity. The dependent variable is the monovalent cation concentration ([M+]). The parameters include a “standard affinity” at 1 M M+ (K0), the apparent number of phosphodiester bonds on the substrate interacting with the protein (Z), and the fraction of phosphodiester bonds in the nucleic acid that thermodynamically associate with a monovalent ion (ϕ). When divalent cations are varied in the absence of monovalent ions, the slope is distinguished by replacing ϕ with φ. Standard affinity values were converted to energetic values using Gibbs free energy definitions and assuming equilibrium conditions (Equation 5), for which R is the gas constant (1.987 cal K−1 mol−1) and T is the temperature (300 K for our assays).
| (4) |
| (5) |
Model building
Crystal structures of AtPRORP1 (PDB ID: 4g23) and AtPRORP2 (PDB ID: 5diz) and yeast tRNAPhe (PDB ID: 1ehz) were used to model the elbow region of pre-tRNA bound to the proteins. Initial models were obtained using ZDOCK server (Pierce et al. 2014) and these were processed through iterative rounds of manual adjustment by PyMOL (The PyMOL Molecular Graphics System, Version 1.5, Schrödinger, LLC). The model amino acid or nucleotide geometry regularization and use of allowed side chain rotomers were corrected with Coot (Emsley et al. 2010). The coordinates of these models are available upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael J. Howard for supplying the WT AtPRORP1 enzyme preparation. We thank David R. Engelke for helpful discussions and Nancy Wu for helpful comments on this manuscript. This work was supported in part by the National Institute of General Medical Sciences, National Institutes of Health (R01 GM055387 to C.A.F. and R01 GM117141 to M.K.); the Endowment for the Basic Sciences–Endowment for the Development of Graduate Education (EBS–EDGE) Award to B.P.K.; and by an American Heart Association pre-doctoral fellowship (16PRE29890011 to A.K.).
Author contributions: B.P.K. and A.K. designed the mutagenesis experiments for AtPRORP1 and AtPRORP2, respectively. B.P.K., A.K., K.J.K., A.J.L.D., A.Z.T., A.S., N.J., and M.J.H. purified mutants and performed assays. A.K. and M.K. performed the modeling experiments. B.P.K. and C.A.F. designed the remaining experiments with AtPRORP1. B.P.K. performed these experiments. All authors wrote or edited the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.061457.117.
REFERENCES
- Anantharaman V, Aravind L. 2006. The NYN domains: novel predicted RNAses with a PIN domain-like fold. RNA Biol 3: 18–27. [DOI] [PubMed] [Google Scholar]
- Barkan A, Rojas M, Fujii S, Yap A, Chong YS, Bond CS, Small I. 2012. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLOS Genet 8: e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley MD, Lewis PA, Sullivan GE. 1981. Ion effects on the lac repressor–operator equilibrium. Biochemistry 20: 3842–3851. [DOI] [PubMed] [Google Scholar]
- Bonnard G, Gobert A, Arrivé M, Pinker F, Salinas-Giegé T, Giegé P. 2016. Transfer RNA maturation in Chlamydomonas mitochondria, chloroplast and the nucleus by a single RNase P protein. Plant J 87: 270–280. [DOI] [PubMed] [Google Scholar]
- Brillante N, Gößringer M, Lindenhofer D, Toth U, Rossmanith W, Hartmann RK. 2016. Substrate recognition and cleavage-site selection by a single-subunit protein-only RNase P. Nucleic Acids Res 44: 2323–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li X, Gegenheimer P. 1997. Ribonuclease P catalysis requires Mg2+ coordinated to the pro-RP oxygen of the scissile bond. Biochemistry 36: 2425–2438. [DOI] [PubMed] [Google Scholar]
- Chen X, Feng F, Qi W, Xu L, Yao D, Wang Q, Song R. 2016. Dek35 encodes a PPR protein that affects cis-splicing of mitochondrial nad4 intron 1 and seed development in maize. Mol Plant 10: 427–441. [DOI] [PubMed] [Google Scholar]
- Crary SM, Niranjanakumari S, Fierke CA. 1998. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37: 9409–9416. [DOI] [PubMed] [Google Scholar]
- Day-Storms JJ, Niranjanakumari S, Fierke CA. 2004. Ionic interactions between PRNA and P protein in Bacillus subtilis RNase P characterized using a magnetocapture-based assay. RNA 10: 1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deHaseth PL, Lohman TM, Record MT Jr. 1977. Nonspecific interaction of lac repressor with DNA: an association reaction driven by counterion release. Biochemistry 16: 4783–4790. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. 1985. Hydrogen bonding and biological specificity analysed by protein engineering. Nature 314: 235–238. [DOI] [PubMed] [Google Scholar]
- Gobert A, Gutmann B, Taschner A, Gößringer M, Holzmann J, Hartmann RK, Rossmanith W, Giegé P. 2010. A single Arabidopsis organellar protein has RNase P activity. Nat Struct Mol Biol 17: 740–744. [DOI] [PubMed] [Google Scholar]
- Gobert A, Pinker F, Fuchsbauer O, Gutmann B, Boutin R, Roblin P, Sauter C, Giegé P. 2013. Structural insights into protein-only RNase P complexed with tRNA. Nat Commun 4: 1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guckian KM, Schweitzer BA, Ren RX, Sheils CJ, Tahmassebi DC, Kool ET. 2000. Factors contributing to aromatic stacking in water: evaluation in the context of DNA. J Am Chem Soc 122: 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace NR, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Gutmann B, Gobert A, Giegé P. 2012. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev 26: 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, Pfeiffer T, Zuleeg T, Limmer S, Ciesiolka J, Feltens R, Hartmann RK. 2001. Exploring the minimal substrate requirements for trans-cleavage by RNase P holoenzymes from Escherichia coli and Bacillus subtilis. Mol Microbiol 41: 131–143. [DOI] [PubMed] [Google Scholar]
- Hoang C, Ferré-D'Amaré AR. 2002. Cocrystal structure of a tRNA Ψ55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell 107: 929–939. [DOI] [PubMed] [Google Scholar]
- Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W. 2008. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474. [DOI] [PubMed] [Google Scholar]
- Howard MJ, Lim WH, Fierke CA, Koutmos M. 2012. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci 109: 16149–16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ, Liu X, Lim WH, Klemm BP, Fierke CA, Koutmos M, Engelke DR. 2013. RNase P enzymes: divergent scaffolds for a conserved biological reaction. RNA Biol 10: 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ, Klemm BP, Fierke CA. 2015. Mechanistic studies reveal similar catalytic strategies for phosphodiester bond hydrolysis by protein-only and RNA-dependent ribonuclease P. J Biol Chem 290: 13454–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ, Karasik A, Klemm BP, Mei C, Shanmuganathan A, Fierke CA, Koutmos M. 2016. Differential substrate recognition by isozymes of plant protein-only ribonuclease P. RNA 22: 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA III, Phillips S, Edgell MH, Gillam S, Jahnke P, Smith M. 1978. Mutagenesis at a specific position in a DNA sequence. J Biol Chem 253: 6551–6560. [PubMed] [Google Scholar]
- Imai T, Nakamura T, Maeda T, Nakayama K, Gao X, Nakashima T, Kakuta Y, Kimura M. 2014. Pentatricopeptide repeat motifs in the processing enzyme PRORP1 in Arabidopsis thaliana play a crucial role in recognition of nucleotide bases at TψC loop in precursor tRNAs. Biochem Biophys Res Commun 450: 1541–1546. [DOI] [PubMed] [Google Scholar]
- Karasik A, Shanmuganathan A, Howard MJ, Fierke CA, Koutmos M. 2016. Nuclear protein-only ribonuclease P2 structure and biochemical characterization provide insight into the conserved properties of tRNA 5′ end processing enzymes. J Mol Biol 428: 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kawabata M, Hisano K, Kazama T, Matsuoka K, Sugita M, Nakamura T. 2012. Identification and characterization of the RNA binding surface of the pentatricopeptide repeat protein. Nucleic Acids Res 40: 2712–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JC, Fierke CA. 2002. The affinity of magnesium binding sites in the Bacillus subtilis RNase P•pre-tRNA complex is enhanced by the protein subunit. Biochemistry 41: 9545–9558. [DOI] [PubMed] [Google Scholar]
- Kurz JC, Niranjanakumari S, Fierke CA. 1998. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry 37: 2393–2400. [DOI] [PubMed] [Google Scholar]
- Lai LB, Bernal-Bayard P, Mohannath G, Lai SM, Gopalan V, Vioque A. 2011. A functional RNase P protein subunit of bacterial origin in some eukaryotes. Mol Genet Genomics 286: 359–369. [DOI] [PubMed] [Google Scholar]
- Latt SA, Sober HA. 1967. Protein-nucleic acid interactions. II. Oligopeptide-polyribonucleotide binding studies. Biochemistry 6: 3293–3306. [DOI] [PubMed] [Google Scholar]
- Mao G, Chen TH, Srivastava AS, Kosek D, Biswas PK, Gopalan V, Kirsebom LA. 2016. Cleavage of model substrates by Arabidopsis thaliana PRORP1 reveals new insights into its substrate requirements. PLoS One 11: e0160246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AM, Horton NC, Lusetti S, Reich NO, Perona JJ. 1999. Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry 38: 8430–8439. [DOI] [PubMed] [Google Scholar]
- Marvin MC, Engelke DR. 2009. Broadening the mission of an RNA enzyme. J Cell Biochem 108: 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62. [DOI] [PubMed] [Google Scholar]
- Niranjanakumari S, Kurz JC, Fierke CA. 1998. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res 26: 3090–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole N, Hattori M, Andres C, Iida K, Lurin C, Schmitz-Linneweber C, Sugita M, Small I. 2008. On the expansion of the pentatricopeptide repeat gene family in plants. Mol Biol Evol 25: 1120–1128. [DOI] [PubMed] [Google Scholar]
- Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, et al. 2001. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen 6: 429–440. [DOI] [PubMed] [Google Scholar]
- Pavlova LV, Gößringer M, Weber C, Buzet A, Rossmanith W, Hartmann RK. 2012. tRNA processing by protein-only versus RNA-based RNase P: kinetic analysis reveals mechanistic differences. ChembioChem 13: 2270–2276. [DOI] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. 2014. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30: 1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinker F, Schelcher C, Fernández-Millán P, Gobert A, Birck C, Thureau A, Roblin P, Giegé P, Sauter C. 2017. Biophysical analysis of Arabidopsis protein-only RNase P alone and in complex with tRNA provides a refined model of tRNA binding. J Biol Chem 292: 13904–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record MT Jr, Lohman ML, deHaseth P. 1976. Ion effects on ligand-nucleic acid interactions. J Mol Biol 107: 145–158. [DOI] [PubMed] [Google Scholar]
- Record MT Jr, Anderson CF, Lohman TM. 1978. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q Rev Biophys 11: 103–178. [DOI] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragón A. 2010. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468: 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, Hsieh J, Day-Storms JJ, Fierke CA, Walter NG. 2005. The 5′ leader of precursor tRNAAsp bound to the Bacillus subtilis RNase P holoenzyme has an extended conformation. Biochemistry 44: 16130–16139. [DOI] [PubMed] [Google Scholar]
- Schmitz-Linneweber C, Small I. 2008. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci 13: 663–670. [DOI] [PubMed] [Google Scholar]
- Shi H, Moore PB. 2000. The crystal structure of yeast phenylalanine tRNA at 1.93 A resolution: a classic structure revisited. RNA 6: 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Wang Z, Koo BK, Patel A, Cascio D, Collins K, Feigon J. 2012. Structural basis for telomerase RNA recognition and RNP assembly by the holoenzyme La family protein p65. Mol Cell 47: 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Peeters N. 2000. The PPR motif—a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci 25: 46–47. [DOI] [PubMed] [Google Scholar]
- Sugita C, Komura Y, Tanaka K, Kometani K, Satoh H, Sugita M. 2014. Molecular characterization of three PRORP proteins in the moss Physcomitrella patens: nuclear PRORP protein is not essential for moss viability. PLoS One 9: e108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner A, Weber C, Buzet A, Hartmann RK, Hartig A, Rossmanith W. 2012. Nuclear RNase P of Trypanosoma brucei: a single protein in place of the multicomponent RNA–protein complex. Cell Rep 2: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. 2012. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res 40: 11583–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczyk D, Gößringer M, Rossmanith W, Zatsepin TS, Oretskaya TS, Hartmann RK. 2016. Analysis of the cleavage mechanism by protein-only RNase P using precursor tRNA substrates with modifications at the cleavage site. J Mol Biol 428: 4917–4928. [DOI] [PubMed] [Google Scholar]
- Walker SC, Engelke DR. 2006. Ribonuclease P: the evolution of an ancient RNA enzyme. Crit Rev Biochem Mol Biol 41: 77–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y, Hayashi S, Kobayashi K, Hirayama T, Nakamura T. 2013. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One 8: e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Li Q, Yan C, Liu Y, Liu J, Yu F, Wang Z, Long J, He J, Wang HW, et al. 2013. Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature 504: 168–171. [DOI] [PubMed] [Google Scholar]
- Zhang J, Férre-D'Amaré AR. 2016. The tRNA elbow in structure, recognition and evolution. Life (Basel) 6: E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.