

Abstract
The insulin-induced gene 1 protein (Insig1) inhibits the cholesterol biosynthesis pathway by retaining transcription factor SREBP in the endoplasmic reticulum, and by causing the degradation of HMGCR, the rate-limiting enzyme in cholesterol biosynthesis. Liver-specific microRNA miR-122, on the other hand, enhances cholesterol biosynthesis by an unknown mechanism. We have found that Insig1 mRNAs are generated by alternative cleavage and polyadenylation, resulting in specific isoform mRNA species. During high cholesterol abundance, the short 1.4-kb Insig1 mRNA was found to be preferentially translated to yield Insig1 protein. Precursor molecules of miR-122 down-regulated the translation of the 1.4-kb Insig1 isoform mRNA by interfering with the usage of the promoter-proximal cleavage–polyadenylation site that gives rise to the 1.4-kb Insig1 mRNA. These findings argue that precursor miR-122 molecules modulate polyadenylation site usage in Insig1 mRNAs, resulting in down-regulation of Insig1 protein abundance. Thus, precursor microRNAs may have hitherto undetected novel functions in nuclear gene expression.
Keywords: microRNA, Insig1, cholesterol, polyadenylation site, mRNA isoforms
INTRODUCTION
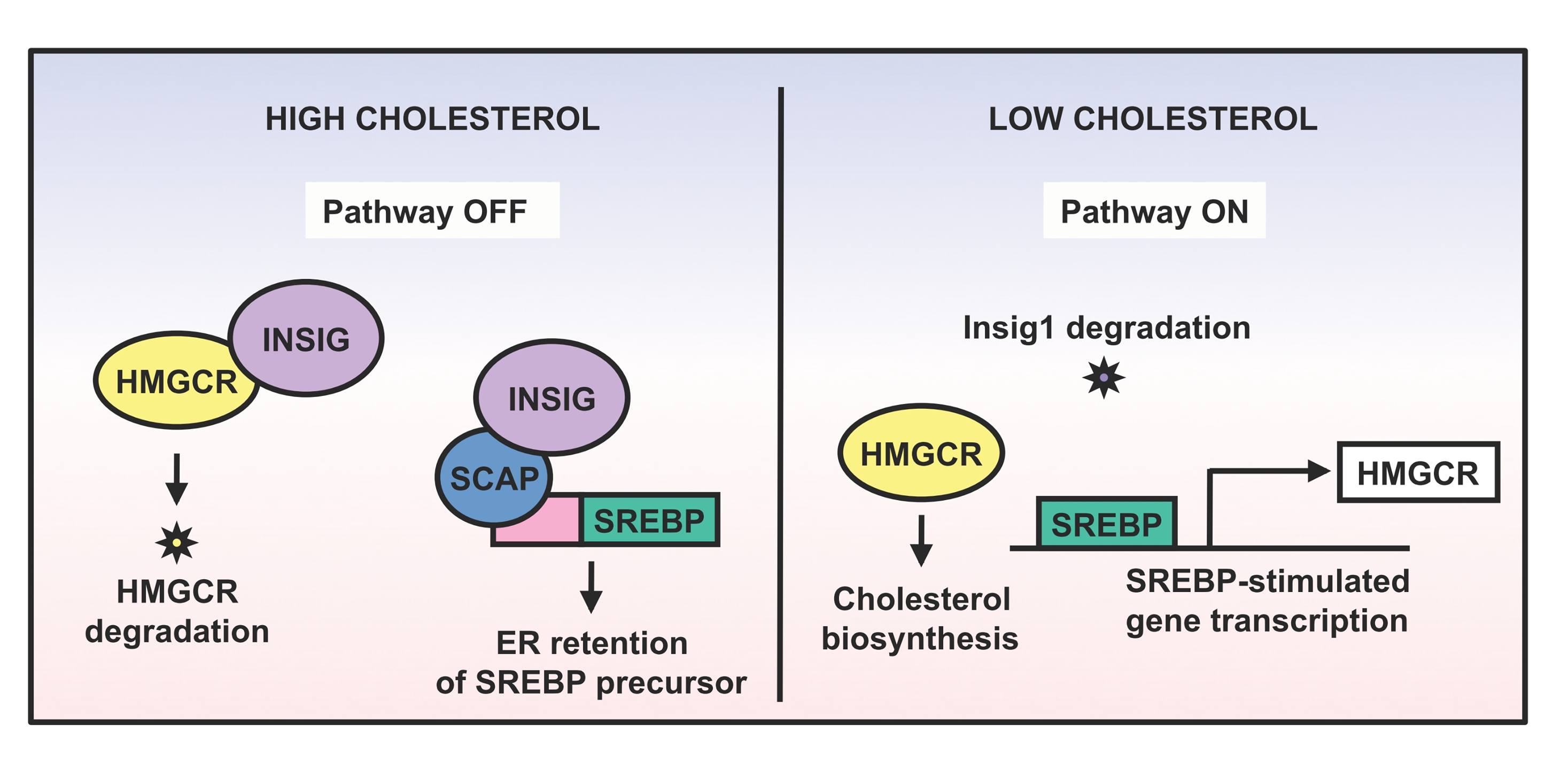
Lipid metabolism contributes vital functions to many cell processes, ranging from steroid hormone biosynthesis, to cholesterol homeostasis, to modulating effective antiviral responses (Espenshade and Hughes 2007). Accordingly, the cholesterol biosynthesis pathway is tightly regulated on multiple levels. A central inhibitor of the cholesterol biosynthesis pathway is the insulin-induced gene Insig1 (Supplemental Fig. S1). When sterols are abundant, Insig1 binds two sterol-sensing ER membrane proteins: 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis, and sterol regulatory element binding protein cleavage-activating protein (SCAP) (Yang et al. 2002; Sever et al. 2003). Insig1-bound HMGCR is targeted for ubiquitination and degradation (Sever et al. 2003), while Insig1–SCAP sequesters the sterol regulatory element binding protein (SREBP) in the endoplasmic reticulum (Supplemental Fig. S1). The SREBP family of transcription factors stimulates the expression of genes that are involved in the synthesis and uptake of cholesterol as well as in the metabolism of fatty acids, triglycerides, carbohydrates, and phospholipids (for review, see Dong and Tang 2010). When sterol levels decline, Insig1 dissociates from both HMGCR, causing its stabilization, and from SCAP–SREBP, allowing its proteolytic processing and its relocalization to the nucleus where it functions as an active transcription factor (Supplemental Fig. S1). Once sterols accumulate sufficiently, Insig1 protein is stabilized through re-formation of SCAP–SREBP–Insig1 complexes (Gong et al. 2006). As one can predict, overexpression of Insig1 indeed down-regulates cholesterol biosynthesis in mice (Engelking et al. 2004).
Curiously, inactivation of the abundant liver-specific microRNA, miR-122, also inhibits cholesterol biosynthesis in mice and nonhuman primates (Krutzfeldt et al. 2005; Esau et al. 2006; Elmén et al. 2008) by a hitherto unknown mechanism. MicroRNAs normally suppress gene expression by sequence-specific binding to 3′ noncoding regions (NCR) in target mRNAs, leading to mRNA degradation, translational suppression or both (Jackson and Standart 2007; Bartel 2009). To identify target mRNAs for miR-122, Esau et al. (2006) and Krutzfeldt et al. (2005) sequestered miR-122 with antisense oligonucleotides in mouse liver. This treatment did not adversely affect liver function but decreased cholesterol abundance in treated animals. Gene profiling by cDNA microarrays revealed that antisense miR-122-treated animals displayed a decrease in many transcripts coding for enzymes that function in the cholesterol biosynthesis pathway. Interestingly, we noted that many genes that were down-regulated during miR-122 sequestration (Krutzfeldt et al. 2005; Esau et al. 2006) were the same that were down-regulated during overexpression of Insig1 (Engelking et al. 2004), suggesting that miR-122 targets Insig1. Importantly, the 3′NCRs of Insig1 isoform mRNAs contain four putative binding sites for miR-122 (Fig. 1A).
FIGURE 1.
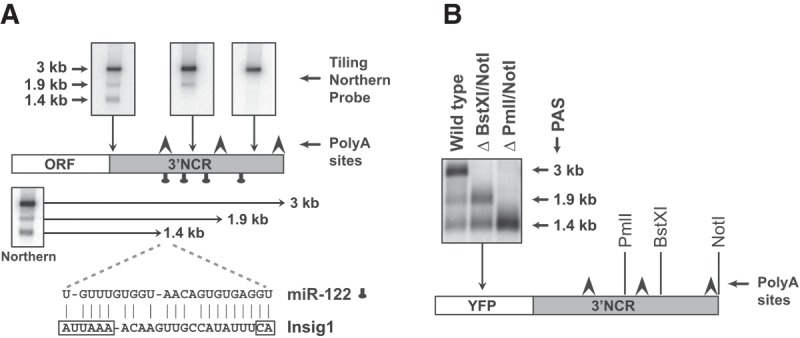
Expression analysis of Insig1 mRNA isoforms. (A) Diagram of Insig1 isoform 3′NCRs. The 1.4-, 1.9-, and 3-kb isoforms are revealed by Northern blot analyses (top). The locations of putative polyadenylation sites (arrows) and seed match sequences for miR-122 (tags) are indicated. The diagram shows the predicted interaction of miR-122 with the AUUAAA-polyadenylation and CA-cleavage sequence motif that generates the 1.4-kb Insig1 isoform mRNA. (B) Insig1 polyadenylation site usage in YFP-Insig1-3′NCR chimeric mRNAs. Size and integrities of chimeric mRNAs were analyzed in Northern blots using hybridization probes against YFP.
In this study, we examined the mechanism by which miR-122 regulates Insig1 mRNA expression. We found that Insig1 expression is regulated by alternative cleavage and polyadenylation to produce three major mRNA isoforms. The shortest mRNA is 1.4 kb in length, is preferentially expressed during sterol abundance, and is the major mRNA template for the synthesis of Insig1 protein. Remarkably, the precursor miR-122 specifically suppressed the abundance of the 1.4-kb Insig1 isoform mRNA, and indirectly its subsequent translation, through modulation of the promoter proximal polyadenylation site (PAS) usage. These results point to a specific role for pre-miR-122 in the suppression of Insig1 1.4-kb mRNA expression and suggest more general roles for nuclear pre-microRNAs in gene expression.
RESULTS
Generation of Insig1 mRNAs by usage of distinct cleavage–polyadenylation sites
We noted that functional inactivation of miR-122 by antisense oligonucleotides (Krutzfeldt et al. 2005; Esau et al. 2006) and overexpression of Insig1 (Engelking et al. 2004) in the liver of mice resulted in the down-regulation of a similar set of genes. This observation suggested that Insig1 could be targeted by miR-122. Inspection of the 3′NCR of Insig1 revealed the presence of four putative binding sites for miR-122 (Fig. 1A; Supplemental Fig. S2; Bartel 2009). Northern blot analyses revealed that three polyadenylation sites (PAS) are used to generate Insig1 mRNAs that are 1.4, 1.9, and 3 kb in length (Fig. 1A,B; Supplemental Fig. S2). These Insig1 isoforms were also expressed to similar abundances in liver 5B and non-liver HeLa cells (Supplemental Fig. S3). Curiously, one predicted miR-122 target site involves base-pair interactions between miR-122 with the promoter-proximal PAS (Fig. 1A), while the other three predicted miR-122 binding sites reside within the 1.9- and 3-kb Insig1 3′NCRs (Fig. 1A; Supplemental Fig. S2). Also, 3′ RACE analysis with RNA isolated from liver Huh7 cells revealed the existence of Insig1 isoform mRNAs with distinct 3′ NCRs (data not shown). Clearly, the 3-kb isoform was the predominant RNA species in proliferating Huh7 cells (Fig. 1A). To substantiate that the Insig1 isoform mRNAs resulted from alternate cleavage/polyadenylation, a chimeric gene was constructed that consisted of the yellow fluorescent protein (YFP) coding region fused to the entire Insig1 3′NCR (Fig. 1B). The chimeric gene was transfected into Huh7 cells and RNA expression studied by Northern blot analysis. The results showed that the 3′NCR of Insig1 was sufficient to generate the three predicted YFP isoform mRNAs (Fig. 1B). Deletion of the two 3′ proximal PAS sites greatly enhanced the abundance of Insig1 mRNA generated from the promoter-proximal PAS, suggesting that usage of these PAS is regulated (Fig. 1B).
To examine whether usage of PAS in Insig1 is modulated, we monitored the stabilities of the Insig1 mRNA isoforms during low and high abundances of sterols. Briefly, Huh7 cells were incubated with medium lacking sterols for 14 h and subsequently incubated with complete medium (Fig. 2A). Western blot analysis showed that Insig1 protein abundance increased at 4 h after addition of sterols and mevalonate, especially in the presence of the proteasome inhibitor MG132 (carbobenzoxy-Leu-Leu-leucinal) (Fig. 2A, lower panel; Lee and Goldberg 1998). In addition, the abundance of HMGCR increased under this condition, arguing that the known proteasomal degradation of Insig1 and HMGCR can be monitored in cultured Huh7 cells. Thus, this experimental protocol allows the studying of sterol-dependent regulation of Insig1 protein production. Next, the production of the Insig1 isoform mRNAs was examined. Figure 2B shows that addition of sterols leads to an overall decrease of all three Insig1 isoform mRNAs (Fig. 2B), especially at 4 h after addition when Insig1 protein was clearly accumulating (Fig. 2A, lower panel). Interestingly, the 1.4-kb Insig1 mRNA abundance decreased at a much slower rate than the 1.9- and 3-kb mRNAs after add-back of sterols (Fig. 2B, lower panel). To examine which Insig1 isoform mRNA is translated, the association of Insig1 mRNAs with ribosomes was inspected in sucrose-gradient analyses. The polysomal profiles in Figure 2C show that the 3-kb Insig1 isoform mRNA was the predominant polysome-associated mRNA species during sterol depletion. In contrast, the 1.4-kb Insig1 isoform was the predominant polysomal mRNA after add-back of sterols (Fig. 2C, lower panel), suggesting that the 1.4-kb mRNA is efficiently translated during high abundance of sterols. These results suggest that 1.4-kb Insig1 mRNA, generated at the promoter-proximal PAS, is the Insig1 isoform mRNA that is most efficiently translated during high sterol abundance.
FIGURE 2.
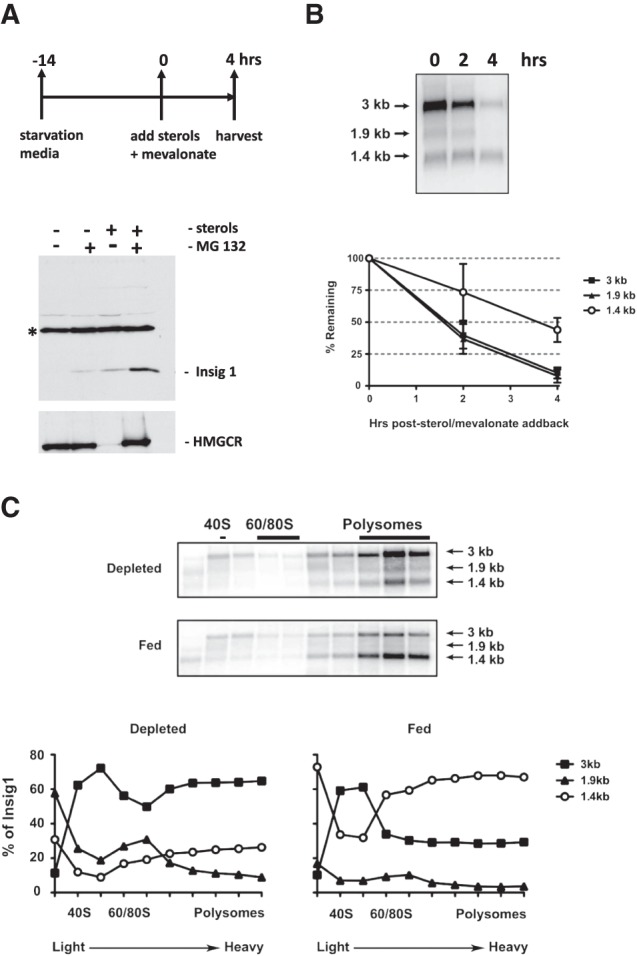
Insig1 isoform abundances in response to sterol abundance. (A) Cells were depleted of sterols for 14 h (O/N) in media containing lipoprotein-deficient serum, lovastatin, and low levels of mevalonate. At time zero, sterols and mevalonate were added back. (Lower panel) Western blot to visualize Insig1 and HMGCR protein under conditions of sterol starvation and during addback, in the presence and absence of the proteasome inhibitor, MG132. The band marked by an asterisk reacts nonspecifically with the Insig1 antibody and serves as an internal loading control. (B) Stabilities of Insig1 mRNA isoforms during depletion and following add-back of sterol/mevalonate. Huh7 cells were depleted of sterols for 14 h. At time zero, sterols and sterol intermediates were added and RNA was harvested at the indicated times. Insig1 mRNA isoform abundances were measured by Northern blot analyses. Insig1 isoform RNA abundance was normalized to actin mRNA at each time point (lower panel). Autoradiographs were quantitated using ImageQuant (GE Healthcare). RNA abundance at time zero was set to 100%. Error bars represent standard error of the mean. (C) Polysomal distribution of Insig1 mRNA isoforms. Cells were either depleted of sterols, or depleted and re-fed for four hours. Lysates were prepared and analyzed by sucrose-gradient analysis, as described in Materials and Methods. The relative abundances of the three Insig1 mRNA isoforms in each fraction under sterol-depleted and sterol-fed conditions were measured in Northern blots. (Lower panels) Representative Northern blot displaying Insig1 isoform RNAs in each gradient fraction. Autoradiographs were quantitated using ImageQuant (GE Healthcare). The data are representative of at least three independent replicates.
Differential regulation of Insig1 mRNA isoform expression by miR-122
Next, we tested whether Insig1 mRNA PAS usage is regulated by miR-122 via noncanonical microRNA–mRNA interactions. First, the steady-state abundance of miR-122 during low and high sterol conditions was examined. In untreated liver cells, the majority of miR-122 consisted of 21-, 22-, and 23-nt miR-122 species (Fig. 3A). Quantitation of Northern blots showed that the steady-state abundances of each mature species did not change during low and high sterol conditions (Fig. 3A, middle panel). While we have been unable to detect pre-miR-122 in Huh7 cells under any condition, the abundance of the primary miR-122 (pri-miR-122) was statistically significant (P-value of 0.0567 after sterol add-back for 2 h; legend to Fig. 3) diminished during high sterol abundance (Fig. 3A, right panel), suggesting that the synthesis of pri-miR-122 is slowed or the turnover of pri-miR-122 is enhanced.
FIGURE 3.

Effects of miR-122 abundance on Insig1 mRNA isoform abundances. (A) (Left panel) Northern blot for mature miR-122. miR-122 is expressed mainly as three isofroms of 21, 22, and 23 nt in length. Cells were depleted of sterols overnight and re-fed with sterols and mevalonate for 2 and 4 h, and mature (middle panel) and precursor (right panel) miR-122 abundance measured by Northern blot. The differences of abundance of the mature microRNAs after sterol add-back were statistically not significant (all P-values were larger than 0.57). The differences in abundance between pri-miR-122 from 0 to 2, and from 0 to 4 h after sterol add-back were significant, with P-values of 0.0567 and 0.097, respectively. (B–D) Cells were transfected with antisense locked nucleic acids (LNA) against miR-122, miR-106b/miR-93, or miR-25/miR-92 and depleted of sterols overnight. At time zero, cells were re-fed with sterols and mevalonate and total RNA harvested at the indicated time points. Insig1 mRNA isoforms were visualized by Northern blot analysis and quantitated using normalization to actin mRNA. RNA abundances from cells transfected with antisense (as) miR-106b/93 LNA at time zero were set to one. The data are representative of at least three independent replicates. Error bars represent standard error of the mean.
To test the idea that newly synthesized miR-122 or its precursors have roles in the regulation of Insig1, Huh7 cells were first transfected with antisense, locked nucleic acids (LNA) to sequester miR-122 or control LNAs to sequester miR-106b/93 or miR-25/92, using conditions that sequester more than 90% of the microRNAs (Elmén et al. 2008; Machlin et al. 2011). Cells were then depleted of sterols and Insig1 isoform abundances were measured after sterol add-back (Fig. 3B–D). Northern blot analyses showed that sequestration of miR-122 did not significantly affect the abundance of the 3-kb Insig1 mRNA (Fig. 3B), slightly decreased 1.9-kb Insig1 mRNA abundance (Fig. 3C), but specifically enhanced 1.4-kb Insig1 mRNA abundance (Fig. 3D). Similarly, sequestration of miR-122 enhanced the abundance of the 1.4-kb, but decreased the abundance of the 1.9-kb Insig1 isoforms in chimeric YFP-Insig1 3′NCR mRNAs (Fig. 4A), arguing that miR-122 mediated regulation is conferred by the 3′NCR of Insig1. In contrast, little effects on Insig1 isoform abundances were observed in the presence of control LNAs. These data argue that miR-122 inhibited promoter-proximal PAS usage in Insig1 mRNA.
FIGURE 4.
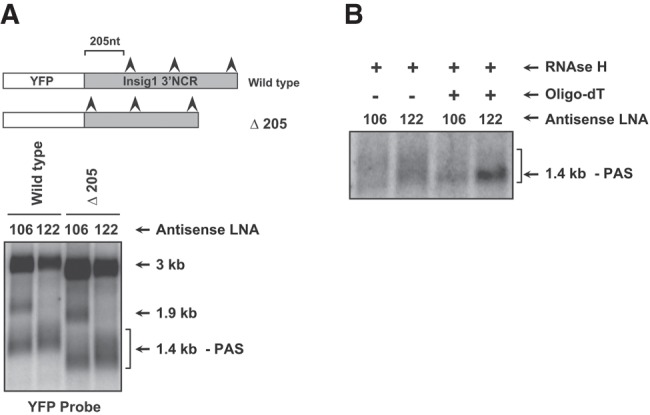
Effects of miR-122 abundance on the 3′ terminal polyadenosine length of Insig1 isoforms. (A) Effects of miR-122 and miR-106b/93 sequestration on Insig1 isoform RNA abundances. Cells were transfected simultaneously with indicated antisense LNA oligonucleotides and the YFP-Insig1-3′NCR encoding plasmids and allowed to recover for 28 h. Cells were then depleted of sterols for 14 h and RNA was harvested. Insig1 isoforms were examined in a Northern blot using a hybridization probe against the YFP open reading frame. Plasmid Δ205 lacks the first 205 nt of the Insig1 3′NCR. (B) Poly(A) tail length measurements in Insig1 isoform mRNAs. Cells were treated as in panel A. Purified mRNA was hybridized to oligo(dT) and treated with RNase H. Shown is a Northern blot using a hybridization probe against the YFP open reading frame.
Inhibition of polyadenylation of 1.4-kb Insig1 mRNA by miR-122
Inspection of the Northern blot in Figure 4A indicates that miR-122 sequestration generated a 1.4-kb Insig1 isoform mRNA that migrated more slowly in the polyacrylamide gel than the 1.4-kb Insig1 isoform that was detected in cells in which miR-106 was sequestered. To substantiate more carefully this observation, we monitored the migration of a chimeric Insig1 mRNA in which the 205 nt upstream of the 1.4 kb PAS were deleted. Indeed, Figure 4A shows that miR-122 sequestration affected the migration of the deleted, chimeric mRNA, yielding longer mRNAs of heterogeneous lengths. To examine whether sequestration of miR-122 enhances polyadenylation at the 1.4-kb PAS, RNA was isolated from miR-122 LNA- and miR-106 LNA-treated cells, hybridized to oligo(dT), and treated with RNase H. Figure 4B shows that the larger Insig1 1.4-kb mRNA species collapsed to a distinct homogeneous species in the presence of RNase H and oligo(dT). In contrast, the polyadenylation at the 1.4-kb PAS was not affected by the presence of LNAs directed against miR-106b/93. These results argue that miR-122 sequestration allowed cleavage and polyadenylation at the 1.4-kb PAS.
Precursor miR-122 modulates the 1.4-kb PAS usage in Insig1
The above data suggest that miR-122 inhibits Insig1 1.4 PAS usage in the nucleus. While it has been shown that some microRNAs, such as miR-29b, locate to the nucleus (Hwang et al. 2007), miR-122 lacks the characteristic hexameric sequence motif that directs miR-29b to the nucleus. In situ hybridization located miR-122 mostly to the cytoplasm with some minor nuclear staining (data not shown); however, this minor nuclear fraction could be functional. Therefore, effects of overexpression of mature miR-122 and pre-miR-122 on the expression of chimeric YFP-3′NCR Insig1 RNAs (Fig. 5A) were examined. Expression of mature miR-122 was performed through transfection of duplex miR-122 mimetics (Machlin et al. 2011). The mimetic-derived mature miR-122 down-regulated YFP synthesis (Fig. 5B). Interestingly, transfection of pre-miR-122 also down-regulated YFP-3′NCR Insig1 expression in a dose-dependent manner (Fig. 5C). Furthermore, pre-miR-122 down-regulated YFP-3′NCR Insig1 expression also during add-back of sterols (Supplemental Fig. S4). To exclude that the precursor miR-122 mimetics were processed to yield mature miR-122 molecules, precursor miR-122 molecules were generated that contained nine substituted deoxynucleotides around the Dicer cleavage sites to prevent these pre-miR-122 molecules from being cleaved into mature miR-122 in cells (Cox et al. 2013). These Dicer-resistant pre-miR-122 (pre-miR-122-dNx9) (Cox et al. 2013) down-regulated YFP-3′NCR Insig1 expression consistently (Fig. 5D). These data indicate that both mature miR-122 and DICER-resistant pre-miR-122 regulate the relative abundance of the translation-competent 1.4-kb Insig1 mRNA isoform by inhibiting the usage of the 1.4-kb PAS.
FIGURE 5.
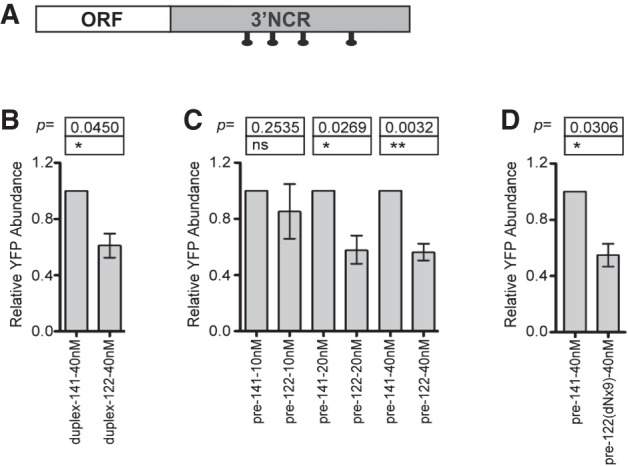
Effects of miR-122 precursor miRNA on Insig1 isoform translation. Cells were transfected with YFP:Insig1 3′ NCR with precursor hairpin or duplex mimetic miR-122 or miR-141 RNA at the indicated concentrations. Shown is the fold change in YFP protein relative to miR-141-transfected cells at each concentration. (A) Diagram of reporter mRNA with marking of putative miR-122 binding sites (B) Effects of duplex mimetic miR-122. (C) Dose-responsiveness of pre-miR-122 effects on YFP:Insig1. (D) Effect of Dicer-resistant pre-miR-122 (pre-miR-122-dNx9). The data are representative of three independent replicates. Error bars represent standard error of the mean; ns, not significant; (*) P < 0.05, (**) P < 0.01, Student's t-test.
DISCUSSION
We provided evidence that miR-122 targets Insig1 expression by controlling alternate Insig1 mRNA isoform expression. In support of a functional miR-122 binding site at the promoter-proximal PAS in Insig1, Luna and colleagues reported a binding site for Argonaute, an essential component of the miR-122/RISC, upstream of the 1.4-kb PAS (Luna et al. 2017). Here, we have defined a novel mechanism by which Insig1 protein accumulation can be modulated through microRNA-mediated usage of an alternative polyadenylation site. Specifically, both mature mir-122 and precursor miR-122 molecules target the promoter-proximal PAS in Insig1, thereby inhibiting the generation of 1.4-kb Insig1 mRNA isoforms that are the main mRNA templates for protein synthesis during high cholesterol abundance. Whether the few detected nuclear mature miR-122 molecules are functional in PAS modulation or whether ectopic expression of mature miR-122 causes an artificial increase in nuclear miR-122, needs to be determined. It is possible that the DICER-resistant pre-miR-122 and associated proteins bind to the 1.4-kb polyadenylation signal and antagonize the binding of components of the polyadenylation machinery. Such a mechanism could involve a direct competition, additional protein recruitment, or modulation of Insig1 mRNA structure. Alternatively, the pre-miR-122/RISC complex could allow polyadenylation at the 1.4-kb PAS, followed by a rapid deadenylation. Pre-mRNA cleavage and polyadenylation requires assembly of a large protein complex on the 3′NCR, guided by recognition of an A(A/U)UAAA hexameric sequence upstream and a U/GU-rich sequence downstream from the cleavage site (Shi et al. 2009). Thus, pre-miR-122 may also modulate polyadenylation machinery components that may dictate PAS usage (Takagaki et al. 1996; Kubo et al. 2006). Clearly, a defined in vitro system is needed to distinguish between these possibilities.
It is curious that miR-122 also targets two modulators of polyadenylation, nocturnin (Kojima et al. 2010) and the cytoplasmic polyadenylation element binding protein CPEB (Burns et al. 2011). Nocturnin is a circadian deadenylase that is important in lipid metabolism (Green et al. 2007). Interestingly, pri-miR-122 and pre-miR-122, but not mature miR-122, are also expressed in a circadian manner (Gatfield et al. 2009). CPEB plays a role in activating dormant mRNAs for translation through post-transcriptional lengthening of polyadenosine tails (Richter 2007). Thus, down-regulation of CPEB by miR-122 could contribute to loss of poly(A) tails and destabilization of specific mRNAs.
Alternative polyadenylation is very common in the transcriptome, with greater than 50% of all mRNAs exhibiting alternative 3′ end processing (Tian et al. 2005). However, the mechanisms regulating alternative poly(A) site use are unknown for most mRNAs. Like miRNA expression, alternative polyadenylation is highly tissue- and developmentally regulated (Zhang et al. 2005; Wang et al. 2008; Ji and Tian 2009; Wilusz and Spector 2010). Our observations set a paradigm in which pre-microRNAs can modulate the turnover fate of target mRNAs by targeting the polyadenylation site usage, a mechanism that may be more prevalent in mRNAs that carry long 3′ NCRs enriched with microRNA binding sites. Shortening of 3′NCR can stabilize mRNAs and lead to a 40-fold up-regulation of protein production (Mayr and Bartel 2009). Thus, pre-miRNA-induced changes in polyadenylation site usage may have profound consequences on a variety of cell processes.
MATERIALS AND METHODS
Oligonucleotides and DNA constructs
Sequences of LNA and DNA oligonucleotides are provided in Supplemental Table S1. Antisense miR-122 LNAs were kindly provided by Santaris Pharma (Denmark) (Elmén et al. 2008) and antisense miR-106b/93 and miR-25/92a LNA were purchased from Integrated DNA Technologies. Because miR-106b and miR-93 are very similar in sequence, LNAs are predicted to inhibit both of these miRNAs. The same is true for miR-25 and miR-92a. All DNA primers and precursor hairpin miRNA were purchased from the Stanford PAN Facility and Dharmacon.
The YFP plasmid pcDNA-CL encodes Venus YFP. The sequences of the human Insig1 3′ NCR were inserted downstream from the YFP open reading frame using the EcoRV and NotI sites and the primers indicated in Supplemental Table S1. Polyadenylation sites were removed through deletion of the sequence between NotI and SapI. All other 3′ NCR deletions were performed using PCR with the primers indicated in Supplemental Table S1, followed by blunt-end ligation. The 1.4-kb PAS was mutated from AUAAAA to CUCAAA by site-directed mutagenesis using the QuikChange II XL kit (Stratagene) according to the manufacturer's protocol.
Cell culture and transfection
Unless otherwise noted, all reagents were obtained from Sigma-Aldrich. Huh7 human liver hepatoma cells were propagated in DMEM containing 10% fetal bovine serum, 200 µM l-glutamate, and 1× nonessential amino acids (GIBCO). Sterol and isoprenoid depletion media contained DMEM with 200 µM l-glutamate, 1× nonessential amino acids (GIBCO), 5% lipoprotein-deficient serum (Biomedical Technologies), 50 µM lovastatin (US Biological), and 100 µM sodium mevalonate. Sodium mevalonate was prepared from mevalonic acid lactone as described in Ye et al. (2003). Sterol and mevalonate addback media is the same as depletion media with added 10 mM sodium mevalonate, 10 µg/mL cholesterol, and 1 µg/mL 2,5-hydroxycholesterol.
For chimeric YFP:Insig1 RNA expression studies, 3 × 105 cells were plated in 60-mm dishes the day prior to transfection. YFP plasmids were transfected at 200 ng per 60-mm plate and LNA at a final concentration of 25 nM using Lipofectamine 2000 (Invitrogen) according to the manufacturer's directions. Cells were then allowed to recover in complete media for 28 h, washed 1× in phosphate buffered saline (PBS), and then sterols were depleted by overnight incubation. RNA was then harvested for analysis. To examine endogenous Insig1 RNA abundance, 6 × 105 cells were transfected with 25 nM LNA and immediately starved overnight, followed by sterol addback for 0, 2, or 4 h before RNA extraction. For YFP protein expression studies, 60-mm dishes of Huh7 cells were transfected with 1 μg of YFP plasmid along with precursor hairpins or LNAs and Lipofectamine 2000, and allowed to recover for 28 h in complete media. Cells were then washed with PBS and incubated in complete media or starved overnight, followed by sterol addback prior to protein harvesting.
RNA isolation and Northern analysis
Total RNA was isolated from Huh7 cells using TRIzol (Invitrogen), following the manufacturer's recommendations. Endogenous human Insig1 was measured by Northern blot containing 8 µg of total RNA. YFP reporter RNAs were measured after purification of 15 µg of total RNA on OligoTex-beads (Qiagen). RNA was resolved by electrophoresis in 1% to 1.2% agarose gels and hybridized to random-primed, 32P-labeled probes as described in Machlin et al. (2011), with the exception that Ultrahyb hybridization buffer (Ambion) was used for overnight hybridization.
3′RACE
Total RNA from Huh7 cells was reverse transcribed using an anchored oligo(dT) primer (Supplemental Table S1). cDNA was then amplified using Insig1 forward primers and the anchor reverse primer (Supplemental Table S1). PCR products were cloned into pCR2.1 TOPO (Invitrogen) and then sequenced to determine the polyadenylation sites.
RNase H assay
Approximately 20 µg of total RNA was used as starting material for each reaction. Total RNA was isolated with TRIzol, followed by extraction with acid phenol/chloroform and chloroform. RNA was precipitated, washed in 70% ethanol, resuspended, and then treated with RQ1 DNase (Promega), purified with an RNeasy kit (Qiagen), and mRNA was isolated using Oligotex (Qiagen) and precipitated. For RNase H assays, RNA was resuspended in annealing buffer (1 mM EDTA [pH 8] [Ambion], 0.2 M KCl) with and without 100 pmol of oligo(dT21), heated to 95°C for 3 min, and allowed to anneal at room temperature for 20 min. RNaseH buffer and RNase H (New England Biolabs) were then added, and reactions were incubated at 37°C for 45 min. Reactions were then adjusted to 0.5% SDS, 10 mM EDTA, and 250 µg/mL proteinase K (New England Biolabs), and were incubated at 37°C for 30 min. RNA was purified by acid phenol chloroform and chloroform extraction and analyzed by 1.2% agarose–formaldehyde gel electrophoresis and Northern blot analysis.
Polysome analysis
One 15-cm plate of Huh7 cells was either depleted of sterols overnight, or depleted and re-fed for 4 h. Cells were treated for 3 min with cycloheximide (100 μg/mL) at 37°C, washed 2× in cold PBS containing 100 μg/mL cycloheximide, and lysed for 10 min on ice in gradient buffer (150 mM KCl, 15 mM Tris–HCl, pH 7.5, 15 mM MgCl2, 100 μg/mL cycloheximide, 1 mg/mL heparin) containing 1% Triton X-100. Lysates were cleared with a 5 min spin at 8400g at 4°C, then layered onto 10% to 60% sucrose gradients composed of the above gradient buffer. Gradients were spun in an SW41 ultracentrifuge rotor for 2 h 45 min at 35,000 rpm at 4°C. Of note, 750 μL fractions were collected using the Isco Retriever II/UA-6 detector system. Fractions were treated immediately with 10 µL proteinase K (20 mg/mL, New England Biolabs) and 0.5% SDS for 1 h at 37°C, and then RNA was isolated using acid–phenol chloroform extraction, followed by ethanol precipitation.
Western blotting
Cells were washed once with PBS and lysed in RIPA buffer (50 mM Tris pH 7.5, 0.1% SDS, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, protease inhibitor [Roche]). Protein samples (∼40 μg) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to Immobilon P membranes (Millipore). Western blots were performed using anti-GFP primary antibody (Roche) for detecting YFP protein expression. The anti-Insig1 primary antibody was purchased from Biovision. Blots were treated by alkaline phosphatase secondary antibodies (Invitrogen) and developed using enhanced chemifluorescence according to the manufacturer's instructions (Amersham GE Healthcare). Signal was detected using a Storm Phosphoimager and quantitated with ImageQuant (GE Healthcare); or blots were processed with horseradish peroxidase-conjugated secondary antibodies and developed using Pierce ECL Western Blot Substrate (Amersham GE Healthcare), according to the manufacturer's instructions, and exposed to Biomax Light Films.
DATA DEPOSITION
Human Insig1 numbering is based on Genbank accession GI: 38327531. Polyadenylation sites are either verified by 3′RACE or predicted by Aceview.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Navdar Sever for helpful advice and Karla Kirkegaard and Susan Lynch for critical reading of the manuscript. We express gratitude to members of the Sarnow laboratory for stimulating discussions. We are grateful to Joe Luna for sharing Ago-CLIP data results. We are grateful for funding from the Alberta Heritage Foundation for Medical Research (to K.L.N.) and the National Institutes of Health (AI 069000) to P.S.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.063099.117.
REFERENCES
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DM, D'Ambrogio A, Nottrott S, Richter JD. 2011. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EM, Sagan SM, Mortimer SA, Doudna JA, Sarnow P. 2013. Enhancement of hepatitis C viral RNA abundance by precursor miR-122 molecules. RNA 19: 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XY, Tang SQ. 2010. Insulin-induced gene: a new regulator in lipid metabolism. Peptides 31: 2145–2150. [DOI] [PubMed] [Google Scholar]
- Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U, et al. 2008. LNA-mediated microRNA silencing in non-human primates. Nature 452: 896–899. [DOI] [PubMed] [Google Scholar]
- Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, Liang G. 2004. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J Clin Invest 113: 1168–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. 2006. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3: 87–98. [DOI] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. 2007. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 41: 401–427. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepää AL, Oresic M, Esau CC, Zdobnov EM, et al. 2009. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23: 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. 2006. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab 3: 15–24. [DOI] [PubMed] [Google Scholar]
- Green CB, Douris N, Kojima S, Strayer CA, Fogerty J, Lourim D, Keller SR, Besharse JC. 2007. Loss of Nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc Natl Acad Sci 104: 9888–9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. 2007. A hexanucleotide element directs microRNA nuclear import. Science 315: 97–100. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Standart N. 2007. How do microRNAs regulate gene expression? Sci STKE 2007: re1. [DOI] [PubMed] [Google Scholar]
- Ji Z, Tian B. 2009. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS One 4: e8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Gatfield D, Esau CC, Green CB. 2010. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase Nocturnin in mouse liver. PLoS One 5: e11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. 2005. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438: 685–689. [DOI] [PubMed] [Google Scholar]
- Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H. 2006. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res 34: 6264–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. 1998. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol 18: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna JM, Barajas JM, Teng K, Sun HL, Moore MJ, Rcie CM, Darnell RB, Goshal K. 2017. Argonaute CLIP defines a deregulated miR-122 -bound transcriptome that correlates with patient survival in human liver cancer. Mol Cell 67: 400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, Sagan SM. 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci 108: 3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD. 2007. CPEB: a life in translation. Trends Biochem Sci 32: 279–285. [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. 2003. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11: 25–33. [DOI] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR III, Frank J, Manley JL. 2009. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell 33: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL. 1996. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 87: 941–952. [DOI] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS. 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, Spector DL. 2010. An unexpected ending: noncanonical 3′ end processing mechanisms. RNA 16: 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. 2002. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110: 489–500. [DOI] [PubMed] [Google Scholar]
- Ye J, Wang C, Sumpter R Jr, Brown MS, Goldstein JL, Gale M Jr. 2003. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci 100: 15865–15870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lee JY, Tian B. 2005. Biased alternative polyadenylation in human tissues. Genome Biol 6: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}