

Abstract
In budding yeast, inactivating mutations within the 40S ribosomal subunit decoding center lead to 18S rRNA clearance by a quality control mechanism known as nonfunctional 18S rRNA decay (18S NRD). We previously showed that 18S NRD is functionally related to No-Go mRNA Decay (NGD), a pathway for clearing translation complexes stalled on aberrant mRNAs. Whereas the NGD factors Dom34p and Hbs1p contribute to 18S NRD, their genetic deletion (either singly or in combination) only partially stabilizes mutant 18S rRNA. Here we identify Asc1p (aka RACK1) and Rps3p, both stable 40S subunit components, as additional 18S NRD factors. Complete stabilization of mutant 18S rRNA in dom34Δ;asc1Δ and hbs1Δ;asc1Δ strains indicates the existence of two genetically separable 18S NRD pathways. A small region of the Rps3p C-terminal tail known to be subject to post-translational modification is also crucial for 18S NRD. We combine these findings with the effects of mutations in the 5′ → 3′ and 3′ → 5′ decay machinery to propose a model wherein multiple targeting and decay pathways kinetically contribute to 18S NRD.
Keywords: RNA decay, RNA quality control, RPS3, ribosome, translation
INTRODUCTION
Accurate and efficient flow of information from nucleic acids to proteins is essential for all life. Central to this process is protein synthesis, which requires the coordinated action of myriad components including mRNAs and rRNAs. As with any complex manufacturing process, tight quality control is crucial for both ensuring functional products and eliminating defective machine parts. Therefore, numerous mechanisms exist to ensure the overall integrity of the translation machinery (Shoemaker and Green 2012). In eukaryotes, the best understood quality control pathways are those that eliminate mRNAs that contain a premature stop codon (subject to nonsense mediated decay; NMD) (Amrani et al. 2006), lack a stop codon altogether (subject to non-stop decay; NSD) (Frischmeyer et al. 2002; van Hoof et al. 2002; Saito et al. 2013; Horikawa et al. 2016), or have some structural feature that leads to ribosome stalling (subject to no-go decay; NGD) (Doma and Parker 2006; Tsuboi et al. 2012). Each of these pathways involves specific factors that recognize and target the defective mRNA for degradation by the general mRNA decay machinery. Decay is often initiated via endonucleolytic cleavage of the mRNA at or adjacent to the ribosome stall site, followed by 5′ → 3′ and 3′ → 5′ degradation by Xrn1p and the exosome, respectively (Gatfield and Izaurralde 2004; Doma and Parker 2006; Dimitrova et al. 2009; Eberle et al. 2009; Tsuboi et al. 2012).
Other eukaryotic quality control pathways monitor and target rRNA. Previous work from our laboratory examined the fate of S. cerevisiae rRNAs containing inactivating mutations in either the 18S rRNA decoding center or the 25S rRNA peptidyl-transferase center (LaRiviere et al. 2006). These mutant rRNAs are synthesized, processed, and assembled into ribosomal subunits similar to wild-type rRNAs. The functionally defective mature subunits, however, are cleared by mechanistically distinct pathways known, respectively, as 18S and 25S nonfunctional rRNA decay (18S NRD and 25S NRD). Large ribosomal subunits containing defective 25S rRNAs fail to form stable 80S monosomes (LaRiviere et al. 2006) and localize to perinuclear foci (Cole et al. 2009). Elimination of these particles by 25S NRD can occur in the absence of ongoing translation (Cole et al. 2009), requires the DNA damage repair factors, Mms1p and Rtt101p, and involves ubiquitination of 60S subunit proteins (Fujii et al. 2009, 2012). Therefore, 25S NRD appears to occur via a mechanism unrelated to mRNA quality control. In contrast, 18S NRD shares many similarities with NGD. Mutant 18S rRNAs exhibit diffuse cytoplasmic localization and cosediment with 40S subunits, 80S monosomes and, to a lesser extent, polysomes (LaRiviere et al. 2006; Cole et al. 2009). Further, 18S NRD does not occur in the presence of translation elongation inhibitors and is substantially reduced in yeast strains lacking the known NGD factors DOM34 and HBS1 (Cole et al. 2009). Structurally related to eRF1 and eRF3, the Dom34p:Hbs1p heterodimer recognizes the A-site of stalled ribosomes and functions to both dissociate the ribosomal subunits and initiate decay of the associated mRNA (Lee et al. 2007; Passos et al. 2009; Shoemaker et al. 2010; van den Elzen et al. 2010, 2014; Becker et al. 2011; Pisareva et al. 2011; Shoemaker and Green 2011; Tsuboi et al. 2012; Guydosh and Green 2014; Hilal et al. 2016).
Whereas translation inhibitors completely abrogate 18S NRD, elimination of either DOM34 or HBS1 only slows its kinetics (Cole et al. 2009); therefore, additional factors must contribute. Although we previously demonstrated that NMD factors are not required for 18S NRD (Cole et al. 2009), other candidates include proteins involved in nascent peptide-dependent translation arrest (PDTA) (Dimitrova et al. 2009; Kuroha et al. 2010) or ribosome quality control (RQC) (Brandman et al. 2012). PDTA targets mRNAs containing rare codons or encoding stretches of positively charged amino acids, whereas RQC mediates the degradation of nascent peptide chains associated with stalled ribosomes. One factor known to participate in NGD, PDTA, and RCQ is the WD-repeat protein Asc1p (Dimitrova et al. 2009; Brandman et al. 2012). Asc1p (aka RACK1 in mammals) is a stoichiometric component of the small ribosomal subunit located in the vicinity of the mRNA exit channel (Coyle et al. 2009; Ben-Shem et al. 2010). ASC1 deletion increases the ability of ribosomes to read through rare codons and stretches encoding positively charged amino acids (Kuroha et al. 2010; Brandman et al. 2012; Letzring et al. 2013). Deletion of ASC1 also increases ribosome frameshifting at CGA repeats (Wolf and Grayhack 2015). Although these observations led to a model wherein Asc1p somehow promoted ribosomal stalling (Kuroha et al. 2010; Inada 2013; Letzring et al. 2013), more recent data suggest Asc1p functions instead to target stalled ribosomes for quality control (Sitron et al. 2017). Asc1p may also promote endonucleolytic cleavage of NGD substrates (Ikeuchi and Inada 2016).
Another set of factors implicated in NGD, PDTA, and NSD are the Ski proteins. Ski2p, Ski3p, and Ski8p form the Ski complex, which binds tightly to ribosomes stalled on mRNA 3′ ends (van Hoof et al. 2002; Wang et al. 2005; Schmidt et al. 2016). The central component Ski2p is a DExH-box helicase located near the mRNA entrance channel where it is well positioned to feed the mRNA 3′ end into the exosome (Schmidt et al. 2016). Exosome recruitment is mediated by Ski7p, which bridges the exosome to Ski3p and Ski8p (Wang et al. 2005; Kowalinski et al. 2016; Schmidt et al. 2016). We previously showed that elimination of SKI7 in combination with HBS1 completely abrogates 18S NRD (Cole et al. 2009), but we did not examine the requirement of other Ski proteins.
The major goal of this study was to identify additional 18S NRD factors and elucidate their genetic and mechanistic relationships. Here we show that both ASC1 and SKI2 contribute to the rate of mutant 18S decay, and we identify a small region of Rps3p, an essential 40S subunit protein physically residing between Asc1p and the mRNA entrance channel, as crucial for 18S NRD.
RESULTS
A simplified system for monitoring 18S NRD
The system we use to monitor 18S NRD employs a galactose-inducible (GAL7 promoter) URA+ plasmid encoding the entire 35S pre-rRNA (Fig. 1A). Benign sequence tags within the 18S and 25S regions allow for specific Northern blot detection of plasmid-derived rRNAs, which, when fully induced, account for only ∼1% of total rRNA in BY4741 yeast (LaRiviere et al. 2006). Introduction of an A to C mutation at position 1755 in the decoding center (equivalent to A1492C in E. coli 16S rRNA) renders the 40S subunit incapable of carrying out efficient elongation and therefore subject to NRD (LaRiviere et al. 2006; Cole et al. 2009). Whereas wild-type 18S rRNA (18S:WT) has no discernible decay over a 6-h time course (data not shown), mutant 18S rRNA (18S:A1492C3) decays with a half-life of <100 min (LaRiviere et al. 2006; Cole et al. 2009).
FIGURE 1.

(A) Diagram of rDNA plasmid reporter (top) and summary of experimental design (bottom). The reporter contains sequence tags for Northern blot detection of plasmid-derived 18S and 25S rRNAs. (B) Time course analysis of tagged 18S and 25S rRNAs in parental, dom34Δ, and hbs1Δ yeast strains. (C) Same as B, but for dom34Δ, ski7Δ, dom34Δ;ski7Δ, and hbs1Δ;ski7Δ strains. (B,C) Tagged 18S:tagged 25S ratios (18S/25S) were normalized to the T = 0 time point; error bars represent standard error of the mean (n = 3). 18S:A1492C half-life is indicated on the right. (D–F) Single time-point analyses of tagged 18S rRNAs. Error bars represent standard error of the mean (n = 3). (D) Unpaired t-test was used for significance testing against parental strain 18S:A1492C levels. (E) One-way ANOVA with planned comparisons was used for significance testing comparing 18S/25S in parental strain to single mutants or in dom34Δ strain to double mutants. (ns) Not significant. (F) One-way ANOVA with planned comparisons was used for significance testing comparing 18S/25S in all mutant strains against the parental strain.
In previous studies, we monitored 18S NRD by normalizing 18S:A1492C to endogenous SCR1 RNA after correcting for cell growth (LaRiviere et al. 2006; Cole and LaRiviere 2008; Cole et al. 2009). However, knowledge that 18S NRD is an entirely post-ribosome synthesis process (LaRiviere et al. 2006) raised the possibility that the tagged wild-type 25S rRNA (25S:WT) derived from the same 35S pre-rRNA transcript as 18S:WT or 18S:A1492C might be a better (or at least equivalent) normalization control. An added advantage of normalizing tagged 18S to tagged 25S:WT is that there is no need to correct for cell growth or variable plasmid copy number. To test the reliability of this 18S:25S ratio approach, we grew parental BY4741 yeast harboring either the 18S:WT or 18S:A1492C plasmid (both paired with 25S:WT) to mid-log phase in synthetic complete minus uracil media (SC-ura) plus raffinose, induced tagged pre-rRNA expression for 90 min with galactose, turned off transcription by adding glucose, then collected samples over time (Fig. 1B). Our results demonstrate that normalizing tagged 18S:WT or 18S:A1492C to the tagged 25S:WT yielded similar findings as normalizing to endogenous SCR1 RNA. That is, whereas there was no apparent decay of 18S:WT, 18S:A1492C had a half-life of 76 min in the parental strain (Figs. 1B, 2B). The 18S:25S ratio also reproduced previous findings that individual deletion of either DOM34 or HBS1 resulted in partial 18S:A1492C stabilization (Fig. 1B; Cole et al. 2009).
FIGURE 2.

(A) Single time-point analysis of tagged 18S and 25S rRNAs. One-way ANOVA with planned comparisons was used for significance testing comparing the parental strain and each single deletion 18S:A1492C/25S:WT ratio. Unpaired t-test was used to compare the parental strain and dom34Δ;asc1Δ 18S:A1492C/25S:WT ratios. Error bars represent standard error of the mean (n = 3). (B) Time course analysis of 18S:A1492C and 18S:WT decay in dom34Δ and asc1Δ single and double deletion strains. Representative Northern blots of tagged 18S:A1492C and 25S rRNAs and graphs summarizing multiple (n = 3) biological replicates. Normalization and error bars as in Figure 1B and C. (C) Single time-point analysis of tagged 18S:A1492C rRNA in parental and asc1Δ strains harboring indicated plasmids. One-way ANOVA with planned comparisons was used for significance testing of all strains against the “Parental strain + empty vector” strain. Error bars represent standard error of the mean (n = 3).
Because time courses are inherently low throughput, we also tested the feasibility of monitoring a single time point. After inducing expression for 90 min, cells were immediately harvested and subjected to Northern analysis. As expected, 18S:A1492C was substantially lower than 18S:WT in the parental BY4741 yeast, with the decrease being less drastic, but still statistically significant (P = 0.0022, unpaired t-test), in the dom34Δ strain (Fig. 1D). Thus, a single-time-point assay proved sufficient as an initial mutant screen.
DOM34 paralogs are not involved in 18S NRD
Whereas deletion of DOM34, HBS1, or both slows 18S:A1492C decay, small molecule translation inhibitors completely abrogate decay (Cole et al. 2009); this suggests the existence of a second, kinetically separable 18S NRD pathway. Consistent with the multiple-pathway hypothesis, we previously showed that double deletion of HBS1 and SKI7 completely stabilizes 18S:A1492C (Fig. 1C; Cole et al. 2009). Since Hbs1p and Dom34p form a heterodimer and their simultaneous deletion had no additive effect on 18S NRD (Cole et al. 2009), we reasoned that double deletion of DOM34 and SKI7 would also completely stabilize 18S:A1492C. Unexpectedly, however, no synthetic effect was apparent in the dom34Δ;ski7Δ strain (Figs. 1C, 4). One possible explanation for this result was the existence of a cross-functional Dom34p paralog. Dom34p consists of three domains: N (1–131 amino acids), M (136–268 amino acids), and C (271–370 amino acids), with M and C serving as the binding sites for Hbs1p (Chen et al. 2010; van den Elzen et al. 2010). S. cerevisiae contains two genes of unknown function, YCL001W-A and YCL001W-B, and a multiple sequence alignment showed both having high similarity with the M and C domains of Dom34p (data not shown). However, no decrease in 18S NRD efficiency was observed in either a YCL001W-A or YCL001W-B knockout strain, and when either deletion was combined with the DOM34 deletion, there was no enhancement of the dom34Δ phenotype (Fig. 1E). Thus, we conclude that neither YCL001W-A nor YCL001W-B contribute to 18S NRD.
FIGURE 4.

Time course analysis of 18S:A1492C and 18S:WT in single and double deletion strains. Representative Northern blots of tagged 18S:A1492C and 25S rRNAs and graphs summarizing multiple (n = 2–3) biological replicates. Normalization and error bars as in Figure 1B and C. 18S:A1492C data from the parental and asc1Δ single deletion strains are the same as in Figure 1B (parental) and Figure 2B (asc1Δ).
Asc1p contributes to 18S NRD
To identify additional 18S NRD factors, we performed a small screen in strains lacking proteins previously implicated in other degradation pathways (Fig. 1F). Among these, only the ASC1 knockout diminished 18S NRD (Figs. 1F, 2A). Substantially more 18S:A1492C was observed in the asc1Δ strain than the parental strain, with the level being comparable to the dom34Δ strain. In the dom34Δ;asc1Δ double-deletion strain, 18S:A1492C levels were indistinguishable from 18S:WT (Fig. 2A). Time course data confirmed that, whereas deletion of either ASC1 or DOM34 alone resulted in a twofold increase in 18S:A1492C half-life, deletion of both led to its complete stabilization (Fig. 2B). ASC1 was also synthetic with HBS1 (Fig. 4), and in no strain was 18S:WT detectably degraded (Figs. 2B, 4). Although the dom34Δ;asc1Δ and hbs1Δ;asc1Δ strains grew more slowly than the parental strain, neither grew more slowly than the asc1Δ single deletion strain (Fig. 3B). Therefore, slower cell growth could not account for the observed synthetic effects.
FIGURE 3.
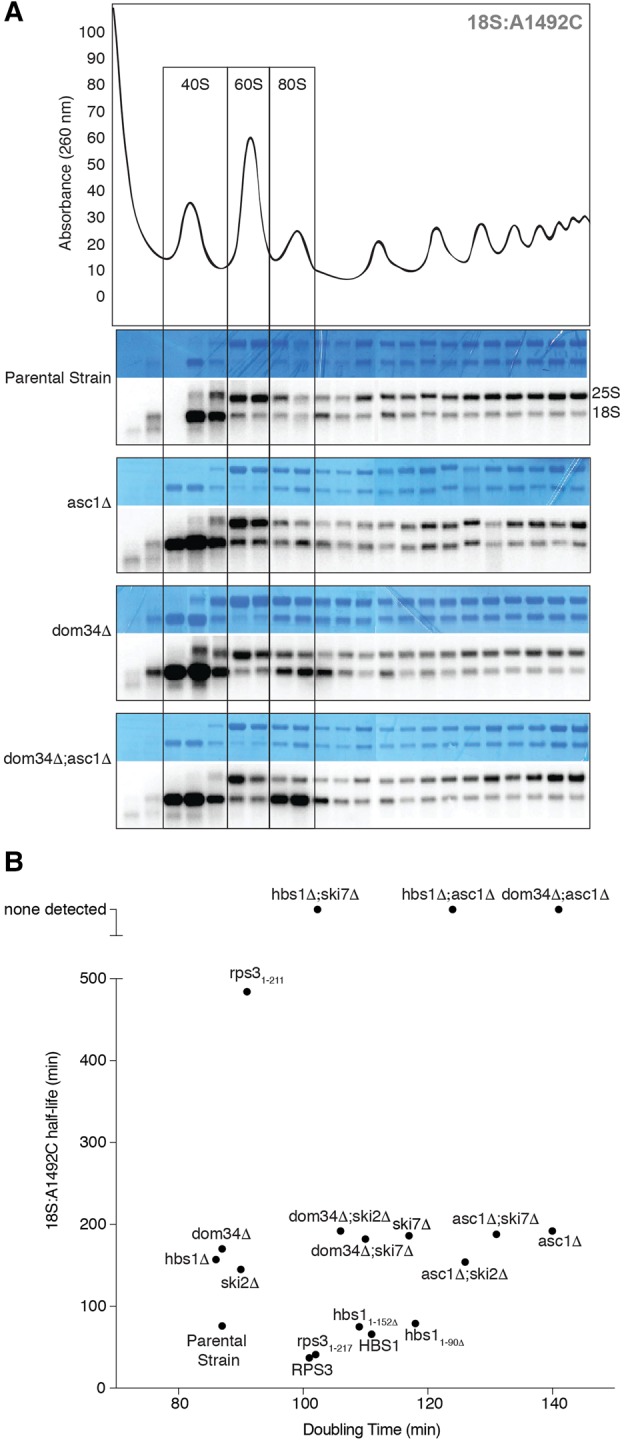
(A) Polysome profiles. (Top) Representative sucrose gradient trace for the parental strain. (Bottom) Methylene Blue stains and Northern blots of sucrose gradient fractions for the parental, asc1Δ, dom34Δ, and asc1Δ;dom34Δ strains harboring the 18S:A1492C plasmid. Boxes show positions of 40S, 60S, and 80S ribosomes. (B) Scatter plot of mean 18S:A1492C half-lives (n = 3) versus mean growth rates (doubling time; n = 3) of various yeast strains used in current study.
To confirm that the 18S NRD defect observed in the asc1Δ strain was due to the loss of the ASC1 protein and not the snR24 snoRNA that derives from the ASC1 intron, we transformed the asc1Δ strain with different plasmids containing the ASC1 gene with or without the intron, both under control of the endogenous ASC1 promoter. The intron-less version completely restored 18S NRD (Fig. 2C). Further, when transformed into the parental strain, neither plasmid enhanced 18S:A1492C decay. Thus, we conclude that Asc1p is an 18S NRD factor, and its endogenous levels are sufficient for optimal 18S NRD. Taken together, these data indicate the existence of two genetically separable pathways contributing to 18S NRD kinetics: one involving DOM34 and HBS1, and another involving ASC1.
Increased nonfunctional 80S monosomes in dom34Δ;asc1Δ lysates
In a wild-type background, 18S:A1492C rRNA predominantly cosediments on sucrose gradients with 40S subunits (Fig. 3A; LaRiviere et al. 2006). This suggests highly efficient resolution of stalled 80S monosomes containing the 18S:A1492C mutation. Since both Asc1p and Dom34p have been implicated in targeting and resolving stalled ribosomes (Shoemaker et al. 2010; Tsuboi et al. 2012; Sitron et al. 2017), we next examined sucrose gradients of asc1Δ, dom34Δ, and dom34Δ;asc1Δ. The sedimentation pattern in asc1Δ lysates revealed a slight increase in the amount of 18S:A1492C cosedimenting with 80S monosomes, and this increase was more noticeable in dom34Δ lysates. This effect was further amplified in the dom34Δ;asc1Δ double-mutant (Fig. 3A). While there was some increase in bulk 80S monosomes in both the dom34Δ and dom34Δ;asc1Δ profiles (data not shown), this increase was much less pronounced than the change in 80S cosedimentation of 18S:A1492C in the dom34Δ;asc1Δ lysate (Fig. 3A). Thus, cells lacking both ASC1 and DOM34 are substantively impaired in their ability to resolve nonfunctional 80S ribosomes.
Variable effects of Ski proteins on 18S NRD
Having identified a new ASC1-dependent pathway contributing to 18S NRD, we next tested whether ASC1 was synthetic with SKI7. However, we could detect no difference in the rate of 18S:A1492C decay between the asc1Δ single deletion and asc1Δ;ski7Δ double deletion strain (Fig. 4). To investigate whether the Ski complex itself contributes to 18S NRD, we examined 18S:A1492C decay kinetics in ski2Δ strains (Fig. 4). Deletion of SKI2 alone had no effect on cell doubling time (Fig. 3B), but it slowed 18S:A1492C decay to a similar extent as deletion of either DOM34 or ASC1 alone. No further decrease in decay rate was observed, however, in either a dom34Δ;ski2Δ or asc1Δ;ski2Δ strain (Fig. 4) despite decreased growth rates of these double-mutant strains (Fig. 3B). Thus, while both SKI7 and SKI2 contribute to the rate of 18S:A1492C decay, neither is synthetic with DOM34 or ASC1. Further, there is no clear relationship between cell doubling time and the rate of 18S:A1492C decay.
The Rps3p C-terminal tail
On the ribosome, the binding sites for Dom34p:Hbs1p and Asc1p are separated by >75 Å (Becker et al. 2011; Hilal et al. 2016). Whereas Dom34p:Hbs1p interact with the A-site at the interface between the large and small subunits, Asc1p resides on the opposite (solvent-exposed) surface of the small subunit in the general area of the mRNA exit channel. Physically linking these two sites is a single protein: Rps3p (Fig. 5A). The body of Rps3p consists of an N-terminal type II KH domain (three-stranded β-sheet backed by three α-helices; amino acids 1–88) attached via a nine-amino acid linker to a central RRM-like domain (four-stranded β-sheet backed by two α-helices; amino acids 98–189); together these form part of the mRNA entrance channel adjacent to the A-site. Intriguingly, cryo-EM studies indicate that the body of Rps3p contacts the N-terminal 90-amino acid globular domain of Hbs1p, which is attached via a 62-amino acid flexible linker to the GTPase core (Becker et al. 2011; Hilal et al. 2016). A 50-amino acid C-terminal tail extends from the body of Rps3p along the outer surface of the small ribosomal subunit and contacts the fourth WD repeat in Asc1p (Ben-Shem et al. 2010). This network of structural contacts between Hbs1p, Rps3p, and Asc1p suggested to us that RPS3 might be a component of the 18S NRD pathway.
FIGURE 5.

(A, left) Cryo-EM structure of the yeast 80S ribosome in complex with Dom34p:Hbs1p (orange:red), P-site tRNA (yellow), and nonstop mRNA (black) (PDB: 5M1J) (Hilal et al. 2016). Also highlighted are Asc1p (green) and Rps3p (purple). (Right) Close-up of the Hbs1p:Rps3p:Asc1p interaction. Positions of Rps3p point mutations at the Hbs1p:Rps3p interface are shown in yellow. Note that amino acids 3–225 of Rps3p have been resolved (full-length: 240). (B, left) Plasmid shuffle experiment showing growth of strains (10-fold dilution series) harboring plasmids expressing wild-type RPS3 or indicated point mutations on a LEU−URA− or LEU−5-FOA plate. (Right) Single time-point analyses of tagged 18S and 25S rRNAs. For all strains, 18S:A1492C/25S:WT ratio was normalized to the 18S:WT/25S:WT ratio. Error bars represent standard error of the mean. One-way ANOVA with planned comparisons was used for significance testing comparing all rps3 variant strains against the wild-type RPS3 strain; only rps31-211 was statistically different (P < 0.0001). (C) Diagram of Hbs1p domains (top) and time course analysis of 18S:A1492C in hbs1 strains (bottom). Data normalization as in Figure 1B and C. Representative Northern blots and graph of time course data with indicated 18S:A1492C rRNA half-lives. Error bars represent standard error of the mean (n = 2).
Because RPS3 encodes an essential protein, it was impossible to monitor 18S NRD in an RPS3 knockout strain. We therefore implemented a 5-FOA plasmid shuffle approach in a rps3Δ background to test the effects of various rps3 mutations (Figs. 5B, 6A). Validating this approach, a LEU+ plasmid encoding wild-type RPS3 complemented the knockout strain, whereas the empty LEU+ vector did not. For point mutations, we chose positions that were highly conserved across all eukaryotes (data not shown) and were previously proposed to make specific interactions with the Hbs1p amino-terminal domain (Fig. 5A; Becker et al. 2011). All point mutations tested complemented the rps3Δ strain, indicating that none of the amino acids we mutated were required for viability. All four mutations also had 18S:A1492C/25:WT ratios similar to wild-type RPS3 (Fig. 5B), suggesting that the mutations fail to compromise the interaction between Rps3p and Hbs1p or that the interaction might be dispensable for 18S NRD.
FIGURE 6.

(A) Plasmid shuffle experiment showing growth of strains (10-fold dilution series) harboring plasmids expressing wild-type RPS3 or indicated C-terminal truncation variant on a LEU−URA− or LEU−5-FOA plate. (B) Time course analysis of 18S rRNA in rps3 yeast strains. Representative Northern blots of tagged 18S and tagged 25S rRNA and graphs summarizing multiple (n = 3) biological replicates. Normalization and error bars as in Figure 1B and 1C. (C) Multiple sequence alignment of RPS3 protein sequences.
To more rigorously test the functionality of the Rps3p:Hbs1p interaction, we next mutated HBS1. Earlier work had shown that an hbs1 protein variant lacking the entire N-terminal domain (amino acids 1–152) retains its ability to bind to ribosomes in complex with Dom34p (Becker et al. 2011). When we complemented the hbs1Δ strain with plasmids encoding either full-length HBS1 (1–611) or hbs1 lacking either the first 90 or 152 amino acids (hbs11–90Δ and hbs11–152Δ, respectively), we observed no difference in the rate of 18S:A1492C decay (Fig. 5C). Therefore, we conclude that the interaction of the Hbs1p N terminus with Rps3p is dispensable for 18S NRD.
We next examined the Asc1p:Rps3p interaction. The Rps3p C-terminal tail (amino acids 190–240) exhibits much lower sequence conservation than the N-terminal domains (data not shown). To test the essentiality of the tail, we made a series of C-terminal truncation mutants. Whereas rps31–200 proved inviable, cells expressing either rps31–211 or rps31–217 grew equally well as cells expressing full-length RPS3 (amino acids 1–240) (Figs. 3B, 6A). Thus, amino acids 1–211 of S. cerevisiae Rps3p are sufficient for both survival and wild-type cell growth. We did, however, observe a significant difference between the two truncation mutants with regard to 18S NRD efficiency: Whereas yeast expressing rps31–217 degraded 18S:A1492C at a rate indistinguishable from cells expressing the full-length protein, 18S:A1492C decay in rps31–211 yeast was >11-fold slower (Fig. 6B). The magnitude of this effect is so far the largest we have observed for any single mutation or gene deletion tested (this study; Cole et al. 2009). Thus, a small region of the Rps3p C-terminal tail is crucial for efficient 18S NRD. Of the six amino acids (KEEEPI), the first three (KEE) are highly conserved across eukaryotic species (Fig. 6C).
DISCUSSION
Here we identified Asc1p, Ski2p, and Rps3p as factors contributing to 18S:A1492C rRNA decay kinetics. We found that ASC1 is synthetic with DOM34 and HBS1, but not with SKI7 (Figs. 2B, 4). Further, whereas deleting SKI2 slowed 18S:A1492C rRNA decay similarly to deletion of ASC1, DOM34, or HBS1, no synthetic effects were detectable upon combining a SKI2 deletion with deletion of ASC1 or DOM34 (Fig. 4). Finally, we found that mutant 18S rRNA is substantially stabilized upon deletion of a six-amino acid region within the C-terminal tail of Rps3p (Fig. 6B), implicating Rps3p as a central player in targeting nonfunctional 40S subunits for preferential elimination by 18S NRD. We synthesize these findings with previous data to propose a model wherein 18S NRD is the result of multiple independent targeting and decay pathways (Fig. 7).
FIGURE 7.

Proposed model depicting the contributions of multiple independent targeting and decay pathways to 18S NRD. A stalled ribosome harboring mutant 18S rRNA (blue splatter) can be marked for decay by two separate pathways involving either Asc1p or Dom34p:Hbs1p. Once marked (possibly by covalent modification of the Rps3p C-terminal tail), 40S ribosomes are disassembled and 18S rRNA degraded by 5′ → 3′ and 3′ → 5′ decay pathways.
18S NRD, NGD, PDTA, and RQC: different outcomes of ribosome stalling
The data in this paper strengthen and extend our previous findings that 18S NRD is functionally related to the mRNA and protein quality control pathways associated with ribosome stalling (this study; Cole et al. 2009). NGD, PDTA, 18S NRD, and RQC were all discovered independently by examining the fate of mRNAs (NGD and PDTA) or rRNAs (18S NRD) with features that inhibit protein production, or by a genome-wide screen for deletions that overactivate the heat-shock stress response pathway (RQC) (Doma and Parker 2006; LaRiviere et al. 2006; Cole et al. 2009; Dimitrova et al. 2009; Kuroha et al. 2010; Brandman et al. 2012). Subsequent investigation of the identified RQC genes led to the finding that this increased stress response was due to accumulation of aberrant nascent peptides associated with stalled ribosomes (Brandman et al. 2012; Choe et al. 2016). Evidence accumulating since these initial discoveries suggests that all four pathways are simply alternate outcomes of the same initiating event—a stalled or slowly elongating ribosome (Shoemaker and Green 2012; Brandman and Hegde 2016). Whether an individual stall event leads to all or only a subset of these outcomes may depend on both the nature of the stall and the kinetic stability of the stalling event (i.e., whether the ribosome is completely halted or is simply moving slowly).
A recently proposed unifying model (Brandman and Hegde 2016) suggests the following order of events for the resolution of stalled ribosomes: (i) recognition and targeting of the stalled ribosome by Asc1p, Hel2p (an E3 ubiquitin ligase), and the recently identified RQT complex (Brandman et al. 2012; Matsuo et al. 2017; Sitron et al. 2017); (ii) subunit dissociation by Dom34p, Hbs1p, and Rli1p (mammalian ABCE1) (Shoemaker et al. 2010; Pisareva et al. 2011; Shoemaker and Green 2011; Tsuboi et al. 2012); (iii) degradation of the associated aberrant mRNA (Frischmeyer et al. 2002; van Hoof et al. 2002; Doma and Parker 2006; Tsuboi et al. 2012); (iv) assembly of the remaining RQC components onto the 60S subunit (Brandman et al. 2012; Shao et al. 2015); (v) CAT-tailing by Rqc2p and/or ubiquitination of the nascent peptide chain by Ltn1p (another E3 ubiquitin ligase) (Bengtson and Joazeiro 2010; Shao and Hegde 2014; Shen et al. 2015; Shao et al. 2015; Osuna et al. 2017); and (vi) extraction and degradation of the ubiquitinated nascent peptide (Brandman et al. 2012; Defenouillère et al. 2013; Verma et al. 2013; Kostova et al. 2017; Osuna et al. 2017). Missing from this model is the fate of the 40S subunit. In cases where ribosome stalling was not due to any specific 40S dysfunction, it would seem reasonable that the subunit would simply be released to return to the translationally active pool. However, when stalls are due to a specific 40S defect (such as the decoding center mutation used here), the defective subunit is ultimately dismantled and the 18S rRNA decayed. Since ASC1 and DOM34:HBS1 are additive for 18S:A1492C kinetics (Figs. 2B, 4), our data suggest that initial targeting of the 40S subunit for decay occurs prior to or concurrent with 80S ribosome dissociation. However, the lack of an 18S NRD defect in the ltn1Δ strain (Fig. 1F) suggests that the 40S subunit fate is not tied to the process of dismantling of the nascent peptide-60S complex.
Multiple kinetic contributors to 18S NRD targeting and decay
While we were successful in identifying additional 18S NRD factors (Asc1p, Rps3p, and Ski2p), some of the single- and double-mutant results were confounding. For example, while the deletion of SKI2 partially stabilizes mutant 18S rRNA, it is not synthetic with either DOM34 or ASC1 (Fig. 4). Furthermore, whereas a HFY1200 strain lacking XRN1 exhibits decreased 18S NRD kinetics, this same deletion in a BY4741 background is without apparent consequence (Cole et al. 2009). Finally, we previously observed no substantial decrease in 18S NRD kinetics in a temperature-sensitive exosome strain (Cole et al. 2009). To explain these apparent inconsistencies, we propose a model wherein multiple parallel and sequential pathways kinetically contribute to 18S NRD (Fig. 7). In this model, Asc1p and the Dom34p:Hbs1p heterodimer sit at the top of the pathway where they function independently to target nonfunctional 40S subunits for decay. Once targeted, 18S rRNA decay likely involves subunit disassembly and both endonucleolytic and exonucleolytic activities, with the relative kinetic contributions of 5′ → 3′ decay by Xrn1p and 3′ → 5′ decay by the exosome being highly dependent on strain background and growth conditions (Cole et al. 2009; Merrikh 2012).
Whereas the half-lives of NGD, NSD, and PDTA mRNAs range from 2 to 9 min in wild-type yeast (Frischmeyer et al. 2002; Doma and Parker 2006; Sweet et al. 2012; Tsuboi et al. 2012), the half-life of 18S:A1492C rRNA ranges from 41 to 96 min (this study; LaRiviere et al. 2006; Cole et al. 2009). Since the 18S:A1492C half-lives are of similar magnitude to the doubling time of wild-type yeast (87 min), a simple explanation could have been that 18S NRD is somehow tied to the cell cycle. However, we observed no consistent relationship between 18S:A1492C decay kinetics and cell growth rate over multiple strain backgrounds (Fig. 3B). What can account for the 10-fold slower kinetics of 18S NRD compared to defective mRNA decay? Is 18S NRD targeting slow, with nonfunctional 40S subunits going through multiple rounds of initiation prior to being tagged for decay, or is targeting efficient and decay slow? What steps are involved in decay? Does decay first require 40S disassembly (i.e., removal of some or all of the proteins) to allow for 18S rRNA decay, or is 18S rRNA decay initiated within the intact subunit with protein disassembly occurring concomitant with rRNA degradation? While the available data do not address these questions directly, the observation of no 18S:A1492C decay in the presence of elongation inhibitors (Cole et al. 2009) suggests that targeting of nonfunctional 40S subunits to 18S NRD is relatively inefficient and may require multiple rounds of initiation. Slow targeting would also be consistent with the variable kinetic effects of eliminating individual components of the degradation machinery.
A central role for RPS3
Rps3p (a.k.a. uS3 in the recently adopted systematic ribosomal protein nomenclature [Ban et al. 2014]) is an essential component of the small ribosomal subunit. Along with Rps2p (uS5) and Rps30p (eS30), Rps3p forms part of the mRNA entrance channel (Ben-Shem et al. 2010). The type II KH and RRM-like domains of Rps3p are structurally conserved from bacteria to humans, whereas the C-terminal tail is more conserved in eukaryotes (data not shown). Proteomics studies in S. cerevisiae have identified C-terminal tail amino acids K200, T207, K212, S221, and T231 as sites of acetylation, phosphorylation, succinylation, and/or ubiquitination (Peng et al. 2003; Albuquerque et al. 2008; Seyfried et al. 2008; Holt et al. 2009; Soulard et al. 2010; Weinert et al. 2013; Fang et al. 2014). If 18S NRD consists of independent targeting and decay pathways, then post-translation modification of the Rps3p tail could serve as the mark that connects targeting to decay. Indeed, the significant decrease in 18S:A1492C rRNA half-life (>11-fold) observed upon removing Rps3p amino acids 212–217 (Fig. 6B) suggests a central role for one or more of these residues.
In S. cerevisiae, Drosophila, and human ribosomes, the Rps3p/RPS3 tail interacts with the WD40 blade IV of Asc1p/RACK1 (Ben-Shem et al. 2010; Anger et al. 2013), but functional relevance had been lacking. Recent studies in mammalian cells investigating ribosome read-through on poly(A) stretches now suggest a regulatory link between Asc1p and Rps3p (Juszkiewicz and Hegde 2017; Sundaramoorthy et al. 2017). RACK1 (yeast ASC1) and ZNF598 (yeast HEL2) are both involved in ubiquitination of stalled ribosomes (Saito et al. 2015; Juszkiewicz and Hegde 2017; Sundaramoorthy et al. 2017). HEL2 was previously identified as a genetic component of the S. cerevisiae RQC pathway where it, along with ASC1, acts upstream of LTN1 (Brandman et al. 2012; Letzring et al. 2013; Sitron et al. 2017). Both mammalian ZNF598 and RACK1 facilitate RPS3 (uS3) ubiquitination, although ZNF598 primarily participates in RPS10 (eS10) and RPS20 (uS10) ubiquitination, whereas RACK1 primarily participates in RPS2 (uS5) and RPS3 ubiquitination. Furthermore, inhibitors of translation elongation and activators of the unfolded protein response pathway also result in RPS3 ubiquitination, with the ubiquitination site occurring on the yeast equivalent of K212 (Higgins et al. 2015; Juszkiewicz and Hegde 2017; Sundaramoorthy et al. 2017). Another recent study showed that yeast Hel2p ubiquitinates both Rps20p (uS10) K6/K8 and Rps3p K212 in stalled ribosomes (Matsuo et al. 2017). Collectively, these findings may explain why loss of ASC1 only partially stabilizes 18S:A1492C, whereas deletion of RPS3 amino acids 212–217 has a much stronger effect. It seems likely that Asc1p and Hel2p converge to ubiquitinate Rps3p K212, which in turn serves to target the mutant 18S rRNA for decay.
Multiple pathways for resolving stalled ribosomes across domains
The existence of multiple pathways in eukaryotes for detecting and eliminating functionally defective ribosomes parallels the situation in prokaryotes. Although the decoding center A1492C and G530U mutations are known to inactivate the 16S rRNA decoding center (Powers and Noller 1990, 1993; Yoshizawa et al. 1999; Ogle et al. 2001), these mutant rRNAs are not subject to preferential degradation in E. coli (Paier et al. 2015). Nonetheless, E. coli does harbor three functionally redundant pathways for rescuing ribosomes stalled on truncated mRNAs: the trans-translation pathway, ArfB, and ArfA (Keiler et al. 1996; Karzai et al. 1999; Chadani et al. 2010, 2011). In the trans-translation pathway, tmRNA/SmpB recognizes the empty A-site and acts as both a tRNA and mRNA to allow continued elongation and normal termination at the tmRNA stop codon. ArfB and ArfA act as backups to the tmRNA/SmpB system. ArfB, a release factor 2 (RF2) homolog, binds to the stalled ribosome and initiates peptidyl-tRNA hydrolysis and ribosome release. ArfA accomplishes the same thing, but does so by binding within the empty mRNA channel and directly recruiting RF2 to the A-site. Of particular note for our findings, the ArfA and ArfB pathways were discovered only upon tmRNA/SmpB inactivation (Chadani et al. 2010, 2011). The existence of so many redundant mechanisms in bacteria to rescue stuck ribosomes suggests that ribosome stalling is a pervasive problem. Indeed, it has been estimated that 2%–4% of all translating ribosomes in E. coli are in need of rescue at any given time (Ito et al. 2011; Keiler 2015). Given the greater complexity of their translation machinery, one might predict ribosome stalling to be even more problematic in eukaryotes. Thus, it would not be surprising if more pathways for resolving stalled translation complexes await discovery.
MATERIALS AND METHODS
Yeast strains and plasmids
All strains and plasmids used in this study are listed in Supplemental Table S1. The dom34Δ;asc1Δ, hbs1Δ;asc1Δ, asc1Δ;ski7Δ, and asc1Δ;ski2Δ mutants were constructed by transforming a dom34Δ, hbs1Δ, ski7Δ, or ski2Δ strain with dsDNA encoding asc1Δ::NATMX and colony selection on G418;ClonNat plates. The dom34Δ;ski7Δ and dom34Δ;ski2Δ mutants were constructed by transforming a ski7Δ or ski2Δ strain with dsDNA encoding dom34Δ::NATMX and colony selection on G418;ClonNat plates. Homologous recombination at the ASC1 or DOM34 locus was confirmed by PCR. rps3 mutant strains were constructed by plasmid shuffling. Briefly, a RPS3/rps3Δ::KANMX diploid strain was transformed with a RPS3/URA3+ plasmid, sporulated, and selected on SC-ura;G418 plates to obtain a haploid rps3Δ::KANMX strain harboring the URA3+ plasmid. LEU2+ plasmids encoding rps3 variants were then transformed into the rps3Δ::KANMX;URA3+ haploid strain; rps3 variants that had lost the URA3+ plasmid were then selected on SC-leu;5-FOA plates. To generate hbs1 N-terminal deletion strains, a hbs1Δ haploid strain was transformed with plasmids encoding either HBS1, hbs11-90Δ, or hbs11-152Δ; all variants were expressed under the endogenous HBS1 promoter.
Single time-point assay
Strains harboring pSC40-WT (18S:WT;25S:WT) or pSC40-A1492C (18S:A1492C;25S:WT) plasmids were grown at 30°C in SC-ura media plus 2% raffinose to mid-log phase (OD600 = 0.4–0.5). Following addition of 20% galactose (final concentration = 2%) to induce rRNA transcription, cells were incubated at 30°C for an additional 90 min. Pre-warmed SC-ura media plus 2% galactose was added as necessary to maintain OD600 = 0.5. Cells were then harvested by centrifugation and flash frozen in liquid nitrogen or a dry ice/EtOH mixture.
Pulse-chase analysis
Pulse-chase analysis was performed as previously described (Cole and LaRiviere 2008) with some modifications. Strains harboring pSC40-WT or pSC40-A1492C plasmids were grown at 30°C in SC-ura media plus 2% raffinose to mid-log phase (OD600 = 0.4–0.5). Twenty percent of galactose was added to a final concentration of 2% to induce rRNA transcription, and cells were incubated for an additional 90 min. Fifty percent of glucose was then added to a final concentration of 2% and the first-time point (T = 0) was immediately harvested; subsequent time points were taken at indicated intervals post glucose addition. Samples were pelleted by centrifugation, and then flash frozen in liquid nitrogen or a dry ice/EtOH mixture. Prewarmed SC-ura media plus 2% glucose was added as necessary to maintain an OD600 of 0.5 for the duration of the time course.
Northern blot analysis
For each sample, 2.0–2.5 µg total RNA was electrophoresed on a 1% agarose–formaldehyde gel and transferred to a nitrocellulose membrane as previously described (Cole and LaRiviere 2008; Cole et al. 2009). Total RNA was detected via staining with Methylene Blue (Molecular Research). Membranes were hybridized with 32P-end-labeled probes FL125 (anneals to plasmid-derived 18S rRNA) and FL126 (anneals to plasmid-derived 25S rRNA) for 12–24 h at 42°C in ExpressHyb (BD Biosciences). Bands were visualized using a Typhoon Phosphorimager (Molecular Dynamics) and quantified via ImageQuant software (GE Lifesciences). Signal from the tagged-18S rRNA was normalized to the signal from the tagged-25S rRNA. Curve fitting was done using GraphPad Prism 7 software.
Sucrose gradients
The parental wild-type, dom34Δ, asc1Δ, and dom34Δ;asc1Δ strains were transformed with the pSC40-A1492C plasmid and grown to mid-log in SC-ura media plus 2% raffinose (80 mL culture volume per gradient). After a 90-min induction with 2% galactose, cycloheximide was added to a final concentration of 0.1 mg/mL. Cells were immediately chilled on ice, harvested, and lysed in the presence of glass beads and lysis buffer (20 mM Tris–HCl pH = 8.0, 140 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 1% Triton X-100, 0.1 mg/mL cycloheximide, 1 mg/mL heparin). Individual lysates (12 A260 units) were layered onto 5%–47% sucrose gradients (sucrose, 20 mM Tris–HCl pH = 8.0, 140 mM KCl, 5 mM MgCl2, 0.5 mM DTT, 0.1 mg/mL cycloheximide, 0.5 mg/mL heparin) and spun in an ultracentrifuge (35K rpm, 160 min, SW41 rotor). Individual fractions were then collected and subjected to Northern blot analysis.
Multiple sequence alignments
A multiple sequence alignment of the DOM34, YCL001W-A, or YCL001W-B protein sequences was performed using ClustalW (v1.83). A multiple sequence alignment of RPS3 protein sequences across various species (from E. coli to H. sapiens) was also performed using ClustalW (v1.83).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Andrei Korostelev, Dr. Nicholas Rhind, and Dr. Elisabet Mandon for their invaluable input, and members of the Moore laboratory for many helpful discussions. This research was supported by the Howard Hughes Medical Institute (M.J.M. was an HHMI Investigator when the research was conducted), Moderna Therapeutics, and the National Institute of General Medical Sciences of the National Institutes of Health under award number F31GM109753 (K.A.L.). The content is solely the responsibly of the authors and does not necessarily represent the official view of the National Institutes of Health.
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.061671.117.
Although 18S:A1755C would be a more accurate name for this mutation in S. cerevisiae, we originally chose to dub it 18S:A1492C in our first NRD paper (LaRiviere et al. 2006) to call particular attention to the fact that this position is equivalent to E. coli 16S rRNA nucleotide A1492, about which the effects of mutations on ribosome decoding were well understood. In the time since, multiple papers using our plasmids have continued to use the 18S:A1492C nomenclature (e.g., Fujii et al. 2009, 2012; van den Elzen et al. 2010). Therefore, to avoid compounding any confusion, we will retain the 18S:A1492C nomenclature here.
REFERENCES
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. 2008. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 7: 1389–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Sachs MS, Jacobson A. 2006. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7: 415–425. [DOI] [PubMed] [Google Scholar]
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. 2013. Structures of the human and Drosophila 80S ribosome. Nature 497: 80–85. [DOI] [PubMed] [Google Scholar]
- Ban N, Beckmann R, Cate JHD, Dinman JD, Dragon F, Ellis SR, Lafontaine DLJ, Lindahl L, Liljas A, Lipton JM, et al. 2014. A new system for naming ribosomal proteins. Curr Opin Struct Biol 24: 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Armache JP, Jarasch A, Anger AM, Villa E, Sieber H, Motaal BA, Mielke T, Berninghausen O, Beckmann R. 2011. Structure of the no-go mRNA decay complex Dom34–Hbs1 bound to a stalled 80S ribosome. Nat Struct Mol Biol 18: 715–720. [DOI] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. 2010. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467: 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Jenner L, Yusupova G, Yusupov M. 2010. Crystal structure of the eukaryotic ribosome. Science 330: 1203–1209. [DOI] [PubMed] [Google Scholar]
- Brandman O, Hegde RS. 2016. Ribosome-associated protein quality control. Nat Struct Mol Biol 23: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, et al. 2012. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151: 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadani Y, Ono K, Ozawa SI, Takahashi Y, Takai K, Nanamiya H, Tozawa Y, Kutsukake K, Abo T. 2010. Ribosome rescue by Escherichia coli ArfA (YhdL) in the absence of trans-translation system. Mol Microbiol 78: 796–808. [DOI] [PubMed] [Google Scholar]
- Chadani Y, Ono K, Kutsukake K, Abo T. 2011. Escherichia coli YaeJ protein mediates a novel ribosome-rescue pathway distinct from SsrA- and ArfA-mediated pathways. Mol Microbiol 80: 772–785. [DOI] [PubMed] [Google Scholar]
- Chen L, Muhlrad D, Hauryliuk V, Cheng Z, Lim MK, Shyp V, Parker R, Song H. 2010. Structure of the Dom34-Hbs1 complex and implications for no-go decay. Nat Struct Mol Biol 17: 1233–1240. [DOI] [PubMed] [Google Scholar]
- Choe YJ, Park SH, Hassemer T, Körner R, Vincenz-Donnelly L, Hayer-Hartl M, Hartl FU. 2016. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 531: 191–195. [DOI] [PubMed] [Google Scholar]
- Cole SE, LaRiviere FJ. 2008. Chapter 12. Analysis of nonfunctional ribosomal RNA decay in Saccharomyces cerevisiae. Methods Enzymol 449: 239–259. [DOI] [PubMed] [Google Scholar]
- Cole SE, LaRiviere FJ, Merrikh CN, Moore MJ. 2009. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell 34: 440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle SM, Gilbert WV, Doudna JA. 2009. Direct link between RACK1 function and localization at the ribosome in vivo. Mol Cell Biol 29: 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defenouillère Q, Yao Y, Mouaikel J, Namane A, Galopier A, Decourty L, Doyen A, Malabat C, Saveanu C, Jacquier A, et al. 2013. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci 110: 5046–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova LN, Kuroha K, Tatematsu T, Inada T. 2009. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J Biol Chem 284: 10343–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Mühlemann O, Jensen TH. 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55. [DOI] [PubMed] [Google Scholar]
- Fang NN, Chan GT, Zhu M, Comyn SA, Persaud A, Deshaies RJ, Rotin D, Gsponer J, Mayor T. 2014. Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat Cell Biol 16: 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC. 2002. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295: 2258–2261. [DOI] [PubMed] [Google Scholar]
- Fujii K, Kitabatake M, Sakata T, Miyata A, Ohno M. 2009. A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev 23: 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Kitabatake M, Sakata T, Ohno M. 2012. 40S subunit dissociation and proteasome-dependent RNA degradation in nonfunctional 25S rRNA decay. EMBO J 31: 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E. 2004. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature 429: 575–578. [DOI] [PubMed] [Google Scholar]
- Guydosh NR, Green R. 2014. Dom34 rescues ribosomes in 3′ untranslated regions. Cell 156: 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins R, Gendron JM, Rising L, Mak R, Webb K, Kaiser SE, Zuzow N, Riviere P, Yang B, Fenech E, et al. 2015. The unfolded protein response triggers site-specific regulatory ubiquitylation of 40S ribosomal proteins. Mol Cell 59: 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal T, Yamamoto H, Loerke J, Bürger J, Mielke T, Spahn CMT. 2016. Structural insights into ribosomal rescue by Dom34 and Hbs1 at near-atomic resolution. Nat Commun 7: 13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa W, Endo K, Wada M, Ito K. 2016. Mutations in the G-domain of Ski7 cause specific dysfunction in non-stop decay. Sci Rep 6: 29295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Inada T. 2016. Ribosome-associated Asc1/RACK1 is required for endonucleolytic cleavage induced by stalled ribosome at the 3′ end of nonstop mRNA. Sci Rep 6: 28234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T. 2013. Quality control systems for aberrant mRNAs induced by aberrant translation elongation and termination. Biochim Biophys Acta 1829: 634–642. [DOI] [PubMed] [Google Scholar]
- Ito K, Chadani Y, Nakamori K, Chiba S, Akiyama Y, Abo T. 2011. Nascentome analysis uncovers futile protein synthesis in Escherichia coli. PLoS ONE 6: e28413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Hegde RS. 2017. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol Cell 65: 743–750.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai AW, Susskind MM, Sauer RT. 1999. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA). EMBO J 18: 3793–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC. 2015. Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol 13: 285–297. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271: 990–993. [DOI] [PubMed] [Google Scholar]
- Kostova KK, Hickey KL, Osuna BA, Hussmann JA, Frost A, Weinberg DE, Weissman JS. 2017. CAT-tailing as a fail-safe mechanism for efficient degradation of stalled nascent polypeptides. Science 357: 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E, Kögel A, Ebert J, Reichelt P, Stegmann E, Habermann B, Conti E. 2016. Structure of a cytoplasmic 11-subunit RNA exosome complex. Mol Cell 63: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Akamatsu M, Dimitrova L, Ito T, Kato Y, Shirahige K, Inada T. 2010. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep 11: 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. 2006. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell 24: 619–626. [DOI] [PubMed] [Google Scholar]
- Lee HH, Kim YS, Kim KH, Heo I, Kim SK, Kim O, Kim HK, Yoon JY, Kim HS, Kim DJ, et al. 2007. Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol Cell 27: 938–950. [DOI] [PubMed] [Google Scholar]
- Letzring DP, Wolf AS, Brule CE, Grayhack EJ. 2013. Translation of CGA codon repeats in yeast involves quality control components and ribosomal protein L1. RNA 19: 1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, et al. 2017. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun 8: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrikh CN. 2012. Characterization of new factors in the 18S nonfunctional ribosomal RNA decay pathway in S. cerevisiae: a dissertation. University of Massachusetts Medical School; GSBS Dissertations and Theses. Paper 613. http://escholarship.umassmed.edu/gsbs_diss/613. [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. [DOI] [PubMed] [Google Scholar]
- Osuna BA, Howard CJ, Kc S, Frost A, Weinberg DE. 2017. In vitro analysis of RQC activities provides insights into the mechanism and function of CAT tailing. eLife 6: e27949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paier A, Leppik M, Soosaar A, Tenson T, Maiväli Ü. 2015. The effects of disruptions in ribosomal active sites and in intersubunit contacts on ribosomal degradation in Escherichia coli. Sci Rep 5: 7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos DO, Doma MK, Shoemaker CJ, Muhlrad D, Green R, Weissman J, Hollien J, Parker R. 2009. Analysis of Dom34 and its function in no-go decay. Mol Biol Cell 20: 3025–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. 2003. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21: 921–926. [DOI] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CUT, Pestova TV, Pisarev AV. 2011. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J 30: 1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Noller HF. 1990. Dominant lethal mutations in a conserved loop in 16S rRNA. Proc Natl Acad Sci 87: 1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Noller HF. 1993. Evidence for functional interaction between elongation factor Tu and 16S ribosomal RNA. Proc Natl Acad Sci 90: 1364–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Hosoda N, Hoshino SI. 2013. The Hbs1-Dom34 protein complex functions in non-stop mRNA decay in mammalian cells. J Biol Chem 288: 17832–17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Horikawa W, Ito K. 2015. Inhibiting K63 polyubiquitination abolishes no-go type stalled translation surveillance in Saccharomyces cerevisiae. PLoS Genet 11: e1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Kowalinski E, Shanmuganathan V, Defenouillère Q, Braunger K, Heuer A, Pech M, Namane A, Berninghausen O, Fromont-Racine M, et al. 2016. The cryo-EM structure of a ribosome-Ski2-Ski3-Ski8 helicase complex. Science 354: 1431–1433. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, Xu P, Duong DM, Cheng D, Hanfelt J, Peng J. 2008. Systematic approach for validating the ubiquitinated proteome. Anal Chem 80: 4161–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Hegde RS. 2014. Reconstitution of a minimal ribosome-associated ubiquitination pathway with purified factors. Mol Cell 55: 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Brown A, Santhanam B, Hegde RS. 2015. Structure and assembly pathway of the ribosome quality control complex. Mol Cell 57: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, et al. 2015. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347: 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R. 2011. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci 108: E1392–E1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R. 2012. Translation drives mRNA quality control. Nat Struct Mol Biol 19: 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. 2010. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330: 369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitron CS, Park JH, Brandman O. 2017. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA 23: 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulard A, Cremonesi A, Moes S, Schütz F, Jenö P, Hall MN. 2010. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell 21: 3475–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ. 2017. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell 65: 751–760.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J. 2012. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol 10: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Kuroha K, Kudo K, Makino S, Inoue E, Kashima I, Inada T. 2012. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol Cell 46: 518–529. [DOI] [PubMed] [Google Scholar]
- van den Elzen AMG, Henri J, Lazar N, Gas ME, Durand D, Lacroute F, Nicaise M, van Tilbeurgh H, Séraphin B, Graille M. 2010. Dissection of Dom34-Hbs1 reveals independent functions in two RNA quality control pathways. Nat Struct Mol Biol 17: 1446–1452. [DOI] [PubMed] [Google Scholar]
- van den Elzen AMG, Schuller A, Green R, Séraphin B. 2014. Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J 33: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania RS, Kolawa NJ, Deshaies RJ. 2013. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2: e00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lewis MS, Johnson AW. 2005. Domain interactions within the Ski2/3/8 complex and between the Ski complex and Ski7p. RNA 11: 1291–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. 2013. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4: 842–851. [DOI] [PubMed] [Google Scholar]
- Wolf AS, Grayhack EJ. 2015. Asc1, homolog of human RACK1, prevents frameshifting in yeast by ribosomes stalled at CGA codon repeats. RNA 21: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa S, Fourmy D, Puglisi JD. 1999. Recognition of the codon-anticodon helix by ribosomal RNA. Science 285: 1722–1725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.