

Abstract
In patients with connective tissue disease, vascular injury induced by primary or secondary vasculitis syndromes can lead to organ dysfunction due to the loss of nutrient supply from the blood. Such vasculitis syndromes can be refractory to treatment and fatal. The nomenclature and the definition of vasculitis syndromes have recently been revised, and clinical practice guidelines for diseases associated with vasculitis syndrome are evolving. The present review provides an overview of vasculitis syndromes from the viewpoint of diagnosis and treatment.
Keywords: diagnosis, nomenclature, therapy, vasculitis syndrome
1. Introduction
Systemic vasculitis is divided into two main categories: primary vasculitis syndrome and secondary vasculitis syndrome. The former is a type of vasculitis caused by inflammation of the blood vessels, while the latter is induced by the underlying conditions, including connective tissue disease, tumors, infection, and drug allergy. Primary vasculitis syndrome and secondary vasculitis syndrome associated with connective tissue disease are included within the concept of systemic autoimmune disease. The etiology of all vasculitis disorders is elusive. Primary vasculitis syndromes are classified according to the pathologically confirmed size of the affected vessels. Treatment plans are designed to suit the respective illnesses. The Chapel Hill Consensus Conference (CHCC) in 1994 formed the basis for categorization of the aforementioned diseases.1 However, nearly 20 years later, the CHCC met again to define primary and secondary vasculitis syndromes that were not otherwise defined at their 1994 meeting. Another purpose of the conference was the reexamination and unification of the nomenclature of new types of vasculitis syndromes and their associated illnesses (Table 1).2 This review provides an overview of this new nomenclature.
Table 1.
Definition of vasculitides adopted by the 2012 Chapel Hill Consensus Conference on the nomenclature of vasculitis
| Large‐vessel vasculitis (LVV) | |
| Takayasu arteritis (TAK) | Vasculitis affecting large arteries more often than other vasculitides. Large arteries are the aorta and its major branches. Any size artery may be affected |
| Giant cell arteritis (GCA) | |
| Medium‐vessel vasculitis (MVV) | |
| Polyarteritis nodosa (PAN) | Vasculitis predominantly affecting medium arteries defined as the main visceral arteries and their branches. Any size artery may be affected. Inflammatory aneurysms and stenoses are common |
| Kawasaki disease (KD) | |
| Small‐vessel vasculitis (SVV) | Vasculitis predominantly affecting small vessels, defined as small intraparenchymal arteries, arterioles, capillaries, and venules. Medium arteries and veins may be affected |
| ANCA‐associated vasculitis (AAV) | |
| Microscopic polyangiitis (MPA) | Necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (ie, capillaries,venules, arterioles, and small arteries), associated with myeloperoxidase (MPO) ANCA or proteinase 3 (PR3) ANCA. Not all patients have ANCA. Add a prefix indicating ANCA reactivity, eg, MPO‐ANCA, PR3‐ANCA, ANCA negative |
| Glanulomatosis with polyangiitis (GPA)a (former Wegener's glanulomatosis) | |
| Eosinophilic granulomatous with polyangiitis (EGPA)a (former Churg‐Strauss syndrome) | |
| Immune complex vasculitis | |
| Antiglomerular basement membrane (Anti‐GBM) diseasea (former Goodpasture syndrome) | Vasculitis with moderate to marked vessel wall deposits of immunoglobulin and/or complement components predominantly affecting small vessels (ie, capillaries, venules, arterioles, and small arteries). Glomerulonephritis is frequent |
| Cryoglobulinemic vasculitis (CV) | |
| IgA vasculitis (IgAV)a (former Henoch‐Schönlein purpura) | |
| Hypocomplementemic urticarial vasculitis (HUV) (Anti‐C1q vasculitis) | |
| Variable vessel vasculitis (VVV) | |
| Behçet's disease (BD) | Vasculitis with no predominant type of vessel involved that can affect vessels of any size (small, medium, and large) and type (arteries, veins, and capillaries) |
| Cogan's syndrome (CS) | |
| Single organ vasculitis (SOV) | |
| Cutaneous leukocytoclastic angiitis | Vasculitis in arteries or veins of any size in a single organ that has no features that indicate that it is a limited expression of a systemic vasculitis. The involved organ and vessel type should be included in the name (eg, cutaneous small vessel vasculitis, testicular arteritis, central nervous system vasculitis). Vasculitis distribution may be unifocal or multifocal (diffuse) within an organ. Some patients originally diagnosed as having SOV will develop additional disease manifestations that warrant redefining the case as one of the systemic vasculitides (eg, cutaneous arteritis later becoming systemic polyarteritis nodosa, etc.) |
| Cutaneous arteritis | |
| Primary CNS vasculitis | |
| Isolated aortitis | |
| Others | |
| Vasculitis associated with systemic disease | |
| Lupus vasculitis | Vasculitis that is associated with and may be secondary to (caused by) a systemic disease. The name (diagnosis) should have a prefix term specifying the systemic disease (eg, rheumatoid vasculitis, lupus vasculitis, etc.) |
| Rheumatoid vasculitis | |
| Sarcoid vasculitis | |
| Relapsing polychondritis vasculitis | |
| Others | |
| Vasculitis associated with probable etiology | |
| Hepatitis C virus‐associated cryoglobulinemic vasculitis | Vasculitis that is associated with a probable specific etiology. The name (diagnosis) should have a prefix term specifying the association (eg, hydralazine‐associated microscopic polyangiitis, hepatitis B virus‐associated vasculitis, hepatitis C virus‐associated cryoglobulinemic vasculitis, etc.) |
| Hepatitis B virus‐associated vasculitis | |
| Syphilis‐associated aortitis | |
| Drug‐associated immune complex vasculitis | |
| Drug‐associated ANCA‐associated vasculitis | |
| Cancer‐associated vasculitis | |
| Others | |
The disease that a nomenclature was changed to (New nomenclatures are expressed in a bold‐face.) Revised from reference 2.
2. Concept of Vasculitis Syndrome for Diagnosis
The pathophysiologic basis of the clinical features of primary vasculitis is related to the fact that blood vessels supply oxygen to maintain the function of solid organs and the interstitium. As a result of inflammation in blood vessels, bleeding caused by the breakdown of vascular structures (secondary to inflammation) or interruption of blood flow can lead to organ dysfunction. This occurs in the local area of the vasculitic lesion as well as in the visceral organs and interstitium in the peripheral perfusion area. With regard to diagnosis of primary vasculitis syndrome, it is important to systematize individual localized symptoms, which, at first glance, appear to be independent. The clinician should assess whether or not such individual localized symptoms are the results of ischemia caused by the inflammation of blood vessels or due to symptoms that arise as a result of bleeding. Hence, rather than keeping a diagnosis of subdivided individual vasculitis in mind, it is more important for clinicians to understand the diagnostic process of the vasculitis syndrome (Figure 1) and the clinical symptoms of vasculitis.
Figure 1.
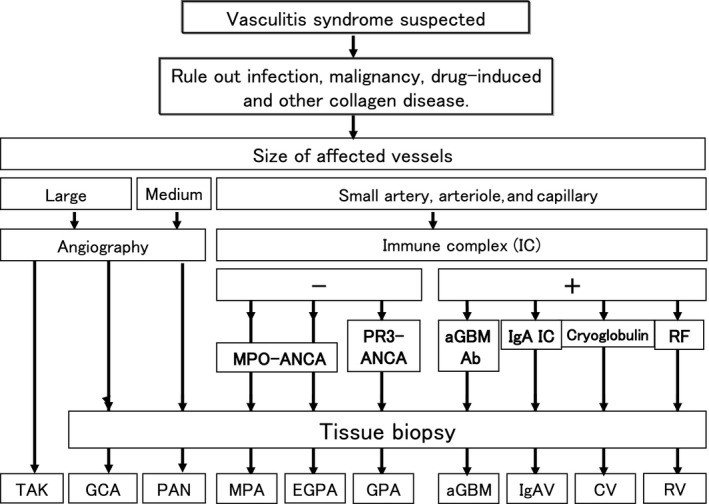
Approach to the diagnosis of vasculitis syndrome. TAK, Takayasu arteritis; GCA, giant cell arteritis; PAN, polyarteritis nodosa; MPA, microscopic polyangiitis; EGPA, eosinophilic granulomatosis with polyangiitis; GPA, granulomatosis with polyangiitis; aGBM, antiglomerular basement membrane disease; IgAV, IgA vasculitis; CV, cryoglobulinemic vasculitis; RV, rheumatoid vasculitis. Revised from reference 3. Kawasaki disease is excluded in this diagnostic procedure because the diagnostic procedure for Kawasaki disease is based solely on characteristic clinical signs and symptoms
3. Diagnosis of Vasculitis Syndromes
Clinical features of primary vasculitis syndromes can be classified largely into two categories: systemic signs/symptoms caused by inflammation and localized visceral signs/symptoms specific to the affected organs (Table 2).3 Signs/symptoms that could evoke the possibility of vasculitis syndrome are described as follows.
Table 2.
Visceral manifestations of large‐/medium‐vessel arteritis and small‐vessel vasculitis
| I. Visceral manifestation of large‐/medium‐sized vessel | |
| Common carotid artery | Dizziness, headache, syncope |
| Maxillary artery | Jaw claudication |
| Ophthalmic artery | Visual loss, blindness |
| Subclavian artery | Numbness & cold sensation of upper extremities, easy fatigability, difference in blood pressure between right and left arm, pulse deficit |
| Renal artery | Hypertension, renal failure |
| Mesenteric artery | Ischemic colitis (abdominal pain melena etc.) |
| Coronary artery | Angina pectoris, myocardial infarction |
| Pulmonary artery | Cough, bloody sputum, dyspnea |
| II. Visceral manifestation of small‐sized vessel arteritis | |
| Skin | Livedo reticularis, subcutaneous nodules, purpura, skin ulcer, finger/toe‐tip necrosis |
| Peripheral nerve | Mononeuritis multiplex |
| Muscles | Myalgia |
| Joints | Arthralgia |
| Kidney | Necrotizing crescentic glomerulonephritis |
| Gastrointestinal tract | Gastrointestinal ulcer, gastrointestinal hemorrhage |
| Heart | Myocarditis, arrhythmia |
| Lung | Interstitial pneumonitis, pulmonary alveolar hemorrhage |
| Serous membrane | Pericarditis, pleuritis |
| Eye | Retinal hemorrhage (visual loss), scleritis |
Revised from Ozaki et al. 3.
3.1. Systemic manifestations
Fever of unknown origin (FUO): In many cases, the patient experiences a high, spiking fever of 38‐39°C.
Weight loss caused by inflammation.
Weakness, general malaise.
Arthralgia, muscle pains: This systemic manifestation indicates that vasculitis has spread systemically, while localized manifestations result from insufficient supply of local blood flow caused by the inflammation of blood vessels. Histological study with biopsy is recommended in cases of localized myalgia.
The aforementioned systemic symptoms are frequently seen among elderly people with malignant tumors. Therefore, during the process of diagnosis for vasculitis syndrome, it is recommended to also conduct screening examinations for malignant tumors.4
3.2. Local symptoms
Local symptoms are featured by the simultaneous (or sequential) appearance of symptoms from different affected organs in a patient with vasculitis syndrome.
Visceral Signs/Symptoms of Large‐ and Medium‐Vessel Vasculitis. As large to medium‐sized blood vessels run from the aorta to the organs, the signs/symptoms of vasculitis result from injury to organs supplied by the affected vessels. These signs/symptoms include pulse deficit, jaw claudication, loss of vision, and acute abdomen. Especially in large‐vessel vasculitis, clinicians should be aware of the possibility that any size of artery may be affected, because a decrease in aortic blood pressure can result in compromise of blood flow to all downstream arteries.
Visceral signs/symptoms of Small‐Vessel Vasculitis. In cases with skin rash, the so‐called palpable purpura is a distinguishing feature that frequently develops in the lower limbs. Mononeuritis multiplex is a clinical manifestation of vasculitis of medium to small arteries that feed the affected nerves. In the early stage, it may manifest as sensory disturbance, such as hyperesthesia or hypoesthesia, and, when progressive, leads to motor disturbance that may result in drop hand or foot. The clinical features of small‐vessel vasculitis in the kidney include those of nephritis, such as hematuria, proteinuria, and cylindruria. In some cases, pulmonary alveolar hemorrhage caused by arteriolitis or venulitis in the lungs develops with bloody and foamy sputum.
3.3. Laboratory findings and imaging studies
General Laboratory Findings: Acute inflammatory markers, such as erythrocyte sedimentation rate (ESR), CRP, and white blood cell (WBC) count, may be present. In cases with eosinophilic granulomatosis with polyangiitis (EGPA), a significant increase in eosinophil is observed. At times, high level of plasma renin activity or HBV antigen positivity can be seen in a patient with polyarteritis nodosa (PAN).
Urinalysis: Even in its early stage, proteinuria, erythruria, leukocyturia, and cylindruria can be found in patients with microscopic polyangiitis (MPA). However, in PAN and granulomatosis with polyangiitis (GPA), there are cases in which abnormal urinary findings appear over time rather than immediately.
Biochemical markers: In MPA, PAN, and GPA, renal dysfunction, such as elevation of serum creatinine, a rise in BUN, and a decline in creatinine clearance, can be observed. As interstitial pneumonia is one of representative clinical features in MPA, a rise in KL‐6 can also be seen.
Antineutrophil cytoplasmic antibody (ANCA): In MPA and EGPA, it is common (50%‐80% of cases) to detect myeloperoxidase (MPO)‐ANCA (perinuclear [p] staining pattern with an indirect immunofluorescent [IIF] assay, p‐ANCA). Antineutrophil cytoplasmic antibody testing is usually negative in PAN (<20% ANCA positive). In GPA, proteinase 3 (PR3)‐ANCA (cytoplasmic (c) staining pattern with IIF assay, c‐ANCA) is positive (90%). In recent years, genetic factors,5 biological environment factors (the induction of neutrophil extracellular traps [NETs] from neutrophil caused by bacterial infections),6 scientific environment factors (antithyroid drugs, such as propylthiouracil,7 atmospheric environmental chemicals such as silica) have been presumed as causes of ANCA positivity. Detailed history taking via an interview is highly important to distinguish primary vasculitis from genetic or environmental factor‐associated vasculitis in a patient with positive ANCA and equivocal clinical features of vasculitis.
Antiglomerular basement membrane (GBM) antibody: Especially in cases exhibiting RPGN and/or alveolar hemorrhage, it is essential to suspect anti‐GBM disease. Serum anti‐GBM antibody is specific to laboratory findings for anti‐GBM disease. Although there are various methods to detect anti‐GBM antibody, such as the IIF assay, hemagglutination assay, radioimmunoassay (RIA) method, and enzyme‐linked immunoassay (ELISA) method, the RIA and ELISA method have superior sensitivity (>95%) and specificity (>97%).8
Imaging study: Plain radiographs of the chest and paranasal sinuses, ultrasonography, CT, MRI, and thermography have been used to confirm abnormal vascular wall structure and blood flow dysfunction. If acute‐phase Takayasu disease (TAK) is present, aortic wall thickening will be densely stained with gadolinium on contrast‐enhanced MRI. As MRA can clearly outline whether or not there are any irregularities, contractions, or occlusion of the vascular walls, this imaging modality is often useful for interpretation of symptoms related to ischemia. Further, it may be possible to localize aortic inflammation using 18F‐fluorodeoxyglucose‐positron emission tomography (FDG‐PET). Increased glycolytic metabolism in inflammatory tissues or malignant tumors is the rationale behind the common use of FDG‐PET.9 Diagnostic imaging studies of the thorax (X‐rays, CT, MRI) may show interstitial infiltrative shadows in the lung field in a patient with MPA. Granulomatous lesions can also be accompanied by infiltrative shadows in patients with EGPA and GPA. PAN can cause multiple microaneurysms and/or contractions in association with inflammation of medium‐sized and small arteries. Aneurysms can be frequently detected in the branches of the abdominal aorta (eg, renal, mesenteric, and hepatic arteries) and can be confirmed via angiography. However, aneurysms are usually not detected during the acute phase. In some cases, it is possible to detect impaired blood flow using MRA or the ultrasonic Doppler method. However, in Japan, some physicians tend to avoid conventional angiography because of its invasiveness and because PAN can typically be diagnosed via biopsy.
Physiological tests: electrocardiography (ECG), pulse‐wave detection, electroencephalography (EEG), electromyography (EMG), and nerve conduction studies are important for the diagnosis of vasculitis syndrome and to classify its severity. EMG and nerve conduction studies are crucial for the diagnosis of latent vasculitis syndrome and to identify appropriate biopsy sites.
3.4. Tissue biopsy
With regard to small‐vessel vasculitis, it is essential to test for ANCA and immune complexes. However, these are definitive factors for diagnosis; rather, tissue biopsy is the most important diagnostic method for vasculitic syndrome. If it is necessary to differentiate ANCA‐associated vasculitis (AAV) from immune complex‐mediated vasculitis, it is preferable to prepare the materials for IIF staining in advance before conducting the tissue biopsy. Because the primary lesions of TAK are in the aorta and those of Kawasaki disease (KD) are in the coronary arteries, it is not possible to perform a biopsy for diagnosis of these diseases. Hence, in such cases, diagnostic imaging study plays a central role in diagnosis. For other primary vasculitis syndromes, biopsy of the affected vessels is the most useful method for diagnosis.
Some considerations with regard to tissue biopsy for systemic vasculitis are as follows:
Giant cell arteritis (GCA): A biopsy of the temporal artery is essential for diagnosis. As noncontiguous segmental lesions develop in GCA, it is crucial to prepare serial sections of biopsy specimens.10
PAN: Although the typical histological finding of PAN is necrotizing angiitis, which is characterized by fibrinoid necrosis in the tunica media, old and new lesions are often observed within the same tissues. By definition, PAN does not include inflammation of the arterioles, venules, and capillaries, meaning that PAN is not associated with glomerulonephritis.
MPA: The typical histological finding of MPA is necrotizing angiitis in arterioles and capillaries in the kidneys, indicating necrotizing crescentic glomerulonephritis.
GPA: A finding of necrotizing granulomatous lesions with giant cells in lesion sites of the upper respiratory tract (eg, nose, paranasal sinuses, and soft palate) is useful for early diagnosis. Various patterns of glomerulonephritis can also be found in kidney biopsy specimens.
EGPA: Biopsy specimens from peripheral nerves, muscles, and lungs with infiltration show vasculitides and granulomas with prominent invasion with eosinophils.
Anti‐GBM Disease: Diffuse crescentic glomerulonephritis is a distinctive feature of this disease. IIF staining can show deposition of IgG and C3 along the glomerular capillary walls.
IgA vasculitis (IgAV): Necrotizing angiitis can be observed with blood vessels in the area from the papillary layer to the reticular layer of the skin. Using immunofluorescent staining, in accordance with the areas where necrotizing angiitis has occurred, deposition of IgA in areas from the vascular endothelial cells to the vascular lumen can be observed.
As mentioned above, to diagnose a typical vasculitis syndrome, clinicians must collect patient information via interview, physical examinations, laboratory testing, imaging studies, and histopathological examination of biopsy specimens. This allows establishment of a diagnosis of vasculitis syndrome based on the classification criteria. After diagnosis, treatment should commence as soon as possible. However, it may require considerable time to establish a definitive diagnosis of vasculitis syndrome. During the diagnosis process, some patients may experience worsening in their condition. Therefore, in cases of vasculitis syndrome, rapid testing and treatment is necessary, hospitalization should be considered to increase the speed of this process. Further, referral or transfer to specialized centers is highly recommended if vasculitis syndrome is suspected.
4. Treatment of Vasculitis
To prevent irreversible organ failure caused by the progression of vasculitis, it is important to use immunosuppressant drugs to reduce inflammation as soon as possible. It is also critical to maintain blood flow through the affected vessels.
In most cases, the first‐line drug for the treatment of vasculitis syndrome is a corticosteroid. Some of the treatment methods for typical vasculitis syndrome are listed below:
4.1. Treatment of large‐vessel vasculitis
Corticosteroid is effective for most patients with TAK and GCA. Strong anti‐inflammatory effect has enabled a physician to begin administration with steroids in the early stages of the disease. However, some cases of vasculitis are resistant to corticosteroids. Therefore, attempts have been made to develop new methods of treatment using immunosuppressants to control such cases of steroid‐resistant vasculitis (Figure 2).11
Figure 2.
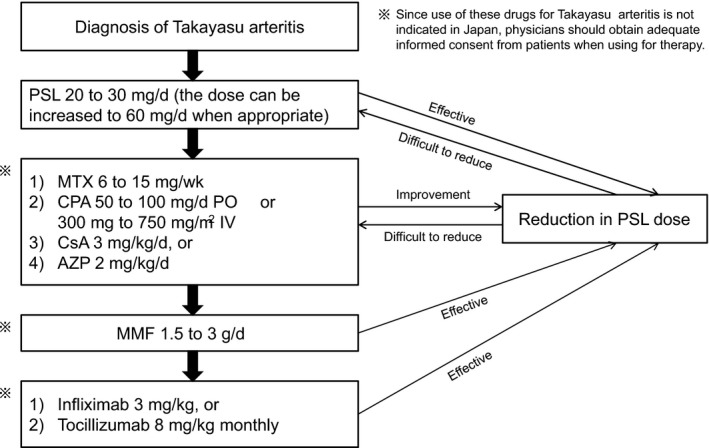
Protocol for medical treatment of Takayasu arteritis. PSL, prednisolone; MTX, methotrexate; PO, oral administration; IV, intravenous injection; CsA, cyclosporin A; AZP, azathioprine; MMF, mycophenolate mofetil. Revised from reference 3
4.2. Treatment for medium‐vessel vasculitis
Polyarteritis nodosa is the representative medium‐vessel vasculitis. Based on the classification of vasculitis syndrome by the CHCC in 1994, MPA was separated from PAN. Since then, the number of reported cases of PAN has decreased. The response to corticosteroid therapy is better with PAN than with MPA. However, in patients with organ failure, combination therapy with immunosuppressive agents is desirable (Figure 3).
Figure 3.
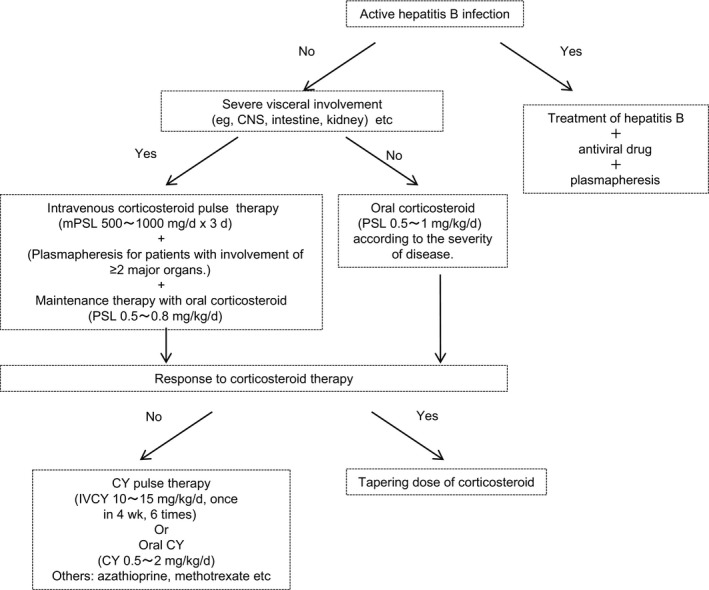
Induction therapy for remission in polyarteritis nodosa. Revised from reference 3. mPSL, methylprednisolone; PSL, prednisolone; IVCY, intravenous cyclophosphamide; CY, cyclophosphamide
4.3. Treatment for small‐vessel vasculitis
Microscopic polyangiitis is a representative of small‐vessel vasculitis with necrotizing angiitis of small vessels (capillaries and arterioles). In many cases, lungs and kidneys are the major target organs. The incidence of ANCA positivity is high (~80%) in patients with MPA. In Japan, cases of MPO‐ANCA are more predominant than those of PR3‐ANCA.12 In Western countries, there are more positive cases of proteinase 3 (PR3)‐ANCA in AAV. Treatment reports regarding AAV in Western countries mainly involve cases with positive PR3‐ANCA.13 Three groups of the Ministry of Health and Labor's Research on Measures for Intractable Diseases Project in Japan jointly established the clinical practice guidelines for ANCA‐associated vasculitis with MPO‐ANCA‐positive vasculitis. Especially in the JMAAV protocol, AAV is classified into three separate categories: mild cases (renal limited type, pulmonary fibrosis type), severe cases (systemic vasculitis, interstitial pneumonitis with glomerulonephritis, and RPGN type), and most severe cases (diffuse alveolar hemorrhage, intestinal perforation type). They used three different protocols for remission induction therapy using immunosuppressive agents according to the degree of severity. The main drugs used in the combination therapy are corticosteroids and cyclophosphamide.14 Furthermore, in Japan, other than the aforementioned standardized drugs for remission induction therapy, various treatments (eg, intravenous immunoglobulin therapy for cases with peripheral nerve involvement caused by steroid‐resistant EGPA15; or treatment with the anti‐CD20 antibody, rituximab,16 in GPA or MPA that is resistant to the standard therapy with cyclophosphamide and corticosteroids) are permitted under the medical care insurance system.
Vasculitis syndrome is a rare disease of unknown etiology. In addition to advances in knowledge regarding the pathogenesis of vasculitis, we believe that large multicenter clinical trials in Japan could help define the etiology and optimal treatment for vasculitis syndrome.
Conflict of Interest
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
Okazaki T, Shinagawa S, Mikage H. Vasculitis syndrome—diagnosis and therapy. J Gen Fam Med. 2017;18:72–78. https://doi.org/10.1002/jgf2.4
References
- 1. Jennette JC, Folk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: the proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. [DOI] [PubMed] [Google Scholar]
- 2. Jennette JC, Folk RJ, Bacon K, et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. [DOI] [PubMed] [Google Scholar]
- 3. Ozaki S, Ando M, Isobe M, et al. Guideline for management of vasculitis syndrome (JCS2008). Circ J. 2011;75:474–503. [DOI] [PubMed] [Google Scholar]
- 4. Fain O, Hamidou M, Cacoub P, et al. Vasculitides associated with malignancies: analysis of sixty patients. Arthritis Rheum. 2007;57:1473–80. [DOI] [PubMed] [Google Scholar]
- 5. Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA‐associated vasculitis. N Engl J Med. 2012;367:214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flint SM, McKinney EF, Smith KG. Emerging concepts in the pathogenesis of antineutrophil cytoplasmic antibody‐associated vasculitis. Curr Opin Rheumatol. 2015;27:197–203. [DOI] [PubMed] [Google Scholar]
- 7. Nakazawa D, Tomaru U, Suzuki A, et al. Abnormal conformation and impaired degradation of propylthiouracil‐induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody‐associated vasculitis. Arthritis Rheum. 2012;64:3779–87. [DOI] [PubMed] [Google Scholar]
- 8. Greco A, Rizzo MI, De Virgilio A, et al. Goodpasture's syndrome: a clinical update. Autoimmun Rev. 2015;14:246–53. [DOI] [PubMed] [Google Scholar]
- 9. Lee KH, Cho A, Choi YJ, et al. The role of 18F‐fluorodeoxyglucose–positron emission tomography in the assessment of disease activity in patients with Takayasu arteritis. Arthritis Rheum. 2012;64:866–75. [DOI] [PubMed] [Google Scholar]
- 10. Nordborg E, Nordborg C. Giant cell arteritis: strategies in diagnosis and treatment. Curr Opin Rheumatol. 2004;16:25–30. [DOI] [PubMed] [Google Scholar]
- 11. Chatterjee S, Flamm SD, Tan CD, Rodriguez ER. Clinical diagnosis and management of large vessel vasculitis: Takayasu arteritis. Curr Cardiol Rep. 2014;16:499–508. [DOI] [PubMed] [Google Scholar]
- 12. Furuta S, Chaudhry AN, Hamano Y, et al. Comparison of phenotype and outcome in microscopic polyangiitis between Europe and Japan. J Rheumatol. 2014;41:325–33. [DOI] [PubMed] [Google Scholar]
- 13. Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68:310–7. [DOI] [PubMed] [Google Scholar]
- 14. Ozaki S, Atsumi T, Hayashi T, et al. Severity‐based treatment for Japanese patients with MPO‐ANCA associated vasculitis: JMAAV study. Mod Rheumatol. 2012;22:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koike H, Akiyama K, Saito T, et al. Intravenous immunoglobulin for chronic residual peripheral neuropathy in eosinophilic granulomatosis with polyangiitis (Churg‐Strauss syndrome): a multicenter, double‐blind trial. J Neurol. 2015;262:752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nagafuchi H, Atsumi T, Hatta K, et al. Long‐term safety and efficacy of rituximab in 7 Japanese patients with ANCA‐associated vasculitis. Mod Rheumatol. 2015;25:603–8. [DOI] [PubMed] [Google Scholar]