

Abstract
Autophagy plays an important role in the maintenance of normal heart function. However, the role of autophagy in the inulin resistant and diabetic heart is not well understood. Furthermore, the upstream signaling and the downstream targets involved in cardiac autophagy regulation during obesity and type 2 diabetes mellitus (T2DM) are not fully elucidated. The aim of this study was to measure autophagic flux and to dissect the upstream and downstream signaling involved in cardiac autophagy regulation in the hearts of obese T2DM mice. Our study demonstrated that cardiac autophagic flux is suppressed in the heart of obese diabetic (ob/ob) mice due to impaired autophagosome formation. We showed that suppression of autophagy was due to sustained activation of mTOR as we could restore cardiac autophagy by inhibiting mTOR. Moreover, the novel finding of this study is that while IGF-1 receptor-mediated Akt activation contributes to cardiac hypertrophy, it is not involved in mTOR activation and autophagy suppression in obesity and T2DM. In contrast, inhibition of ERK signaling abolished mTOR activation and restored autophagy in the heart of obese diabetic (ob/ob) mice. The study identifies mechanisms regulating cardiac autophagy in obesity and T2DM that are mediated by ERK/mTOR but are distinct from Akt. The findings are of significant importance as they demonstrate for the first time the contribution of IGF-1 receptors (IGF-1R) and Akt signaling in cardiac hypertrophy but not in cardiac autophagy regulation in in obesity and T2DM.
Graphical abstract
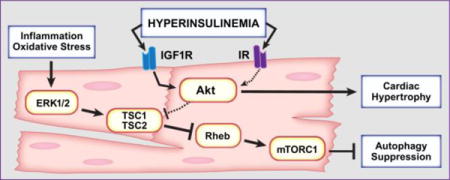
1. Introduction
Insulin resistance, T2DM and the associated hyperinsulinemia promote the development diabetic cardiomyopathy that is independent of coronary artery disease and hypertension [1]. Diabetic cardiomyopathy is a major cause of morbidity and mortality in the developed countries and its prevalence is rising due to the epidemic of obesity and T2DM [2]. Several mechanisms have been shown to contribute to the development of diabetic cardiomyopathy, including impaired insulin signaling, mitochondrial dysfunction, oxidative stress, impaired calcium homeostasis and altered myocardial autophagy [3].
Autophagy is a regulated intracellular process that eukaryotic cells employ to degrade long-lived proteins, macromolecular aggregates, and damaged organelles [4]. Under basal conditions, autophagy plays a house-keeping role to maintain cellular homeostasis, however, in response to nutritional stress such as starvation, autophagy is activated to degrade and recycle cellular components for energy supply and cell survival. In contrast, chronic activation of autophagy can lead to cell death. Autophagy plays an important role in the maintenance of normal heart function and morphology [5, 6]. Disruption of autophagy by ablation of autophagy-related 5 (Atg5), results in age-dependent cardiac dysfunction and worsens cardiac remodeling in response to pressure overload in mice [7, 8]. Furthermore, cardiac-specific ablation of the class III PI3K/vacuolar protein sorting 34 (Vps34), a critical regulator of autophagy, reduces autophagy leading to cardiac hypertrophy and dysfunction [9]. Moreover, autophagy declines with aging, which may account for increased incidence of heart disease with advanced age [10].
The role of autophagy in the insulin resistant/diabetic heart is not completely understood. Diabetes is characterized by loss of insulin action as a result of insulin deficiency or insulin resistance. Since insulin signaling is a known inhibitor of autophagy, it has been postulated that autophagy may be increased in diabetic hearts [11]. However, insulin deficiency caused by type 1 diabetes inhibited cardiac autophagy and this inhibition plays an important role in mitigating diabetes-induced cardiac dysfunction [12, 13]. The mechanisms underlying autophagy inhibition in type 1 diabetic hearts involved the mammalian target of rapamycin complex 1 (mTORC1) activation and AMP-activated protein kinase (AMPK) inhibition. In contrast, changes in autophagy in hearts of humans or mouse models with T2DM is less consistent and more controversial. As reviewed recently [14], there are a number of reports showing increased/decreased/unchanged autophagy in the hearts of humans or animals with T2DM [15– 19]. Furthermore, the role of insulin signaling in the regulation of cardiac autophagy in obesity and T2DM is not understood.
In this study, we investigated the mechanisms underlying the increase in Akt/mTOR activation and the suppression of cardiac autophagy in (ob/ob) mice, an established mouse model of diabetic cardiomyopathy that exhibit myocardial insulin resistance [20–22]. We tested the hypothesis that systemic hyperinsulinemia activated Akt/mTOR signaling through IGF-1R and suppressed cardiac autophagic flux in (ob/ob) mice. Our findings reveal a previously unrecognized role for IGF-1R in the activation of Akt and the regulation of cardiomyocyte hypertrophy in (ob/ob) hearts. This study also revealed that suppression of cardiac autophagy in (ob/ob) mice is mediated by ERK and may not involve Akt signaling. These findings have implications for pharmacological efforts directed at manipulating IGF-1R and ERK signaling for the treatment of cardiac hypertrophy and the restoration of cardiac autophagy in obesity and type 2 diabetes.
Materials and methods
1.1. Animal models, diets and care
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication no. 85-23, revised 1996) and was approved by the Institutional Animal Care and Use Committee of the University of Utah. Mice lacking IR selectively in cardiomyocytes (CIRKO) were generated using cre/loxP recombination as previously described [23]. Leptin-deficient (ob/ob) mice and littermate C57BL6/J controls were purchased from The Jackson Laboratory (Bar Harbor, ME). For feeding studies, 7-week old male C57Bl/6J mice consumed normal chow diet (NCD) (Research Diets Inc., New Brunswick, NJ) containing (kilocalories) 10% fat, 70% carbohydrate and 20% protein or high-fat diet (HFD) containing (kilocalories) 45% fat, 35% carbohydrate, and 20% for 14 weeks. Wild-type and (ob/ob) mice lacking one allele of IGF-1R in cardiomyocytes were generated by crossing WT and (ob/ob) mice with mice lacking IGF-1R specifically in cardiomyocytes generated on a mixed background [24].
1.2. Autophagic flux assessment
Autophagic flux was measured by assessing LC3II, LC3I, ATG3 and p62/SQSTM1 accumulation by western-blots in the presence or absence of the lysosomal acidification inhibitor chloroquine (CQ). For DIO, mice, random fed mice were injected i.p. with CQ (50 mg/kg body weight) 4 hours before harvesting the hearts. For (ob/ob) mice and their corresponding controls, a dose of 75 mg/g of lean mass of CQ was used. Lean mass was measured by a Bruker NMR minispec (Bruker Biospin Corporation, Billerica, MA). This adjustment was made because we observed high mortality rate in (ob/ob) mice receiving 50 mg/kg body weight of CQ, possibly due to the increase in fat mass.
1.3. Rapamycin and PD98059 treatment
Rapamycin was used to acutely inhibit mTORCl and determine the role of this complex in the autophagy regulation. Wild-type and (ob/ob) mice were injected i.p. with 2 mg/kg body weight rapamycin dissolved in dimethyl sulfoxide (DMSO) or equivalent volumes of DMSO as previously described [25] and the hearts were harvested 4 hours later. The MAPK inhibitor PD98059 was used to chronically suppress ERK1/2 signaling and assess autophagy and cardiac function. Wild-type and (ob/ob) mice were injected i.p. with 10 mg/kg body weight/day PD98059 (MAPK inhibitor) dissolved in dimethyl sulfoxide (DMSO) or equivalent volumes of DMSO as previously described [26] and the hearts were harvested 4 days later.
1.4. Echocardiography measurements
Cardiac function was monitored at rest using transthoracic echocardiography as previously described [27]. Measurements were performed in lightly anesthetized (isoflurane) mice with a Vevo 2100 high-resolution echocardiography machine equipped with a 22–55 MHz transducer (VisualSonic, Toronto, ON). All acquisitions were performed by an investigator blinded to genotype and analyzed using the VevoStrain software (VisualSonic, Toronto, ON).
1.5. Western blot analysis
Total proteins were extracted from frozen heart muscle as previously described [28]. See the supplemental materials for details. A list of used antibodies is provided in Supplemental Table 1.
1.6. Histology and morphometric analysis
Hearts were snap frozen in OCT, sectioned at 10μm, and stained with wheat germ agglutinin (WGA, Alexa 488) and DAPI (Alexa 405). Cardiomyocyte diameter was measured counting a minimum of 300 cells containing a central nucleus on 3 non-consecutive sections per animal. Cardiomyocyte cross-sectional area was measured 5um-thick heart sections stained with WGA (Alexa fluor 488). Images were taken using CellSens Dimension viewing software (Version 1.1.3, Olympus). The area was assessed on fibers with circular shapes in a blinded fashion by quantitative image analysis using CellSens Dimension software. The images were acquired with an XM10 Olympus fluorescence camera.
1.7. Circulating insulin and IGF-1 levels
Blood was collected from the left ventricle of fully anesthetized mice during euthanasia and serum was separated by centrifugation (12,000 g; 15 minutes; 4°C). Both serum insulin (Crystal Chem, Downers Grove, IL) and IGF1 (Life Technologies, Carlsbad, CA) levels were measured by enzyme-linked immunosorbent assay (ELISA) following instructions provided by the manufacturer.
1.8. Statistical analysis
All data are expressed as means ± standard errors of the means (SEM). Unpaired Student’s t test was used to analyze differences relative to wild-type controls. One-way or Two-way ANOVA was performed to analyze differences by genotype and by treatment, followed by bonferroni protected least significant difference test. Statistical calculations were performed using the GraphPad Prism software (GraphPad, San Diego, CA). For all analyses, a p value of <0.05 was considered significantly different.
2. Results
2.1. Altered cardiac autophagic flux in obesity and type 2 diabetes
To study the regulation of cardiac autophagy in obesity, we measured autophagic flux in the hearts of both genetic (ob/ob) and dietary (high-fat diet (HFD) models of murine obesity. We found that obesity resulted in altered autophagic flux in the hearts of both (ob/ob) and HFD mice. Surprisingly, genetic obesity altered autophagy at the level of autophagosome formation but not at their clearance step as evidenced by reduced LC3II levels both at baseline and after inhibition of lysosomal function by chloroquine (CQ) (Figures 1A and 1B). In contrast, HFD altered autophagosome clearance as supported by high levels of LC3II and LC3II/LC3I ratios at baseline and the absence of a further increase with CQ (Supplemental Figure 1). Consistent with reduced autophagy in (ob/ob) hearts, p62 (also called SQSTM1), which is known to be degraded by autophagy, was increased in (ob/ob) hearts both at baseline and following CQ treatment (Figure 1C). In contrast, p62 levels were unchanged in the hearts of HFD mice (Supplemental Figure 1), highlighting important differences in autophagy regulation between these two mouse models of obesity. It is worth noting that despite an increase in CQ-mediated accumulation of LC3II in (ob/ob) hearts, p62 levels remained increased. This suggest the existence of an additional mechanism responsible for p62 accumulation in (ob/ob) hearts that requires further investigation.
Figure 1. Reduced cardiac autophagic flux in leptin-deficient (ob/ob) mice.
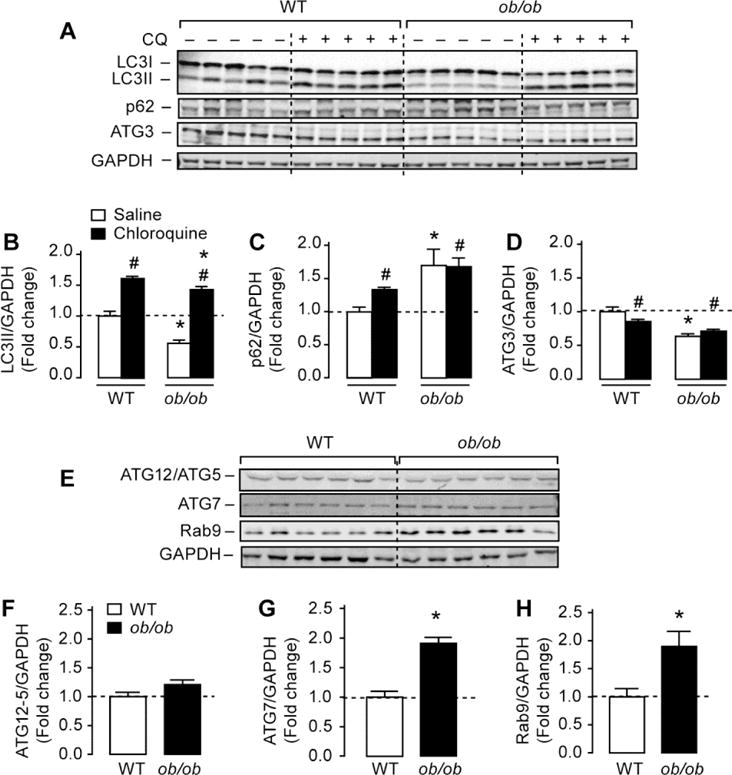
(A) Western-blots of microtubule associated protein 1 light chain 3 (LC3) I and LC3II, p62/SQSTM1, autophagy related 3 (ATG3) and glyceraldehyde dehydrogenase (GAPDH) in whole heart homogenates from wild-type and (ob/ob) mice. Wild-type and (ob/ob) mice received an intraperitoneal injection of saline or 75 mg/g lean mass chloroquine (CQ) for 4 hours before sacrifice. (B), (C) and (D) The corresponding densitometry of LC3II/GAPDH, p62/GAPDH and ATG3/GAPDH expressed as fold-change relative to wild-type saline. (E) Representative western-blots of autophagy-related protein 12 (ATG12)/ATG5, ATG7, ras-related protein rab9 and GAPDH in whole heart homogenates from wild-type and (ob/ob) mice. (F), (G) and (H) The corresponding densitometry of ATG12/ATG5, ATG7 and Rab9 expressed as fold-change relative to wild-type values. Data are means ± SEM. N=5–6 mice per group. * P < 0.05 versus wild-type under the same treatment condition; # P < 0.05 versus saline within the same genotype.
To explore potential mechanisms that may underlie autophagy inhibition in (ob/ob) hearts, we examined the expression of key autophagy molecules involved in autophagosome maturation and LC3 lipidation. As shown in Figures 1A and 1D, protein levels of the ubiquitin-like conjugated enzyme ATG3, which is important for the lipidation of LC3I to LC3II [29], was reduced by almost 40% in (ob/ob) hearts. The regulation of cardiac ATG3 content with obesity is likely to occur via post-transcriptional mechanisms as its mRNA expression was not altered (data not shown). Contrary to ATG3, obesity was associated with a ~2-fold increase in ATG7 and Rab9 proteins whereas the content of ATG12/5 complex was unaltered (Figures 1E–H), suggesting the existence of a compensatory elevation in alternative autophagy pathways. Taken together, these observations support the conclusion that defective autophagy in the hearts of genetically obese mice is caused by altered autophagososme formation.
2.2. Obesity-induced cardiac hypertrophy is insensitive to the inhibition of lysosomal function
Because it was previously shown that decreased autophagy can induce cardiac dysfunction in mice [7], we asked whether the inhibition of autophagosome formation and their clearance correlated with changes in left ventricular (LV) dimension and performance. WT and (ob/ob) mice were injected with saline or CQ for 4 hours before assessing LV diameters and fractional shortening (FS). Although we observed a modest increase in LV wall thickness in (ob/ob) hearts treated with saline or with CQ relative to equivalently treated WT, CQ had no noticeable effects on LV performance with the exception of a modest but significant reduction in LVIDs, which may suggest decreased ventricular filling (Supplemental Table 2).
2.3. Increased Akt, mTOR and ERK signaling in (ob/ob) hearts
Because autophagy is negatively regulated by the insulin/Akt/mTOR axis [30, 31], we sought to determine if this pathway is up-regulated in (ob/ob) hearts. Consistent with previous reports by us [21] and by others [22], we observed increased Akt/mTOR signaling in (ob/ob) hearts as evidenced by increased Akt phosphorylation at Ser473 and at Thr308 residues (Figures 2A and 2B) and mTOR phosphorylation at Ser2448 (Figures 2C and 2D). Similarly, phosphorylation of ERK1/2 at Thr202/Tyr204 and ribosomal S6 at Ser235/236 were elevated in (ob/ob) hearts compared to WT hearts (Figure 2). The ATG1 homologue ULK-1 may regulate autophagy downstream of mTOR [32] and our results clearly demonstrated that mTOR activation increased ULK-1 phosphorylation at Ser757 in (ob/ob) hearts, thus preventing its activation (Figures 2C and 2D). Finally, as AMPK is another regulator of autophagy, we measured its phosphorylation at Thr172 and found no difference between the groups. Our results suggest that autophagy suppression in (ob/ob) hearts may be caused by the activation of Akt/mTOR/ULK-1 signaling.
Figure 2. Enhanced Akt/mTOR and ERK1/2 signaling in (ob/ob) hearts.
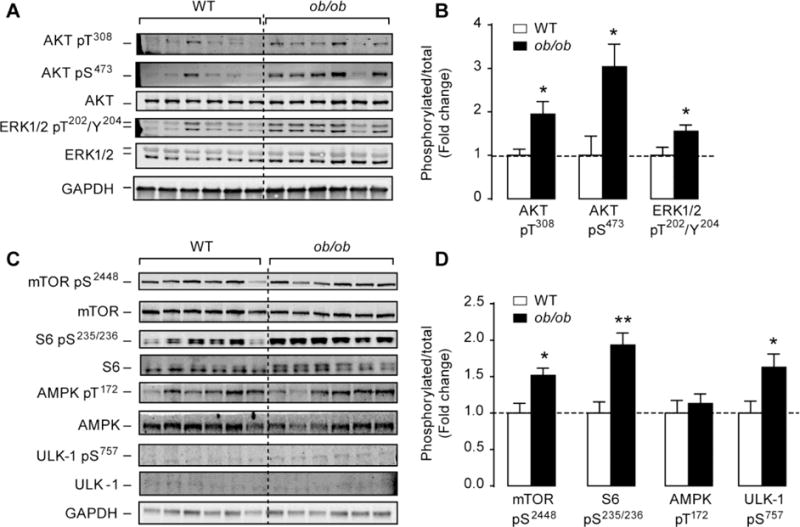
(A) and (C) Western-blots of phospho(Thre308 and Ser473) Akt, total Akt, phospho(Thr202/Tyr204) ERK1/2, total ERK1/2, phospho(Ser2448) mTOR, total mTOR, phospho(Ser235/236) S6, total S6, phospho(Thr172) AMPK, total AMPK, phosphor(Ser757) ULK-1, total ULK-1 and GAPDH in whole heart homogenates from wild-type and (ob/ob) mice. (B) and (D) The corresponding densitometry of phosphor/total Akt, ERK1/2, mTOR, S6, AMPK and ULK-1 expressed as fold-change relative to wild-type values. Data are means ± SEM. N=6 mice per group. * P < 0.05; ** P < 0.005 versus wild-type controls.
2.4. Inhibition of mTORC1 by rapamycin restored cardiac autophagy in (ob/ob) mice
Though the suppression of autophagy in (ob/ob) hearts was associated with enhanced Akt/mTOR signaling, this does not prove causality. Thus, to directly determine the implication of mTOR signaling in the inhibition of autophagy in (ob/ob) hearts, we inhibited mTORCl signaling by rapamycin. As depicted in Figures 3 A–D, rapamycin abolished the activation of mTORCl downstream targets P70S6 kinase and ribosomal S6 in WT and (ob/ob) hearts without affecting Akt phosphorylation. Similarly, rapamycin attenuated ULK-1 phosphorylation on Ser757 in (ob/ob) hearts but had no effect on ERK1/2 phosphorylation (Figures 3A, 3E and 3F). To determine if mTORC1 inhibition restored autophagy in (ob/ob) hearts, we measured autophagy markers and showed that LC3II levels and LC3II/LC3I ratios were increased in (ob/ob) mice treated with rapamycin when compared to DMSO-treated (ob/ob) mice (Figures 3G–I). Similarly, the reduction in ATG3 protein content in (ob/ob) hearts were completely normalized to WT levels by rapamycin treatment (Figures 3A and 3K). It is noteworthy to mention that the level of autophagy was not enhanced in the hearts of WT mice receiving rapamycin. These data suggest that activation of mTOR signaling mediates autophagy inhibition in (ob/ob) hearts. Interestingly, p62 levels were attenuated by rapamycin treatment in (ob/ob) hearts but the levels were not completely normalized to WT values, suggesting additional mechanisms involved in p62 regulation (Figures 3A and 3J).
Figure 3. Inhibition of mTOR signaling by rapamycin restored cardiac autophay in (ob/ob) mice.
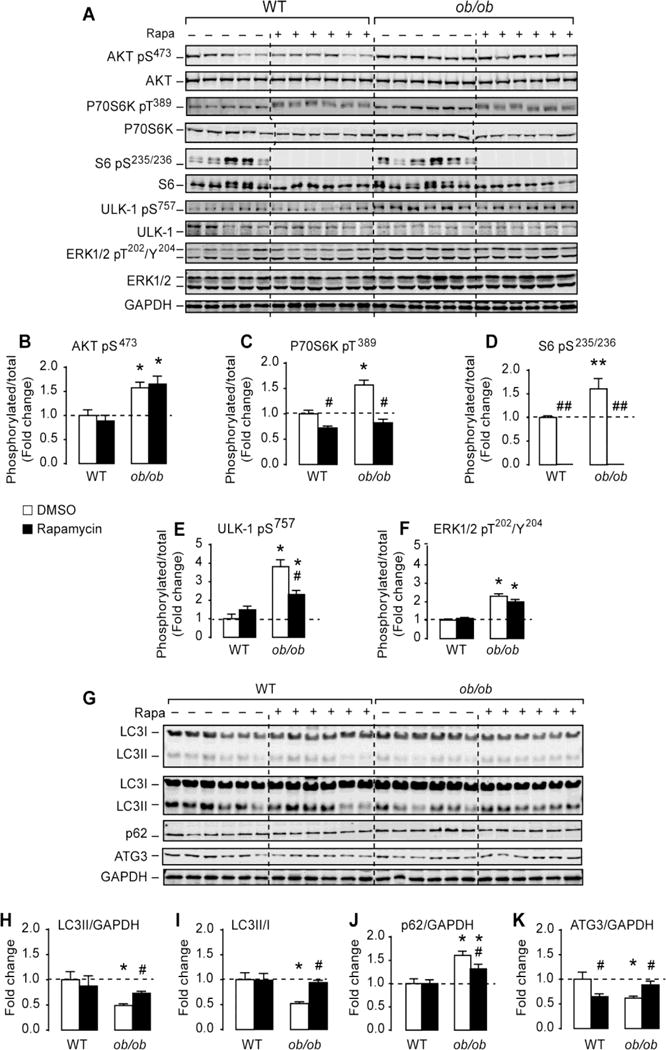
(A) Western-blots of Akt, mTOR and ERK signaling in heart homogenates of wild-type and (ob/ob) mice treated with DMSO or rapamycin (rapa). (B)–(F) The corresponding densitometry of phospho/total for the proteins shown in (A) expressed as fold change relative to wild-type DMSO group. (F) Western-blots of LC3 I and LC3II (low and high exposure), p62/SQSTM1, ATG3 and GAPDH in whole heart homogenates from wild-type and (ob/ob) mice treated with DMSO or rapamycin. (G)–(J) The corresponding densitometry of LC3II/GAPDH, LC3-II/I ratios, p62/GAPDH and ATG3/GAPDH expressed as fold-change relative to wild-type DMSO. Data are means ± SEM. N=6 mice per group. * P < 0.05; ** P < 0.005 versus wild-type under the same treatment condition; # P < 0.05; ## P < 0.005 versus DMSO within the same genotype.
2.5. Systemic hyperinsulinemia is associated with increased cardiac IGF-1 receptor (IGF-1R) signaling in (ob/ob) mice
Previous studies reported elevated IRS1-PI3K activity and enhanced insulin receptor phosphorylation in (ob/ob) hearts [21, 22]. These findings were also confirmed in the hearts of human subjects with non-insulin dependent diabetes mellitus (NIDDM) free of heart failure [22]. Despite elevated IRS1/PI3K activity insulin failed to increase Akt phosphorylation and glucose uptake in (ob/ob) hearts, consistent with insulin resistance [21, 22]. The increase in IRS1-PI3K activity and the inability to respond to insulin led us to hypothesize that IGF-1R signaling, which is also known to activate IRS1-PI3K, might be activated by systemic hyperinsulinemia in (ob/ob) hearts. Indeed, (ob/ob) mice had more that 26-fold increase in circulating insulin levels when compared to littermate controls (Figure 4A) and consistent with our hypothesis, we showed that IGF-1Rβ phosphorylation at Tyr1135 normalized by total IGF-1Rβ was elevated by ~2-fold in (ob/ob) hearts (Figures 4B and 4C). To determine if hyperinsulinemia and not cardiac insulin resistance per se is causal for the increase in IGF-1R activation, we used mice with cardiac insulin resistance (due to the lack of insulin receptors in cardiomyocytes or CIRKO mice) and normal circulating insulin levels [23] and showed no activation of IGF-1R (Figures 4D and 4E). Similarly, we found no modulation of autophagy in CIRKO hearts (Supplemental Figure 2). Thus systemic hyperinsulinemia may be causal for the activation of IGF-1R in (ob/ob) hearts.
Figure 4. Systemic hyperinsulinemia but not cardiac insulin resistance is associated with increased cardiac IGF-1 receptor (IGF-1R) signaling in (ob/ob) mice.
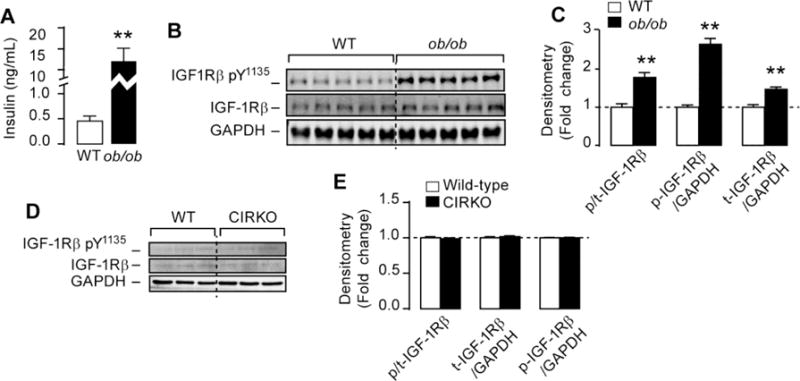
(A) Random fed insulin levels in (ob/ob) mice. (B) and (D) Western-blots of phospho(Tyr1135) IGF-1 receptor β (IGF-1Rβ) total IGF-1Rβ and GAPDH in (ob/ob) and CIRKO hearts respectively. (C) and (E) The corresponding densitometry of phospho/total IGF-1R, total IGF-1R/GAPDH and phospho-IGF-1R/GAPDH in (ob/ob) and CIRKO hearts respectively. Data are means ± SEM. N=3–5 mice per group. ** P < 0.005 versus wild-type mice.
2.6. Partial deletion of IGF-1R reduced Akt phosphorylation and attenuated cardiomyocytes hypertrophy in (ob/ob) mice
To directly test the involvement of IGF-1R in the activation of proximal insulin receptor signaling in (ob/ob) hearts, we generated mice lacking one allele of cardiomyocytes IGF-1R in the (ob/ob) background. We confirmed that IGF-1Rβ phosphorylation at Tyr1135 was significantly attenuated in (ob/ob) heats lacking one allele of the IGF-1R in cardiomyocytes, despite the persistence of systemic hyperinsulinemia (Figures 5A–E). Furthermore, partial deletion of IGF-1R in (ob/ob) hearts had no effect on body weights (Table 1) or circulating IGF-1 (Figure 5F). Interestingly, partial deletion of IGF-1R reduced Akt phosphorylation at Ser473 by 40% and 70% in WT and in (ob/ob) hearts respectively (Figures 5G and 5H). Contrary to Akt, ERK phosphorylation at Thr202/Tyr204 was not affected by IGF-1R partial deletion in WT or (ob/ob) hearts (Figures 5G and 5I). The reduction in Akt, diminished cardiomyocytes hypertrophy in (ob/ob) hearts (Figures 5J and 5K). Consistent with reduced cardiomyocytes diameter and area, lack of one allele of IGF-1R in (ob/ob) mice reduced heart weight/tibia length ratios (Table 1) but did not normalize them to WT values, suggesting the existence of other mechanisms underlying cardiac hypertrophy in these animals. Finally, the reduction of IGF-1R signaling in (ob/ob) hearts had no effect on LV wall dimensions or functional parameters (Table 1).
Figure 5. Partial deletion of IGF-1R in (ob/ob) hearts reduces Akt activation and attenuated cardiomyocytes hypertrophy.
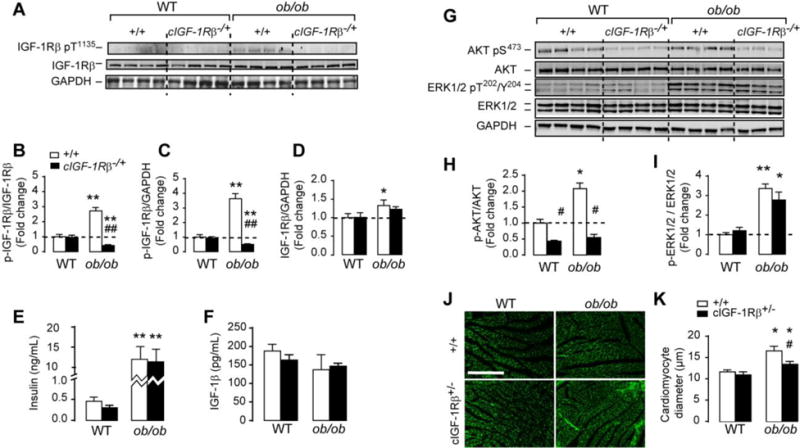
(A) and (C) Western-blots of phospho(Tyr1135) IGF-1 receptor β (IGF-1Rβ) total IGF-1Rβ, phospho(Ser473) Akt, total Akt, phospho(Thr202/Tyr204) ERK1/2, total ERK1/2 and GAPDH in whole heart homogenates from wild-type, cardiac specific IGF-1R heterozygous (cIGF-1R+/−), (ob/ob) and (ob/ob)/cIGF-1R+/− mice. (B)–(D) The corresponding densitometry of phospho/total IGF-1R, total IGF-1R/GAPDH and phospho-IGF-lR/GAPDH. (E) and (F) Circulating insulin and IGF-1 measured at the fed state in wild-type, cIGF-1R+/−, (ob/ob) and (ob/ob)/cIGF-1R+/− mice. (H) and (I) The corresponding densitometry of phosphor/total Akt and ERK1/2. (J) Representative heart sections stained with wheat germ agglutinin (WGA). (K) Quantification of cardiomyocyte diameter (μm). Scale bar in (J) is 200 μm. Data are means ± SEM. N=3–4 mice per group. * P < 0.05; ** P < 0.005 versus wild-type controls; # P < 0.05; ## P < 0.005 versus (ob/ob).
Table 1.
Morphometric parameters and cardiac function at rest in wild-type, cIGF-1R+/−, (ob/ob) and (ob/ob)/cIGF-1R+/− mice
| WT | cIGF-1R+/− | (ob/ob) | (ob/ob)/cIGF-1R+/− | |
|---|---|---|---|---|
| (n=5) | (n=5) | (n=5) | (n=5) | |
| BW (g) | 25.6 ± 1 | 23.8 ± 1.2 | 53.1 ± 2.6* | 51.8 ± 2.* |
| HW (g) | 0.11 ± 0.004 | 0.10 ± 0.003 | 0.14 ± 0.004* | 0.13 ± 0.003* |
| HW/TL | 0.062 ± 0.002 | 0.056 ± 0.002 | 0.08 ± 0.002* | 0.07 ± 0.002*# |
| CCSA (μm2) | 192.9 ± 9.3 | 192.5 ± 15.8 | 266.8 ± 11.8** | 222.9 ± 4.7# |
| LV mass, mg | 83.6 ± 2.6 | 79.9 ± 3.2 | 88.2 ± 1.7 | 94.5 ± |
| LVPWd (mm) | 0.6 ± 0.03 | 0.7 ± 0.07 | 0.8 ± 0.008* | 0.9 ± 0.11* |
| LVPWs (mm) | 0.9 ± 0.05 | 1 ± 0.06 | 1.3 ± 0.11* | 1.3 ± 0.07* |
| LVIDd (mm) | 3.9 ± 0.09 | 3.7 ± 0.2 | 3.7 ± 0.2 | 3.8 ± 0.03 |
| LVIDs (mm) | 2.8 ± 0.1 | 2.9 ± 0.02 | 2.3 ± 0.2 | 2.6 ± 0.1 |
| FS (%) | 26.7 ± 1.9 | 28.6 ± 2.8 | 36.8 ± 2.8* | 31.4 ± 2.6 |
Values are mean ± SEM.
p < 0.05 versus wild-type;
p < 0.05 versus (ob/ob).
BW: body weight; HW: heart weight; HW/TL: heart weight/tibia length; CCSA: cardiomyocyte cress-sectional area; LVPWd: left ventricular posterior wall in diastole; LVPWs: left ventricular posterior wall in systole; LVIDd: left ventricular interior diameter in diastole; LVIDs: left ventricular interior diameter in systole; FS: fractional shortening.
2.7. Persistent activation of mTOR downstream signaling in (ob/ob) hearts with partial reduction of IGF-1R
We expected that the reduction in Akt phosphorylation in (ob/ob) hearts lacking one allele of the IGF-1R will reduce mTOR signaling. Indeed, phosphorylation of tuberous sclerosis 2 (TSC2) by Akt at Ser939, which was elevated in (ob/ob) hearts (consistent with elevated Akt activation), was reduced in (ob/ob) hearts lacking one allele of IGF-1R (Figures 6A and 6B). Interestingly, TSC2 phosphorylation at Ser939 was also reduced in WT hearts lacking one copy of IGF-1R, which is consistent with reduced Akt activation in these hearts as well (Figures 6A and 6B). Similarly, (ob/ob) hearts had a 3-fold increase in TSC2 phosphorylation at Ser664 site by ERK1/2 (Figures 6A and 6C). Although slightly attenuated, TSC2 phosphorylation by ERK1/2 at Ser664 was still elevated by 2-fold in (ob/ob) hearts lacking one allele of IGF-1R, consistent with persistent ERK1/2 activation (Figures 6A and 6C). Although mTOR phosphorylation by Akt (Ser2248) was attenuated (Figures 6A and 6D), downstream targets of mTOR were still activated in (ob/ob)/cIGF-1R+/− hearts as evidenced by increased phosphorylation of P70S6K and S6 (Figures 6A, 6E and 6F). Interestingly, S6 phosphorylation on Ser235/236 was 50% lower in WT hearts lacking one copy of the IGF-1R gene despite no change in mTOR or P70SK phosphorylation (Figures 6A–F). Furthermore, while ULK1 phosphorylation at Ser757 was elevated in WT hearts lacking one allele of IGF-1R when compared to WT controls, the increase in ULK1 phosphorylation observed in (ob/ob) heart was attenuated with partial IGF-1R deletion but remained significantly higher when compared to WT mice (Figures 6A and 6G). Finally, partial deletion of IGF-1R reduced AMPK phosphorylation only in WT hearts but had no effect on (ob/ob) hearts (Figure 6A and 6H). Because mTOR can be activated by amino acids [33] and (ob/ob) mice are hyperphagic, which may increase amino acid uptake, we measured their concentration in the hearts of WT, cIGF-1R+/−, (ob/ob) and (ob/ob)/cIGF-1R+/− mice. We found no significant alterations in the level of branched chain amino acids (BCAA) or any other amino acids (Supplemental Figure 3). Together these data suggest the existence of alternative pathways for mTOR activation in (ob/ob) hearts that are independent of Akt.
Figure 6. Persistent activation of mTOR down-stream signaling and sustained suppression of autophagy in (ob/ob) hearts lacking one allele of IGF-1R.
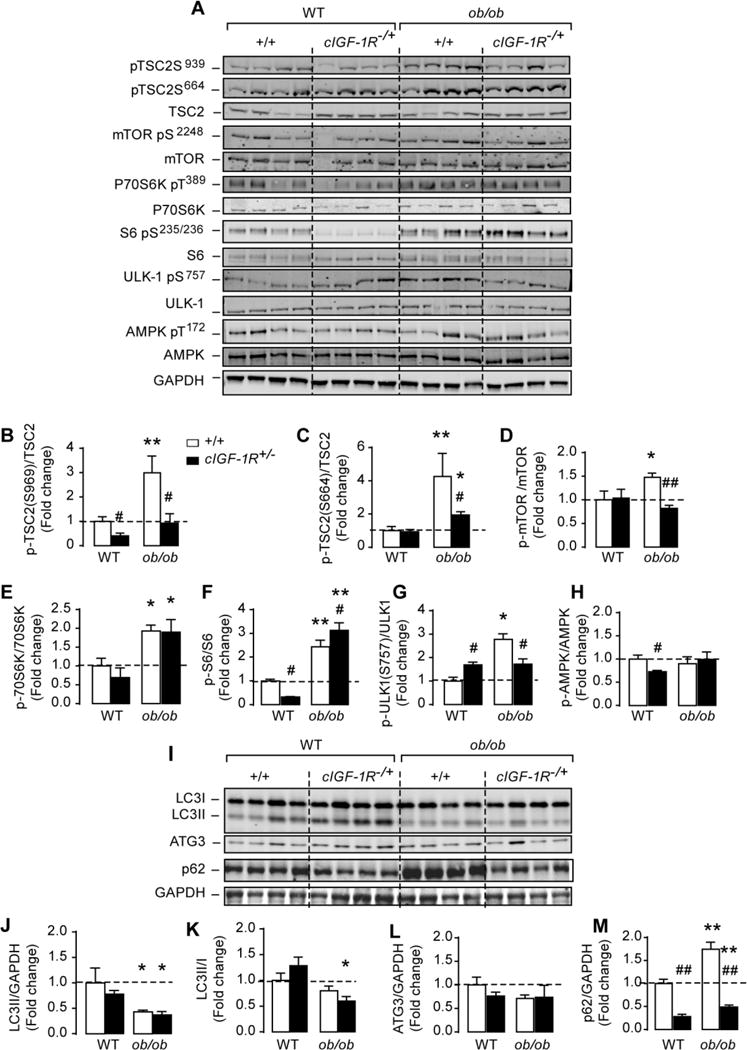
(A) Western-blots of phosphor(Ser939) and (Ser664)TSC2, total TSC2, phospho(Ser2448) mTOR, total mTOR, phospho(Thr389) P70S6K, total P70S6K, phospho(Ser235/236) S6, total S6, phosphor(Ser757) ULK-1, total ULK-1, phospho(Thr172) AMPK, total AMPK and GAPDH in whole heart homogenates from wild-type, cIGF-1R+/−, (ob/ob) and (ob/ob)/cIGF-1R+/− mice. (B)–(H) The corresponding densitometry of phosphor/total mTOR, P70S6K, S6, ULK-1 and AMPK expressed as fold-change relative to wild-type values. (I) Western-blots of LC3 I and LC3II, ATG3, p62/SQSTM1 and GAPDH in whole heart homogenates from wild-type, cIGF-1R+/−, (ob/ob) and (ob/ob)/cIGF-1R+/− mice. (J)–(M) The corresponding densitometry of LC3II/GAPDH, LC3-II/I ratios, ATG3/GAPDH and p62/GAPDH expressed as fold-change relative to wild-type controls. Data are means ± SEM. N=3–4 mice per group. * P < 0.05; ** P < 0.005 versus wild-type controls; # P < 0.05; ## P < 0.005 versus (ob/ob).
2.8. Partial deletion of IGF-1R in (ob/ob) hearts failed to rescue autophagy
To define the contribution of Akt and mTOR signaling in the regulation of cardiac autophagy in obesity, we measured autophagy markers in (ob/ob) hearts lacking one allele of IGF-1R. Despite reduced Akt phosphorylation in (ob/ob)/cIGF-1R+/− hearts, autophagy was still reduced as evidenced by the reduction in LC3II/GAPDH and LC3II/LC3I ratios (Figures 6I–K). While ATG3 content did not change, p62 levels were robustly attenuated with partial IGF-1R deletion in both WT and (ob/ob) hearts (Figures 6I, 6L and 6M). Together, these data demonstrate that autophagy suppression in (ob/ob) hearts may be mediated by mTOR downstream signaling pathways but may not involve Akt.
2.9. Inhibition of ERK signaling resorted autophagy in (ob/ob) hearts
Because we observed a persistent increase in ERK1/2 phosphorylation, activation of mTOR downstream signaling and reduced autophagy in (ob/ob)/cIGF-1R+/− hearts, we reasoned that ERK1/2 may be responsible for mTOR activation and cardiac autophagy suppression in (ob/ob) mice. Indeed, ERK1/2 can directly phosphorylate TSC2 on Ser664 leading to disruption of the TSC1-TSC2 complex, TSC2 inactivation and mTOR activation [34]. To directly test this possibility, we treated WT and (ob/ob) mice with the MAPK inhibitor PD98059 daily for 4 days and then assessed autophagy. As depicted in Figure 7A and 7B, PD98059 treatment reduced ERK1/2 phosphorylation and TSC2 Ser664 phosphorylation in (ob/ob) hearts. Although MAPK inhibition diminished Akt phosphorylation at Ser473, it is still higher in (ob/ob) mice treated with PD98059 (Figures 7A and 7B). Inhibition of ERK1/2 in (ob/ob) mice enhanced LC3II/LC3I ratios and restored ATG3 levels (Figures 7C and 7D). However, inhibition of ERK by PD98059 for 4 days had no effect on left ventricular dimensions, heart rate or fractional shortening (Supplemental Table 3). Taken together, these data suggest that cardiac autophagy suppression in (ob/ob) mice is mediated by ERK/mTOR axis.
Figure 7. Inhibition of ERK1/2 signaling reduced mTOR activation and restored autophagy in (ob/ob) hearts.
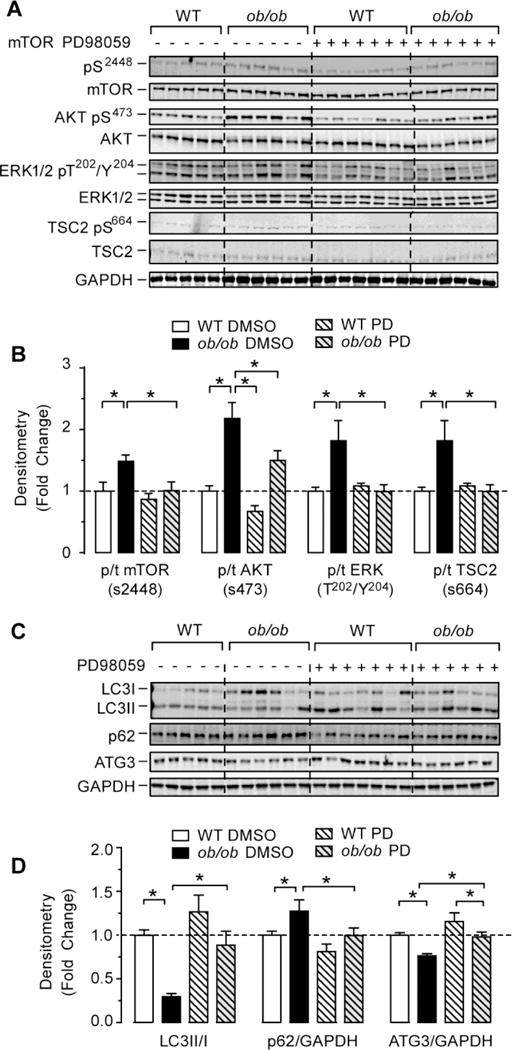
(A) Western-blots of phospho(Ser2448) mTOR, total mTOR, phospho(Ser473) Akt, total Akt, phospho(Thr202/Tyr204) ERK1/2, total ERK1/2, phosphor(Ser664) TSC2, total TSC2 and GAPDH in whole heart homogenates from wild-type, and (ob/ob) mice treated with the MEK inhibitor PD98059 (PD) or DMSO control. (B) The corresponding densitometry of phosphor/total mTOR, Akt, ERK1/2 and TSC2 expressed as fold-change relative to wild-type values. (C) Western-blots of LC3 I and LC3II, p62/SQSTM1, ATG3 and GAPDH in whole heart homogenates from wild-type and (ob/ob) mice treated with PD or DMSO. (D) The corresponding densitometry of LC3-II/I ratios, p62/GAPDH and ATG3/GAPDH expressed as fold-change relative to wild-type DMSO. Data are means ± SEM. N=5–7 mice per group. * P < 0.05; ** P < 0.005 as indicated.
3. Discussion
Growth factors such as insulin and IGF-1 signal through their receptors to inhibit cardiac autophagy [35, 36]. This inhibition is believed to be mediated by the PI3K/Akt/mTOR pathway in the case of insulin and by AMPK/mTOR pathway in the case of IGF-1. Given the inhibitory effect of insulin signaling on myocardial autophagy, it was hypothesized that autophagy would be enhanced in insulin resistant hearts. However, accumulating evidence suggest that auophagy is rather decreased in the heart with obesity, insulin resistance and T2DM [37–40]. The upstream and downstream signals involved in cardiac autophagy suppression in obesity and T2DM are not well understood. Furthermore, whether the suppression of cardiac autophagy has beneficial or detrimental functional consequences in obesity and T2DM is largely unknown. Thus, we undertook this study to define upstream signals and downstream targets of insulin/IGF-1 receptor signaling that are involved in cardiac autophagy suppression with obesity and T2DM. Our study demonstrated that hyperinsulinemia mediated-IGF-1 receptor signaling contributes to cardiomyocyte hypertrophy through the activation of Akt. In contrast, this pathway may not regulate cardiac autophagy in obesity and T2DM. Indeed, our work provides evidence that autophagy suppression in the heart with obesity and T2DM may be largely mediated mTOR signaling through alternative upstream pathways such as ERK signaling.
Studies involving autophagy assessment in the hearts of mice with obesity and T2DM have been controversial and the results are not consistent. This may be due in part to the metabolic complexity of these conditions and to the existence of compensatory pathways that can influence the level of cardiac autophagy in different mouse models. In this study, we used the (ob/ob) mouse, a genetic model of obesity and T2DM and a dietary HFD mouse model. Leptin-deficient (ob/ob) mice have been shown to manifest similar abnormalities in myocardial insulin signaling similar to those seen in humans with T2DM with preserved systolic function [22], providing clinical significance for this study. Humans with T2DM and (ob/ob) mice had elevated circulating insulin levels, increased cardiac IRS-1/PI3K activity and impaired cardiac glucose uptake [21, 22]. Using (ob/ob) mice we showed that autophagic flux is impaired in the heart and this is due to alterations in autophagosome formation rather than a defect in their clearance by lysosomes. This is different from the diet-induced obese (DIO) mouse model, where the defect in cardiac autophagy appears to be caused by an impairment in lysosomal clearance [37, 38, 40]. The basis for this difference is not entirely known but may involve the induction of mTOR signaling in (ob/ob) hearts versus no change or a reduction in this pathway in DIO mice [38, 40]. However, other DIO studies with longer durations and higher fat content in the diet have reported higher Akt and mTOR signaling in the heart [16, 37], similar to what we observed in (ob/ob) hearts. Our DIO model is similar to the one used by Jaishy et al. [38] and our results on autophagy are consistent with their results demonstrating a defect in autophagosome turnover.
Nutrient sensing pathways such as mTOR and AMPK are critical regulators of autophagy in cells [32]. Therefore, we examined if these pathways are involved in reducing autophagy in (ob/ob) hearts. While the activity of AMPK was unchanged, Akt/mTOR signaling was elevated in (ob/ob) hearts as evidenced by the phosphorylation of downstream targets such as P70S6K and S6. To investigate if activation of mTOR is responsible for autophagy suppression, we inhibited mTORCl with rapamycin and evaluated autophagy in (ob/ob) hearts. Rapamycin treatment completely restored autophagy in (ob/ob) hearts, implicating this pathway in the regulation of autophagy. Similar restoration of cardiac autophagy was previously observed in DIO mice treated with rapamycin [16], suggesting that mTOR activation may be a common cause for autophagy suppression in the heart with severe obesity and T2DM.
After confirming the involvement of mTOR in autophagy suppression, we next sought to determine upstream signaling converging to mTOR in (ob/ob) hearts. We reasoned that Akt activation could be responsible for mTOR activation and autophagy suppression. Thus, we first investigated possible mechanisms involved in Akt activation in the hearts of obese T2DM mice. Our study is the first to show that hyperinsulinemia activates Akt through IGF-1R signaling in the (ob/ob) hearts. Indeed, insulin receptors and IGF-1R have significant overlap and IGF-1 was shown to activate both IGF-1R and insulin receptors to control growth and metabolism in the heart [41]. Thus, we found that higher circulating insulin levels (but not IGF-1 levels) activated IGF-1R in (ob/ob) hearts and that reduction of IGF-1R content reduced Akt activation despite persistent hyperinsulinemia in (ob/ob)/cIGF-1R+/− mice. It is worth noting that Akt activation was also diminished in WT mice lacking one copy of IGF-1R gene, highlighting the role of this receptor in Akt activation at baseline. However, despite the reduction in Akt phosphorylation, mTOR downstream targets S6K and S6 remained elevated in (ob/ob)/cIGF-1R+/− mice. These results are in contrast with a recent study showing that lack of Akt2 reduced HFD-induced mTOR activation and autophagy impairment in the heart [37]. As this study involved whole body Akt2 knockout mice that were protected from HFD-induced obesity, it is not clear if reduced mTOR activation and autophagy suppression was caused by the lack of Akt2 per se or by the lack of obesity and thus the improvement in the metabolic milieu. Furthermore, our study cannot rule out a role for insulin/IGF-1 hybrid receptors in the activation of Akt with hyperinsulinemia as these receptors were shown to respond to insulin although with a lower affinity [42]. Thus, it is possible that the combination of higher circulating insulin levels and increased IGF-1R content may have enhanced the activity of hybrid receptors in (ob/ob) hearts.
The main conclusion of this study is that activation of mTORCl and the suppression of cardiac autophagy may occur independently of Akt activation in obesity and T2DM. There are multiple PI3K/Akt-independent activators of mTORCl including BCAA, ERK and NF-kB [33, 43, 44]. However, the involvement of BCAA in mTOR activation in the hearts of (ob/ob) and (ob/ob)/cIGF-1R+/− mice is unlikely as their levels were not altered. In contrast, ERK1/2 phosphorylation remained elevated in in the hearts of (ob/ob) and (ob/ob)/cIGF-1R+/− mice, which could have contributed to mTORCl activation similar to earlier observations in tumor cells [34]. Consistent with this idea, we showed that TSC2 phosphorylation by ERK 1/2 at Ser664 was higher in (ob/ob) and (ob/ob)/cIGF-1R+/− hearts. To directly assess the role of ERK in mTOR activation and autophagy suppression in (ob/ob) mice, we used the MAPK inhibitor PD98059. Treatment with PD98059 abolished ERK1/2 phosphorylation and restored autophagy in (ob/ob) hearts. The exact mechanism underlying ERK1/2 activation in (ob/ob) hearts warrants further investigations but may involve enhanced oxidative stress-activated p38MAPK signaling. Indeed, previous studies have reported increased oxidative stress in (ob/ob) hearts [45, 46].
The importance of autophagy in normal heart function is well established [7–9, 36]. However, the role of autophagy in obesity and type 2 diabetes-associated cardiac dysfunction is less understood. Thus, inhibition of autophagy through Rheb overexpression and mTORC1 activation in the heart increased infarct size following ischemia [16], suggesting a protective role. Similarly, enhancement of autophagy using resveratrol restored cardiac diastolic function, while inhibition of autophagy using chloroquine worsened both cardiac diastolic and systolic function in leptin receptor-deficient (db/db) mice [39]. Furthermore, autophagy insufficiency occurs in sustained cardiac hypertrophy as seen in chronic left ventricular hypertrophy and recent reports suggest that improving autophagy could alleviate cardiac hypertrophy [47]. Therefore, we predicted that autophagy restoration in the hearts of obese T2DM mice will be beneficial for cardiac structure and function and further inhibition will be detrimental. Although we only examined cardiac function at rest using echocardiography, we did not observe significant changes in cardiac dimension or performance in (ob/ob) mice even when lysosomal clearance was inhibited with chloroquine except for a modest reduction in LV filling as indicated by reduced LVIDs. Thus, it is possible that cardiac function may be impaired if these animals are subjected to farther cardiac stress such as ischemia/reperfusion or pressure overload. The major effect observed in (ob/ob)/cIGF-1R+/− mice relative to (ob/ob) mice was the attenuation of cardiomyocyte hypertrophy, which could be the result of decreased Akt-mediated signaling. Indeed, PI3K/Akt activation downstream of insulin or IGF-1 receptors signaling has been involved in physiologic and pathologic cardiac growth [48–50]. It is worth noting that while cardiomyocyte hypertrophy was attenuated in (ob/ob)/cIGF-1R+/− mice, it was not completely normalized to WT levels, suggesting the existence of Akt-independent pathways involved in the modulation of cardiac hypertrophy in obesity and T2DM. Indeed, sustained ERK1/2 activation may have contributed to cardiac hypertrophy in (ob/ob) and (ob/ob)/cIGF-1R+/− mice as MEK-ERK1/2 pathway was shown to induce cardiomyocyte growth in vivo [51]. Furthermore, as (ob/ob) and (ob/ob)/cIGF-1R+/− mice are leptin-deficient and because leptin was shown to reduce cardiac hypertrophy in (ob/ob) mice [52], it is possible that the residual cardiac hypertrophy observed in (ob/ob)/cIGF-1R+/− mice is mediated by the lack of leptin.
5. Conclusion
This study showed that IGF-1R/Akt accounts for some but not all of cardiac growth in mice with obesity and T2DM. However, autophagy suppression in (ob/ob) hearts does remain even whenAkt signaling is decreased. Our study provides evidence that activation of ERK/mTOR axis is causal for the suppression of cardiac autophagy in diabetic mice.
Supplementary Material
Highlights.
Hyperinsulinemia activates cardiac IGF-1 receptors in obese type 2 diabetic mice
Cardiac autophagy is suppressed in obese type 2 diabetic mice through mTOR/ERK signaling
Cardiac hypertrophy in obese type 2 diabetic mice is mediated by IGF-1 receptors/Akt
Acknowledgments
Source of funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants 1-R01-DK-098646-01A1 and R01-DK-099110 and the American Heart Association (AHA) grant 16GRNT30990018 to SB. KMP is supported by AHA postdoctoral fellowship (15POST25360014). JDS is supported by grants 16GRNT31050004 from AHA and 1R03AGO52848-01A1 from the National Institute on Aging. EDA is supported by grants R01DK092065 and R01HL108379 from the NIDDK and the National Heart, Lung, and Blood Institute (NHLBI) respectively. CHS is supported by grant R01HL089592-02 from NHLBI. We would like to thank Mrs. Diana Lim for her assistance in the preparation of the figures. Dr. Sihem Boudina is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors disclose no conflict of interest.
References
- 1.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 2.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 3.Riehle C, Abel ED. Insulin regulation of myocardial autophagy. Circulation journal: official journal of the Japanese Circulation Society. 2014;78:2569–76. doi: 10.1253/circj.cj-14-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 5.Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–65. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. 2007;13:482–91. doi: 10.1016/j.molmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 8.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–6. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 9.Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–8. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 11.Meijer AJ, Codogno P. Macroautophagy: protector in the diabetes drama? Autophagy. 2007;3:523–6. doi: 10.4161/auto.4449. [DOI] [PubMed] [Google Scholar]
- 12.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–8. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–92. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delbridge LM, Mellor KM, Taylor DJ, Gottlieb RA. Myocardial autophagic energy stress responses–macroautophagy, mitophagy, and glycophagy. American journal of physiology Heart and circulatory physiology. 2015;308:H1194–204. doi: 10.1152/ajpheart.00002.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–5. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–46. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta. 2013;1832:1136–48. doi: 10.1016/j.bbadis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French CJ, Tarikuz Zaman A, McElroy-Yaggy KL, Neimane DK, Sobel BE. Absence of altered autophagy after myocardial ischemia in diabetic compared with nondiabetic mice. Coron Artery Dis. 2011;22:479–83. doi: 10.1097/MCA.0b013e32834a3a71. [DOI] [PubMed] [Google Scholar]
- 19.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50:1035–43. doi: 10.1016/j.yjmcc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, et al. Recipes for creating animal models of diabetic cardiovascular disease. Circulation research. 2007;100:1415–27. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 21.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–74. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 22.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. European heart journal. 2010;31:100–11. doi: 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–39. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–43. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–5. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 26.Di Paola R, Crisafulli C, Mazzon E, Genovese T, Paterniti I, Bramanti P, et al. Effect of PD98059, a selective MAPK3/MAPK1 inhibitor, on acute lung injury in mice. International journal of immunopathology and pharmacology. 2009;22:937–50. doi: 10.1177/039463200902200409. [DOI] [PubMed] [Google Scholar]
- 27.McKellar SH, Javan H, Bowen ME, Liu X, Schaaf CL, Briggs CM, et al. Animal model of reversible, right ventricular failure. The Journal of surgical research. 2015;194:327–33. doi: 10.1016/j.jss.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 29.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Molecular biology of the cell. 2008;19:2092–100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Developmental cell. 2004;7:167–78. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–6. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 32.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 1977;270:174–6. doi: 10.1038/270174a0. [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 35.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energypreserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93:320–9. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest. 2013;123:5319–33. doi: 10.1172/JCI71171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. Journal of molecular cell biology. 2013;5:61–3. doi: 10.1093/jmcb/mjs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, et al. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. Journal of lipid research. 2015;56:546–61. doi: 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11:1146–60. doi: 10.1080/15548627.2015.1051295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andres AM, Kooren JA, Parker SJ, Tucker KC, Ravindran N, Ito BR, et al. Discordant signaling and autophagy response to fasting in hearts of obese mice: Implications for ischemia tolerance. American journal of physiology Heart and circulatory physiology. 2016;311:H219–28. doi: 10.1152/ajpheart.00041.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikeda H, Shiojima I, Ozasa Y, Yoshida M, Holzenberger M, Kahn CR, et al. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J Mol Cell Cardiol. 2009;47:664–75. doi: 10.1016/j.yjmcc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soos MA, Field CE, Siddle K. Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I, but not insulin, with high affinity. The Biochemical journal. 1993;290(Pt 2):419–26. doi: 10.1042/bj2900419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rolfe M, McLeod LE, Pratt PF, Proud CG. Activation of protein synthesis in cardio myocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2) The Biochemical journal. 2005;388:973–84. doi: 10.1042/BJ20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 45.Saraiva RM, Minhas KM, Zheng M, Pitz E, Treuer A, Gonzalez D, et al. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric oxide: biology and chemistry. 2007;16:331–8. doi: 10.1016/j.niox.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N, Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–46. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Cui T. Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy. American journal of physiology Heart and circulatory physiology. 2017;313:H304–H19. doi: 10.1152/ajpheart.00145.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. 2004;279:4782–93. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 49.Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, et al. The conserved phosphoinositi de 3-kinase pathway determines heart size in mice. The EMBO journal. 2000;19:2537–48. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wende AR, O’Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, et al. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Molecular and cellular biology. 2015;35:831–46. doi: 10.1128/MCB.01109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. The EMBO journal. 2000;19:6341–50. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108:754–9. doi: 10.1161/01.CIR.0000083716.82622.FD. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.