

Abstract
Background
Ribosomal proteins are encoded in all genomes of cellular life forms and are, generally, well conserved during evolution. In prokaryotes, the genes for most ribosomal proteins are clustered in several highly conserved operons, which ensures efficient co-regulation of their expression. Duplications of ribosomal-protein genes are infrequent, and given their coordinated expression and functioning, it is generally assumed that ribosomal-protein genes are unlikely to undergo horizontal transfer. However, with the accumulation of numerous complete genome sequences of prokaryotes, several paralogous pairs of ribosomal protein genes have been identified. Here we analyze all such cases and attempt to reconstruct the evolutionary history of these ribosomal proteins.
Results
Complete bacterial genomes were searched for duplications of ribosomal proteins. Ribosomal proteins L36, L33, L31, S14 are each duplicated in several bacterial genomes and ribosomal proteins L11, L28, L7/L12, S1, S15, S18 are so far duplicated in only one genome each. Sequence analysis of the four ribosomal proteins, for which paralogs were detected in several genomes, two of the ribosomal proteins duplicated in one genome (L28 and S18), and the ribosomal protein L32 showed that each of them comes in two distinct versions. One form contains a predicted metal-binding Zn-ribbon that consists of four conserved cysteines (in some cases replaced by histidines), whereas, in the second form, these metal-chelating residues are completely or partially replaced. Typically, genomes containing paralogous genes for these ribosomal proteins encode both versions, designated C+ and C-, respectively. Analysis of phylogenetic trees for these seven ribosomal proteins, combined with comparison of genomic contexts for the respective genes, indicates that in most, if not all cases, their evolution involved a duplication of the ancestral C+ form early in bacterial evolution, with subsequent alternative loss of the C+ and C- forms in different lineages. Additionally, evidence was obtained for a role of horizontal gene transfer in the evolution of these ribosomal proteins, with multiple cases of gene displacement 'in situ', that is, without a change of the gene order in the recipient genome.
Conclusions
A more complex picture of evolution of bacterial ribosomal proteins than previously suspected is emerging from these results, with major contributions of lineage-specific gene loss and horizontal gene transfer. The recurrent theme of emergence and disruption of Zn-ribbons in bacterial ribosomal proteins awaits a functional interpretation.
Background
The core structure and functions of the ribosome, the molecular machine for protein biosynthesis [1,2,3], have been fixed at a very early stage of evolution and apparently were already in place in the last common ancestor (LCA) of all extant cells [4]. This notion is amply supported by the conservation of the sequences of ribosomal RNAs (rRNA) and many ribosomal proteins (r-proteins), along with those of other central components of the translation machinery, in all three superkingdoms of life - Bacteria, Archaea and Eukarya [5,6]. Moreover, in bacteria and archaea, there is also notable conservation of the organization of genes coding for rRNA and r-proteins. Indeed, the r-protein superoperon that includes genes for a varying, but typically large, set of r-proteins is the most conserved gene array in prokaryotic genomes [7,8,9,10].
Genome comparisons have shown that horizontal gene transfer (HGT) is much more common than previously suspected and permeates not only 'operational' genes, but also 'informational' genes [11], including some components of the translation system, for example aminoacyl-tRNA syn-thetases [12,13,14]. Therefore, the issue of the existence and identity of a stable core of prokaryotic genomes that is (practically) free from HGT has become particularly pertinent. Given that rRNA and r-proteins function as a tightly coordinated complex and that the order of the corresponding genes in prokaryotic genomes is partially conserved, it is generally assumed that genes for r-proteins are not subject to HGT or, at least, that horizontal transfer of these genes is rare [6]. Accordingly, rRNA and, to a lesser extent, r-protein sequences have been routinely used as phylogenetic markers [15,16,17]. Individually, most of the r-proteins are small and highly conserved and therefore do not provide particularly suitable material for phylogenetic analysis. However, attempts to construct phylogenetic trees by using a concatenated alignment of multiple r-protein genes resulted in topologies that were generally compatible with the topology of the rRNA tree, which supported the notion that, among r-protein genes, HGT is not common [6]. Paralogy is generally not characteristic of r-protein genes either; most prokaryotic genomes have only one gene for each r-protein. There are, however, several exceptions to this trend, and a recent phylogenetic study on the r-protein S14, which is duplicated in several bacterial genomes, revealed an unexpected tree topology that could be explained only by a combination of HGT and differential gene loss (DGL) "at the heart of the ribosome" [18].
We sought to systematically analyze all cases of duplication of r-protein genes in completely sequenced prokaryotic genomes, with the aim of reconstructing their evolutionary history and, in particular, assessing the contributions of HGT and DGL. We found that DGL following gene duplication probably had the dominant role in shaping the evolutionary patterns of these r-protein genes, but many instances of probable HGT were also identified. In addition, we observed an unexpected phenomenon of consistent disruption of Zn-ribbon modules in r-proteins that have undergone gene duplication.
Results and discussion
Duplications of r-protein genes: C+ and C- versions
To identify duplications of r-protein genes, we checked the clusters of orthologous groups (COGs) [19] for all 54 ribosomal proteins of the large and small ribosomal subunits on the case-by-case basis. Four r-proteins (L31, L33, L36, S14) are duplicated in several bacterial genomes and six proteins (L11, L28, L7/L12, S1, S15, S18) are so far duplicated in only one genome each (Table 1). The latter six cases appeared to be recent, lineage-specific duplications [20], without indications of any unusual origin of the duplicates such as HGT.
Table 1.
Paralogous genes for ribosomal proteins in bacterial genomes
| r-protein | Genomes containing paralogs | Zn-ribbon present in some forms |
| L36 | Pseudomonas aeruginosa, Vibrio cholerae, Neisseria meningitidis | Yes |
| L31 | Escherichia coli, Pseudomonas aeruginosa, Vibrio cholerae, Neisseria meningitidis, Bacillus | Yes |
| L33 | Bacillus subtilis, Lactococcus lactis, Mycoplasma pneumoniae, Mycoplasma genitalium, Ureaplasma urealyticum | Yes |
| S14 | Bacillus subtilis, Lactococcus lactis, Streptococcus pyogenes, Mycobacterium tuberculosis | Yes |
| S18 | Mycobacterium tuberculosis | Yes |
| L28 | Mycobacterium tuberculosis, Streptomyces coelicolor | Yes |
| S1 | Synechocystis sp. | No |
| S15 | Haemophilus influenzae | No |
| L11 | Bacillus halodurans | No |
| L7/L12 | Synechocystis sp. | No |
In contrast, the paralogous pairs of the former four r-proteins showed considerable divergence, with each of the paralogs showing much greater sequence similarity to the corresponding r-proteins from other species. This observation suggested that each of these duplications occurred on only one occasion during evolution, with the extant distribution of the duplicates resulting from a combination of HGT and DGL. To gain insight into the evolutionary trajectories of these r-proteins, we examined their multiple alignments and the genomic context of their genes, and performed phylogenetic analyses for each of them. Surprisingly, we observed the same distinctive pattern of amino acid variation for all these four r-proteins. Each of them comes in two types, the first type containing a pattern of two pairs of conserved cysteines (one of which is replaced by a histidine in some of the L36 sequences), and the second type, in which this pattern is completely or partially eliminated by substitution of the cysteines with amino acids that cannot chelate metal cations (Figures 1,2,3,4). We designated these two types of r-proteins C+ and C-, respectively. Typically, when two paralogous genes for an r-protein gene were present in a bacterial genome, one of the two versions was of the C+ variety and the other of the C-variety (Tables 1,2).
Figure 1.
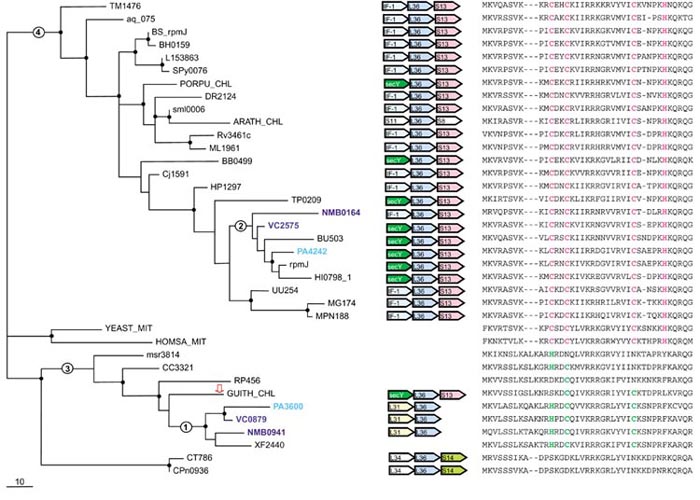
Phylogenetic tree, conserved gene context and multiple alignment of L36 ribosomal proteins. A maximum-likelihood unrooted tree was built using the MOLPHY program. The same program was used to compute bootstrap probabilities. Those branches that were supported by bootstrap probability greater than 70% are marked by small black circles. Gene names of organisms that have duplications of this protein are highlighted by different colors. The red-outlined arrowindicates a protein that probably has been subject to HGT (see text). Those branches whose alternative placements were assessed using the RELL method are indicated by circles with numbers (see Table 3). The scale bar (10) indicates the number of substitutions per 100 sites. Conserved genes in the neighborhood of the L36 gene are shown by colored arrows (center of figure). White arrows indicate adjacent genes that encode translation-system proteins but whose context is not conserved in genomes of distant species. Orthologous genes are shown in the same color. Genes are denoted by systematic names adopted for the respective genomes; a key is given in Table 2. Gene name abbreviations: IF-1, translation initiation factor IF-1; secY, preprotein translocase subunit SecY; L36, S13, S11, S3, S14, L31, L34, ribosomal proteins. A partial multiple alignment of L36 protein sequences is shown on the right (the complete multiple alignment used for the tree construction contained 41 positions); cysteines and histidines of the Zn-ribbon are shown in magenta. Remnants of this motif in sequences that do not have all four conserved residues of the Zn-ribbon are shown in green.
Figure 2.
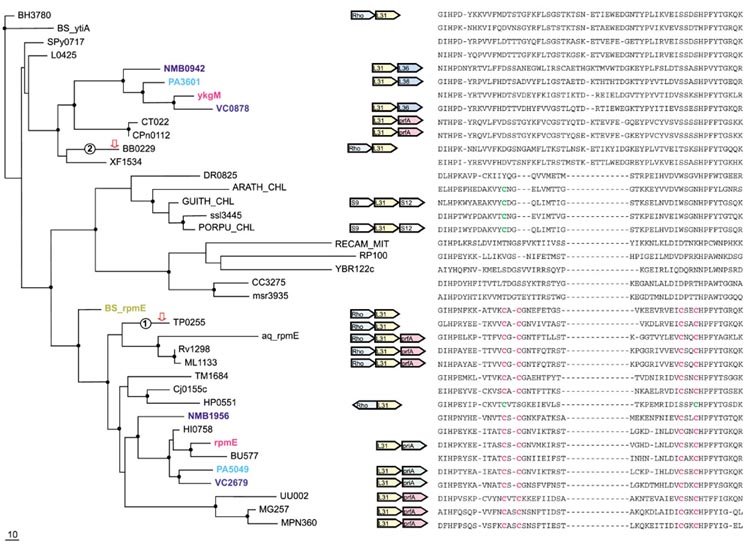
Phylogenetic tree, conserved gene context and multiple alignment of L31 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: Rho, transcription termination factor Rho; prfA, peptide chain release factor 1; priA, primosomal protein N'; L31, S9, S12, L36, ribosomal proteins. A partial multiple alignment of L31 protein sequences is shown on the right (the complete multiple alignment used for the tree construction contained 96 positions).
Figure 3.
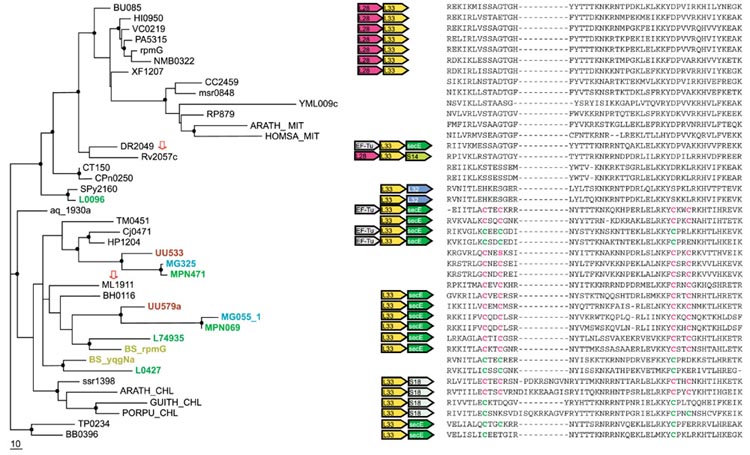
Phylogenetic tree, conserved gene context and multiple alignment of L33 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: EF-Tu, elongation factor EF-Tu; secE, preprotein translocase secE subunit; L28, L33, S14, S18, ribosomal proteins. A partial multiple alignment of L33 protein sequences is shown on the right (the complete multiple alignment used for tree construction contained 58 positions). The sequence of UU579a was translated from the complete genome sequence of Ureaplasma urealyticum using the TBLASTN program [44].
Figure 4.
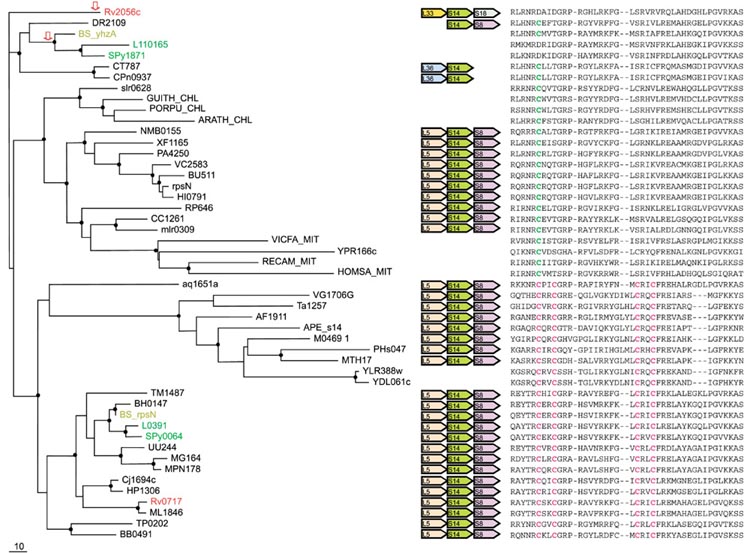
Phylogenetic tree, conserved gene context and multiple alignment of S14 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: S18, S14, S8, L5, L36, L33, ribosomal proteins. A partial multiple alignment of S14 protein sequences is shown on the right (the complete multiple alignment used for tree construction contained 103 positions). Mitochondrial S14 protein from Vicia faba (GI: 134068) was included in the tree instead of a sequence from Arabidopsis, in which it was not detected. The sequence APE_s14 was translated from the complete genome sequence of Aeropyrum pernix using the TBLASTN program [44].
Table 2.
Distributions of Zn-ribbons in seven ribosomal proteins in sequenced bacterial and organellar genomes
| Species | Prefix used in gene names | Taxon | L36 | L33 | L31 | L32 | L28 | S14* | S18 |
| C+, C- or both (B) forms | |||||||||
| Escherichia coli | None (genes designated With systematic Four-letter names) | Gamma-proteobacteria | + | - | B | - | - | - | - |
| Buchnera sp. | BU | + | - | - | - | - | - | - | |
| Haemophilus influenzae | HI | + | - | - | - | - | - | - | |
| Pseudomonas aeruginosa | PA | B | - | B | - | - | - | - | |
| Vibrio cholerae | VC | B | - | B | - | - | - | - | |
| Xylella fastidiosa | XF | - | - | - | - | - | - | - | |
| Neisseria meningitidis | NM | Beta-proteobacteria | B | - | B | - | - | - | - |
| Helicobacter pylori | HP | Epsilon-proteobacteria | + | - | + | - | - | + | - |
| Campylobacter jejuni | Cj | + | - | + | - | - | + | - | |
| Caulobacter crescentus | CC | Alpha-proteobacteria | - | - | - | - | - | - | - |
| Mesorhizobium loti | Msr, mlr | - | - | - | - | - | - | - | |
| Rickettsia prowazekii | RP | - | - | - | - | - | - | - | |
| Bacillus subtilis† | BS | Gram-positive bacteria, | + | - | B | + | - | B | - |
| Bacillus halodurans† | BH | Bacillus-Clostridium group | + | + | - | + | - | + | - |
| Lactococcus lactis | L | + | B | - | - | - | B | - | |
| Streptococcus pyogenes | SPy | + | - | - | + | - | B | - | |
| Mycoplasma pneumoniae | MPN | + | + | + | + | - | + | - | |
| Mycoplasma genitalium | MG | + | + | + | + | - | + | - | |
| Ureaplasma urealyticum | UU | + | + | + | + | - | + | - | |
| Mycobacterium tuberculosis | Rv | Actinomycetales | + | + | + | - | - | B | B |
| Mycobacterium leprae | ML | + | - | + | - | - | + | + | |
| Aquifex aeolicus | Aq_ | Aquifecales | + | + | + | + | - | + | + |
| Thermotoga maritima | TM | Thermotogales | + | + | + | + | + | + | + |
| Deinococcus radiodurans | DR | Thermus-Deinococcus group | + | - | - | + | - | - | - |
| Thermus thermophilus | Not included in trees | + | + | NA† | + | NA | + | - | |
| Treponema pallidum | TP | Spirochaetales | + | - | + | + | + | + | + |
| Borrelia burgdorferi | BB | + | - | - | + | - | + | - | |
| Chlamydophyla pneumoniae | CPn | Chlamydiales | - | - | - | + | - | - | - |
| Chlamydia trachomatis | CT | - | - | - | + | - | - | - | |
| Synechocystis PCC6803 | sml, ssr | Cyanobacteria | + | + | - | - | - | - | - |
| Arabidopsis thaliana, chloroplast | None, see Methods and materials | Eukaryota, Viridiplantae | + | + | - | - | - | - | - |
| Guillardia theta, chloroplast | None, see Methods and materials | Eukaryota, Cryptophyta | - | - | - | - | - | - | - |
| Porphyra purpurea, chloroplast | None, see Methods and materials | Eukaryota, Rhodophyta | + | - | - | - | - | - | - |
| Reclinomonas americana, mitochondrion | None, see Methods and materials | Eukaryota, core jakobids | NA | NA | - | + | - | - | NA |
| Homo sapiens, mitochondrion | None, see Methods and materials | Eukaryota, Chordata | + | - | NA | + | - | - | - |
| Saccharomyces cerevisiae, mitochondrion | None, see Methods and materials | Eukaryota, Fungi | + | - | - | + | - | - | - |
| Arabidopsis thaliana, mitochondrion | None, see Methods and materials | Eukaryota, Viridiplantae | NA | - | NA | NA | - | -* | - |
*S14 was not detected among the available protein sequences from Arabidopsis and the sequence from Vicia faba was used in all analyses (Figure 4). †NA, sequence not available.
In light of these unexpected findings, we examined all the remaining multiple alignments of r-proteins (regardless of the existence of paralogs) for the possible presence of the C+/- pattern. Three additional C+/- r-proteins, namely S18, L32, and L28 (Figures 5,6,7), were identified. An apparent lineage-specific duplication of S18 was detected in Mycobacterium tuberculosis and, as now could have been predicted, one of the paralogs was of the C+ variety, whereas the other one was C- (Figure 5). Only L28 is an exception, in that the lineage-specific duplication, also in M. tuberculosis, involves two C- proteins (Figure 6; however, see discussion below). Phylogenetic analysis was undertaken for all seven r-proteins that display the C+/- pattern, in combination with comparison of their genomic context. When attempting to infer evolutionary scenarios from this data, we assumed that the presence of a Zn-ribbon was the ancestral state of each of these r-proteins and only disruption, but not convergent emergence of the Zn-ribbon, occurred during their evolution. These assumptions appear to be justified because, in almost all cases, different stages in the disruption of the Zn-ribbon were detected, from replacement of only one cysteine residue to complete elimination (Figures 1,2,3,4,5,6,7). All r-protein sequences are short, which often renders the results of phylogenetic analysis inconclusive. Therefore, whenever possible, we sought not to rely in our analysis on phylogenetic tree topology alone, but to integrate the information from the trees, shared derived characters (synapomorphies) identified in multiple sequence alignments, and genomic context (gene order).
Figure 5.
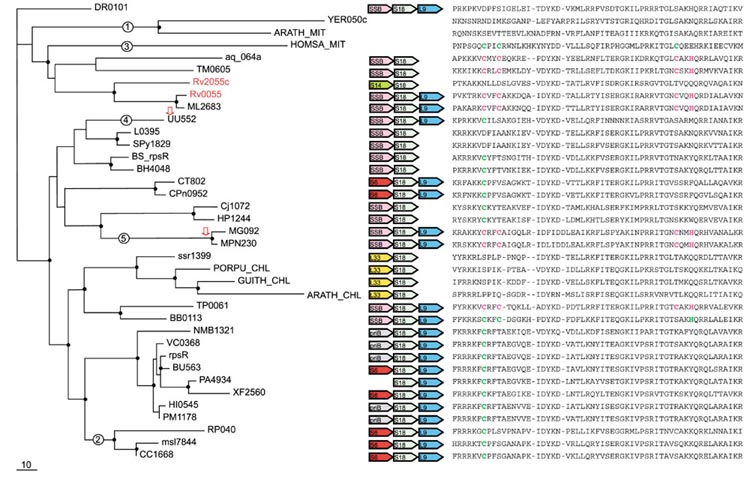
Phylogenetic tree, conserved gene context and multiple alignment of S18 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: SSB, single-strand-binding protein; priB, primosomal protein N; S18, S14, S6, L9, L33, ribosomal proteins. A partial multiple alignment of S18 protein sequences is shown on the right (the complete multiple alignment used for the tree construction contained 71 positions). The sequence ARATH_MIT was translated from the expressed sequence tag (EST) sequence (GI: 12083223).
Figure 6.
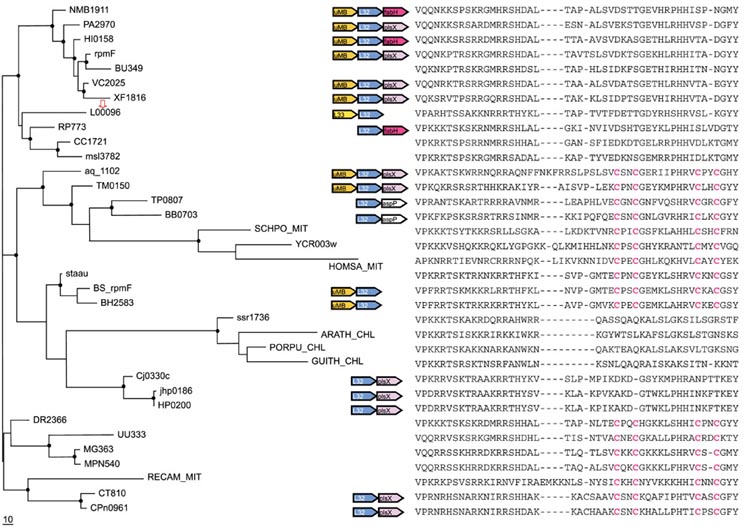
Phylogenetic tree, conserved gene context and multiple alignment of L32 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: fabH, 3-oxoacyl-(acyl-carrier protein) synthase; plsX, fatty acid/phospholipid synthesis protein; aspP, acyl carrier protein; uMB, predicted metal-binding, possibly nucleic acid-binding protein (COG1399); L32, L33, ribosomal proteins. A partial multiple alignment of L32 protein sequences is shown on the right (the complete multiple alignment used for the tree reconstruction had 58 positions). Mitochondrial L32 protein from Schizosaccharomyces pombe (GI:7493310) was additionally included in the tree. The L32 protein from Staphylococcus aureus (GI: 13700928) was included in the tree instead of the sequence from S. pyogenes (SPy2159), which appears to be truncated.
Figure 7.
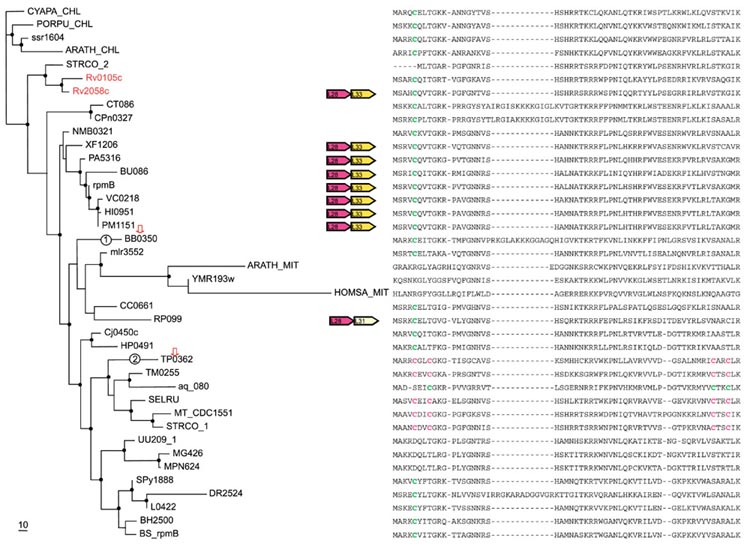
Phylogenetic tree, conserved gene context and multiple alignment of L28 ribosomal proteins. Designations are as in Figure 1. Gene name abbreviations: L28, L31, L33, ribosomal proteins. A partial multiple alignment of L28 protein sequences is shown on the right (the complete multiple alignment used for the tree construction contained 71 positions).
L36 (RpmJ)
The maximum likelihood phylogenetic tree, the multiple alignment, and the conserved genomic contexts for the r-protein L36 are shown in Figure 1. This is a small protein with only 41 aligned positions. Nevertheless, three major branches of the L36 tree are strongly supported by bootstrap analysis, the large C+ cluster and two smaller C- clusters (Figure 1). The C+ L36 sequences contain a 'CXXC..CXXXH' motif that forms a metal-binding Zn-ribbon as shown by NMR analysis [21] of this protein. The C+ cluster includes most of the bacterial sequences as well as sequences from chloroplasts and mitochondria. With the sole exception of the Arabidopsis chloroplast, all genomes that encode a C+ L36 protein contain the conserved gene pair L36-S13 preceded by either the secY gene or the IF-1 gene (Figure 1). This partial conservation of the genomic context further supports the monophyly of the C+ cluster. The first, larger C-cluster consists mostly of proteobacterial proteins. Proteins of this cluster retain from one to three residues of the Zn-ribbon and also contain a distinct three-residue insert, a synapomorphy that supports the monophyly of this cluster (Figure 1). The L31-L36 gene pair is present in three proteobacterial species with this type of L36, whereas in other proteobacteria, the gene for the C- L36 is not adjacent to any r-protein genes. Unexpectedly, the Guillardia chloroplast genome contains the SecY-(C-)L36-S13 triad characteristic of the C+ cluster. The second C- group so far includes only chlamydial proteins and is characterized by complete elimination of the Zn-ribbon residues and a one-residue insert compared to the C+ cluster.
Three proteobacteria (Neisseria meningitidis, Pseudomonas aeruginosa, Vibrio cholerae) encode both C+ and C- forms of L36. Given the presence of the C- form of L36 in all three subdivisions of proteobacteria and the presence of paralogs in beta and gamma subdivisions, it appears that the duplication of the L36 that gave rise to the two forms occurred, at the latest, at the onset of proteobacterial divergence. A comparison of the likelihoods of different tree topologies using the RELL method (see Materials and methods) suggests that the duplication occurred even earlier, prior to the divergence of the main bacterial lineages, because the likelihoods of the topologies supporting the monophyly of the two proteobacterial clusters in the L36 tree (2 and 3 in Figure 1) was found to be low (Table 3). The possibility remains that the duplication dates back only to the divergence of proteobacteria, but was followed by a major acceleration of evolution, particularly in the C- cluster. However, this interpretation does not seem to be supported by the relatively short branch lengths in this part of the tree.
Table 3.
Log-likelihood analysis of possible placements of selected branches of maximum likelihood trees for the analyzed ribosomal proteins
| Tree* | Difference in log-likelihood† | Standard error‡ | RELL-BP§ |
| L36 original | 0.0 | NA | 0.7797 |
| 1→2 | -45.7 | 11.8 | 0.0000 |
| 2→3 | -29.1 | 10.9 | 0.0024 |
| 2→1 | -42.7 | 10.9 | 0.0000 |
| 3→4 | -3.1 | 3.6 | 0.2179 |
| L31 original | 0.0 | NA | 0.9949 |
| 1→2 | -36.7 | 12.6 | 0.0007 |
| 2→1 | -36.6 | 14.6 | 0.0044 |
| S18 original | 0.0 | NA | 0.9594 |
| 1→2 | -6.7 | 3.9 | 0.0327 |
| 2→1 | -20.3 | 8.9 | 0.0027 |
| 3→2 | -16.7 | 7.1 | 0.0052 |
| S18 original | 0.0 | NA | 0.8344 |
| 4→5 | -7.1 | 6.1 | 0.0838 |
| 5→4 | -6.7 | 5.8 | 0.0818 |
| L28 original | 0.0 | NA | 0.9550 |
| 1→2 | -18.9 | 10.5 | 0.0331 |
| 2→1 | -19.3 | 9.4 | 0.0119 |
*The numbers refer to local rearrangements of the tree as indicated on the corresponding figures. †Difference of the log-likelihoods relative to the best tree. ‡ Standard error of difference in log-likelihood. §Bootstrap probability (BP) of the given tree calculated using the RELL method (resampling of estimated log-likelihoods) [44]). NA, standard error estimate is not applicable for the aximum likelihood tree.
Regardless of the exact position of the duplication in the tree, multiple, alternative losses of the C+ and C- forms of L36 seem to have occurred during bacterial evolution. In particular, all alpha-proteobacteria have the C- L36, whereas proteins from mitochondria, which evolved from alpha-proteobacteria [22], have the C+ form. Probably the ancestor of mitochondria encoded both forms of L36, with C- form lost in ancient mitochondria and C+ form lost in alpha-proteobacteria after their divergence from the mitochondrial ancestor.
An enigmatic observation is the presence of a proteobacterial-type C- L36 in the genomic context characteristic of the C+ cluster (including the chloroplast of the red alga Porphyra purpurea) in the Guillardia theta chloroplast genome (Figure 1). Given the presence of the C+ form of L36 in all other sequenced chloroplast genomes and in cyanobacteria, it appears practically certain that the ancestor of chloroplasts had a C+ L36 in the SecY-L36-S13 context. If that is the case, the ancestral L36 gene was probably displaced 'in situ', without a change of the genomic context, by a C- L36 gene that was introduced into the Guillardia chloroplast via horizontal transfer, probably from mitochondria.
L31 (RpmE)
The gene for the r-protein L31 is also duplicated in some proteobacteria and in Bacillus subtilis, all of which have both the C+ and the C- forms (Figure 2, Table 2). Similarly to the L36 case, the tree consists of three major branches, one of which includes C+ forms and the other two consisting of C- forms. In the L31 tree, the two C- clusters appear to form distinct clades, one of which includes proteobacteria, several species of Gram-positive bacteria, Chlamydia and the spirochete Borrelia burgdorferi, and is supported by a clear-cut synapomorphy, an 11-13 amino-acid insert (Figure 2). Thus, in the case of L31, the loss of the Zn-ribbon appears to be polyphyletic, with the C- form in cyanobacteria-chloroplasts, alpha-proteobacteria, and Deinococcus probably derived from the C+ form independently (Figure 2). The species pattern in this secondary C- cluster is difficult to explain without postulating at least two HGT events, one between cyanobacteria and alpha-proteobacteria, and another one between one of these lineages (most likely, cyanobacteria) and Deinococcus.
The most likely evolutionary scenario for L31 involves an ancient duplication antedating the divergence of the major bacterial lineages followed by multiple losses. However, as with L36, a duplication at the base of proteobacterial evolution followed by horizontal acquisition of the C- form by B. subtilis could not be ruled out. In addition to the probable HGT in the secondary C- cluster, two independent cases of 'gene displacement in situ' seem to have occurred during evolution of L31. The first case involves the two spirochetes, Treponema pallidum and B. burgdorferi, that have the same gene context, Rho-L31, but differ in that B. burgdorferi has the C- form as opposed to the C+ form in T. pallidum. The different positions of the two spirochetes in the L31 tree are convincingly supported by sequence synapomorphies, bootstrap values, and the RELL analysis (Figure 2, Table 3). Furthermore, a phylogenetic tree for the Rho protein unequivocally supports the expected clustering of the spirochetes (data not shown) ruling out HGT of an entire operon. Thus, displacement in situ of the C+ form of L31 in B. burgdorferi by a proteobacteria-type C- form seems to be the most plausible explanation for the observed evolutionary pattern. A similar displacement appears to have taken place in B. halodurans compared to B. subtilis (Figure 2).
L33 (RpmG)
Evolution of the r-protein L33 seems to follow the same scenario, with an early duplication and elimination of the Zn-ribbon in one of the paralogs, with subsequent differential gene loss. This model is supported by the tree topology and sequence synapomorphies, and also by conserved operon organization, which is different for the C+ and C- forms (Figure 3). A notable aspect of the evolution of L33 is the probable secondary duplication(s) in Gram-positive bacteria leading to the presence of paralogous C+ forms in several genomes from this lineage (Figure 3). An interesting case in point is Lactococcus lactis, which has three paralogous L33 genes, one of which is C- and apparently the product of the postulated ancient duplication, whereas the other two are of the C+ variety and presumably originate from the secondary duplication. In Ureaplasma urealyticum, B. subtilis and L. lactis, the apparent secondary duplication was followed by incipient disruption of the Zn-ribbon. However, an alternative explanation of the pattern of C+ L33 distribution in Gram-positive bacteria could involve HGT - for example, acquisition of the gene from epsilon-proteobacteria by the mycoplasmal lineage (Figure 3). The direction of possible HGT in this case is suggested by the fact that, in epsilon-proteobacteria, the L33 gene is in the characteristic, conserved context, whereas no such context is seen in the mycoplasmas (Figure 3).
As with other C+/- r-proteins, isolated occasions of probable HGT were detected for L33. In particular, Deinococcus radiodurans encodes a C- protein, but has genomic context (EF-Tu, L33, secE) identical to that in Aquifex aeolicus, Thermotoga maritima, and epsilon-proteobacteria, which all encode the C+ version of L33 (Figure 3). The phylogenetic tree for the SecE protein showed statistically supported clustering of D. radiodurans with A. aeolicus and epsilon-proteobacteria (data not shown), in agreement with the identical genomic context. Thus, displacement in situ appears to be the best explanation for the presence of the C- form of L33 in D. radiodurans.
A rare case of probable xenologous displacement of the C- form with the C+ version is seen in M. leprae when compared to M. tuberculosis (Figure 3). Among all r-proteins, L33 is the only case when the two mycobacteria do not group together in phylogenetic trees (Figures 1,2,3,4,5,6,7 and data not shown). The ancestral mycobacterium most likely encoded the C- form because M. tuberculosis has the L28-L33 gene pair typical of the genomes that encode the C- version of L33, whereas M. leprae does not have any conserved context around the L33 gene (Figure 3). Moreover, in the tree for the L28 protein, the two mycobacteria confidently group together (see below). Thus, at a relatively recent stage of evolution, after the divergence from M. tuberculosis, M. leprae probably acquired a C+ form of L33 by HGT (possibly from Gram-positive bacteria), with subsequent elimination of the ancestral C- form.
S14 (RpsN)
The main aspects of the evolution of r-protein S14 were described by Brochier and colleagues [18]. However, the relationship between the C+ and C- forms is not considered in their work. Unlike the other C+/C- r-proteins, S14 is universally present in ribosomes from all three superkingdoms, which presents unequivocal evidence that the C+ state is ancestral because this is the form found in archaea and eukaryotes (Figure 4). It has been shown that the cysteines in S14 are indeed involved in Zn-binding and the formation of a Zn-ribbon domain [23].
Paralogous C+ and C- versions of S14 are seen in B. subtilis, L. lactis, Streptococcus pyogenes, and M. tuberculosis (Figure 4). The phyletic distribution of the C+ and C- forms of S14 among bacteria closely resembles the distribution of the L33 forms (compare Figures 4 and 3), with the exception of the cyanobacteria/chloroplast lineage that, in the case of S14 belongs to the C- cluster. However, a distinctive feature of S14 is the conservation of the genomic context between proteobacteria, which have the C- form, and those bacteria and archaea that have the C+ version (Figure 4). Displacement in situ of the C+ form by the C- form in proteobacteria appears to be the most plausible explanation of this evolutionary pattern.
S18 (RpsR)
Paralogous genes for S18 were detected only in M. tuberculosis. Although the two M. tuberculosis proteins belong to the same branch, which indicates a lineage-specific duplication (probably occurring prior to the divergence of M. tuberculosis and M. leprae, with one of the paralogs lost in the latter), the Rv2055 protein is of the C+ type, whereas Rv2055 is of the C- type (Figure 5). Thus, it appears that, over a comparatively short evolutionary span, all metal-chelating amino acids in the latter protein have been substituted (Figure 5). In the case of S18, the C- form is a strong majority, with the C+ forms scattered around the tree (Figure 5). The leitmotif of evolution of this r-protein seems to be independent disruption of Zn-ribbons on many occasions. At face value, there seems to be no evidence of an ancient duplication. However, the alpha-proteobacterial and mitochondrial branches do not cluster together in the S18 tree (Figure 5), which is fully supported by the RELL analysis (Table 3). An early duplication, with subsequent differential gene loss, seems to be the best explanation for this tree topology as discussed above for L36, but in the case of S18, this scenario is confounded by the apparent secondary loss of the Zn-ribbon in the mitochondrial proteins (Figure 5). This chain of events is supported by the varying degree of the Zn-ribbon disruption in the mitochondria of different eukaryotes, with only one cysteine lost in humans, but all of them eliminated in yeasts and Arabidopsis (Figure 5).
In addition, a clear-cut case of HGT was detected that involves three closely related bacterial species of the order Mycoplasmatales - U. urealyticum and two mycoplasmas. The mycoplasmas have a C+ form of S18, which forms an unexpected but strongly supported cluster with the C- proteins from epsilon-proteobacteria, whereas U. urealyticum has a C- form, which belongs to the cluster of Gram-positive bacteria, as generally expected of the mycoplasmas (Figure 5). The RELL test supported the respective positions of the U. urealyticum and the mycoplasmas in the tree (Table 3). Given this topology, it seems most likely that, in the mycoplasmas, the C- form, which was probably present in the ancestor of the Gram-positive bacteria, was replaced with a C+ version, possibly of proteobacterial origin (Figure 5). As with other r-proteins, the displacement seems to have occurred in situ, without a change in the operon structure (Figure 5).
L32 (RpmF)
The emerging picture of evolution of L32 resembles, in several respects, that for S18. None of the available genomes encodes paralogous forms of L32, but the separation of alpha-proteobacteria and mitochondria (Figure 6), again, is most compatible with an ancient duplication-differential loss scenario. In this case, all mitochondrial L32-proteins are C+ forms, but the mitochondrial protein from Reclinomonas americana does not cluster with those from crown-group eukaryotes. It appears that, in one of these mitochondrial lineages, the original L32 gene has been displaced by that from a different bacterial lineage; with Reclinomonas being an early-branching eukaryote, it remains unclear which lineage has the ancestral version. The C- versions do not form a single clade in the L32 tree, which indicates that the Zn-ribbon might have been eliminated independently on at least two or three occasions during evolution (Figure 6).
A case of apparent xenologous gene displacement (displacement with an ortholog from a distant lineage [24]) is detectable among the Gram-positive bacteria. Most bacteria of this lineage encode a C+ L32 protein, but L. lactis has a C- form that falls within the proteobacterial clade (Figure 6). Displacement of the typical Gram-positive form of L32 with a C- proteobacterial form can be confidently inferred and, in this case, is accompanied by a change in genome context (Figure 6).
L28 (RpmB)
Paralogous forms of this r-protein are seen in two species of actinomycetes, M. tuberculosis and Streptomyces coelicolor. M. tuberculosis clearly has a lineage-specific duplication of the C- form: in contrast, S. coelicolor has distinct C- and C+ forms and a closely related C+ form was detected also in Mycobacterium CDC1551 (Figure 7). The proteins from alpha-proteobacteria and mitochondria form a distinct clade of C- forms in the L28 tree. Notably, the two spirochetes, B. burgdorferi and T. pallidum, have, respectively, a C- form and a C+ form which belong to different clusters as supported by the RELL analysis (Table 3). The C+ forms of L28 comprise a small cluster, which includes representatives of diverse bacterial lineages (Figure 7). Thus, the most likely scenario for the evolution of this r-protein seems to involve several independent disruptions of the Zn-ribbon, followed by HGT in spirochetes and actinomycetes, with or without displacement of the original L28 gene, respectively. The less plausible possibility is an ancient duplication, a single disruption of the Zn-ribbon, and the loss of the C+ form in most lineages, and of the C- form in a few.
Conclusions
Recent comparisons of prokaryotic genomes revealed a more dynamic picture of evolution than previously envisaged, with major contributions from horizontal gene transfer and differential gene loss. However, information processing systems in general, and the translation system in particular, are considered to be much less prone to these evolutionary processes than metabolic and signal transduction systems [25]. To a considerable extent, these notions are supported by phylogenetic analysis of several components of the translation and transcription systems that typically follow the "standard model" of evolution [12], with the first major bifurcation separating the bacterial and the archaeo-eukaryotic branches, and representatives of the major branches of bacteria and archaea forming coherent clusters [26,27]. However, notable deviations from this pattern were detected in phylogenetic analyses of aminoacyl-tRNA synthetases and some translation factors [12,13,14].
In the case of r-proteins, which function together as parts of a complex molecular machine, HGT and DGL a priori might seem to be particularly unlikely. This notion is supported by the lack of any indications of exchange of r-protein genes between bacteria and archaea, which probably reflects the major functional difference between bacterial and archaeal ribosomes. However, within the bacterial superkingdom, a different picture seems to be emerging. Phylogenetic analysis of S14 [18] first indicated, and the evolutionary study of six more r-proteins described here confirmed, that both HGT and DGL have been important in the evolution of bacterial r-proteins. The evolutionary patterns revealed by the present analysis appear to point to DGL as the major factor that has affected the evolution of r-proteins subsequent to ancient duplications, with HGT emerging, in each case, as an additional force resulting in a further increase in the complexity of evolutionary scenarios. In retrospect, the relatively common occurrence of HGT during evolution of r-protein genes might not be particularly surprising because compatibility of several r-proteins from distant species has been demonstrated in replacement experiments [28,29]. In particular, replacement of E. coli S18 (C+ type) with a C- form from chloroplast did not affect ribosome assembly and function [28].
The recurring pattern of early emergence and subsequent repeated disruption of Zn-ribbons in seven bacterial r-proteins and its connection to duplication are the most unexpected findings of this study. The correlation between the presence of a Zn-ribbon in a r-protein and gene duplication is indeed notable: Zn-ribbons were detected in seven bacterial r-proteins and, for six of these, gene duplication was also observed, of the total of ten duplicated r-proteins (Table 1). The persistence of this theme strongly suggests a common underlying teleology. Examination of the distribution of C+ and C- forms of r-protein among bacterial lineages does not reveal any strict regularity, but two trends deserve to be mentioned: first, thermophilic bacteria (Aquifex and Thermotoga) almost always have a C+ form (with the single exception of the partially disrupted Zn-ribbon in L28 from Aquifex); and second, alpha-proteobacteria always have a C- form (Table 2). Examination of the available r-protein sequences from another thermophile, Thermus thermophilus, also shows a preponderance of C+ forms of r-proteins, with the sole exception of S18. In particular, T. thermophilus has C+ forms of L33 and S14, in contrast to the C- form seen in its mesophilic relative, D. radiodurans (Table 2). Taken together, these observations seem to suggest that the dominance of C+ forms in bacterial thermophiles is adaptive and might contribute to the stability of the ribosome at high temperatures. Notably, seven archaeo-eukaryote-specific r-proteins of hyperthermophilic archaea - S27E, L34E, L24E, L37AE, L37E, L40E, L44E - contain Zn-ribbons and, in none of these cases, was the C+/- pattern observed (data not shown). Given that all C+/- r-protein, with the exception of S14, are bacteria-specific, it seems likely that, at a certain early stage in bacterial evolution, subsequent to the divergence from the archaeo-eukaryotic lineage, several Zn-ribbon proteins have been recruited for ribosome-associated functions. This might have been associated with a thermophilic stage in the early evolution of bacteria. The possible cause of complete elimination of the C+ forms of r-proteins from alpha-proteobacteria remains a mystery. The presence of both C+ and C- forms in mitochondria that have been derived from alpha-proteobacteria suggests that the exclusive loss of the C+ forms in the free-living bacteria of this lineage is due to some unknown environmental pressure.
Unique functions of paralogous r-proteins that are present in many bacteria (Table 1) are not understood. It seems possible that some of these proteins might assume functions distinct from their role in ribosome structure and translation [30], for example, regulation of the expression of the second paralog at the level of translation. Autogenous translation regulation by r-proteins is a well-known phenomenon [31,32] which, among the C+/- proteins, has been demonstrated for yeast S14 [33].
It seems to be potentially significant that in several bacteria two or more C+/- r-proteins form distinct operons, for example S18-S14-L33-L28 in M. tuberculosis, L36-L31 in proteobacteria, L36-S14 in chlamydia, L33-L32 in L. lactis and S. pyogenes, and L33-S18 in Synechocystis and chloroplasts (Figures 1,2,3,4,5,6,7). Curiously, the small protein whose gene is adjacent to the L32 gene in several bacterial genomes (COG1399; Figure 6), although not present in all bacteria and not known to be a ribosome-associated protein, also displays the C+/- pattern (data not shown). Thus, some of the C+/- r-proteins might be linked at the levels of function and regulation of expression.
Several of the probable cases of HGT that involve C+/- r-proteins appear to have occurred by the displacement in situ route, that is, without disruption of the local gene order. At first glance, incorporation of an incoming alien gene into the recipient genome in the exact same place of the resident orthologous gene seems to be extremely unlikely. The only plausible explanation is that the corresponding gene arrangements confer a substantial selective advantage upon the bacteria that have them and, accordingly, displacements that result in a disruption of the operon organization are eliminated by purifying selection. On some, or even all, occasions, displacement in situ might have occurred via a two-stage mechanism, whereby the acquired alien gene is initially incorporated in a different place in the recipient genome, and subsequently displaces the resident gene by intra-genomic recombination. Evidence for such a two-stage mechanism has been presented previously in a different context, when considering gene fusions that involve horizontally transferred genes [34]. Displacement in situ is likely to be particularly prominent in the case of r-operons because these are the most conserved gene arrays in prokaryotic genomes, but this phenomenon has been noticed also during the analysis of other operons (M.V. Omelchenko, K.S.M. and E.V.K., unpublished observations).
One of the driving forces behind the differential evolution of the C+ and C- forms of r-proteins, and in particular HGT, could be antibiotic resistance. Although there is no direct evidence of a role of Zn-ribbons in antibiotic resistance, S14, which is part of the peptidyl-transferase center [2], interacts with different antibiotics and interaction with puromycin has also been demonstrated for S18 [35].
The present analysis of C+/- r-proteins raises interesting functional questions and shows that the evolution of r-proteins, at least in bacteria, substantially deviates from straightforward vertical inheritance and includes multiple instances of DGL and HGT. The C+/- pattern and the duplication of the corresponding r-protein genes provide the framework for detecting these events even in cases when phylogenetic trees alone do not offer sufficient support for a specific evolutionary scenario. It cannot be ruled out that DGL and HGT are even more common in the evolution of the ribosome, but their identification for other r-proteins will require additional data and more sophisticated phylogenetic analyses.
Materials and methods
Sequence data
Amino acid sequences of r-proteins from completely sequenced prokaryotic genomes were extracted from the Genome division of the Entrez retrieval system [36,37]. The analyzed genomes included those of bacteria: Escherichia coli, Buchnera sp., Haemophilus influenzae, Pseudomonas aeruginosa, Vibrio cholerae, Xylella fastidiosa, Neisseria meningitidis, Helicobacter pylori, Campylobacter jejuni, Caulobacter crescentus, Mesorhizobium loti, Rickettsia prowazekii, Bacillus subtilis, Bacillus halodurans, Lactococcus lactis, Streptococcus pyogenes, Mycoplasma pneumoniae, Mycoplasma genitalium, Ureaplasma urealyticum, Mycobacterium tuberculosis, Mycobacterium leprae, Aquifex aeolicus, Thermotoga maritima, Deinococcus radiodurans, Thermus thermophilus, Treponema pallidum, Borrelia burgdorferi, Chlamydophyla pneumoniae, Chlamydia trachomatis, Synechocystis PCC6803; archaea: Archaeoglobus fulgidus, Halobacterium sp. NRC-1, Methanococcus jannaschii, Methanobacterium thermoautotrophicum, Thermoplasma acidophilum, Pyrococcus horikoshii, Aeropyrum pernix; chloroplasts from Arabidopsis thaliana (ARATH_CHL), Guillardia theta (GUITH_MIT), Porphyra purpurea (PORPU_MIT); mitochondrial r-proteins from Reclinomonas americana (RECAM_MIT), Homo sapiens (HOMSA_MIT), Saccharomyces cerevisiae, Arabidopsis thaliana (ARATH_MIT). In some cases, additional sequences were included (see figure legends).
Each sequence set of orthologous proteins as defined in the COG database [19] was aligned using the ClustalW program [38], with subsequent manual validation and correction. Evolutionary distances were calculated using the Dayhoff PAM model as implemented in the PROTDIST program of the PHYLIP package [39]. Distance trees were constructed using the least-square method [40] as implemented in the FITCH program of PHYLIP [39]. Maximum likelihood trees were constructed by using the ProtML program of the MOLPHY package [41], with the JTT-F model of amino acid substitutions [41,42], to optimize the least-square trees with local rearrangements. Bootstrap analysis was performed for each maximum likelihood tree as implemented in MOLPHY using the Resampling of Estimated Log-Likelihoods (RELL) method [41,43]. Alternative placements of selected clades in maximum-likelihood trees were compared by using the rearrangement optimization method as implemented in the ProtML program [41].
Acknowledgments
Acknowledgements
We thank Yuri I. Wolf for help and advice with phylogenetic tree construction, and I. King Jordan for critical reading of the manuscript. Kira Makarova is supported by the Microbial Genome Program, Office of Biological and Environmental Research, DOE (DE-FG02-98ER62583).
References
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- Brimacombe R. The bacterial ribosome at atomic resolution. Structure Fold Des. 2000;8:R195–R200. doi: 10.1016/s0969-2126(00)00510-4. [DOI] [PubMed] [Google Scholar]
- Poole A, Jeffares D, Penny D. Early evolution: prokaryotes, the new kids on the block. Bioessays. 1999;21:880–889. doi: 10.1002/(SICI)1521-1878(199910)21:10<880::AID-BIES11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Muller EC, Wittmann-Liebold B. Phylogenetic relationship of organisms obtained by ribosomal protein comparison. Cell Mol Life Sci. 1997;53:34–50. doi: 10.1007/PL00000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann S, Martin W. Phylogeny of 33 ribosomal and six other proteins encoded in an ancient gene cluster that is conserved across prokaryotic genomes: influence of excluding poorly alignable sites from analysis. Int J Syst Evol Microbiol. 2000;50:1655–1663. doi: 10.1099/00207713-50-4-1655. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Mori H, Itoh T, Gojobori T. Genome plasticity as a paradigm of eubacteria evolution. J Mol Evol. 1997;44:S57–S64. doi: 10.1007/pl00000052. [DOI] [PubMed] [Google Scholar]
- Itoh T, Takemoto K, Mori H, Gojobori T. Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol Biol Evol. 1999;16:332–346. doi: 10.1093/oxfordjournals.molbev.a026114. [DOI] [PubMed] [Google Scholar]
- Lathe WC, 3rd, Snel B, Bork P. Gene context conservation of a higher order than operons. Trends Biochem Sci. 2000;25:474–479. doi: 10.1016/s0968-0004(00)01663-7. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Kondrashov AS, Koonin EV. Genome alignment, evolution of prokaryotic genome organization and prediction of gene function using genomic context. Genome Res. 2001;11:356–372. doi: 10.1101/gr.gr-1619r. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF, Handy J. Evolutionary anomalies among the aminoacyl-tRNA synthetases. Curr Opin Genet Dev. 1998;8:630–636. doi: 10.1016/s0959-437x(98)80030-0. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases - analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710. [PubMed] [Google Scholar]
- Woese CR, Olsen GJ, Ibba M, Soll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Lilburn TG, Parker CT, Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Philippe H, Moreira D. The evolutionary history of ribosomal protein RpS14: horizontal gene transfer at the heart of the ribosome. Trends Genet. 2000;16:529–533. doi: 10.1016/s0168-9525(00)02142-9. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Makarova KS, Spouge JL, Wolf YI, Koonin EV. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001;11:555–565. doi: 10.1101/gr.166001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hard T, Rak A, Allard P, Kloo L, Garber M. The solution structure of ribosomal protein L36 from Thermus thermophilus reveals a zinC-ribbon-like fold. J Mol Biol. 2000;296:169–180. doi: 10.1006/jmbi.1999.3433. [DOI] [PubMed] [Google Scholar]
- Kurland CG, Andersson SG. Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev. 2000;64:786–820. doi: 10.1128/mmbr.64.4.786-820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiboli P, Triantafillidou D, Franceschi F, Choli-Papadopoulou T. Studies on the Zn-containing S14 ribosomal protein from Thermus thermophilus. Eur J Biochem. 1998;256:136–141. doi: 10.1046/j.1432-1327.1998.2560136.x. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes - quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc Natl Acad Sci USA. 1995;92:2441–2445. doi: 10.1073/pnas.92.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Archaea and the prokaryote-to-eukaryote transition. Microbiol Mol Biol Rev. 1997;61:456–502. doi: 10.1128/mmbr.61.4.456-502.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weglohner W, Junemann R, von Knoblauch K, Subramanian AR. Different consequences of incorporating chloroplast ribosomal proteins L12 and S18 into the bacterial ribosomes of Escherichia coli. Eur J Biochem. 1997;249:383–392. doi: 10.1111/j.1432-1033.1997.00383.x. [DOI] [PubMed] [Google Scholar]
- Uhlein M, Weglohner W, Urlaub H, Wittmann-Liebold B. Functional implications of ribosomal protein L2 in protein biosynthesis as shown by in vivo replacement studies. Biochem J. 1998;331:423–430. doi: 10.1042/bj3310423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool IG. Extraribosomal functions of ribosomal proteins. Trends Biochem Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- Lindahl L, Zengel JM. Expression of ribosomal genes in bacteria. Adv Genet. 1982;21:53–121. doi: 10.1016/s0065-2660(08)60297-7. [DOI] [PubMed] [Google Scholar]
- Allen T, Shen P, Samsel L, Liu R, Lindahl L, Zengel JM. Phylogenetic analysis of L4-mediated autogenous control of the S10 ribosomal protein operon. J Bacteriol. 1999;181:6124–6132. doi: 10.1128/jb.181.19.6124-6132.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Woolford JL., Jr Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA. Mol Cell Biol. 1999;19:826–834. doi: 10.1128/mcb.19.1.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Kondrashov AS, Koonin EV. Interkingdom gene fusions. Genome Biol. 2000;1:research0013.1–0013.13. doi: 10.1186/gb-2000-1-6-research0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann-Liebold B, Uhlein M, Urlaub H, Muller EC, Otto A, Bischof O. Structural and functional implications in the eubacterial ribosome as revealed by protein-rRNA and antibiotic contact sites. Biochem Cell Biol. 1995;73:1187–1197. doi: 10.1139/o95-128. [DOI] [PubMed] [Google Scholar]
- Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics. 1999;15:536–543. doi: 10.1093/bioinformatics/15.7.536. [DOI] [PubMed] [Google Scholar]
- Entrez retrieval system http://www.ncbi.nlm.nih.gov:80/PMGifs/Genomes/org.html
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Fitch WM, Margoliash E. Construction of phylogenetic trees. Science. 1967;155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- Adachi J, Hasegawa M. MOLPHY: Programs for molecular phylogenetics. In Computer Science Monographs 27 Tokyo: Institute of Statistical Mathematics; 1992.
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Kishino H, Miyata T, Hasegawa M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J Mol Evol. 1990;31:151–160. [Google Scholar]
- Natale DA, Galperin MY, Tatusov RL, Koonin EV. Using the COG database to improve gene recognition in complete genomes. Genetica. 2000;108:9–17. doi: 10.1023/a:1004031323748. [DOI] [PubMed] [Google Scholar]