

Summary
Mesenchymal stem cells (MSCs) were officially named more than 25 years ago to represent a class of cells from human and mammalian bone marrow and periosteum that could be isolated and expanded in culture while maintaining their in vitro capacity to be induced to form a variety of mesodermal phenotypes and tissues. The in vitro capacity to form bone, cartilage, fat, etc., became an assay for identifying this class of multipotent cells and around which several companies were formed in the 1990s to medically exploit the regenerative capabilities of MSCs. Today, there are hundreds of clinics and hundreds of clinical trials using human MSCs with very few, if any, focusing on the in vitro multipotential capacities of these cells. Unfortunately, the fact that MSCs are called “stem cells” is being used to infer that patients will receive direct medical benefit, because they imagine that these cells will differentiate into regenerating tissue‐producing cells. Such a stem cell treatment will presumably cure the patient of their medically relevant difficulties ranging from osteoarthritic (bone‐on‐bone) knees to various neurological maladies including dementia. I now urge that we change the name of MSCs to Medicinal Signaling Cells to more accurately reflect the fact that these cells home in on sites of injury or disease and secrete bioactive factors that are immunomodulatory and trophic (regenerative) meaning that these cells make therapeutic drugs in situ that are medicinal. It is, indeed, the patient's own site‐specific and tissue‐specific resident stem cells that construct the new tissue as stimulated by the bioactive factors secreted by the exogenously supplied MSCs. Stem Cells Translational Medicine 2017;6:1445–1451
Keywords: Medicinal signaling cells, Mesenchymal stem cells, MSCs, Regenerative medicine
Introduction
Mesenchymal stem cells (MSCs) were officially named more than 25 years ago 1 to represent a class of cells from human 2 and mammalian bone marrow and periosteum 3 that could be isolated and expanded in culture while maintaining their in vitro capacity to be induced to form a variety of mesodermal phenotypes and tissues (Fig. 1, The Mesengenic Process). The in vitro capacity to form bone, cartilage, fat, etc., became an assay for identifying this class of multipotent cells 9 and around which several companies (including Osiris Therapeutics, which my colleagues and I started,) were formed in the 1990s to medically exploit the regenerative capabilities of MSCs. Initially, the driving concept that a multipotent progenitor or “stem cell” existed in adult marrow was not only challenged, but was actively disregarded, especially by the orthopedic industry. Fast‐forward to today and there are hundreds of clinics 10 and hundreds of clinical trials 11 using human MSCs (hMSCs) with very few, if any, focusing on the in vitro multipotential capacities of these cells.
Figure 1.
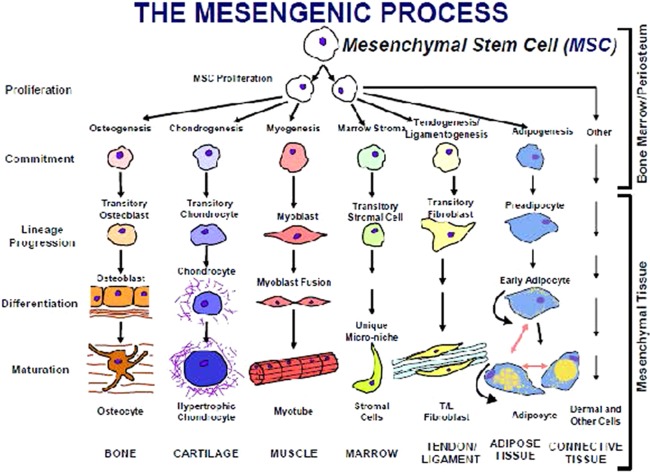
The mesengenic process. This hypothesis was originally verbalized in crude form in 1988 4, refined as a figure in 1990 5, 6, 7 and 1991 1, with its current format published in 1994 8. All of the proposed lineage pathways to bone, cartilage, muscle, etc., have been verified by us and others using inductive cell culture conditions. There are no tissue‐engineered products initiated with human MSCs that are approved and in use medically at this time. Abbreviation: MSC, mesenchymal stem cell.
Unfortunately, the fact that MSCs are called “stem cells” is being used to infer that patients will receive direct medical benefit, because they imagine that these cells will differentiate into the regenerating tissue‐producing cells (i.e., these “stem cells” will be incorporated into and these differentiated cells will fabricate the diseased or missing tissue). Such a stem cell treatment will presumably cure the patient of their medically relevant difficulties ranging from osteoarthritic (bone‐on‐bone) knees to various neurological maladies, including dementia. I long ago urged, in print, that we change the name of MSCs to Medicinal Signaling Cells 12 to more accurately reflect the fact that these cells home in on sites of injury or disease and secrete bioactive factors 13 that are immunomodulatory and trophic 14 (regenerative), meaning that these cells make therapeutic drugs 15 that are medicinal. It is, indeed, the patient's own site‐specific and tissue‐specific resident stem cells that construct the new tissue as stimulated by the bioactive factors secreted by the exogenously supplied MSCs 16, 17.
History of MSCs from a Caplan Perspective
In the early 1970s into the 1980s, my colleagues and I published a number of papers based on the culturing of stage 24, embryonic chick limb bud mesodermal cells (ECLBMCs) that were observed to differentiate into cartilage, muscle, and bone under certain culture conditions 18, 19, 20, 21, 22. These in vitro studies were correlated with a variety of in vivo studies that focused on the cellular and molecular events associated with the formation of embryonic limb bone 23, 24, cartilage 25, and muscle 26 in which several very prominent dogmas‐of‐the‐day were challenged. For example, the concept that “cartilage is replaced by bone” led to the implication that if one could form cartilage in culture from embryonic mesodermal progenitor cells, one could observe the transition of that new cartilage into bone. Moreover, the endochondral replacement of cartilage by bone implied that the cartilage so formed in culture would differentiate into hypertrophic cartilage, which would calcify and provide the calcified matrix for subsequent bone formation. Although we documented that the ECLBMCs formed cartilage in culture 22, 25, 26 and that hypertrophic chondrocytes could be identified by the production of type X collagen, the only mineral that formed in culture was observed in the noncartilage, connective tissue valleys between mounds of cartilage 23, 24. Reducing the initial plating densities of the freshly isolated ECLBMCs (where no cartilage formed) allowed us to observe the differentiation of a maximum number of cells into calcified matrix‐producing osteoblasts 27.
Only when we went back to the developing chick embryo and carefully completed rigorous histology of the mid‐diaphysis of the developing embryonic tibia, did we firmly establish that the new bone that formed came from a progenitor cell layer (stacked cell layer) outside and away from the already formed and expanding cartilage core (or cartilage model as it was called 28) 29, 30. Importantly, the hypertrophic cartilage core was replaced by invading vasculature and then marrow, not bone. Moreover, others 31, 32 clearly showed that these embryonic hypertrophic chondrocytes could be isolated, cultured, and maintained for many weeks in vitro, documenting that hypertrophic chondrocytes were not “programmed” to die (i.e., their demise was due to the nutrient and oxygen deprivation by a collar of calcified bone that was outside and away from the cartilage core) 29.
In addition, our early pioneering studies on the synthesis of proteoglycans of cartilage by the ECLBMC cells in culture with Vincent Hascall's 33, 34 group brought us into areas of detailing the extracellular matrix (ECM) of first cartilage 35, 36, then muscle 37, 38 and bone and played an important role in our current interest in the basement membrane surrounding all blood vessels (to be discussed below).
We spent considerable time and effort in optimizing these stage 24 ECLBMC‐cultures which, incidentally, we never called mesenchymal or mesodermal “stem cells,” although the evidence strongly suggested that they were multipotent. During this same time period and especially in the early 1980s, Marshal Urist and others were isolating molecular agents from the matrix of demineralized bone 39, 40, 41. The phenomenological basis for such efforts stemmed from implantations of demineralized bone pieces into muscle or subcutaneous pockets in rodents, which eventually caused bone to de novo form from host cells 39. Urist coined the term “bone morphogenetic proteins (BMPs)” to summarily refer to the bioactive agents released from demineralized bone matrix that could cause de novo bone to form in nonosseous tissues, such as muscle, or subcutaneously.
Stimulated by the public lectures and publications of Urist and because of a talented postdoctoral fellow, Glenn Syftestad, who had worked in Dr. Urist's lab at UCLA before coming to my lab in 1981, we joined the race to purify the BMPs. Our first approach was to take high salt extracts of demineralized bone exactly as published by Urist and to put them on cultures of stage 24 ECLBMCs arranged to just form bone 42, 43. To our great surprise, these extracts caused the cells to form mounds of cartilage. We named the presumed active agents as chondrogenic stimulating activity, which we purified, and the university filed patents 44, 45, which for reasons that could be challenged, they stopped maintaining. In the mid‐1980s and certainly by 1987, it became known that Dr. John Wozney and his colleagues at Genetics Institute, Inc. (Cambridge, MA) had cloned BMP2 and had patented the BMP‐family of molecules 46, 47. The race for the BMPs was over, and my colleagues and I had failed to win, much less “place or show.”
In one of the demineralized bone implantation systems, Dr. Hari Reddi purified one member of this BMP‐family and, importantly, characterized the in vivo temporal events caused by these factors 48, 49, 50. These temporal events involved the invasion of the implant and cell division of host mesenchymal cells followed by their differentiation into cartilage which became hypertrophic and which was replaced by vascularized and marrowized bone 50. Using Dr. Reddi's histology slides of these subcutaneous implantation specimens, which he generously provided, I suggested that the temporal sequence of cartilage replacement by bone was identical to that which we described in the developing embryonic chick tibia 5—essentially, that the implanted demineralized bone particles were surrounded by mesenchymal progenitor cells, which were attracted to the demineralized particles and formed cartilage. Since the implant was walled‐off, encysted by a layer of these mesenchymal cells comparable to the stack cell layer of the embryonic chick tibia, all blood vessels were excluded. The blood vessels outside the layer of surrounding and encysting mesenchymal cells caused the bottom layer of encircling cells to differentiate into a layer of osteoblasts, which fabricated a layer of osteoid that became mineralized. The deprivation of nutrients and oxygen caused the encased chondrocytes to form hypertrophic cartilage (Reddi documented the production of type X collagen) whose cells expired, releasing large quantities of vascular endothelial growth factor (VEGF), which caused the external vasculature to invade just as occurs in the mid‐dyaphasis of the embryonic chick tibia 5, 29, 30. These invading vessels brought a fresh supply of mesenchymal progenitor cells, which then formed vascularized and marrowized bone.
Without going into details, the central fact that comes from the above is that upon jamming the demineralized bone into muscle or the subcutaneous sites, the release and clustering of mesenchymal progenitor cells could be documented in these adult rodent hosts. In concert with these facts was the realization that adult bone marrow contained the same or similar primitive osteochondral progenitors 51, 52, 53, 54, 55, 56, 57. The presence of these mesenchymal progenitors could be deduced from many avenues of exploration: (a) since the days of Aristotle, bone marrow was known to enhance orthopedic/bone healing 51; (b) in modern terms, Connolly et al. 52, 53 and more recently Hernigou 58, documented the direct osteochondral potency of bone marrow or bone marrow aspirates; (c) Friedenstein et al., as rediscovered and popularized by Owen, showed that clones of adherent osteogenic progenitor cells could be isolated and propagated in culture from adult marrow 54, 55, 56, 57; and last (d) Owen herself imagined a crude mesenchymal lineage comparable to that described for descendants of hematopoietic stem cell (HSC) 57. It is important to stress that in the 1980s and early 1990s, the dogma‐of‐the‐day was that the only stem cells that existed in the adult body were HSCs.
The above facts (especially the demineralized bone implantations into adult hosts) led Dr. Stephen Haynesworth and me to see if we could isolate and purify the mesenchymal progenitor cells from adult human bone marrow 1, 59, 60, 61, 62, 63. At that time, we were not aware of the work of Friedenstein and of Owen, which was lucky because we had the ECLBMC system, which was quite different from the culture conditions of Friedenstein and Owen. We had long before optimized this ECLBMC system, in particular by optimally choosing the batch of fetal bovine serum (FBS) used to culture these chick embryonic cells 64. This lucky batch of serum was later shown to be optimal for the attachment, propagation, and maintenance of the multipotency of the culture adherent cells from human adult marrow 65, 66. Indeed, one in 10–20 batches of FBS was shown to be optimal for marrow‐derived hMSCs by the ECLBMC culture assay system, which eventually was replaced by other criteria 66. This assayed batch of FBS allowed MSCs to optimally attach to the culture dishes, to expand to form colonies (referred to as colony form units/fibroblast, CFU‐f by Friedenstein 55, that could be counted to give MSC titers, which ranged from 1 in 10,000 marrow cells in newborns to 1 in 2 million marrow cells in 80‐year‐old marrow donors 67. Given all of the above, I named these propagated cells that were multipotent in culture assay: MSCs 1.
MSCs: Various Names Mean the Same
Given the historic outline above, various names for these culture adherent and passaged adult marrow‐derived, multipotent mesenchymal cells came to mind:
Marrow Stromal Cells
The term “stroma” is an older morphological term meaning from connective tissue or the structural component of tissue. As defined by Owen in 1988 57, these are fibroblastic cells that adhere to plastic and expand, forming colonies (CFU‐f) that are osteogenic. One could also envision that bone marrow stroma was a unique scaffold that supports different lineage arms of hematopoiesis. Such a three‐dimensional connective tissue scaffold does not exist in marrow, although the vision of such a specialized framework is enchanting.
Multipotent Stromal Cells
MSCs can be multipotent, as documented in various culture circumstances.
Mesodermal Stem Cells
Because of our studies of ECLBMC cells, this term was highly favored, especially because all of the induced or bioactive factor‐treated cells and tissue formed in culture were of mesodermal (middle layer of the embryo) origin.
MSCs
I chose this term because mesenchyme is a type of tissue characterized by loosely associated cells that lack polarity and are surrounded by a large ECM. Because of their in vitro multipotency and clonability 68, I, provocatively, called them “stem cells” to especially appeal to the orthopedic community. As defined by hematologists, all stem cells must be capable of serial transplantation and unlimited doublings. Indeed, there are published reports that support this definition 69, 70.
Mesenchymal Stromal Cell
A group of scientists at an international meeting termed the MSC as a “stromal” cell because they did not favor the stem cell classification and imagined, incorrectly, that the origin of MSCs, from a variety of tissues, was the connective tissue layer of that tissue 9.
Medicinal Signaling Cell
Because the function of MSCs in vivo is secretory and primarily functional at sites of injury, disease, or inflammation, I now favor this terminology 12.
The New Science: MSCs Are Derived from Pericytes
Central to the renaming strategy is the fact that most, if not all, MSCs are derived from the differentiation of perivascular or mural cells, pericytes 71. The studies of Dr. Bruno Péault and colleagues 72 clearly document that pericytes isolated from a variety of tissues give rise to MSCs, as identified by cell surface antigens and their in vitro multipotency. Importantly, MSCs can be isolated from every vascularized tissue 73 and even from menstrual flow 74, 75 (i.e., broken blood vessels release the perivascular cells that differentiate into MSCs). The perivascular location as the origin of MSCs and their functional capacity to be immunomodulatory and trophic (including fabricating and secreting antibiotic proteins 76) challenges the “stromal” name and origin of the MSCs 9, 77, 78.
Based on the above, we have assembled the new and current information on the pericyte MSC (pMSC) into a poster, which has a number of interesting and unusual pieces of information not previously appreciated 79. These include the fact that each separate tissue‐specific stem cell is both in communication with its underlying vascular endothelial cells and neighboring specific pericyte/MSC [Universal Stem Cell Niche]. These pMSCs are specific to each stem cell, including a chemically different pMSC next to the active versus quiescent HSC in marrow 80, 81. In every tissue examined in detail, the marrow, neural tissue 82, liver 83, heart 84, etc., tissue‐specific stem cells are next to its specific pMSC on a blood vessel. These observations further support the concept that all pMSCs have both MSC‐common and MSC‐unique chemical and functional features. In the in vitro multipotency assays, the assay must be optimized for each tissue specific MSC. For example, hMSCs of marrow were shown to be induced in culture into the chondrogenic lineage by TGF‐β 85, while fat‐derived hMSCs require both TGF‐β and BMP‐6 86. The main in vivo functional differences of MSCs from different tissues or organs remain largely unknown, even though the major therapeutic functionality of MSCs at various sites of disease or injury are very similar when comparing these different MSCs 87.
Changing Names
Since the main functionality in vivo of MSCs 88 is not multipotency and, thus, not as a stem cell 89, 90, 91, I propose that its name be changed. The precedent for changing medical terms is not new. For example, names of many diseases have been changed: ablepsy was changed to blindness; ague to malarial fever; American plague to yellow fever; anasarca to generalized massive edema; aphonia to laryngitis; aphtha to thrush in infants; and apoplexy to paralysis due to stroke 92. Of course, there is great stigma associated with the accepted names for some diseases; multiple sclerosis was once called hysterical paralysis when people believe this was caused by stress linked with oedipal fixations. Chronic fatigue syndrome is a serious ailment, yet 85% of clinicians view it as a psychiatric disorder; activists are currently trying to change the name to remove the bias and stigma. There is no stigma associated with the term MSC except, for me, the implied promise that it is a true “stem cell,” which it is not in vivo.
It has been argued, because MSC science and clinical use is so strong and, indeed, positive with almost 700 clinical trials listed on clinicaltrials.gov, that the MSC nomenclature should remain. The problem is not with the “mesenchymal” part of the name; it is the “stem cell” part of the name that is the issue. As outlined in our poster, the pMSC functions quite differently from the released pericyte that forms an activated, site‐specific MSC. Infused auto‐ or allogenic MSCs appear to home in on active vascular sites of injury or inflammation 93. At such disease sites, the MSC rarely or never differentiate into the tissue at that site 13, 88, but they secrete bioactive factors (some of the names of these factors we know 94) and their therapeutic effects can be analyzed as site‐specific clinical outcome parameters. Outcomes for graft‐versus‐host disease, acute myocardial infarct, low back pain, osteoarthritic knees, tendonitis, and aspects of inflammatory bowel disease or Crohn's disease have been reported (www.mesoblast.com). Again, for emphasis, these MSC‐effects are medicinal.
MSCs Are Not Stem Cells
The science and commercialization of adult MSCs were enhanced by the popularization of embryonic stem cells (ESCs) and made more attractive by President Bush's prohibition of the use and study of ESCs 95. This popularization of ESCs also served as a disadvantage because all “stem cells” have been viewed by the public as being pluripotent or multipotent. Thus, the infusion of hMSCs in an osteoarthritic knee is imagined to contribute directly into the regeneration of cartilage tissue by the infused MSCs forming functional chondrocytes that fabricate functional cartilage tissue. The infusion into cardiac patients of hMSCs assumes that these cells will directly convert into functional heart muscle cells to replace the cells that die from the ischemia of the heart attack. And so on and so on: stem cells directly convert into the diseased or injured tissue in question. Although we, in this field, all have our own favorite explanation for the mechanisms that govern the observed positive therapeutic outcomes, the in vivo effects of infused hMSCs are best described as medicinal and most likely not associated with the infused cells differentiating into regenerative or replacement tissue 96, 97, 98, 99.
These stem cell misconceptions have led some practitioners in the United States and worldwide to advertise the availability of stem cell‐treatments (i.e., MSCs can cure the blind, make the lame walk, and make old tissue young again 10). I, of course, want the MSC nomenclature to remain in use, but not as stem cells. Perhaps we should call them magic signaling cells, more strategic cells, maxi secreting cells, most sensitive cells, main secreting cells, or message secreting cells. I propose to change the name of MSCs to reflect our new understanding that they do not function in the body as progenitors for tissues, neither in the normal steady‐state nor in disease or injury circumstances; they are not stem cells.
MSCs and Metastasis
Last, we recently published a treatise which documents that the pMSC actively binds to and pulls circulating melanoma cells into the marrow of bone 100. This grab/pull mechanism for melanoma metastasis is counter to the current concept that metastatic cells secrete digestive enzymes that allow the melanoma to erode its way into bone. We further hypothesized that the laminin identity in the basement membrane ECM of the blood vessels plays both an active and permissive role in the extravasation of melanoma into bone. Thus, the melanoma must pass through the endothelial cell layer, its basal lamina or basement membrane and past the dense covering of mural cells. The active pMSC not only facilitates this extravasation, but is actively and molecularly controlling this translocation from the circulation into the marrow of bone. Clearly, the pMSC is not medicinal in this context even though its differentiated progeny, the MSC, can provide powerful medicinal benefit given other circumstances. Last, the pMSC is not multipotent nor does it, itself, cause tumors to form. The pMSC is corrupted by the cancer cell; it does not corrupt normal cells to become cancerous.
Conclusion
It should be permissible for the person who named the MSCs to drop the stem cell nomenclature because it is scientifically and therapeutically misleading. In 2010, I proposed that we call them medicinal signaling cells 12. That is what these do, and the culture plasticity of most mesenchymal cells (we can induce adult human chondrocytes to make a bone or fat in cell cultures 101) means that the stem cell moniker is inappropriate. I was wrong. I take back the name that I gave these hugely important cells. Call them MSCs, but please, not stem cells.
Disclosure of Potential Conflicts of Interest
Case Western Reserve University receives royalties from Osiris Therapeutics, Inc. (now by Mesoblast Limited) which they share with AIC for MSC technology transfered in 1992. The authors indicated no other potential conflicts of interest.
Acknowledgments
I thank the National Institutes of Health and the Virginia and David Baldwin Fund for their financial support.
References
- 1. Caplan AI. Mesenchymal stem cells. J Orthop Res 1991;9:641–650. [DOI] [PubMed] [Google Scholar]
- 2. Haynesworth SE, Goshima J, Goldberg VM et al. Characterization of cells with osteogenic potential from human marrow. Bone 1992;13:81–88. [DOI] [PubMed] [Google Scholar]
- 3. Nakahara H, Bruder SP, Goldberg VM et al. In vivo osteochondrogenic potential of cultured cells derived from the periosteum. Clin Orthop Relat Res 1990;259:223–232. [PubMed] [Google Scholar]
- 4. Caplan AI. Biomaterials and bone repair. Biomaterials 1988;87:15–24.
- 5. Caplan AI. Cartilage begets bone versus endochondral myelopoiesis. Clin Orthop Relat Res 1990;261:257–267. [PubMed] [Google Scholar]
- 6. Caplan AI. Cell delivery and tissue regeneration. J Control Release 1990;11:157–165. [Google Scholar]
- 7. Caplan AI. Stem cell delivery vehicle. Biomaterials 1990;11:44–46. [PubMed] [Google Scholar]
- 8. Caplan AI. The mesengenic process. Clin Plast Surg 1994;21:429–435. [PubMed] [Google Scholar]
- 9. Horwitz EM, Le Blanc K, Dominici M et al. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005;7:393–395. [DOI] [PubMed] [Google Scholar]
- 10. Turner L, Knoepfler P. Selling stem cells in the USA: Assessing the direct‐to‐consumer industry. Cell Stem Cell 2016;19:154–157. [DOI] [PubMed] [Google Scholar]
- 11.ClinicalTrials.Gov. Mesenchymal stem cells (MSCs) clinical trials. Available at https://clinicaltrials.gov/. Accessed February 1, 2017.
- 12. Caplan AI. What's in a name? Tissue Eng Part A 2010;16:2415–2417. [DOI] [PubMed] [Google Scholar]
- 13. da Silva Meirelles L, Fontes AM, Covas DT et al. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev 2009;20:419–427. [DOI] [PubMed] [Google Scholar]
- 14. Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem 2006;98:1076–1084. [DOI] [PubMed] [Google Scholar]
- 15. Caplan AI, Correa D. The MSC: An injury drugstore. Cell Stem Cell 2011;9:11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol 2012;12:383–396. [DOI] [PubMed] [Google Scholar]
- 17. Caplan AI. Adult mesenchymal stem cells: When, where, and how. Stem Cells Int 2015;2015:628767. doi:10.1155/2015/628767. Epub 2015 Jul 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caplan AI, Zwilling E, Kaplan NO. 3‐acetylpyridine: Effects in vitro related to teratogenic activity in chicken embryos. Science 1968;160:1009–1010. [DOI] [PubMed] [Google Scholar]
- 19. Caplan AI, Rosenberg MJ. Interrelationship between poly (ADP‐Rib) synthesis, intracellular NAD levels, and muscle or cartilage differentiation from mesodermal cells of embryonic chick limb. Proc Natl Acad Sci USA 1975;72:1852–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caplan AI. The molecular control of muscle and cartilage development. In: 39th Annual Symposium of the Society for Developmental Biology, Ed. S Subtelney and U Abbott, Alan R. Liss, Inc., New York, pp. 37–68 (1981).
- 21. Caplan AI, Koutroupas S. The control of muscle and cartilage development in the chick limb: The role of differential vascularization. J Embryol Exp Morphol 1973;29:571–583. [PubMed] [Google Scholar]
- 22. Caplan AI. Molecular basis for limb morphogenesis. In: Proceedings of the 5th International Conference on Birth Defects, Ed. J Littlefield and J DeGrouchy, Excerp. Medica. Amsterdam‐Oxford 1978;208–220.
- 23. Osdoby P, Caplan AI. First bone formation in the developing chick limb. Dev Biol 1981;86:147–156. [DOI] [PubMed] [Google Scholar]
- 24. Osdoby P, Caplan AI. Characterization of a bone‐specific alkaline phosphatase in chick limb mesenchymal cell cultures. Dev Biol 1981;86:136–146. [DOI] [PubMed] [Google Scholar]
- 25. Caplan AI, Stoolmiller AC. Control of chondrogenic expression in mesodermal cells of embryonic chick limb. Proc Natl Acad Sci USA 1973;70:1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenberg MJ, Caplan AI. Nicotinamide adenine dinucleotide levels in cells of developing chick limbs: Possible control of muscle and cartilage development. Dev Biol 1974;38:157–164. [DOI] [PubMed] [Google Scholar]
- 27. Caplan AI, Syftestad G, Osdoby P. The development of embryonic bone and cartilage in tissue culture. Clin Orthop Relat Res 1983;174:243–263. [PubMed] [Google Scholar]
- 28. Fell HB. The histogenesis of cartilage and bone in the long bones of the embryonic fowl. J Morphol 1925;40:417–459. [Google Scholar]
- 29. Caplan AI. Bone development and repair. Bioessays 1987;6:171–175. [DOI] [PubMed] [Google Scholar]
- 30. Caplan AI, Pechak, David G. The cellular and molecular embryology of bone formation. In: Peck W, ed. Bone and Mineral Research. New York: Elsevier, 1987:117–184.
- 31. Schmid TM, Linsenmayer TF. Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J Cell Biol 1985;100:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solursh M, Jensen KL, Reiter RS et al. Environmental regulation of type X collagen production by cultures of limb mesenchyme, mesectoderm, and sternal chondrocytes. Dev Biol 1986;117:90–101. [DOI] [PubMed] [Google Scholar]
- 33. Caplan AI, Hascall VC. Structure and development changes in proteoglycans. In: Naftolin F, Stubblefield PG, eds. Dilatation of the Uterine Cervix. New York: Raven Press, 1980:79–98.
- 34. Caplan AI. Cartilage. Sci Am 1984;251:84–94. [DOI] [PubMed] [Google Scholar]
- 35. Hascall VC, Oegema TR, Brown M et al. Isolation and characterization of proteoglycans from chick limb bud chondrocytes grown in vitro. J Biol Chem 1976;251:3511–3519. [PubMed] [Google Scholar]
- 36. De Luca S, Caplan AI, Hascall VC. Biosynthesis of proteoglycans by chick limb bud chondrocytes. J Biol Chem 1978;253:4713–4720. [PubMed] [Google Scholar]
- 37. Carrino DA, Caplan AI. Isolation and preliminary characterization of proteoglycans synthesized by skeletal muscle. J Biol Chem 1982;257:14145–14154. [PubMed] [Google Scholar]
- 38. Carrino DA, Caplan AI. Proteoglycans produced by skeletal muscle in vitro and in vivo. Prog Clin Biol Res 1982;110:379–389. [PubMed] [Google Scholar]
- 39. Urist MR. Bone: Formation by autoinduction. Science 1965;150:893–899. [DOI] [PubMed] [Google Scholar]
- 40. Urist MR, Dowell TA, Hay PH et al. Inductive substrates for bone formation. Clin Orthop Relat Res 1968;59:59–96. [PubMed] [Google Scholar]
- 41. Urist MR, Mikulski A, Lietze A. Solubilized and insolubilized bone morphogenetic protein. Proc Natl Acad Sci USA 1979;76:1828–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Syftestad GT, Caplan AI. A fraction from extracts of demineralized adult bone stimulates the conversion of mesenchymal cells into chondrocytes. Dev Biol 1984;104:348–356. [DOI] [PubMed] [Google Scholar]
- 43. Syftestad GT, Lucas PA, Caplan AI. The in vitro chondrogenic response of limb‐bud mesenchyme to a water‐soluble fraction prepared from demineralized bone matrix. Differentiation 1985;29:230–237. [DOI] [PubMed] [Google Scholar]
- 44. Caplan AI, Syftestad GT, inventors; Patent, assignee. Bone protein purification process. 1986. Patent No. 4,608,199.
- 45. Caplan AI, Syftestad GT, inventors; Patent, assignee. Process of adapting soluble bone protein for use in stimulating osteoinduction. July 5, 1986. Patent No. 4,620,327.
- 46. Wozney J, Rosen V, Celeste A et al. Novel regulators of bone formation: Molecular clones and activities. Science 1988;242:1528–1534. [DOI] [PubMed] [Google Scholar]
- 47. Celeste AJ, Iannazzi JA, Taylor RC et al. Identification of transforming growth factor beta family members present in bone‐inductive protein purified from bovine bone. Proc Natl Acad Sci USA 1990;87:9843–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reddi AH. Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat Biotech 1998;16:247–252. [DOI] [PubMed] [Google Scholar]
- 49. Reddi AH, Huggins C. Biochemical sequences in the transformation of normal fibroblasts in adolescent rats. Proc Natl Acad Sci USA 1972;69:1601–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reddi AH. Bone and cartilage differentiation. Curr Opin Genet Dev 1994;4:737–744. [DOI] [PubMed] [Google Scholar]
- 51. Cooper B. The origins of bone marrow as the seedbed of our blood: From antiquity to the time of Osler. Proc (Bayl Univ Med Cent) 2011;24:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Connolly JF, Guse R, Tiedeman J et al. Autologous marrow injection as a substitute for operative grafting of tibial nonunions. Clin Orthop Relat Res 1991;266:259–270. [PubMed] [Google Scholar]
- 53. Connolly J, Guse R, Lippiello L et al. Development of an osteogenic bone‐marrow preparation. J Bone Joint Surg Am 1989;71:684–691. [PubMed] [Google Scholar]
- 54. Friedenstein AJ, Petrakova KV, Kurolesova AI et al. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 1968;6:230–247. [PubMed] [Google Scholar]
- 55. Friedenstein AJ, Deriglasova UF, Kulagina NN et al. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol 1974;2:83–92. [PubMed] [Google Scholar]
- 56. Owen M, Friedenstein AJ. Stromal stem cells: Marrow‐derived osteogenic precursors. Ciba Found Symp. 1988;136:42–60. [DOI] [PubMed] [Google Scholar]
- 57. Owen M. Marrow stromal stem cells. J Cell Sci Suppl 1988;10:63–76. [DOI] [PubMed] [Google Scholar]
- 58. Hernigou P. Autologous bone marrow grafting of avascular osteonecrosis before collapse. Rev Rhum (Engl Ed) 1995;62:650–651.
- 59. Caplan AI, Haynesworth SE, inventors; Patent assignee. Method for enhancing the implantation and differentiation of marrow‐derived mesenchymal cells. 1993. Patent No. 5,197,985.
- 60. Caplan AI, Haynesworth SE, inventors; Patent assignee. Method for treating connective tissue disorders. 1993. Patent No. 5,226,914.
- 61. Caplan AI, Haynesworth SE, inventors; Patent assignee. Human mesenchymal stem cells. 1996. Patent No. 5,486,359.
- 62. Gerson SL, Caplan AI, Haynesworth SE, inventors; Patent, assignee. Transduced mesenchymal stem cells. 1997. Patent No. 5,591,625.
- 63. Caplan AI, Haynesworth SE, inventors; Patent, assignee. Monoclonal antibodies for human osteogenic cell surface antigens. July 1, 1997. Patent No. 5,643,736.
- 64. Caplan AI. Muscle, cartilage and bone development and differentiation from chick limb mesenchymal cells. In: Vertebrate Limb and Somite Morphogenesis. Ed. DA Ede, JR Hinchliffe and M Balls, Cambridge University Press, Cambridge, England, 1977; 199–213.
- 65. Osdoby P, Caplan AI. The possible differentiation of osteogenic elements in vitro from chick limb mesodermal cells. I. Morphological evidence. Dev Biol 1976;52:283–299. [DOI] [PubMed] [Google Scholar]
- 66. Lennon DP, Haynesworth SE, Bruder SP et al. Human and animal mesenchymal progenitor cells from bone marrow: Identification of serum for optimal selection and proliferation. In Vitro Cell Dev Biol Anim 1996;32:602–611. [Google Scholar]
- 67. Haynesworth SE, Caplan AI. Diminution of the number of mesenchymal stem cells as a cause for skeletal aging. In: Buckwalter JA, Goldberg VM, Woo SLY, eds. Musculoskeletal Soft‐Tissue Aging: Impact on Mobility. Rosemont, IL: American Academy of Orthopedic Surgeons, 1994:79–87.
- 68. Pittenger MF, Mackay AM, Beck SC et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999;284:143–147. [DOI] [PubMed] [Google Scholar]
- 69. Dennis JE, Carbillet JP, Caplan AI et al. The STRO‐1+ marrow cell population is multipotential. Cells Tissues Organs 2002;170:73–82. [DOI] [PubMed] [Google Scholar]
- 70. Dennis JE, Caplan AI. Advances in mesenchymal stem cell biology. Curr Opin Orthop 2004;15:341–346. [Google Scholar]
- 71. Caplan AI. All MSCs are pericytes? Cell Stem Cell 2008;3:229–230. [DOI] [PubMed] [Google Scholar]
- 72. Crisan M, Yap S, Casteilla L et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008;3:301–313. [DOI] [PubMed] [Google Scholar]
- 73. Nombela‐Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol 2011;12:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alcayaga‐Miranda F, Cuenca J, Luz‐Crawford P et al. Characterization of menstrual stem cells: Angiogenic effect, migration and hematopoietic stem cell support in comparison with bone marrow mesenchymal stem cells. Stem Cell Res Ther 2015;6:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Khoury M, Alcayaga‐Miranda F, Illanes SE et al. The promising potential of menstrual stem cells for antenatal diagnosis and cell therapy. Front Immunol 2014;5:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Krasnodembskaya A, Song Y, Fang X et al. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL‐37. Stem Cells 2010;28:2229–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. da Silva Meirelles L, de Deus Wagatsuma VM, Malta TM et al. The gene expression profile of non‐cultured, highly purified human adipose tissue pericytes: Transcriptomic evidence that pericytes are stem cells in human adipose tissue. Exp Cell Res 2016;349:239–254. [DOI] [PubMed] [Google Scholar]
- 78. da Silva Meirelles L, Malta TM, de Deus Wagatsuma VM et al. cultured human adipose tissue pericytes and mesenchymal stromal cells display a very similar gene expression profile. Stem Cells Dev 2015;24:2822–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Somoza RA, Correa, D , Caplan, AI. Roles for mesenchymal stem cells as medicinal signaling cells. Nat Protoc. Vol 11 No 1. 2015. [Epub ahead of print]. Available at http://www.nature.com/nprot/posters/msc/index.html.
- 80. Kunisaki Y, Bruns I, Scheiermann C et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013;502:637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sá da Bandeira D, Casamitjana J, Crisan M. Pericytes, integral components of adult hematopoietic stem cell niches. Pharmacol Ther 2017;171:104–113. [DOI] [PubMed] [Google Scholar]
- 82. Tavazoie M, Van der Veken L, Silva‐Vargas V et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell 2008;3:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bautch VL. Stem cells and the vasculature. Nat Med 2011;17:1437–1443. [DOI] [PubMed] [Google Scholar]
- 84. Chong James JH, Chandrakanthan V, Xaymardan M et al. Adult cardiac‐resident MSC‐like stem cells with a proepicardial origin. Cell Stem Cell 2011;9:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yoo JU, Barthel TS, Nishimura K et al. The chondrogenic potential of human bone‐marrow‐derived mesenchymal progenitor cells. J Bone Joint Surg Am 1998;80:1745–1757. [DOI] [PubMed] [Google Scholar]
- 86. Diekman BO, Rowland CR, Lennon DP et al. Chondrogenesis of adult stem cells from adipose tissue and bone marrow: Induction by growth factors and cartilage‐derived matrix. Tissue Eng Part A 2010;16:523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Geevarghese A, Herman IM. Pericyte‐endothelial crosstalk: Implications and opportunities for advanced cellular therapies. Transl Res 2014;163:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guimarães‐Camboa N, Cattaneo P, Sun Y et al. Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 2017;20:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Deasy BM, Gharaibeh BM, Pollett JB et al. Long‐term self‐renewal of postnatal muscle‐derived stem cells. Mol Biol Cell 2005;16:3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tao H, Han Z, Han ZC et al. Proangiogenic features of mesenchymal stem cells and their therapeutic applications. Stem Cells Int 2016;2016:1314709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Overturf K, al‐Dhalimy M, Ou CN et al. Serial transplantation reveals the stem‐cell‐like regenerative potential of adult mouse hepatocytes. Am J Pathol 1997;151:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- 92. Kay M. Lexemic change and semantic shift in disease names. Cult Med Psychiatry 1979;3:73–94. [DOI] [PubMed] [Google Scholar]
- 93. Lin P, Correa D, Kean TJ et al. Serial transplantation and long‐term engraftment of intra‐arterially delivered clonally derived mesenchymal stem cells to injured bone marrow. Mol Ther 2014;22:160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp Mol Med 2013;45:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Murugan V. Embryonic stem cell research: A decade of debate from Bush to Obama. Yale J Biol Med 2009;82:101–103. [PMC free article] [PubMed] [Google Scholar]
- 96. Couzin J, Vogel G. Renovating the heart. Science 2004;304:192–194. [DOI] [PubMed] [Google Scholar]
- 97. Phinney DG, Isakova IA. Mesenchymal stem cells as cellular vectors for pediatric neurological disorders. Brain Res 2014;1573:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Maes C, Kobayashi T, Selig MK et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 2010;19:329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Boyle AJ, Schulman SP, Hare JM. Stem cell therapy for cardiac repair. Ready for the next step. Circulation 2006;114:339–352. [DOI] [PubMed] [Google Scholar]
- 100. Correa D, Somoza RA, Lin P et al. Mesenchymal stem cells regulate melanoma cancer cells extravasation to bone and liver at their perivascular niche. Int J Cancer 2016;138:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tallheden T, Dennis JE, Lennon DP et al. Phenotypic plasticity of human articular chondrocytes. J Bone Joint Surg Am 2003;85‐A(suppl 2):93–100. [DOI] [PubMed] [Google Scholar]