

Abstract
Down syndrome (DS) is a genetic disorder caused by trisomy 21 (T21). Over the past two decades, the use of mouse models has led to significant advances in the understanding of mechanisms underlying various phenotypic features and comorbidities secondary to T21 and even informed the design of clinical trials aimed at enhancing the cognitive abilities of persons with DS. In spite of its success, this approach has been plagued by all the typical limitations of rodent modeling of human disorders and diseases. Recently, several laboratories have succeeded in producing T21 human induced pluripotent stem cells (T21‐iPSCs) from individuals with DS, which is emerging as a promising complementary tool for the study of DS. Here, we describe the method by which we generated 10 T21‐iPSC lines from epithelial cells in urine samples, presumably from kidney epithelial origin, using nonintegrating episomal vectors. We also show that these iPSCs maintain chromosomal stability for well over 20 passages and are more sensitive to proteotoxic stress than euploid iPSCs. Furthermore, these iPSC lines can be differentiated into glutamatergic neurons and cardiomyocytes. By culturing urine‐derived cells and maximizing the efficiency of episomal vector transfection, we have been able to generate iPSCs noninvasively and effectively from participants with DS in an ongoing clinical trial, and thus address most shortcomings of previously generated T21‐iPSC lines. These techniques should extend the application of iPSCs in modeling DS and other neurodevelopmental and neurodegenerative disorders, and may lead to future human cell‐based platforms for high‐throughput drug screening. Stem Cells Translational Medicine 2017;6:1465–1476
Keywords: Down syndrome, Trisomy 21, Induced pluripotent stem cells, Induced pluripotent stem cell‐derived glutamatergic neurons, Induced pluripotent stem cells‐derived Cardiomyocytes, Urine derived cells
Significance Statement.
In this study, we describe a method for derivation, differentiation, and characterization of trisomy 21 (T21) induced pluripotent stem cell (iPSC) lines from urine‐derived epithelial cells using nonintegrating episomal vectors. We show that these iPSCs maintain chromosomal stability for 20 passages, can be differentiated into glutamatergic neurons and cardiomyocytes, and are more sensitive to proteotoxic stress than euploid iPSCs. We have been able to generate iPSCs noninvasively and effectively from participants with Down syndrome (DS) in an ongoing clinical trial, and thus address most shortcomings of previously generated T21 iPSC lines. These techniques should improve the quality and extend the application of iPSCs in modeling DS and may lead to future human cell‐based platforms for high‐throughput drug screening in translational preclinical studies, and should help streamline cell‐banking applications.
Introduction
Down syndrome (DS) is caused by the presence of an extra copy of chromosome 21 (HSA21), which is also known as trisomy 21 (T21). DS is the most common genetically defined cause of intellectual disability, occurring in 1 out of 700 live births 1. DS also includes several other neurological phenotypes such as increased incidence of seizure disorder in relation to the general population, motor (including oculomotor) dysfunction, and virtually universal incidence of a neuropathology indistinguishable from Alzheimer's disease by age 40 years 2. In addition, DS is associated with significantly increased occurrence of various non‐neurological comorbidities, such as congenital heart defects, gastrointestinal tract malformations, leukemia, and hypothyroidism 3, 4, 5, 6.
Over the past 25 years, several mouse models of DS have been produced to aid in elucidating the molecular mechanisms underlying various phenotypic features and comorbidities secondary to T21 [7]. Much of the focus of the research in these animals has been on the neurobiology of DS. Results from such research have driven the design of half a dozen clinical trials aimed at enhancing the cognitive skills of individuals with DS 7. Many DS researchers have long recognized, however, the intrinsic lack of specificity and incompleteness of nonhuman systems in emulating certain distinctive features of human biology. This has led to a search for robust, diverse, and ethically sound sources of human cells.
In 2006, the landmark publication by Takahashi and Yamanaka 8 provided the pathway toward a new era of human cellular disease modeling. It showed that introducing a set of defined reprogramming factors (OCT4, KLF4, SOX2, and c‐MYC; now known as the OSKM factors) into somatic cells was sufficient to generate induced pluripotent stem cells (iPSCs). In addition to its great potential in regenerative medicine, in vitro models derived from iPSCs offer a complementary strategy to animal modeling for elucidating the etiology and pathophysiology of many human diseases and disorders, including DS.
In recent years, several trisomy 21 iPSC (T21‐iPSC) lines have been developed 9. However, all but one of these studies used fibroblasts as starting cells, with the exception being a study that used cells harvested from amniotic fluid 10. Establishing a fibroblast culture requires that a dermal sample be obtained from a skin biopsy. Although this is only a mildly invasive procedure, it is still incongruous with the growing consensus that an ideal source for iPSCs should provide cells easily and noninvasively, especially when one is procuring cells from vulnerable populations, such as children and individuals with intellectual disability. The work by Zhang and coworkers 11, 12, describing a detailed protocol for iPSCs generation using cells isolated from urine (presumably exfoliated renal epitheliocytes), provided the potential blueprint for a noninvasive, simple, and safe source for iPSC generation 13, 14, 15, 16 that could be applicable to research in DS.
Another important issue to consider is the selection of a proper delivery method for the reprogramming factors. The T21‐iPSC lines currently described in the literature have been primarily generated through integrative delivery systems 9, 17. Although this method is simple and efficient, the random integration of the vectors into host cell genomes can lead to insertion of mutagenesis in the obtained iPSCs 18. These concerns about genome integrity in the process of generation of iPSCs have led to the development of nonintegrative methods for factor delivery such as episomal DNA vectors 19. So far, the only successful example of the use of a nonintegrative approach using oriP/Epstein‐Barr nuclear antigen‐1 (oriP/EBNA1)‐based episomal vector for the generation of T21‐iPSCs was the one described by Briggs et al. 20 in fibroblasts from a mosaic DS patient.
In the present study, we describe the efficient generation of integration‐free urine‐derived iPSC lines from individuals with DS. Furthermore, we show that these T21‐iPSCs are more sensitive to proteotoxic stress than disomic control cells, and can robustly differentiate into valuable lineage‐specific cell types such as glutamatergic neurons and cardiomyocytes. The effective and noninvasive nature of this method for generating T21‐iPSCs from individuals with DS should greatly enhance and extend the application of iPSCs in modeling DS and other neurodevelopmental and neurodegenerative disorders, and may lead to future human cell‐based platforms for high‐throughput drug screening.
Materials and Methods
Ethical Statement
The urine sample donors for this study and their parents/legal guardians have signed written informed consents to provide urine samples for T21‐iPSC generation and further differentiation. The experimental protocol involving urine sample donation was reviewed and approved by the University Hospitals Cleveland Medical Center's Institutional Review Board (IRB) as part of the larger protocol of an ongoing clinical trial (IRB number: 06‐14‐41; NCT02304302 at http://www.clinicaltrials.gov). In order to adapt and troubleshoot the iPSC generation protocol and the cytogenetic methods used here, we first produced one control euploid cell line from a urine sample donated by one of the authors, which was not terminally differentiated or further characterized, and was later discarded. To confirm the pluripotent potential of the transfected cells, teratomas were produced by subcutaneous injection of iPSCs in immunocompromised NOD scid IL2Rg mice (NSG) at the Athymic Animal & Xenograft Core, which is part of the Case Western Reserve University's Comprehensive Cancer Center. The animal protocol for these experiments were reviewed and approved by Case Western Reserve University's Institutional Animal Care and Use Committee.
Collection and Expansion of the Urine‐Derived Epithelial Cells
Putative renal epithelial cells were collected and expanded from urine with minor modifications from methods previously described 11, 12. Briefly, urine samples were collected from 10 individuals with DS and one individual without DS and centrifuged at 400g for 10 minutes. The pellet was washed in buffer containing 1X Dulbecco's phosphate‐buffered saline (DPBS) with Penicillin/Streptomycin (PS, Hyclone, Pittsburgh, PA, www.gelifesciences.com), 0.5 µg/ml Amphotericin B (Sigma‐Aldrich, St. Louis, MO, www.sigmaaldrich.com). The pellet was resuspended with primary culture media containing Renal Cell Growth Medium (REGM, Lonza, Basel, Switzerland, www.lonza.com), 10% FBS (Gibco, Waltham, MA, www.thermofisher.com), PS, and 0.5 µg/ml Amphotericin B. Cells were then transferred to 12‐well tissue culture dishes coated with 1% gelatin solution (Gibco). During the first 3 days, 1 ml of primary culture medium was added. Starting on the fourth day, most of the medium was aspirated and 1.5 ml of REGM was added. Then, this procedure was repeated every other day until the T21 urine‐derived cells had expanded enough to cover above 70% of the plate.
Transfection Efficiency Test
Expanded urine‐derived T21 cells (5 × 105 cells) were transferred to gelatin‐coated 100‐mm dishes and cultured with REGM for 2 days. On the day of transfection, the cells were detached using 0.25% trypsin‐Ethylenediaminetetraacetic acid (trypsin‐EDTA Gibco) and dissociated into single cells by pipetting up and down. Five micrograms of pmax green fluorescent protein (GFP) vector (1 µg/µl, Lonza) was mixed with 0.6–1.2 × 106 urine‐derived T21 cells in P1 or P3 solution and transferred to 16 wells in Nucleocuvette strips. Electroporation was performed with Amax 4D‐Nucleofector using P1 or P3 Primary Cell 4D‐Nucleofector X kit. Seven different programs (CM‐102, DC‐100, EA‐104, EL‐110, EDE‐100, CM‐113, and DS‐109) in P1 and P3 solutions were tested according to the manufacturer's instructions (Lonza). The pmax‐GFP vector transfected urine‐derived T21 cells from each program was transferred into 12‐well plates and incubated overnight. The number of GFP positive cells, live cells, and total cells were counted to obtain the percentages of GFP positive and live cells.
Generation of T21‐iPSCs from Urine‐derived T21 Cells with Episomal Vectors
Dissociated single urine‐derived T21 cells (6 × 105 cells) were electroporated with 4.2 µl of Episomal iPSC Reprogramming vectors (Invitrogen, Waltham, MA, http://www.thermofisher.com/) using an Amax 4D‐Nucleofector device with P1 solution and the program EA‐104. The electroporated cells were transferred to Vitronectin (VTN‐N, Invitrogen) coated 100mm dish in N2B27 media with 3 µM CHIR99021 (Stemgent, Cambridge, MA, www.stemgent.com), 0.5 µM PD0325901 (Stemgent), 0.5 µM A‐83‐01 (Stemgent), 10 ng/ml hLIF (Invitrogen), 10 µM Y‐27632 (Stemgent), 100 ng/ml basic fibroblast growth factor (bFGF, Invitrogen). Half of the media were changed every day until 14 days after transfection and on day 15, media were switched into Essential 8 media (Invitrogen) until embryonic stem cell (ESC)‐like colonies were generated. From day 25 to 30 of transfection, ESC‐like colonies were picked and transferred to VTN‐N coated 12‐well plate for expansion and several passages were performed to establish T21‐iPSC lines. Each cell line was named CWRU1XXXi‐YYT; CWRU (Case Western Reserve University) is the institution name, 1XXX is the deidentified cell line number from each donor (starting at #1001, to prevent any potential confusion with other iPSC lines produced in our institution), YY is the clone number, and T represents trisomy, respectively, according to a proposed nomenclature convention 21.
Teratoma Formation Assay and Histology
NSG mice were used for teratoma formation. T21‐iPSCs were grown to near confluence, treated with Accutase solution (Sigma Aldrich), washed in DPBS, and resuspended in DPBS supplemented with 30% Matrigel (BD Biosciences, San Jose, CA, www.bdbiosciences.com) at densities 2 × 106 cells per 200 microliters. Harvested cells were kept on ice and drawn into 1‐ml syringe immediately before injection. The mice were anesthetized by halothane inhalation (4%–5% for induction, 1%–2% for maintenance). Approximately 2 × 106 cells per injection site were used. Mice were injected in the dorso‐lateral area into the subcutaneous space on both sides. Prior to teratoma removal, mice were killed by cervical dislocation. Teratomas were surgically removed, fixed in 4% formaldehyde and embedded in paraffin. Paraffin‐embedded teratomas were cut into 5‐µm serial sections and stained with hematoxylin and eosin and mounted with glass cover slips. Staining was performed by The Tissue Resources Core Facility at the Case Comprehensive Cancer Center.
Maintenance of T21 and Euploid iPSCs
To maintain iPSC lines, cells were cultured on VTN‐N coated culture dish plates with Essential E8 media. Every 3–5 days, T21‐iPSCs were dissociated with the StemPro EZPassage Disposable Stem Cell Passaging Tool (Invitrogen) or 0.5 mM EDTA treatment for 5 minutes and transferred to a new plate (ratio 1:5 to 1:8). In addition to the T21‐iPSCs generated in this study, Dr. Antony Wynshaw‐Boris (Department of Genetics Case Western Reserve University School of Medicine) kindly shared with us three euploid iPSC lines (BJ4, YH10, 1323‐2). These iPSCs were generated from euploid individuals of comparable ages as our donors with DS, using similar techniques as the ones described here, and were used as controls in experiments aimed at assessing proteotoxic sensitivity, which are described below.
MTT Assay of Metabolic Activity
Disomic and T21 iPSCs (4 × 103 cells), or neuronal cells differentiated from these iPSCs (1.6 × 104), were seeded into 48‐well plates and treated with increasingly higher concentrations (0 to 320 nM for iPSCs or 0 to 1280 nM for neuronal cells) of the heat shock protein 90 (Hsp90) inhibitor 17‐allylamino‐17‐demethoxy‐geldanamycin (17AAG) (Invitrogen, San Diego, CA, http://www.invivogen.com) for 9 days. The 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay was performed using a commercially available MTT assay kit (Invitrogen) according to the manufacturer's instructions. The remaining viable cells with MTT dye uptake were determined by measuring the optical density at 540 nm in a microplate reader. Nonlinear regression fit of the data used for the construction of the dose‐response curves was performed using the four‐parameters variable slope algorithm in GraphPad Prism, version 6 (GraphPad Software Inc., La Jolla, CA).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 30 minutes, then stained with various primary antibodies (Supporting Information Table 1). Primary antibody localization was performed using goat anti‐rabbit and anti‐mouse IgG conjugated to Alexa 488 (1:300, Invitrogen) and goat anti‐rabbit IgG conjugated to Alexa 594 (1:300, Invitrogen). Control protocols for primary and secondary antibodies revealed neither nonspecific staining nor antibody cross‐reactivity. Cells were counter stained with 4′,6‐diamidino‐2‐phenylindole, dihydrochloride (DAPI).
Fluorescence In Situ Hybridization
Fluorescence In Situ Hybridization (FISH) was performed as previously described 22. Briefly, disomic and trisomic iPSCs were harvested followed by 0.1 µg/ml Colcemid (Life Technologies) for 1–2 hours, treated with 75 mM KCl for 20 minutes, and fixed in 3:1 methanol:acetic acid. HSA21 specific probe (XA 21q22, Metasystems, Newton, MA, www.metasystems.org) was used for HSA21 detection.
Spectral Karyotyping Analysis
Spectral karyotyping (SKY) was performed on metaphase spreads of euploid and T21 iPSCs according to the manufacturer's instructions (ASI, Carlsbad, CA) and as previously described 23. Spectral images were acquired and analyzed with a SD 200 Spectral Bio‐imaging System (ASI) attached to a Zeiss Axioskop 2 microscope (Carl Zeiss, Toronto, Ontario, Canada), and analyzed using the SKYVIEW (ver. 1.2; ASI) software.
Differentiation of Glutamatergic Neurons from T21‐iPSCs
Glutamatergic neurons were differentiated from T21‐iPSCs as previously described 24 with some modification. Briefly, T21‐iPSCs were seeded at a density of 6 × 104 cells per cm2 in Essential 8 media. At 50% confluence, the medium was changed to N2B27 with 1 μM LDN193189 (Stemgent) and 10 μM SB432542 (Stemgent) from day 1 to day 7 for neural stem cell induction. For glutamatergic neuron differentiation from neural stem cells, 400 ng/ml Cyclopamine (EMD Millipore) and 10 ng/ml bFGF were added from days 3 to 15. After 12–14 days, cells were detached with Accutase (Invitrogen) treatment and transferred to 10 μg/ml poly‐l‐ornithine (Sigma Aldrich) and 20 μg/ml laminin‐coated plates (Invitrogen) and cultured for an additional 3–5 weeks. BDNF at 10 ng/ml (R&D systems, Minneapolis, MN, www.rndsystems.com) was added 1 week after initiation of neuronal maturation and 10 μg/ml laminin was added every other day to keep mature neurons attached on the plate.
Electrophysiology
Action potentials from cultured neurons derived from T21‐iPSCs were obtained by whole‐cell current‐clamp recordings. Cultures were superfused with artificial cerebral spinal fluid (aCSF—concentrations in mM—120 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d‐glucose, saturated with 95% O2 and 5% CO2) at room temperature. Neurons were identified visually with a CCD camera connected to an inverted phase contrast microscope. Recording micropipettes (5–8 MΩ), pulled from thick‐walled borosilicate glass (1.5 mm outer diameter, 1.0 mm inner diameter, PG52151‐4, WPI Sarasota, FL), were filled with (in mM): 20 KCl, 120 K‐gluconate, 2 MgCl2, 10 HEPES, 4 Na‐ATP, 0.5 EGTA (pH 7.3 with KOH). At least 5 minutes elapsed between seal formation and data collection. All neurons were maintained with seal resistances of 5–15 GΩ and series resistances ≤40 MΩ. Action potentials were evoked by injecting short (800 ms) depolarizing current pulses (in current clamp). Voltage recordings were filtered at 2 kHz (8‐pole Bessel) and digitized at 20 kHz into a Windows‐PC computer using an Axopatch 200B patch‐clamp amplifier, a Digidata 1550A low noise data acquisition system, and the PCLAMP 10.5 software suite (Molecular Devices, Sunnyvale, CA). Offline data analysis was carried out with Clampfit (part of PCLAMP).
Induction of Cardiomyocytes
Cardiomyocytes differentiation was induced using the RPMI+ B27 minus insulin medium method 25 with some modification. Briefly, T21‐iPSCs were seeded at a density of 6 × 104 cells per cm2 in Essential 8 media. At 70% confluence, the medium was changed to differentiation medium #1 including RPMI (Invitrogen) with B27 minus insulin (Invitrogen) supplement for 8 days then changed to differentiation medium #2 including RPMI with B27 plus insulin (Invitrogen) and 2% FBS for 4 to 5 weeks. Then, 6 μM CHIR99021 and 5 μM IWR‐1 (Sigma‐Aldrich) were added on 1–2 days and 5–6 days, respectively.
Quantification of Beating Rates of Cardiomyocyte Clusters
Video was recorded from clusters of beating cardiomyocytes using a Leica AS MDW live cell incubator and imaging system set to 37.0°C, 5% CO2, and 95% air. Video was recorded at low magnification using a ×5 objective, a ×0.55 c‐mount adapter, and an Allied Vision Stingray F‐033B ½′′ CCD high‐speed monochrome camera and acquired at 640 × 480 pixels at 86.38 fps. Video was recorded from two fields of cells for a total of nearly 3 hours. The video included in this analysis was from the middle of the recording session, and includes the 5‐minute period just prior to administering the nonselective beta‐adrenergic agonist isoproterenol, and a second 5‐minute period starting about 11 minutes later, near the peak response to isoproterenol.
Beating patterns of the cardiomyocytes were extracted from the video using algorithms developed internally and written in IgorPro (Ver. 6, Wavemetrics, Oswego, OR). Several simple and more complicated methods were assessed for extracting cellular beating signals from the video. The final algorithm included several components: (a) video regions of interest were manually selected, in this case 20 elliptical regions were selected and processed, with three of those regions shown here; (b) stage 1 of the video processing algorithm created a self‐adaptive movement detecting contrast reference frame (CRF) for each cellular region; (c) stage 2 multiplied each video frame by each CRF and summed the values across pixels to obtain optical motion tracking tracings related to the beating of the cell cluster with each region; (d) the tracings for each region were normalized and used in a beat‐to‐beat interval detection algorithm; (e) the beat‐to‐beat intervals were used to calculate instantaneous and mean beating rates.
Results
Generation of T21‐iPSCs Using Chemically Defined, Integration‐Free Methods
It has been reported that iPSCs can be generated without gene integration into the cells using episomal vectors 19 and that iPSCs can be generated from urine‐derived, RE cells 11, 12. Here, for the first time, we combined these two methods to generate T21‐iPSCs. Urine samples were collected from 10 individuals with DS, comprising 5 females and 5 males (ages 15–29 years). Residual cells were isolated from urine for expansion of urine‐derived T21 cells. In the majority of cases, we found colonies around 4 days after plating and had enough cells for transfection in 20 days (Fig. 1A). We used an Amax 4D‐Nucleofector electrophoresis device for transfection of iPSC episomal vectors. In order to investigate the optimal electrophoresis condition for transfection of episomal vectors in urine‐derived T21 cells, we tested eight different protocols in two different solutions, P1 and P3, with pmax‐GFP vectors. We found that EA‐104 condition in P1 solution showed the best GFP expression efficiency with lowest cell death occurrence (Fig. 1B, 1C). We applied this condition for transfection of iPSC episomal vectors. Cobblestone‐like colonies were observed 25 days after transfection and ESC‐like colonies appeared approximately 50 days and several passages (Fig. 2A). To confirm the properties of these ESC‐like clones, we investigated the expression pattern of each clone with ESC specific markers. Figure 2B shows the five markers, OCT4, SOX2, NANOG, TRA‐1‐60, and SSEA4, being expressed in one of the clones, CWRU1003i‐27T. These T21‐iPSCs formed teratomas consisting of differentiated derivatives of all three primary germ layers (Fig. 2C).
Figure 1.
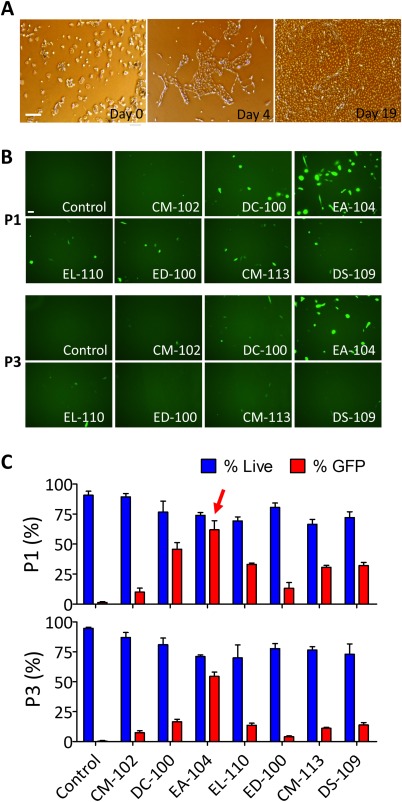
Expansion and transfection efficiency test of cells from urine samples. (A): Representative images of human trisomic urine‐derived cell expansion on days 0, 4, and 19. Scale bar = 100 µm. (B): Representative images of GFP expression 1 day after transfection of pmax‐GFP in urine‐derived T21 cells with indicated solutions and programs. Seven different transfection programs were used in cells transfected in either P1 or P3 solution. Scale bar = 50 µm. (C): Bar graph representation of the survival rate (blue) and efficiency of pmax‐GFP transfection (red) in urine‐derived T21 cells as a function of the transfection program in either P1 (upper panel) or P3 (lower panel) solution. Red arrow in the P1 panel indicates the final program (EA‐104) selected for episomal vector transfection. Efficiencies were calculated as number of GFP positive cells divided by total cell numbers (bar graphs represent means ± SEM); and dead cells were determined by morphology. Abbreviation: GFP, green fluorescent protein.
Figure 2.
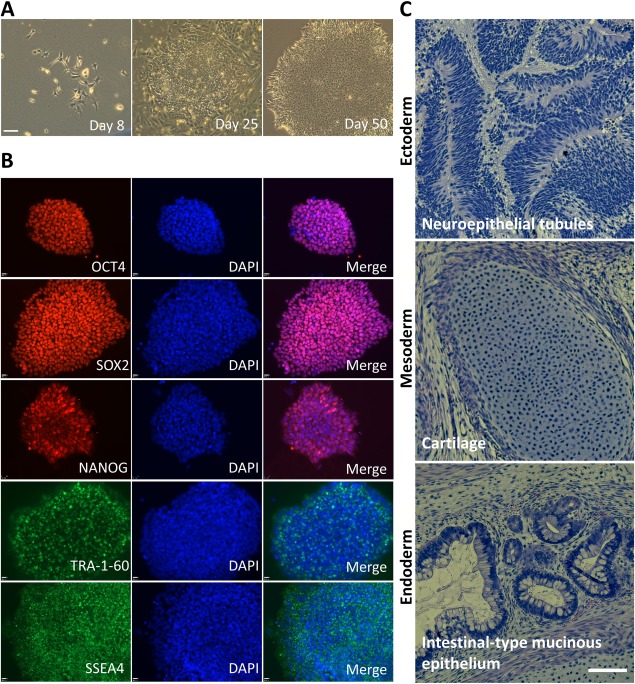
Generation of induced pluripotent stem cells (iPSCs) from urine sample of persons with Down syndrome. (A): Representative phase contrast images of transformation of urine‐derived T21 cells into T21‐iPSCs at 8, 25, 50 days after episomal vector transfection. Scale bar = 100 µm. (B): Representative immunofluorescence staining of pluripotent markers in CWRU1003i‐06T cell line colonies using antibodies against OCT4, SOX2, NANOG, TRA‐1‐60, and SSEA4. Cell nuclei were counterstained with DAPI. Scale bar = 50 µm. (C): Hematoxylin and Eosin staining of teratoma sections of iPSC clone CWRUi1002, P45, 59 days after injection. Scale bar, 100 μm. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole, dihydrochloride.
Using this same protocol, to date, we have generated 10 karyotype‐confirmed T21‐iPSC lines; CWRU1002i, ‐1003i, ‐1005i, ‐1006i, ‐1008i, ‐1011i, ‐1013i, ‐1014i, ‐1019i, ‐1022i, and 1 euploid control line. These results demonstrate that T21‐iPSCs can be generated routinely and reproducibly from expanded urine‐derived T21 cells isolated from urine samples, using an integration‐free method.
Chromosomal Stability of T21‐iPSCs
Because it has been reported that during in vitro maintenance of iPSCs and ESCs there is a chance of loss of the extra chromosome 21 [26] or a gain‐of‐function mutation caused by trisomy 12 and/or trisomy 17 27, 28, 29, we investigated chromosomal stability in the T21‐iPSCs that we generated. First, we examined the number of HSA21 by FISH with HSA21 specific probes. Using this method, we did not observe any HSA21 loss over 20 passages (Fig. 3A). Then, we searched for chromosomal abnormalities other than T21 using SKY, which did not reveal any chromosome abnormality in the disomic iPSC and no aneuploidy other than T21 in the trisomic lines over 20 passages (Fig. 3B, 3C and Supporting Information Figs. 1 and 2). Five of the lines underwent at least 40 passages without any detectable loss of the supernumerary HSA21 as detected by FISH (data not shown). This demonstrates that T21‐iPSCs generated from urine‐derived T21 cells through nonintegrating episomal vectors can maintain their cytogenetic integrity for well over 20 passages.
Figure 3.
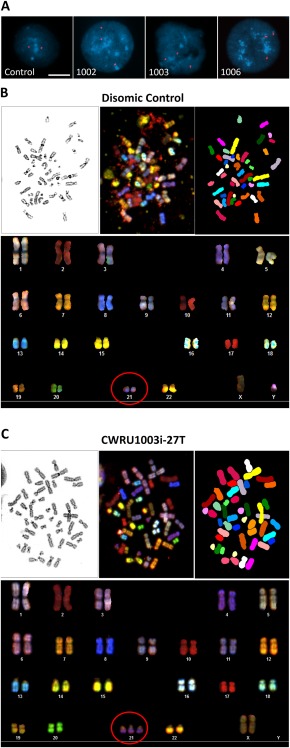
Cytogenetic genotyping of induced pluripotent stem cells (iPSCs). (A): Representative images of nuclei hybridized with fluorescent chromosome 21 probes (FISH) from four individual iPSC clones (Control, CWRU1002i, CWRU1003i, CWRU1006i). Nuclei were counterstained with DAPI, scale bar = 10 µm. Representative spectral karyotyping (SKY) images of chromosomes from disomic control (B) and cell line CWRU1003i (C). The upper panels of (B) and (C) show metaphase spread images using inverted DAPI staining (left), a spectral representation (middle), and SKY pseudocolor classification of chromosomes based on multicolor dye hybridization (right). The lower panels are the resulting karyotype tables of each cell line. The red circles highlight chromosome 21 in disomic or trisomic state.
Differentiation of T21‐iPSCs Into Glutamatergic Neurons
The CWRU1003i‐27T cell line was differentiated into glutamatergic neurons. After 3 weeks of differentiation, TUJ1 positive neurons started to express TBR1 and Glutamate (Fig. 4A). To examine the functional activity of the differentiated neurons, we performed electrophysiological recordings using the whole‐cell patch clamp technique after 120 days of differentiation. As illustrated in Figure 4B, differentiated neurons from T21‐iPSCs showed a capacity to fire action potentials similar to euploid iPSCs.
Figure 4.
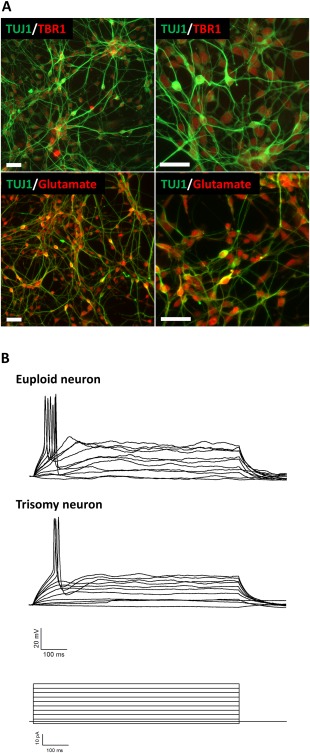
Generation of glutamatergic neurons. (A): Low (left) and high (right) magnification images of cell cultures 20 days after induced pluripotent stem cells (iPSCs) differentiation into glutamatergic neurons. Immunofluorescence staining was performed using antibodies against TUJ1, TBR1, and glutamate, scale bar = 50 µm. (B): Whole‐cell patch‐clamp recordings obtained from the cell soma made in current‐clamp mode. The top set of traces are representative action potentials from neurons derived from euploid iPSCs. The middle set of traces are representative recordings of from neurons that were generated from T21 iPSCs. Stimulus protocol (current steps) is shown at the bottom.
Differentiation of T21‐iPSCs Into Cardiomyocytes
As shown in Figure 5, T21‐iPSCs were successfully differentiated into cardiomyocytes using the insulin deprivation method 25 with some modification. Beating clusters of cardiomyocytes were observed by the 10th day after differentiation. By examining the resulting cells with cardiomyocyte‐specific protein expression markers, we found Troponin T Cardiac Isoform (TNT) and α‐actinin coexpressed cells. Among the α‐actinin positive cells, we observed some atrial and ventricular specific, MLC‐2A and MLC‐2V positive cells, respectively (Fig. 5A). Although one previous report has demonstrated that cardiomyocytes can be generated from T21‐ESCs 30, to our knowledge, this is the first demonstration that cardiomyocytes can be generated from T21‐iPSCs. To investigate the functional properties of the cardiomyocytes, we examined their beating patterns with and without the positive chronotropic agent isoproterenol. Three of the video‐recorded clusters, all derived from T21‐iPSC CWRU1003i‐06T, are shown in Figure 5B, 5C and Supporting Information Video 1. These recordings showed stable 5‐minute average beating rates for the three clusters (204.9 ± 16.1; 167.4 ± 2.4; 260.7 ± 1.9) prior to administration of isoproterenol, which exhibited 37.2%, 50.4%, and 8.6% increases in the average beating rates in response to 5‐μM administration of isoproterenol (Fig. 5D, 5E).
Figure 5.
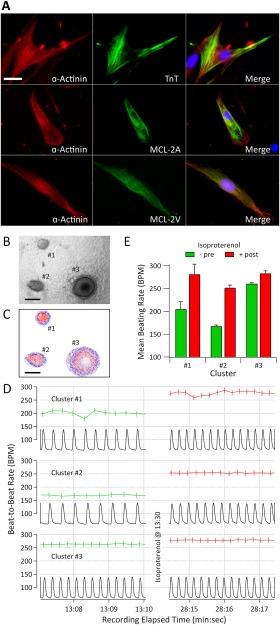
Generation of cardiomyocytes from human T21‐iPS cells. (A): Cardiomyocytes from induced pluripotent stem cell (iPSC) line CWRU1003i‐06T (52 days after differentiation); immunofluorescence staining was performed with the cardiomyocyte specific markers α‐actinin, TNT, MLC‐2A), and MLC‐2V. Cell nuclei were counterstained with DAPI. Scale bar = 25 µm. (B): A section of a single frame of video that contains three representative cardiomyocyte clusters labeled #1, #2, and #3. Scale bar = 200 µm. (C): Representative elliptical red and blue contrast reference frames (CRF) corresponding to each cluster. Within each CRF, bluer is more positive, redder is more negative, and whiter is more neutral. Scale bar = 200 µm. (D): video‐derived, optical motion tracking time tracings (black) from the video detector for the three clusters and the instantaneous beat‐to‐beat rate before (green) and after (red) isoproterenol administration. Cluster #1 exemplifies a cluster with high beat‐to‐beat variability, whereas Clusters #2 and #3 exemplify clusters with much lower variability. (E): Bar graph representing 5‐minute mean ± SD of the beating rates for the three clusters before and after isoproterenol administration. The three clusters showed 37.2%, 50.4%, and 8.6% increases in beating rate, respectively, in response to the administration of this beta‐adrenergic agonist. Abbreviations: MLC‐2A, myosin light chain 2A; MLC‐2V, myosin light chain 2V; TNT, Troponin T Cardiac Isoform.
T21‐iPSC Sensitivity to Proteotoxic Stress
Tang et al. 31 have shown that both aneuploid mouse fibroblasts and human cancer cell lines are more sensitive than euploid control cells to proteotoxic stress induced by the protein folding inhibitor 17‐AAG. Here, we investigated whether the T21‐iPSCs we produced are also more sensitive to 17‐AAG than euploid iPSCs. To this end, we compared cell survival rates between three euploid iPSC lines and three T21 iPSC lines after 9 days of treatment with increasingly higher concentrations of 17‐AAG. Experiments were repeated twice, with three triplicate each. We found that T21‐iPSCs are more sensitive than euploid iPSCs to the proteotoxic effects of 17‐AAG (Fig. 6A). When the individual survival rates (Supporting Information Fig. 3) were combined into genotype‐specific dose‐response curves, we observed a significant shift to the left when compared with euploid iPSCs, as demonstrated by nonlinear regression analysis (Fig. 6A). Accordingly, the half‐maximal inhibitory concentration, or IC50 (±SE), of 17‐AAG for T21 and euploid control iPSCs were 53.09 (±1.04) nM and 97.05 (±1.08), respectively (p < .0001).
Figure 6.
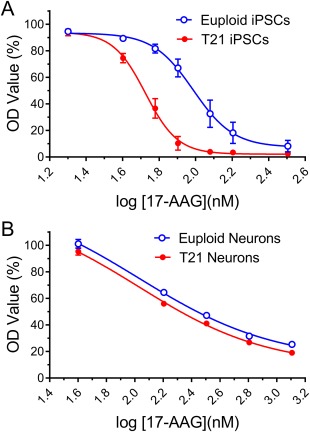
The effect of proteotoxic compounds on iPSCs and neurons. (A): Three euploid cell lines (1323‐2, YH10, BJ4) and three T21‐iPSC lines (CWRU1002i, −1003, −1006), and (B) differentiated neurons from these three euploid‐ and T21‐iPSC lines were treated with the indicated concentration of 17‐AAG. Cell survival rate of each cell lines were determined by MTT assay after 9 days, experiments were run in triplicate and repeated two times. The dose‐response curve for the survival rate of euploid and T21‐iPSCs were calculated exposed to successively higher concentrations of 17‐AAG were calculated. The data are the mean of six independent experiments ± SE, the solid line represents the nonlinear regression fit of the data. Abbreviations: iPSC, induced pluripotent stem cell; T21, trisomy 21.
However, when this experiment was repeated in differentiated neurons from euploid and T21 iPSCs, we found that these postmitotic cells were consistently more resistant to 17‐AAG toxicity (Fig. 6B) than undifferentiated iPSCs. In addition, an IC50 (±SE) of 106.66 (±1.38) nM was the best fit for MTT dye uptake data from neurons of both genotypes, that is, we found no genotype‐dependence for the proteotoxic effects of 17‐AAG in T21 neurons compared with euploid neurons (p = .2713).
Discussion
In the present study, we established that T21‐iPSCs can be generated reliably and efficiently by episomal vector transfection of cultured cells derived from urine samples from individuals with DS. We also demonstrated that these T21‐iPSCs can be differentiated into functional glutamatergic neurons and cardiomyocytes. Using this noninvasive approach, so far, we have generated 10 lines of T21‐iPSCs from participants of an ongoing clinical trial and are continuing to generate more iPSC lines to be used in future basic and translational studies in DS. This technique addresses most shortcomings of T21‐iPSC lines previously described in the literature and should enhance and extend the application of iPSCs in modeling DS and other neurodevelopmental and neurodegenerative disorders.
One of our initial concerns in trying to generate T21‐iPSCs was to determine whether the urine‐derived cells from individuals with DS could expand in vitro as efficiently as euploid cells can. They did grow well, however, they went into senescence earlier than euploid cells (data not shown). In an attempt to overcome this problem, we treated the urine‐derived T21 cells with an anti‐oxidant, an mTOR inhibitor, or a Rho‐associated protein kinase (ROCK) inhibitor 32, 33. However, none of these agents (or combination of such agents) was able to attenuate senescence in these cells (data not shown). Instead, we found that the only effective way to generate T21‐iPSCs from urine‐derived T21 cells was to transfect these cells with episomal vectors within 1‐3 passages. So far, we have generated T21‐iPSC lines from 10 out of 17 urine samples, which represents a 60% efficiency. If we ignore early failures, when we were still optimizing the procedures, the efficiency ratio would climb to approximately 80%.
Another important potential technical obstacle for this project had to do with the genetic stability of urine‐derived T21 cells and T21‐iPSCs, both in terms of preservation of the extra chromosome 21 and the potential emergence of chromosomal abnormalities that might produce a selective advantage to clones of mutated cells. To address this issue, we systematically evaluated the cells cytogenetically by chromosome 21 specific FISH and SKY. SKY provides more reliable results compared to conventional G‐band karyotyping, especially in terms of detection of chromosomal translocations 23, 34.
Careful cytogenetic surveillance of potential genetic drifts of T21‐iPSC cultures is an important quality control component in performing studies involving iPSCs, particularly aneuploid iPSC lines. For example, Maclean et al. 26 reported that a small percentage of T21‐iPSCs can spontaneously lose their extra HSA21; an occurrence that was exploited by these investigators to isolate and grow euploid isogenic iPSC subclones. For the T21‐iPSCs generated in the present study, however, we did not observe any detectable loss of the extra HSA21 over 20 passages. In preliminary experiments, we could not detect euploid cells among T21‐iPSCs even after cells were passaged 65 times. It is possible that the cell source (dermal fibroblasts versus urine‐derived putative epitheliocytes), method used in introducing the reprogramming factors (integrating retrovirus infection versus nonintegrating episomal vector transfection), and/or culture media (coculture with mouse embryonic feeder, MEF, cells versus MEF‐free culture media) may have contributed to the observed differences in chromosomal stability. Another possibility is that the dermal fibroblasts, which were obtained by Maclean et al. from a commercial cell bank (ATCC), might have come from a donor with low levels of mosaicism.
The enhanced chromosomal stability observed in the T21‐iPSCs generated in this study represents a great advantage for future studies involving the systematic characterization of the phenotype of T21‐iPSCs differentiated into various cell types known to be affected by T21. This is because the trisomic state of the resulting differentiated cells is also unlike to change. The technical disadvantage of this enhanced chromosomal stability, however, is our current inability to grow and isolate isogenic euploid control cell clones from these cultures.
We are actively investigating alternative approaches to generate isogenic euploid control cells from these T21‐iPSCs. The first of such approaches involved picking and growing more than 200 clones from high passage T21‐iPSC cultures to try to detect euploid iPSCs resulting from a potential low probability loss of extra HSA21 event. Even after repeating this experiment twice in two different T21‐iPSC lines, we could not identify any euploid iPSC colony (data not shown). Then, we tried to add selective pressure by exposing high passage T21‐iPSC cultures to the Hsp90 inhibitor 17‐AAG. Contrary to a previous report that had failed to identify an increased sensitivity of T21‐iPSCs to this proteotoxic agent 35, as can be seen in Figure 6, we were able to reproduce previous work demonstrating that aneuploid cells are more sensitive than euploid cells to proteotoxic stress 31. However, even after exposing high passage T21‐iPSC cultures to 9 days of treatment with 80 nM 17‐AAG, which killed over 95% of the T21‐iPSC colonies, we still could not detect a single euploid iPSC in the surviving colonies. Interestingly, when treating differentiated euploid and trisomy neurons with 17‐AAG, we found an increased tolerance to this cytotoxic agent and no genotype‐dependence on the cell death rates in these postmitotic cells, which suggests that the 17‐AAG sensitivity in iPSCs and neurons may be associated with cell proliferation capacity.
We are now attempting ZCAN4 transfection, which has been reported to increase the number of euploid cells in T21‐iPSC cultures within a few days 36. We also have established a collaboration with Dr. Wynshaw‐Boris to attempt antibiotic selection for loss of supernumerary HSA21 by introducing the TKNEO gene into one copy of HSA21 [35] and insertion of XIST (the X‐inactivation gene) into the extra HSA21 37. As a practical alternative, we are planning to use euploid control iPSCs derived from control individuals without DS in various experiments, and have received recently IRB approval for an amendment to our original protocol to obtain urine samples and produce euploid control iPSCs.
As mentioned in Introduction, much of the focus of recent DS research has been on the neurobiology of DS. One of the ideas that has emerged from this research has been the so‐called glutamatergic hypothesis, which states that the functional deregulation of the n‐methyl‐d‐aspartate (NMDA) glutamate receptor subtype 38, 39, 40 may affect negatively the cognitive abilities of persons with DS and may also be involved in the neurodegenerative processes associated with DS. Here, we generated cells with morphological characteristics and protein markers of glutamatergic neurons from T21‐iPSCs, and also produced an initial functional assessment of these cells and confirmed that they can generate action potentials (Fig. 4). As we begin the systematic characterization of the phenotype of these T21‐iPSC cultures differentiated into a glutamatergic neuronal fate, we will make use of this virtually unlimited source of human cells to shed light into the molecular mechanisms underlying the hypothesized dysfunction of NMDA receptor activity in T21 glutamatergic neurons.
Because of the simplicity and reliability of the process described here for the generation of T21‐iPSCs it is likely that, by the end of our ongoing clinical trial with the drug memantine, we will have T21‐iPSCs generated from a significant proportion of the 120 participants being recruited in the Cleveland site of this trial. In this study, we are collecting extensive phenotypic information, including clinical history of DS‐related comorbidities, neuropsychological profiles, and response to memantine therapy (NCT02304302 at http://www.clinicaltrials.gov). Therefore, this potentially will result in a sizable biobank of T21‐iPSCs from phenotypically well‐characterized individuals with DS that will be used initially to investigate potential molecular predictors of drug efficacy, but then should eventually evolve into a community resource for various studies on the biology of DS.
We also showed here that cells with cardiomyocyte morphological and protein markers can be generated from T21‐iPSCs. Functionally, cell clusters formed by these differentiated T21‐iPSCs exhibit spontaneous contractions and were sensitive to the beta‐adrenergic agonist isoproterenol. Although there has been a previous report of cardiomyocyte differentiation from T21 ESCs 30, to our knowledge, this is the first time that cardiomyocytes have been produced successfully from T21‐iPSCs. Given that 40% to 50% of all children born with DS display some form of congenital cardiovascular malformation 41, 42, it is easy to argue that this is the main way T21 negatively affects the heart. In spite of the fact that modeling cardiovascular malformations in a meaningful way using iPSCs would be a technically challenging undertaking, the development of heart organoids and other forms of three‐dimensional cell culture techniques may lead eventually to critical insights on the biologically relevant events resulting in endocardial cushion and septal heart defects. Perhaps more amenable to a cell‐based modeling approach would be mild but significant alterations in heart tissue excitability and vascular physiology associated with DS, such as the well‐documented reduced maximum heart rate in persons with DS compared to individuals without DS 43, 44. However, even this more modest goal would require the careful standardization of cell culturing conditions and the development of simple and efficient methods of assessing the physiology and pharmacology of the T21‐iPSCs. For example, the quantification of the contraction frequency of many of the cardiomyocyte clusters that we analyzed (approximately, 300,000 total beats for all clusters recorded in the present study) resulted in a wide range of core basal beating rates, from 40 to 270 bpm. In contrast, in their work using differentiated cardiomyocytes from T21 ESCs, Bosman et al. 40 reported typical basal beating rates of 18 to 24 bpm. It is clear, therefore, that the systematic study of the experimental conditions necessary to produce basal beating rates at more physiological ranges will be essential.
Conclusion
In summary, the techniques reported here offer several advantages compared with previously described methods for producing iPSCs from individuals with DS. First, the epithelial cells that were transformed into iPSCs were harvested by a completely noninvasive method, which is especially important given that we were making iPSCs from a vulnerable population, with limited ability to provide informed consent. Although only mildly invasive, there has been anecdotal reports that a few IRBs or ethical committees have rejected research proposals for the wide‐scale use of skin biopsies in individuals with DS. There has also been anecdotal reports of a significant percentage of persons with DS or their parents/guardians rejecting the procedure, which has limited the establishment of T21‐iPSC banks. Second, because the iPSCs cells are derived from epithelial cells that are not exposed to ultraviolet (UV) rays (such as skin fibroblasts or hair follicles), they are less likely to contain UV‐induced DNA damage and potential genetic mutations. Third, the cells produced here were reprogramed with nonintegrating methods, which also decreases the likelihood that these cells contain mutations that may affect the interpretation of results. Therefore, the methods described here represent a significant qualitative improvement in iPSC technology applied to the modeling of DS, and should be an important step toward the development of human cell‐based platforms for the high‐throughput screening of potential drugs aimed at improving the quality of life of persons with DS. Finally, some of these techniques may also find important applications in the study of other neurodevelopmental and neurodegenerative disorders.
Author Contributions
Y.M.L., B.L.Z., J.J.S.‐M., and M.W.J.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; A.C.S.C.: conception and design, financial support, assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Supporting Information
Acknowledgments
This work was supported by grants from ALANA USA Foundation (Contracts 124124 and 200381), the Alana CWRU/MIT Collaborative Fund, a grant from the Ohio Department of Developmental Disabilities, and charitable contributions from the Awakening Angels Foundation to A.C.S.C. B.L.Z. was supported by a Postdoctoral Fellowship from the Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil (CNPq/MCTI, 202237/2014‐1). We thank Dr. Stefanie Avril for the expert histological identification of tissue types in teratoma slides and Jenifer Mikulan for teratoma slide staining. We thank the participants of our ongoing clinical study (IRB number: 06‐14‐41; NCT02304302 at http://www.clinicaltrials.gov) and their parents/legal guardians for agreeing to provide urine samples for T21‐iPSC generation and further differentiation.
References
- 1. Parker SE, Mai CT, Canfield MA et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004‐2006. Birth Defects Res A Clin Mol Teratol 2010;88:1008–1016. [DOI] [PubMed] [Google Scholar]
- 2. Yates CM, Simpson J, Gordon A et al. Catecholamines and cholinergic enzymes in pre‐senile and senile Alzheimer‐type dementia and Down's syndrome. Brain Res 1983;280:119–126. [DOI] [PubMed] [Google Scholar]
- 3. Nadel L. Down's syndrome: A genetic disorder in biobehavioral perspective. Genes Brain Behav 2003;2:156–166. [DOI] [PubMed] [Google Scholar]
- 4. Patterson D, Costa AC. Down syndrome and genetics ‐ a case of linked histories. Nat Rev Genet 2005;6:137–147. [DOI] [PubMed] [Google Scholar]
- 5. Weijerman ME, van Furth AM, van der Mooren MD et al. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr 2010;169:1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol 2011;164:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costa AC, Scott‐McKean JJ. Prospects for improving brain function in individuals with Down syndrome. CNS Drugs 2013;27:679–702. [DOI] [PubMed] [Google Scholar]
- 8. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 9. Hibaoui Y, Feki A. Concise review: Methods and cell types used to generate down syndrome induced pluripotent stem cells. J Clin Med 2015;4:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu HE, Yang YC, Chen SM et al. Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from Trisomy 21 amniotic fluid cells. Exp Cell Res 2013;319:498–505. [DOI] [PubMed] [Google Scholar]
- 11. Zhou T, Benda C, Duzinger S et al. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol 2011;22:1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou T, Benda C, Dunzinger S et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc 2012;7:2080–2089. [DOI] [PubMed] [Google Scholar]
- 13. Afzal MZ, Strande JL. Generation of induced pluripotent stem cells from muscular dystrophy patients: Efficient integration‐free reprogramming of urine derived cells. J Vis Exp 2015;28:52032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen W, Huang J, Yu X et al. Generation of induced pluripotent stem cells from renal tubular cells of a patient with Alport syndrome. Int J Nephrol Renovasc Dis 2015;8:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Si‐Tayeb K, Idriss S, Champon B et al. Urine‐sample‐derived human induced pluripotent stem cells as a model to study PCSK9‐mediated autosomal dominant hypercholesterolemia. Dis Model Mech 2016;9:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang SZ, Ma LX, Qian WJ et al. Modeling neurological disease by rapid conversion of human urine cells into functional neurons. Stem Cells Int 2016;2016:2452985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Briggs JA, Mason EA, Ovchinnikov DA et al. Concise review: New paradigms for Down syndrome research using induced pluripotent stem cells: Tackling complex human genetic disease. Stem Cells Transl Med 2013;2:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009;27:2667–2674. [DOI] [PubMed] [Google Scholar]
- 19. Yu J, Hu K, Smuga‐Otto K et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009;324:797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Briggs JA, Sun J, Shepherd J et al. Integration‐free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells 2013;31:467–478. [DOI] [PubMed] [Google Scholar]
- 21. Luong MX, Auerbach J, Crook JM et al. A call for standardized naming and reporting of human ESC and iPSC lines. Cell Stem Cell 2011;8:357–359. [DOI] [PubMed] [Google Scholar]
- 22. Meisner LF, Johnson JA. Protocols for cytogenetic studies of human embryonic stem cells. Methods 2008;45:133–141. [DOI] [PubMed] [Google Scholar]
- 23. Padilla‐Nash HM, Barenboim‐Stapleton L, Difilippantonio MJ et al. Spectral karyotyping analysis of human and mouse chromosomes. Nat Protoc 2006;1:3129–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vazin T, Ball KA, Lu H et al. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: A model system to study neurotoxicity in Alzheimer's disease. Neurobiol Dis 2014;62:62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhattacharya S, Burridge PW, Kropp EM et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J Vis Exp 2014;25:52010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maclean GA, Menne TF, Guo G et al. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci USA 2012;109:17567–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Draper JS, Smith K, Gokhale P et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol 2004;22:53–54. [DOI] [PubMed] [Google Scholar]
- 28. Mitalipova MM, Rao RR, Hoyer DM et al. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol 2005;23:19–20. [DOI] [PubMed] [Google Scholar]
- 29. Hanson C, Caisander G. Human embryonic stem cells and chromosome stability. APMIS 2005;113:751–755. [DOI] [PubMed] [Google Scholar]
- 30. Bosman A, Letourneau A, Sartiani L et al. Perturbations of heart development and function in cardiomyocytes from human embryonic stem cells with trisomy 21. Stem Cells 2015;33:1434–1446. [DOI] [PubMed] [Google Scholar]
- 31. Tang YC, Williams BR, Siegel JJ et al. Identification of aneuploidy‐selective antiproliferation compounds. Cell 2011;144:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iglesias‐Bartolome R, Patel V, Cotrim A et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation‐induced mucositis. Cell Stem Cell 2012;11:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011;192:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holland H, Ahnert P, Koschny R et al. Detection of novel genomic aberrations in anaplastic astrocytomas by GTG‐banding, SKY, locus‐specific FISH, and high density SNP‐array. Pathol Res Pract 2012;208:325–330. [DOI] [PubMed] [Google Scholar]
- 35. Li LB, Chang KH, Wang PR et al. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell 2012;11:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amano T, Jeffries E, Amano M et al. Correction of Down syndrome and Edwards syndrome aneuploidies in human cell cultures. DNA Res 2015;22:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang J, Jing Y, Cost GJ et al. Translating dosage compensation to trisomy 21. Nature 2013;500:296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaur G, Sharma A, Xu W et al. Glutamatergic transmission aberration: A major cause of behavioral deficits in a murine model of Down's syndrome. J Neurosci 2014;34:5099–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boada R, Hutaff‐Lee C, Schrader A et al. Antagonism of NMDA receptors as a potential treatment for Down syndrome: A pilot randomized controlled trial. Transl Psychiatry 2012;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Costa AC. The glutamatergic hypothesis for Down syndrome: The potential use of N‐methyl‐D‐aspartate receptor antagonists to enhance cognition and decelerate neurodegeneration. CNS Neurol Disord Drug Targets 2014;13:16–25. [DOI] [PubMed] [Google Scholar]
- 41. Stoll C, Alembik Y, Dott B et al. Study of Down syndrome in 238,942 consecutive births. Ann Genet 1998;41:44–51. [PubMed] [Google Scholar]
- 42. de Rubens Figueroa J, del Pozzo Magana B, Pablos Hach JL et al. [Heart malformations in children with Down syndrome]. Rev Esp Cardiol 2003;56:894–899. [DOI] [PubMed] [Google Scholar]
- 43. Mendonca GV, Pereira FD, Fernhall B. Reduced exercise capacity in persons with Down syndrome: Cause, effect, and management. Ther Clin Risk Manag 2010;6:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guerra M, Llorens N, Fernhall B. Chronotropic incompetence in persons with down syndrome. Arch Phys Med Rehabil 2003;84:1604–1608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information