

Abstract
Diabetes is a major global health issue and the number of individuals with type 1 diabetes (T1D) and type 2 diabetes (T2D) increases annually across multiple populations. Research to develop a cure must overcome multiple immune dysfunctions and the shortage of pancreatic islet β cells, but these challenges have proven intractable despite intensive research effort more than the past decades. Stem Cell Educator (SCE) therapy—which uses only autologous blood immune cells that are externally exposed to cord blood stem cells adhering to the SCE device, has previously been proven safe and effective in Chinese and Spanish subjects for the improvement of T1D, T2D, and other autoimmune diseases. Here, 4‐year follow‐up studies demonstrated the long‐term safety and clinical efficacy of SCE therapy for the treatment of T1D and T2D. Mechanistic studies found that the nature of platelets was modulated in diabetic subjects after receiving SCE therapy. Platelets and their released mitochondria display immune tolerance‐associated markers that can modulate the proliferation and function of immune cells. Notably, platelets also expressed embryonic stem cell‐ and pancreatic islet β‐cell‐associated markers that are encoded by mitochondrial DNA. Using freshly‐isolated human pancreatic islets, ex vivo studies established that platelet‐releasing mitochondria can migrate to pancreatic islets and be taken up by islet β cells, leading to the proliferation and enhancement of islet β‐cell functions. These findings reveal new mechanisms underlying SCE therapy and open up new avenues to improve the treatment of diabetes in clinics. Stem Cells Translational Medicine 2017;6:1684–1697
Keywords: Diabetes, Type 1 diabetes, Stem cell, Platelet, Mitochondria, Stem cell educator, Islet β cell, Immune
Significance Statement.
Abnormalities in multiple immune cells and the deficit of insulin‐producing cells are two crucial common issues in T1D and T2D. Insulin therapy is not a cure. Stem Cell Educator (SCE) therapy is a closed‐loop system that circulates the patient's immune cells, briefly cocultures with adherent cord blood (CB) stem cells in vitro, and returns only the “educated” autologous immune cells to the patient's circulation. We found that the feature of platelets was changed in diabetic subjects after receiving SCE therapy. Notably, mitochondria released by platelets displayed immune tolerance‐, embryonic stem cell‐, and islet β‐cell‐associated markers, leading to the improvement of islet β‐cell functions. Long‐term follow‐up studies demonstrated the safety and clinical efficacy of SCE therapy for the treatment of T1D and T2D.
Introduction
The epidemic of diabetes is a major public health issue worldwide, with prevalence rates exceeding 12.1% of the population in India, 11.6% in China 1, 2, and 9.3% in the United States. Animal and clinical evidence shows that abnormalities in multiple types of immune cells (e.g., T cells, B cells, regulatory T cells [Tregs], monocytes/macrophages [Mo/Mϕs], and dendritic cells [DC]) contribute to the autoimmunity in type 1 diabetes (T1D) 3 and the chronic inflammation that induces insulin resistance in type 2 diabetes (T2D) 4, 5, 6, 7, 8, 9, 10, 11. Thus, overcoming these multiple immune dysfunctions is necessary for both the prevention and treatment of T1D and T2D. Specifically for T1D, a true cure has proven elusive despite intensive research effort using conventional approaches over the past 30 years 3, 12, 13. Several recent clinical trials 13, 14, 15, 16 highlight the challenges in conquering T1D, but their failures provide some valuable lessons about limitations of conventional immune therapy and the future direction of the quest. Specifically, they point to the need for an approach that produces comprehensive immune modulation at both the local pancreatic and systemic levels rather than targeting the pancreatic effects of one or a few components of the immune system.
Deficit of insulin‐producing cells is another crucial common issue for T1D and T2D 17, 18. Although daily insulin injections offer limited control over blood sugar levels, they are not a cure. To overcome the shortage of insulin‐producing cells in diabetic patients, pancreas and islet transplantations have offered potential treatments for independence from insulin injections. However, the scarcity of donors and immune rejections severely hinder their wide application. To date, functional insulin‐producing cells have been generated from embryonic stem (ES) cells and induced pluripotent stem cells (iPSCs) through ex vivo inductions 19, 20, 21, 22. Recent advances in stem cell biology have realized that ES cells, iPSCs, and their derived cells can also cause immune rejections post transplantation 23, 24, 25, in addition to ethical and safety issues. Thus, this compelling need brings a sense of urgency to find a cure for diabetes that can not only overcome the shortage of insulin‐producing β‐cells, but also halt the progression of autoimmunity in T1D and correct multiple immune dysfunctions in T2D in the clinical setting.
CB‐derived multipotent stem cells (CB‐SCs) are a unique type of stem cell identified from human CB 26, 27 and different from other known stem cell types such as mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs) 28, and previously characterized monocyte‐derived pluripotent stem cells 29. Based on their unique properties in immune modulation 27, 30, we developed a new technology, called Stem Cell Educator (SCE) therapy, by using CB‐SCs in a closed‐loop system that circulates a patient's blood through a blood cell separator, briefly co‐cultures the patient's lymphocytes with adherent CB‐SCs in vitro, and returns the “educated” lymphocytes (but not the CB‐SCs) to the patient's circulation 28, 31. A single treatment with SCE therapy provides lasting reversal of autoimmunity, leading to the regeneration of islet β cells and improvement of metabolic control in individuals with longstanding T1D 31, 32 and T2D 33. Specifically, a recent phase I/II study demonstrated the safety and feasibility of SCE therapy in the treatment of alopecia areata (AA) 34, one of the most common T cell‐mediated autoimmune diseases. This trial provided visible evidence that SCE therapy can control autoimmunity and lead to hair regrowth 34. Thus, SCE therapy may revolutionize the clinical treatment of diabetes and other autoimmune‐related diseases without the safety and ethical concerns associated with conventional stem cell‐based approaches. Although SCE therapy appears to be effective, its physiological mechanisms are not fully understood in humans.
We found that platelets were elevated in patients following SCE therapy, prompting further investigation of the possibility platelets may be responsible for long‐lasting clinical effects observed following SCE therapy. We found that platelets display immune tolerance markers, embryonic stem (ES) cell‐associated markers, and human islet β cell‐specific transcription factors that may contribute to the improvement in T1D and T2D pathology and symptoms. These factors are encoded in the platelets' mitochondrial DNA (mitoDNA), which is released from the platelets upon activation. Here we show that mitochondria can migrate into pancreatic islet β cells, resulting in the marked improvement of human islet β‐cell functions. These findings hold the potential for enormous clinical impact on T1D and T2D by paving the way toward the development of novel approaches to treat and prevent diabetes.
Materials and Methods
Patients
Diabetic subjects recruited from three hospitals were enrolled in phase I, phase II, open‐label multi‐center clinical trials. At the Section of Endocrinology at the General Hospital of Jinan Military Command (Jinan, Shandong, China) and the first affiliated hospital of Hebei Medical University, T1D subjects (N = 17) and T2D subjects (N = 26) were enrolled in trials conducted from October 2010 through January 2011 and from August 2011 through October 2013, respectively. At the Endocrinology and Nutrition Service, Hospital Universitario Central de Asturias (Oviedo, Spain), T1D patients (n = 8) were enrolled in a trial conducted from November 27, 2012 through October 1, 2014. With oversight from a planning committee, the principal investigators designed the trial and received ethical approval for the clinical treatment protocol and consent form for each hospital. Patients were qualified for enrollment if they met the diagnosis standards of the American Diabetes Association and if a blood test confirmed the presence of at least one autoantibody to pancreatic islet β cells for T1D subjects. Exclusion criteria included clinically significant liver, kidney, or heart disease; pregnancy; immunosuppressive medication; viral diseases; or diseases associated with immunodeficiency. The primary study endpoints on feasibility and safety of the SCE therapy have been published elsewhere 31, 32, 33. Endpoints relevant to the current study were preliminary evidence of effects of SCE therapy on platelets in T1D and T2D subjects. Baseline blood samples were collected prior to SCE therapy. Four‐year follow‐up studies in Chinese T1D subjects (N = 9) and T2D subjects (N = 21) were performed at the General Hospital of Jinan Military Command. Trial registration: ClinicalTrials.gov numbers, NCT01350219 for T1D trial and NCT01415726 for T2D trial.
Treatment of Human PBMCs with Platelet‐Derived Mitochondria
Human buffy coat blood units were purchased from the New York Blood Center (New York, NY, http://nybloodcenter.org/). Human peripheral blood‐derived mononuclear cells (PBMC) were harvested as previously described 31, 33. As described elsewhere 34, PBMC were stimulated for 2–4 days with Dynabeads coupled with anti‐CD3 and anti‐CD28 antibodies (Life Technologies, Grand Island, NY, https://www.thermofisher.com/us) in the presence of 30 U/ml recombinant human IL‐2 (rIL‐2) (R&D Systems, Minneapolis, MN, www.rndsystems.com/), with or without mitochondria (70 μg/ml), and incubated at 37°C, in 8% CO2. The mitochondria were isolated from PB‐platelets using the Qproteome Mitochondria Isolation kit (Qiagen, Hidden, Germany, https://www.qiagen.com/) according to the manufacturer's recommended protocol. The concentration of mitochondria was determined by the measurement of protein concentration using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, https://www.thermofisher.com/). Cells were collected on day 2 and 4, respectively for cell proliferation assay and flow cytometry.
Treatment of Human Pancreatic Islets with Platelet‐Derived Mitochondria
Freshly isolated human pancreatic islets were purchased from Lonza (San Francisco, CA, www.lonza.com/). In one set of experiments, pancreatic islets were dissociated into single cells with 0.25% trypsin/EDTA for 5 minutes at room temperature. The pancreatic islet cells (1 × 105 cells/ml) seeded at the top inserts were co‐cultured with MitoTracker Deep Red‐labeled platelets (bottom) in Transwell culture system with 0.4 μm pore size (EMD Millipore, Billerica, MA, www.emdmillipore.com/). Islet cells were collected at different time points for kinetic flow cytometry at 1, 2, 4, 6, 20, and 24 hours. After seven days, the treated and untreated islet cells were collected to examine cell viability and viable cell number with a TC20 automated cell counter (Bio‐Rad, Hercules, CA, www.bio-rad.com/).
In another set of experiments, whole pancreatic islets seeded in the top inserts at 125 IEQ per well were treated with platelet‐derived mitochondria (250 μg/ml, bottom well) in 12‐well tissue culture‐treated plates with Transwell inserts (0.4 μm pore size) at 37°C in 8% CO2. Pancreatic islets cultured in pancreatic islet medium (Lonza, San Fransisco, CA, www.lonza.com/) served as controls. The mitochondria were isolated from platelets using the Qproteome Mitochondria Isolation kit (Qiagen, Hidden, Germany, https://www.qiagen.com/) according to the manufacturer's recommended protocol. After 7 days, mitochondrion‐treated and untreated pancreatic islets were collected for multiple functional studies such as flow cytometry, cell viability, and insulin/C‐peptide release.
To determine which molecules contribute to the interaction between mitochondria and islet β cells, the dissociated pancreatic islet cells were initially pre‐treated with 20 μg/ml anti‐CD29 and/or 20 μg/ml anti‐TLR4 Abs at 37°C for 30 minutes followed by adding the MitoTracker Green‐labeled mitochondria (125 mg/ml) isolated from platelets. The islet cells co‐cultured with mitochondria in the absence of blocking Abs served as control. After 5 hours, the mitochondrion‐treated and untreated islets cells were washed and fixed and permeabilized by using PerFix‐nc (no centrifuge assay) kit (Beckman Coulter, Jersey City, NJ, https://www.beckmancoulter.com/) and followed by an intra‐cellular staining with islet β‐cell marker insulin for flow cytometry.
Islet β Cell Function and C‐Peptide Release
Freshly isolated human pancreatic islets (Lonza, San Francisco, CA, www.lonza.com/) at 125 IEQ per well (top insert) were treated with platelet‐derived mitochondria (250 μg/ml, bottom well) in 12‐well tissue culture‐treated plates with Transwell inserts at 37°C in 8% CO2. Untreated pancreatic islets cultured in pancreatic islet medium (Lonza, San Francisco, CA, www.lonza.com/) served as controls. After 7 days, the mitochondrion‐treated and untreated pancreatic islets were collected into 2 ml conical tubes, washed twice with Kreb's buffer (137 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 2.5 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, 0.1% BSA), and then incubated with 1 ml Kreb's buffer for 30 minutes in the absence of glucose at 37°C. After removing the Kreb's buffer, pancreatic islets were incubated with different concentrations of glucose (5.5 and 16.7 mM) and/or 10 μM tolbudamide (Sigma, St. Louis, MO, https://www.sigmaaldrich.com/) in 300 μl Kreb's buffer/well for one hour at 37°C. Supernatants were collected from each group after incubation. C‐peptide levels were examined using the Quantikine ELISA kit (R&D Systems, Minneapolis, MN, www.rndsystems.com/) following the manufacturer's protocol.
Statistical Analysis
An intention‐to treat approach was used, with 36 diabetic patients undergoing SCE therapy. The primary efficacy endpoints were change in platelet numbers between baseline and follow‐up. Statistical analyses of data were performed by the two‐tailed paired Student's t test to determine statistical significance between baseline and follow‐up. Values were given as mean ± SD (standard deviation).
Results
Long‐Term Safety and Clinical Efficacy of SCE Therapy in T1D and T2D Diabetic Subjects
Previous work demonstrated the short‐term safety and clinical efficacy of SCE therapy for the treatment of Chinese T1D 28, 31 and T2D subjects 33 during a year. To define the long‐term safety of SCE therapy, SCE‐treated subjects (total N = 30, including 9 T1D and 21 T2D patients) received the 4‐year follow‐up studies with safety profiles. Serological tests demonstrated that no subjects showed a marked increase in tumor markers including alpha‐fetoprotein (AFP), carcinoembryonic antigen (CEA), cancer antigen (CA) 19–9, CA72–4, CA125, CA15–3, and neuron‐specific enolase (NSE). Chest x‐ray failed to show lung cancer and other abnormalities in these subjects. Ultrasonography did not find tumor formation in livers, gallbladders, pancreata, spleens, and kidneys of these participants. Additionally, no subject experienced any significant adverse events during the 4‐year follow‐up period. Thus, these clinical data confirmed the long‐term safety of SCE therapy.
To determine the long‐term clinical efficacy, we performed 4‐year follow‐up studies in some of these subjects after receiving one treatment with the SCE therapy. Two T1D participants (one female having T1D for 8 months and another male having T1D for 5 months) achieved normal fasting C‐peptide levels and remained completely recovered with no relapse after 4 years (Fig. 1A). The C‐peptide response following a 75‐g oral glucose tolerance test (OGTT) further confirmed the normalization of their islet β cell function (Fig. 1B). To our knowledge, this is the first long‐term clinical study demonstrating that islet β cell function has been completely and safely rescued in recent‐onset T1D subjects after controlling the autoimmunity by the SCE therapy.
Figure 1.
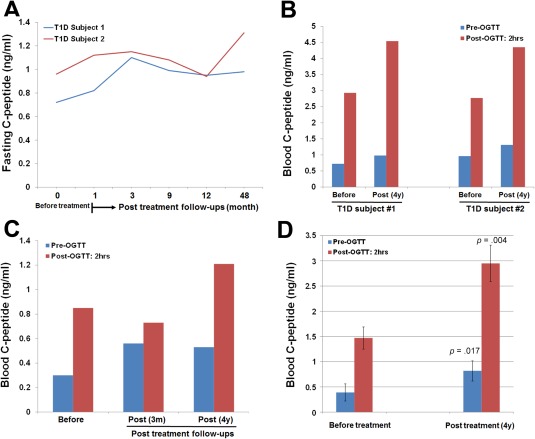
Long‐term follow‐up studies of SCE therapy in Chinese T1D and T2D subjects. All Chinese subjects received one treatment with the SCE therapy. (A): Kinetic examination of fasting C‐peptide levels during the 4‐year follow‐up period. Before receiving a SCE therapy, subject #1 diagnosed with T1D for 8 months with glycated hemoglobin (HbA1C) at 11.3%, islet autoantibodies IA‐2A+ and GAD+; subject 2 diagnosed with T1D for 5 months with HbA1C at 9.4%, islet autoantibodies IA‐2A+ and GAD+ and IAA+. (B): Comparison of C‐peptide levels at a 75‐g oral glucose challenge after 4‐year follow‐up in T1D subjects 1 and 2. (C): Recovered fasting and high glucose‐stimulated C‐peptide levels were retained in subject 3 through the follow‐up at 4 years post‐treatment with a SCE therapy. (D): Comparison of fasting and high glucose‐stimulated C‐peptide levels after 4‐year follow‐up in long‐standing severe T2D subjects (N = 4). Data are presented as mean ± SD. Abbreviation: OGTT, oral glucose tolerance test.
Additionally, subject 3, who had T1D 4 years at the time of treatment, still achieved long‐lasting improvements, including an increase in fasting C‐peptide from 0.3 ng/ml at basal to 0.56 ng/ml at 12 weeks and retained 0.53 ng/ml at 4 years. Glucose‐stimulated C‐peptide levels were increased from 0.85 ng/ml at basal to 1.21 ng/ml at 4 years (Fig. 1C). Other six long‐standing T1D participants showed the decline of fasting C‐peptide at different levels at 4‐year follow‐ups. The data suggest that one treatment with SCE therapy was not enough to control the islet autoimmunity in these patients and additional treatment post 3 months is needed to promote the regeneration of islet β cells.
To explore the long‐term clinical efficacy of SCE therapy in T2D subjects, long‐standing (15–24 years) severe T2D subjects (n = 6) received 4‐year follow‐up studies. The data demonstrated that 4/6 T2D subjects showed a significant increase in fasting C‐peptide from 0.39 ± 0.17 ng/ml at basal to 1.12 ± 0.33 ng/ml at 1 year, and retained normal level 0.82 ± 0.2 ng/ml at 4 years (p = .017). Their C‐peptide levels following OGTT were also markedly increased from 1.47 ± 0.22 ng/ml at basal to 2.95 ± 0.36 ng/ml at 4 years (p = .004) (Fig. 1D). Thus, these data revealed the long‐lasting therapeutic efficacy of SCE therapy in T1D and T2D subjects after receiving one treatment with SCE therapy.
Clinical Efficacy Outcomes in Modulating Platelets by SCE Therapy
To explore clinical mechanisms underlying the above long‐lasting therapeutic effects of SCE therapy, we analyzed the complete blood count (CBC) test results and found that platelets were elevated in diabetic patients following SCE therapy for 1 month, prompting further investigation of the possibility platelets may be responsible for some of the effects observed following SCE therapy. Modulations of platelets were examined in 10 T1D and 16 T2D patients receiving SCE therapy. Age‐ and gender‐matched T1D (n = 7) and T2D (n = 10) subjects who received conventional therapies served as controls, respectively. Clinical results demonstrated that platelet counts were markedly increased in T1D (p = .017) and T2D subjects (p = .027) after receiving the SCE therapy (Fig. 2A). Platelet distribution width (PDW) was significantly increased (Fig. 2B), and mean platelet volume (MPV) was decreased (Fig. 2C) in both T1D and T2D subjects after SCE therapy. No changes were observed in plateletcrit (PCT) following treatment. Clinical data from Caucasian T1D subjects (n = 8) also showed an increase in the platelet count after SCE therapy (p = .04, Fig. 2D). These data suggest that modulations in platelet populations may contribute to the clinical efficacy of SCE therapy for diabetic patients 31, 32, 33.
Figure 2.
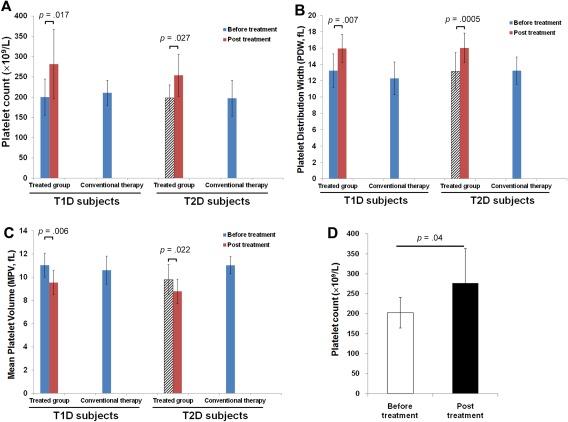
Modulation of platelets by SCE therapy in diabetic patients. (A–C): Clinical data were summarized from the clinical trials in Chinese diabetic subjects. Both T1D (N = 10) and T2D (N = 16) received one treatment with SCE therapy. A complete blood count (CBC) test was performed at baseline (before the treatment) and 1 month after the treatment with SCE therapy. Age‐ and gender matched T1D (N = 7) and T2D (N = 10) subjects receiving conventional therapy served as controls. (A): Increase in the platelet count. (B): Increase in the platelet distribution width (PDW). (C): Decrease in the mean platelet volume (MPV). (D): Increase in the platelet count of Caucasian T1D subjects after receiving one treatment with SCE therapy, N = 8. Data are presented as mean ± SD.
Expression of Immune Tolerance‐Related Markers in Platelets and Mitochondria
To determine the expression of immune tolerance‐related markers in platelets, flow cytometry demonstrated that CB‐derived platelets (Fig. 3A) displayed several co‐inhibitory surface molecules including the programmed death ligand 1 (PD‐L1, CD274), CD270 (a herpes virus entry mediator, HVEM), ICOS (inducible costimulator), and Galectin 9. In contrast, high percentages of platelets expressed PD‐L1 (80.31% ± 10.48%) and CD270 (91.55% ± 5.18%), and almost no platelets expressed ICOS (0.43% ± 0.73%) on the adult peripheral blood (PB)‐platelets (Fig. 3B). Galectin 9 was expressed on PB‐platelets in four individuals (4/8 donors, 54.07% ± 27.59%), but highly positive on CB‐platelets (n = 9 donors, 94.09% ± 7.44%) (Fig. 3A). Intra‐cellular staining showed that they also express the cytokine transforming growth factor β 1 (TGF‐β1) (Fig. 3C, 3D). We also found that 81.31% ± 11.6% of CD41+ CB‐platelets and 89.92% ± 4.12% of CD41+ PB‐platelets display the immune modulation‐associated transcription factor FoxP3 (Fig. 3E). Notably, Western blotting and flow cytometry revealed the expression of autoimmune regulator (AIRE) in CB‐ (Fig. 3F, 3G) and PB‐platelets (Fig. 3H, 3I). Additional flow cytometry showed that platelets displayed high levels of chemokine receptors CXCR4 and CCR3; an intermediate level of CCL2; low levels of CXCL10, CCR4, CCR5, CCR7, CXCR1, CXCR2, CXCR3, and CD62L; and no expression of CXCL1 (Supporting Information Fig. S1).
Figure 3.
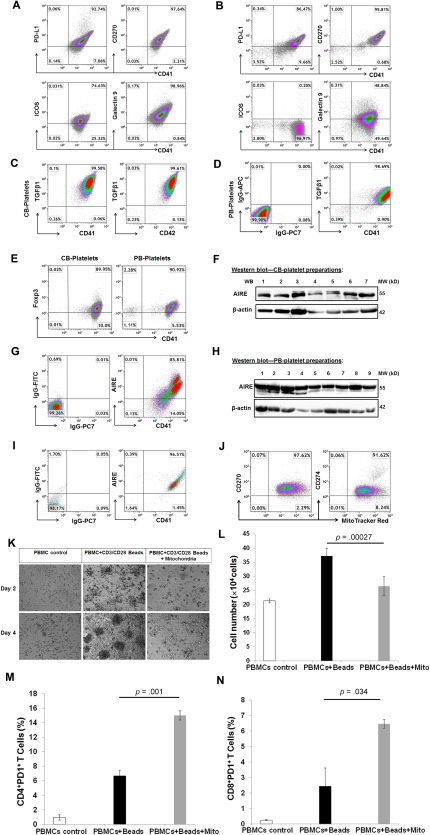
Expression of immune modulation‐related markers in platelets and mitochondria. (A): Flow cytometry showed expression of co‐inhibitory surface markers PD‐L1, CD270, ICOS, and Galectin 9 on CB‐platelets (n = 9). (B): Flow cytometry showed the expression of co‐inhibitory surface markers PD‐L1, CD270, ICOS, and Galectin 9 on PB‐platelets (n = 8). (C): Intra‐cellular staining showed the expression of TGF‐β1 in CB‐platelets (n = 8). (D): Intra‐cellular staining showed the expression of TGF‐β1 in PB‐platelets (n = 4). (E): Intra‐cellular staining showed the expression of FoxP3 in CB‐ and PB‐platelets (n = 9 and 4, respectively). (F): Western blotting showed the expression of AIRE in seven cord blood preparations. (G): Double staining showed the expression of AIRE in CD41+ CB‐platelets (n = 5). (H): Western blotting showed the expression of AIRE in nine adult blood preparations. (I): Double staining showed the expression of AIRE in CD41+ PB‐platelets (n = 3). (J): Flow cytometry showed the expression of CD270 and CD274 (PD‐L1) on CB platelet‐releasing mitochondria labeled with MitoTracker Red. N = 7. (K): Phase contrast microscopy. Human PBMCs were activated with Dynabeads coupled with anti‐CD3 and anti‐CD28 antibodies, 30 U/ml rIL‐2 for 4 days, in absence (middle panels) and presence (right panels) of 70 μg/ml mitochondria. Untreated PBMCs (left right panel) served as control. Original magnification, ×40. (L): Quantification of cell proliferation after the treatment for 2 days. (M): Flow cytometry showed the increase in the percentage of CD4+PD1+ T cells. (N): Flow cytometry showed the increase in the percentage of CD8+PD1+ T cells. Data are presented as mean ± SEM from four experiments. Isotype IgGs serve as negative control for flow cytometry. Abbreviations: PBMC, peripheral blood‐derived mononuclear cells; ICOS, inducible costimulatory.
Platelets are anucleate cells without human genomic DNA. To identify the origin of these immune marker‐related genes, mitochondria were purified from CB‐ and PB‐platelets to be explored for the gene transcriptions of mitochondria DNA (MitoDNA). Real time PCR Array revealed expressions of human T cell anergy and immune tolerance‐related genes (Supporting Information Fig. S2). Flow cytometry further proved that platelet‐releasing mitochondria displayed immune tolerance‐related markers CD270 and CD274 (PD‐L1) (Fig. 3J). To determine the immune modulation of mitochondria on T cells, peripheral blood‐derived mononuclear cells (PBMCs) were treated with allogeneic PB‐platelet‐derived mitochondria in the presence of Dynabeads coupled with anti‐CD3 + anti‐CD28 antibodies + recombinant human IL‐2 (rIL‐2). After ex‐vivo expansion of T cells with this mAb combination for 4 days, there were large numbers of cell clusters with different sizes floating in the supernatant (Fig. 3K, bottom middle panel), it suggestive of significant T cell proliferation. However, this phenomenon was not evident in the presence of mitochondria (Fig. 3K, right panels). The cell number was markedly declined after the combination treatment with mitochondria (Fig. 3L). Next, we examined the expression of an immune checkpoint receptor PD‐1 (programmed death receptor‐1) on T cells, which is the ligand of PD‐L1 (CD274). Notably, the percentage of CD4+PD1+ T cells was increased to 15% ± 0.64% following treatment with mitochondria (Fig. 3M) (p = .001). Further flow cytometry indicated that the percentage of CD8+PD1+ T cells was also improved from 2.43% ± 1.18% to 6.46% ± 0.28% in the presence of mitochondria (Fig. 3N) (p = .034). Thus, these findings demonstrated that platelets and mitochondria have the requisite cellular molecules to act as novel immune modulators and induce immune tolerance.
Platelets Display Human Pancreatic Islet β Cell‐Related Markers
To explore whether platelets contribute to regeneration of islet β cells, we examined human islet β cell‐specific markers 19, 22 including insulin and C‐peptide production, transcription factors (MAFA, PDX1, NKX6.1, NEUROD1, and NGN3), KATP channel proteins (Sur1 and Kir6.2), and glucokinase (GCK). Real time PCR data revealed the clear expressions of insulin, MAFA, and Sur1 mRNAs in CB‐platelets (n = 6); the Kir 6.2 mRNA was displayed in most samples (5/6); a few samples (1/6) were positive for GCK (Fig. 4A, 4B); however, we found no expression or very weak expression of PDX1, NKX6.1, NEUROD1, and NGN3 (Fig. 4B). Interestingly, CB‐platelets displayed the pancreatic islet δ cell‐released hormone somatostatin and ɛ cell marker Ghrelin mRNAs, with very weak or no expression of glucagon and PPY mRNAs (Fig. 4A). Western blotting further demonstrated the expression of MAFA protein in CB‐platelets (Fig. 4C). Using freshly isolated human pancreatic islet cells as positive control (Fig. 4D), flow cytometry confirmed diverse expression of relevant markers including 39.47% ± 20.6% of CD42+Insulin+, 40.74% ± 6.32% of CD42+C‐peptide+, 28.52% ± 18.56% of CD41+MAFA+, 0.24% ± 0.23% of CD42+PDX1+, 0.6% ± 0.42% of CD42+NKX6.1+, and 3.42% ± 0.9% of CD42+Glucagon1+, and 3.87% ± 1.4% of CD41+SST+ in CB‐platelets (Fig. 4E, 4F).
Figure 4.
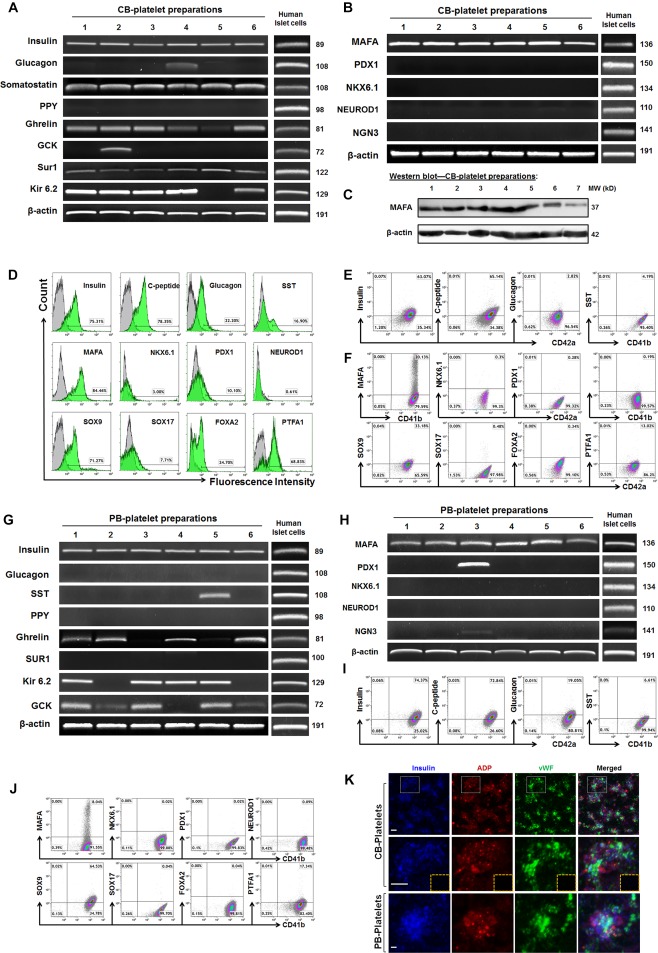
Expression of pancreatic islet β cell‐related markers in platelets. (A): Real time PCR analysis of pancreatic islet‐related hormone products and β‐cell‐related functional markers in CB‐platelets (n = 6). Freshly isolated human islets served as positive controls. (B): Real time PCR analysis of pancreatic islet β‐cell‐related transcription factors in CB‐platelets (n = 6). (C): Western blotting shows the protein expression of an islet β cell‐specific transcription factor MAFA in CB‐platelets. (D): Flow cytometry for human pancreatic islet‐related hormone products in freshly‐isolated human pancreatic islet cells. (E): Flow cytometry for the pancreatic islet‐related hormone products by double staining with platelet markers CD41 or CD42 in CB‐platelets. (F): Flow cytometry for pancreatic islet β‐cell‐related transcription factors by double staining with platelet markers CD41 or CD42 in CB‐platelets (n = 7). (G): Real time PCR analysis of pancreatic islet‐related hormone products and β‐cell‐related functional markers in PB‐platelets (n = 6). (H): Real time PCR analysis of pancreatic islet β‐cell‐related transcription factors in PB‐platelets (n = 6). (I): Flow cytometry for pancreatic islet‐related hormone products by double staining with platelet markers CD41 or CD42 in PB‐platelets (n = 15). (J): Flow cytometry of pancreatic islet β‐cell‐related transcription factors in PB‐platelets (n = 8). (K): Confocal microscopy of human CB‐ and PB‐platelets after triple immunostainings with insulin (blue), dense granule marker ADP (red), and α granule marker vWF (green). Isotype‐matched IgGs served as controls (inserted yellow dashed rectangle). Scale bars, 5 μm. Representative data were from six experiments.
Additionally, flow cytometry revealed low expressions of normal β‐cell development‐related transcription factors (e.g., 23.27% ± 15.14% of CD42+PTFA1+, 1.46% ± 1.05% of CD42+FOXA2+, 0.48% ± 0.08% of CD42+SOX17+) (Fig. 4F). Additional studies in adult human PB‐platelets demonstrated the marker expression and patterns similar to the CB‐platelets (Fig. 4G–4J). In distinction to that, GCK mRNA was displayed in most samples (5/6), but only one sample (1/6) was positive for SST, and none expressed Sur1 in PB‐platelets (Fig. 4G). Confocal microscopy confirmed no typical insulin granules formations in the cytoplasm (Fig. 4K). Thus, these data confirmed that expression of islet β‐cell‐associated markers in human platelets, supporting their potential to promote the differentiation and regeneration of islet β cells.
Human Platelets: The Smallest Cell in Blood Holds the Stemness for Tissue Regenerations
To determine whether platelets contain embryonic stem (ES)‐related self‐renewal markers 35, flow cytometry using human platelets' markers CD41 and CD42 demonstrated that the highly‐purified CB‐platelets (>98% purity) highly display the ES‐associated pluripotent gene markers such as transcription factors octamer‐binding protein 4 (OCT4) and SRY‐box containing gene 2 (SOX2), with little or no expression of NANOG (Fig. 5A, 5B). Using human induced pluripotent stem cells (iPSCs) as positive control (Fig. 5C), similar data were obtained from adult human PB‐platelets (Fig. 5D). The mRNA expression of OCT4, SOX2, Kruppel‐like factor 4 (KLF4), and c‐myelocytomatosis oncogene (C‐MYC) was quantified by real time PCR analysis in CB‐ (Fig. 5E) and PB‐Platelets (Fig. 5G). Western blotting further confirmed the expression of OCT4, SOX2, CRIPTO, GATA‐4, and C‐MYC at protein levels in CB‐platelets (Fig. 5F). Similar data were obtained from PB‐platelets (Fig. 5H). Both NANOG mRNA and protein levels were very low or negative in CB‐platelets (Fig. 5E, 5F) and PB‐platelets (Fig. 5G, 5H). The data imply that platelets hold the stemness' markers that aid in modulating and reprogramming the proliferation and differentiation of adult human cells. To identify the origin of these stemness‐related genes, mitochondria were purified from CB‐ and PB‐platelets to be explored for the gene transcriptions of mitochondria DNA (MitoDNA). The RT2 Profiler PCR Array revealed the expressions of human stem cell‐related transcription factors (Supporting Information Fig. S3) and human stem cell‐associated markers (Supporting Information Fig. S4) in the mitochondria of both human CB‐ and PB‐platelets. The data confirm that the stemness' markers are localized in platelets' mitochondria.
Figure 5.
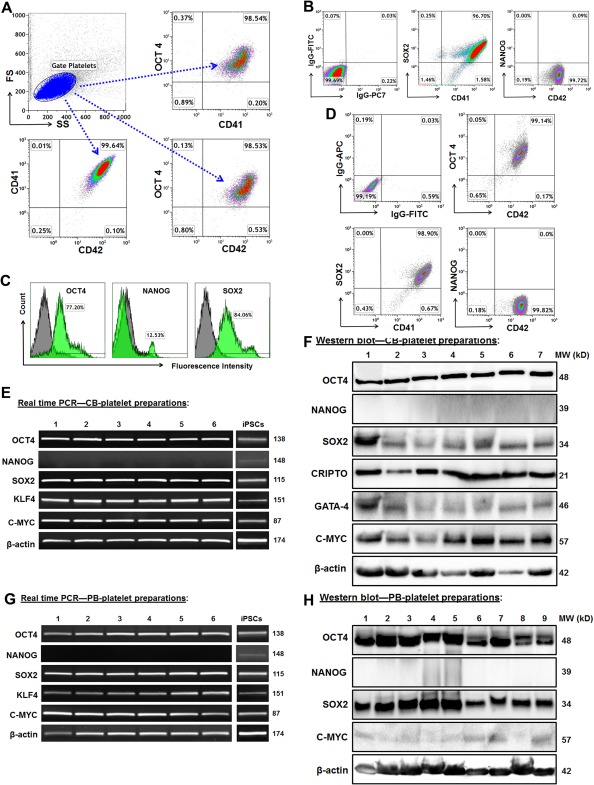
Platelets express human ES cell markers. (A): Analysis of purified CB‐platelets by flow cytometry. The gated platelets in dot plot (top left panel, blue) were analyzed by using platelet markers CD41 and CD42, together with ES marker OCT4. Representative data of those obtained from eight experiments. (B): Flow cytometry after double staining with CD41 and ES markers in CB‐platelets (n = 8). (C): Induced pluripotent stem cells (iPSCs) as positive control express the ES cell markers, with isotype‐matched IgGs as negative controls (grey). (D): Flow cytometry after double staining with CD41 and ES markers in PB‐platelets (n = 4). (E): Gene expressions of ES markers in CB‐platelets are demonstrated by electrophoresis of real time PCR products. Their expressions in iPSCs served as positive controls. (F): Western blot showed the protein expression of ES markers in CB‐platelets. (G): Real time PCR showed gene expressions of ES markers in PB‐platelets. (H): Western blot showed the protein expression of ES markers in PB‐platelets.
Mitochondria Released by Platelets Improve Function and Replication of Human Pancreatic Islet β Cells
To elucidate the interaction between platelets and pancreatic islets, firstly flow cytometry demonstrated that CB‐platelets could release mitochondria (Fig. 6A), and stimulation with different platelet aggregators altered the levels of mitochondria released (adenosine diphosphate (ADP) and arachidonic acid (ARA) increased release, but thrombin reduced release) (Fig. 6B). Using the Transwell co‐culture system to separate human islet cells (top chamber) from CB‐platelets (bottom chamber) (Fig. 6C), notably, kinetic studies by flow cytometry demonstrated that the MitoTracker‐labeled mitochondria released from CB‐platelets (bottom) can pass through the membrane (with a 0.4 μm pore size) of Transwells and are taken up by pancreatic islet cells (top chamber) (Fig. 6D).
Figure 6.
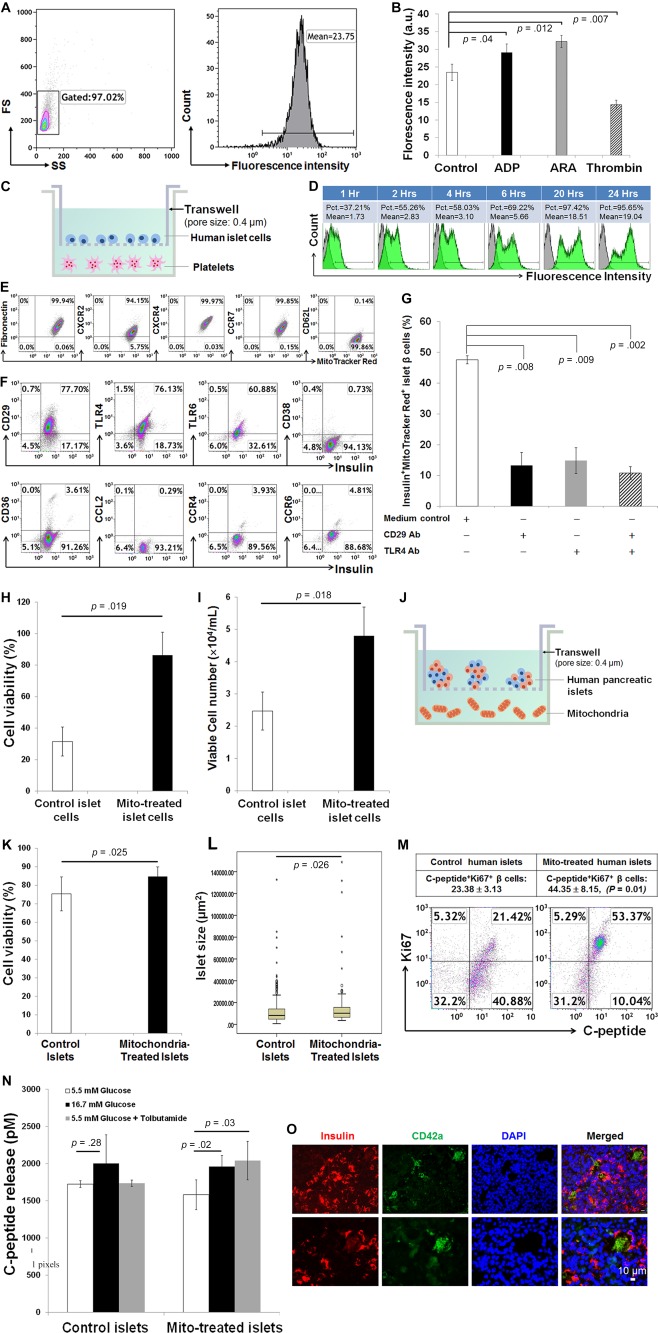
Improve pancreatic islet β‐cell function by the platelet‐derived mitochondria. (A): Flow cytometry showed the basal release of mitochondria from platelets in dot plot (left) and histogram (right). (B): Effects of platelet aggregators on the releasing of mitochondria. Representative data of those obtained from four experiments. (C): Co‐culture of freshly‐isolated human pancreatic single islet cells (top chamber) with platelets labeled with MitoTracker Deep Red (bottom chamber) in Transwells. (D): Kinetic measurements by flow cytometry demonstrated that human pancreatic islet cells take up MitoTracker Deep Red‐labeled mitochondria released from platelets (bottom) in Transwell co‐culture. (E): Expression of chemokine and chemokine receptors on CB‐platelet‐derived mitochondria (n = 11). (F): Expression of adhesion molecules and chemokine receptors on human islet β cells. Representative data of those obtained from three experiments. (G): Migration and the taking up of mitochondria by human islet β cells were markedly declined in the presence of blocking Abs with anti‐CD29 and/or anti‐TLR4. (H): Cell viability was increased after single islet β cells co‐cultured with platelets in Transwells. (I): The number of islet β cells was increased after co‐culture with platelets in Transwells. (J): Co‐culture of freshly‐isolated whole human pancreatic islets (top chamber) with platelets labeled with mitochondria (bottom chamber) in Transwells. The pore size of transmembrane is 0.4 μm. (K): Islet cell viability was increased after co‐cultured with mitochondria. (L): The average islet size was increased after co‐cultured with mitochondria. (M): Flow cytometry indicates that the percentage of Insulin+Ki67+ islet β cells was increased after co‐cultured with mitochondria in Transwells. Mitochondria were purified from CB‐platelets (n = 4). (N): Functional analysis demonstrated that the C‐peptide release from islet β cells was enhanced in the presence of different insulin secretagogues. Data are presented as mean ± SEM (n = 3 experiments). (O): Immunohistochemistry of human pancreatic tissues from diabetic patients (N = 6) showed the migration of platelets into islets, with a formation of platelet clusters (green color, a platelet marker CD42a) at different sizes and close to islet β cells (red color, a β‐cell marker insulin). Scale bar, 10 μm. Abbreviations: ADP, adenosine diphosphate; ARA, arachidonic acid; Pct, percentage.
Next, we clarified molecular mechanisms by which mitochondria migrate to islet β cells. Flow cytometry revealed that CB‐platelet‐derived mitochondria strongly express adhesion molecule and chemokine receptors such as fibronectin, CXC chemokine receptor 2 (CXCR2), CXCR4, and chemokine C‐C motif receptor 7 (CCR7) but are negative for CD62L (Fig. 6E). Human pancreatic islet β cells display the fibronectin ligand CD29 (integrin β1), Toll‐like receptor 4 (TLR4), and TLR6, but they were negative for or only modestly expressed type II transmembrane glycoprotein CD38, thrombospondin receptor CD36, C‐C motif chemokine ligand 2 (CCL2), C‐C motif chemokine receptor 4 (CCR4), and CCR6 (Fig. 6F). To determine which molecules are involved in the interaction between mitochondria and islet β cells, CB‐platelet‐derived mitochondria were co‐cultured with pancreatic islets in the presence of anti‐CD29 and/or anti‐TLR4 antibodies. The uptake of MitoTracker‐labeled mitochondria by islet β cells was markedly reduced (Fig. 6G), indicating that CD29 and TLR4 contribute to the adhesion and uptake of mitochondria by human islet β cells.
To determine the biological effects of platelet‐derived mitochondria on islet β cells, we co‐cultured trypsin/EDTA‐dissociated single pancreatic islet β cells with platelets in Transwells (Fig. 6C). Both cell viability and viable cell numbers were substantially increased after co‐culture for 7 days (Fig. 6H, 6I). We then co‐cultured whole pancreatic islets with CB‐platelet‐derived mitochondria in Transwells for 7 days (Fig. 6J), which also increased islet cell viability (Fig. 6K) and average islet size (p = .026, Fig. 6L). Using the insulin byproduct C‐peptide as indicator for β cells, flow cytometry demonstrated that the percentage of C‐peptide+Ki67+ islet β cells was strikingly enriched following co‐culture with mitochondria (Fig. 6M), and functional analysis demonstrated that the islet β cells exhibited improved C‐peptide release in response to insulin secretagogues (e.g., 16.7 mM high glucose and tolbutamide) (Fig. 6N). These ex vivo studies confirmed that pancreatic islet β‐cell function is improved after treatment with platelet‐derived mitochondria.
To explore the relationship between platelets and islet β cells, the distribution of platelets was examined in pancreatic islets of diabetic subjects. Immunohistochemistry studies demonstrated the migration of platelets into pancreatic islets in the form of single or clusters (Fig. 6O). We also tested the platelet distribution in pancreata of healthy donors (n = 2) and found there were no or only a few platelets scattered in or around pancreatic islets (Supporting Information Fig. S5). It was rare to find the cluster of platelets inside of healthy pancreatic islets. Therefore, platelets may migrate into pancreatic islets and release mitochondria that could be directly taken up by islet β cells, leading to the regeneration of β cells and long‐lasting clinical improvements of islet β‐cell function in diabetic patients after receiving the SCE therapy.
Discussion
Diabetes is a major public health issue worldwide. Abnormalities in multiple types of immune cells contribute to the autoimmunity in T1D and the insulin resistance in T2D. A comprehensive approach is needed to fundamentally address these multiple immune dysfunctions in the clinical setting and find a cure for diabetes. The current study substantiated the long‐lasting clinical efficacy of SCE therapy for the treatment of T1D and T2D subjects, with a complete recovery of islet β‐cell functions after one treatment with SCE therapy for 4 years. Additionally, we found expressions of immune tolerance‐ and ES cell‐associate characteristics in platelets and mitochondria. Pancreatic islet β cells can be reprogrammed to proliferate while maintaining good cell viability and restoring normal β‐cell function by taking up platelet‐releasing mitochondria. This novel mechanism may contribute to the long‐lasting clinical efficacy of SCE therapy in diabetic subjects after receiving one treatment with SCE therapy. Thus, these innovative approaches using platelets to protect and enrich islet β cells may mitigate safety and ethical concerns associated with other approaches that involve the transplantation of insulin‐producing cell surrogates or the viral/drug‐induced transduction of pancreatic cells.
By the time T1D is diagnosed, 70%–80% of total islet β‐cell mass has been lost. Similarly, individuals who have been diagnosed with T2D for more than 10 years also have a shortage of islet β cells due to persistent glucotoxicity, lipotoxicity, chronic metabolic inflammation, oxidative stress, and endoplasmic reticulum stress 36. The shortage of pancreatic islet β cells is a key issue that must be overcome in any cure for T1D and T2D; therefore, promoting β‐cell neogenesis is essential. Current ex vivo studies established that platelet‐derived mitochondria not only display the critical molecules for inducing immune tolerance, but also carry the islet‐specific transcription factor MAFA, the pancreatic progenitor‐associated marker SOX9, and ES‐related self‐renewal markers (e.g., OCT4, SOX2, KLF4, and C‐MYC) 35. Thus, mitochondria function not only as powerhouses, producing 90% of a cell's energy to support human activities of daily living, but also as modulators to induce the immune tolerance, as well as controllers for the tissue regeneration.
Additionally, immunohistochemistry data demonstrated the migration of platelets into pancreatic islets of diabetic subjects. As demonstrated by current ex vivo studies, these islet‐infiltrated platelets could be activated to release mitochondria and enter into islet β cells via fibronectin/CD29, CXCR4/SDF‐1 or other potential chemokines, resulting in the proliferation and recovery of normal β‐cell function. In this regard, current and previous clinical data 31 from the Chinese trials have provided powerful evidence that reversal of autoimmunity by SCE therapy leads to regeneration of islet β cells and improvement of metabolic control in Chinese diabetics. Therefore, these findings advanced our understanding of molecular and cellular mechanisms 28 underlying the long‐lasting (4 years) improvement of islet β‐cell function and metabolic control in diabetic patients after receiving a SCE therapy. This novel mechanism may be applied to the treatment of other degenerative diseases, such as Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis.
Conclusion
Overcoming multiple immune dysfunctions is fundamental for the treatment of T1D. The SCE therapy offers comprehensive immune modulation at both the local and systemic levels in T1D, by using a patient's own immune cells that are “educated” by CB stem cells, thus mitigating the safety and ethical concerns associated with other conventional immunological approaches 28. Current clinical data demonstrated the modulation of platelets in diabetic patients after receiving SCE therapy. Mechanistic studies in platelets and mitochondria revealed high expressions of immune tolerance‐related markers on platelets and released mitochondria. Specifically, there is an expression of autoimmune regulator (AIRE) in both CB‐ and PB‐platelets, which is usually expressed in thymic medullary epithelial cells and play a key role in the deletion of auto‐reactive T cells during T cell differentiation and development 37, 38. Ex vivo data demonstrated the suppression of T‐cell proliferation and the up‐regulation of PD1 expression on CD4+ and CD8+ T cells after the treatment with mitochondria. Thus, the therapeutic potential of SCE therapy can be magnified through in vivo actions of platelets and released mitochondria after the treatment with SCE therapy. SCE therapy delivers not only the long‐lasting recovery of homeostasis in pancreata, but also the comprehensive immune balance for the whole body. Targeting mitochondria and platelets may revolutionize conventional immune therapies and regenerative medicine with current stem cell approaches, by leading the way toward the development of novel therapy to treat human diseases.
Author Contributions
Y.Z.: Conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript; Z.J., E.D.: Conception and design, provision of study material or patients, collection and/or assembly of data, data analysis and interpretation; H.L.: Provision of study material or patients, other (GMP production of stem cell educators for clinical treatments); H.Z.: Provision of study material or patients, collection and/or assembly of data, data analysis and interpretation; W.H., A.J., M.M., H.W.: Other (performed ex vivo studies and flow cytometry); M.P.‐B., Y.Z., Y.L., S.P.‐L., M.A.‐V.: Other (GMP production of stem cell educators for clinical treatments); Q.T., Z.Y.: Collection and/or assembly of data; J.W.: Provision of study material or patients; Q.L.: Administrative support, other (clinical technical support for apheresis); J.Z., Y.L., E.M.R., J.M.G.‐G.: Other (clinical technical support for apheresis); E.M.: Provision of study material or patients; T.M.: Provision of study material (cord blood units); E.G.: Provision of study material (cord blood units), administrative support; J.O.: Conception and design, financial support, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
Dr. Zhao, an inventor of Stem Cell Educator technology, led the clinical study, and has an investment and a fiduciary role in Tianhe Stem Cell Biotechnology Inc. (licensed this technology from University of Illinois at Chicago). Dr. Zhao is an inventor for this technology on platelets and mitochondria that has been submitted for provisional patent application. Dr. Zhao received the governmental funding for clinical trials and also received the funding from Hackensack University Medical Center Foundation for mechanistic studies. Dr. Zhao is the founder and has an investment and an ownership in Tianhe Stem Cell Biotechnology Inc. All other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
Clinical trials in Chinese T1D and T2D subjects were supported by the China Jinan 5150 Program. Spanish clinical trial was supported by the grants from the European Union FEDER funds, Principado de Asturias and FICYT (GRUPIN 14–069). Ex vivo studies were supported by the Hackensack University Medical Center Foundation.
Authored by a member of CBA
References
- 1. Nanditha A, Ma RC, Ramachandran A et al. Diabetes in Asia and the Pacific: Implications for the global epidemic. Diabetes Care 2016;39:472–485. [DOI] [PubMed] [Google Scholar]
- 2. Xu Y, Wang L, He J et al. Prevalence and control of diabetes in Chinese adults. JAMA 2013;310:948–959. [DOI] [PubMed] [Google Scholar]
- 3. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010;464:1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Defuria J, Belkina AC, Jagannathan‐Bogdan M et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T‐cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA 2013;110:5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winer DA, Winer S, Shen L et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winer S, Chan Y, Paltser G et al. Normalization of obesity‐associated insulin resistance through immunotherapy. Nat Med 2009;15:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winer S, Winer DA. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol Cell Biol 2012;90:755762. [DOI] [PubMed] [Google Scholar]
- 8. Liu J, Divoux A, Sun J et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet‐induced obesity and diabetes in mice. Nat Med 2009;15:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246. [DOI] [PubMed] [Google Scholar]
- 10. Talukdar S, Oh DY, Bandyopadhyay G et al. Neutrophils mediate insulin resistance in mice fed a high‐fat diet through secreted elastase. Nat Med 2012;18:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu D, Molofsky AB, Liang HE et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011;332:243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Couzin‐Frankel J. Trying to reset the clock on type 1 diabetes. Science 2011;333:819–821. [DOI] [PubMed] [Google Scholar]
- 13. Herold KC, Vignali DA, Cooke A et al. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 2013;13:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bach JF. Anti‐CD3 antibodies for type 1 diabetes: Beyond expectations. Lancet 2011;378:459–460. [DOI] [PubMed] [Google Scholar]
- 15. Wherrett DK, Bundy B, Becker DJ et al. Antigen‐based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent‐onset type 1 diabetes: A randomised double‐blind trial. Lancet 2011;378:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herold KC, Majzoub JA, Melmed S et al. Endocrinology research‐reflecting on the past decade and looking to the next. Nat Rev Endocrinol 2015;11:672–680. [DOI] [PubMed] [Google Scholar]
- 17. Stewart AF, Hussain MA, Garcia‐ Ocana A et al. Human beta‐cell proliferation and intracellular signaling: Part 3. Diabetes 2015;64:1872–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bernal‐Mizrachi E, Kulkarni RN, Scott DK et al. Human beta‐cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014;63:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pagliuca FW, Millman JR, Gurtler M et al. Generation of functional human pancreatic beta cells in vitro. Cell 2014;159:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agulnick AD, Ambruzs DM, Moorman MA et al. Insulin‐producing endocrine cells differentiated in vitro from human embryonic stem cells function in macroencapsulation devices in vivo. Stem Cells Translational Medicine 2015;4:1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulz TC, Young HY, Agulnick AD et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One 2012;7:e37004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rezania A, Bruin JE, Arora P et al. Reversal of diabetes with insulin‐producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014;32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 23. Boyd AS, Rodrigues NP, Lui KO et al. Concise review: Immune recognition of induced pluripotent stem cells. Stem Cells 2012;30:797–803. [DOI] [PubMed] [Google Scholar]
- 24. Zhao T, Zhang ZN, Rong Z et al. Immunogenicity of induced pluripotent stem cells. Nature 2011;474:212–215. [DOI] [PubMed] [Google Scholar]
- 25. Zhao T, Zhang ZN, Westenskow PD et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015;17:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao Y, Wang H, Mazzone T. Identification of stem cells from human umbilical cord blood with embryonic and hematopoietic characteristics. Exp Cell Res 2006;312:2454–2464. [DOI] [PubMed] [Google Scholar]
- 27. Zhao Y, Mazzone T. Human cord blood stem cells and the journey to a cure for type 1 diabetes. Autoimmun Rev 2010;10:103–107. [DOI] [PubMed] [Google Scholar]
- 28. Zhao Y. Stem cell educator therapy and induction of immune balance. Curr Diab Rep 2012;12:517–523. [DOI] [PubMed] [Google Scholar]
- 29. Zhao Y, Glesne D, Huberman E. A human peripheral blood monocyte‐derived subset acts as pluripotent stem cells. Proc Natl Acad Sci USA 2003;100:2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Y, Lin B, Darflinger R et al. Human cord blood stem cell‐modulated regulatory T lymphocytes reverse the autoimmune‐caused type 1 diabetes in nonobese diabetic (NOD) mice. PloS One 2009;4:e4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao Y, Jiang Z, Zhao T et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood‐derived multipotent stem cells. BMC Med 2012;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Delgado E, Perez‐Basterrechea M, Suarez‐Alvarez B et al. Modulation of autoimmune T‐cell memory by stem cell educator therapy: Phase 1/2 clinical trial. Ebiomedicine 2015;2:2024–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao Y, Jiang Z, Zhao T et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood‐derived multipotent stem cells (CB‐SCs) in stem cell educator therapy: Phase I/II clinical trial. BMC Med 2013;11:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y, Yan B, Wang H et al. Hair regrowth in alopecia areata patients following Stem Cell Educator therapy. BMC Med 2015;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 36. Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011;378:169–181. [DOI] [PubMed] [Google Scholar]
- 37. Mathis D, Benoist C. Aire. Annu Rev Immunol 2009;27:287–312. [DOI] [PubMed] [Google Scholar]
- 38. Metzger TC, Anderson MS. Control of central and peripheral tolerance by Aire. Immunol Rev 2011;241:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information