

Abstract
Growing interest in extracellular vesicles (EVs, including exosomes and microvesicles) as therapeutic entities, particularly in stem cell‐related approaches, has underlined the need for standardization and coordination of development efforts. Members of the International Society for Extracellular Vesicles and the Society for Clinical Research and Translation of Extracellular Vesicles Singapore convened a Workshop on this topic to discuss the opportunities and challenges associated with development of EV‐based therapeutics at the preclinical and clinical levels. This review outlines topic‐specific action items that, if addressed, will enhance the development of best‐practice models for EV therapies. Stem Cells Translational Medicine 2017;6:1730–1739
Keywords: Stem cells, Cellular therapy, Clinical trials, Clinical translation, Extracellular vesicles, Therapeutics, Exosomes, Microvesicles
Significance Statement.
Extracellular vesicles (EVs) have been implicated as important and sometimes sufficient mediators of the effects of stem cells. Best‐practice models must be developed for the rapid development of exosomes, microvesicles, and other EVs as therapeutic entities.
Introduction
Extracellular vesicles (EVs) are membrane bound entities that transmit signals between cells via all major classes of biomolecules 1, 2, 3. The term “EV” encompasses microvesicles, exosomes, oncosomes, and other vesicles 4, 5 that may be variously defined by origin, size, and markers 3, 4. EVs interact with target cells by binding to cell surface receptors, transfer of membrane proteins (e.g., signaling receptors), membrane fusion, endosomal uptake, and cargo extrusion through vesicle‐cell channels 1, 6, 7, 8. The EV protein and RNA compositions generally reflect that of progenitor cells. Some proteins, such as CD9, CD63, CD81, and MHCII, are expressed on many EVs 8, while others may define subtypes 9. EV membrane proteins may target EVs to specific cells 1, 10. Found in every bodily fluid examined, EVs may contain cellular markers from difficult‐to‐access anatomical sites, making them strong candidates as biomarkers of disease 11. Their ability to transport molecules and to target specific cell populations raise possibilities for their development as therapeutics.
EVs, particularly those derived from mesenchymal stem/stromal cells (MSCs), have been shown to have intrinsic therapeutic properties for applications as diverse as wound healing, inflammation, hypertension, cardiovascular disease, brain injury, and cancers 4, 12, 13. EVs also exert immunomodulatory pressures depending on progenitor cell and application 8, 14. The immunostimulatory properties of EVs have led to development of EV vaccine platforms against both infectious agents and tumors 4, while the immunosuppressive property of certain EVs could alleviate immune diseases such as graft‐versus‐host disease (GVHD) 15. These therapeutic potentials position EVs as highly competitive alternatives to stem cells, including stem cells such as MSCs that have proven to be safe in many clinical trials and therapeutically efficacious in several disease indications. EVs are likely to be as safe as or safer than their parental secreting stem cells, although half‐life (of the EVs or of the therapeutic effect) may not be as long. Also, the manufacture, storage, transport and end‐use of nonviable stem cell EV therapeutics, as opposed to viable stem cell therapeutics, are less complex. Together, these factors have helped expedite the prospect of translating stem cell EVs into clinical applications. In anticipation of these continuing, challenging developments, the International Society for Extracellular Vesicles (ISEV) and the Society for Clinical Research and Translation of Extracellular Vesicles Singapore (SOCRATES) convened a Workshop to discuss the most recent advances in EV technology and clinical testing and to develop long‐term perspectives for creating best‐practice models for the therapeutic use of EVs. Here, we share our conclusions and action items to spur the development, testing, and approval of EV‐based therapeutics.
EV Production and Isolation
Best practices for sample collection and analysis of EVs have been treated in position papers and statements from ISEV 16, 17, 18, 19 and a variety of other publications 20, 21, 22, 23, 24, 25, 26. These publications should be consulted for the most complete guidance on isolation and characterization. Here, we summarize these topics in the context of moving toward clinical testing of EV‐based therapeutics.
Methods of EV Production
EV production on an industrial scale must eventually occur in defined or otherwise serum‐free conditions, and the cellular source must be considered carefully. Ideally, similar to the case for cellular therapeutics, xenogenic components would be avoided during the production process. Current strategies are to use either chemically defined media or human platelet lysate (HPL) as a serum replacement. Chemically defined media may not rely on limiting raw materials and allow more control of production conditions, crucial for industrial‐scale manufacture. However, controversy remains as to when the switch to defined media must occur in the progression from basic research to clinical application. Since removing serum may change the phenotype and function of cells and the EVs they produce 27, 28, a culture change would necessitate confirmation that EV properties remain the same across media. HPL is already used for the production of functionally efficient cellular therapeutics 29, 30, 31. Due to interindividual differences in the composition of given HPL preparations, conventionally, platelet units from tens of blood donors are pooled to limit batch‐to‐batch variations 32; even larger pools may be used. Since each given batch has to be qualified for the intended use, larger batch sizes are cheaper to produce than multiple smaller ones. However, considering that pooled HPL may contain unidentified pathogenic components that theoretically might be spread with the product, discussions are ongoing to limit allowed batch sizes. Independent of the batch size, there is limited supply of raw material for HPL due to a naturally restricted number of blood donations worldwide, which may hamper global up‐scaling strategies.
Producer cells must be chosen considering the intended use, as EVs may reflect molecular expression patterns and function(s) of the producer cell. If EVs are to be used as a drug delivery system 33, the method of engineering and loading cargo must be considered. For example, post‐production nucleic acid loading of EVs by electroporation has been reported by some to have low efficiency (0.05%), with nucleic acid aggregates often mistaken for loaded EVs 34. Conversely, synthetic lipid nanoparticles (LNPs) have high nucleic acid encapsulation efficiency (>80%) 35. Fusing these LNPs with EVs in a low pH environment creates a “hybridosome” replete with cargo and, if functionalized with specific membrane proteins, capable of cell targeting and improved biodistribution. However, the possibility that some desirable features of EVs are not transferred to hybridosomes, but are lost upon fusion, requires further investigation.
Isolation and Purification of EVs
Isolation and purification of EVs are linked concepts. Isolation methods are techniques used to separate vesicles from non‐vesicular components. In principle, a pure EV product would contain no non‐vesicular components. However, there is presently no accepted measurement for a pure EV product. Degree of purity, in contrast, is a normalization metric to assess and compare product composition and function across batches or studies. Ratio of protein to particle count is one possible purity metric 36. Sai Kiang Lim proposes a metric based on amount of EV‐specific biomarker per unit weight protein. Such assessment, however limited to EVs from a particular cell source, could facilitate comparison of EV purity among laboratories using similar cells. Certainly, a purity metric could help map therapeutic properties of EVs to specific EV components for elucidation of molecular mechanism of action (MoA). A truly pure EV preparation, although, may not be as useful as an EV preparation with a lesser, defined degree of purity. While purification could result in concentration of the therapeutic effector(s), it is also possible that stringent, harsh purification procedures required to obtain a pure product could result in loss of function, through damage to EV‐intrinsic effectors or even to loss of extrinsic, loosely associated factors that act with EVs to exert function. Indeed, as important as purity will continue to be for molecular studies, potency, reproducibility, and stability measures may be at least as important in therapeutics development.
The optimal EV isolation method depends on the intended therapeutic use, route of administration, starting material (e.g., milk, plasma, urine, cell culture), and desired end product (e.g., CD63+ or total EVs). Several leading technologies are described in Table 1; guidance was also provided by Lener, et al 4. Highest purity of EV subsets is thought to be achieved by flotation and density gradient centrifugation. However, purity is often achieved at the expense of scalability, yield, cost, and therapeutic potency. In recent years, numerous commercial EV isolation kits (e.g., 37) have appeared, most of them based on precipitation using polymers such as polyethylene glycol. (This approach can of course be used without a kit 38) Such kits are easy to use and highly scalable but tend to coprecipitate other large complexes and thus not to be EV‐specific. Immunoaffinity methods (e.g., selecting tetraspanins) are promising but may be cost prohibitive for large industrial scale preparation. Furthermore, unless the binding antibodies or other affinity reagents can be easily removed, EV activity may be impaired by blockage of key molecules. Many of the isolation methods described in Table 1 can be combined [examples in 39] in appropriate workflows. Still, these methods are not necessarily interchangeable with regards to preserving biological properties, again arguing for reliance on potency assays.
Table 1.
Established methods of EV purification
| Method | Scalable? | Advantages | Disadvantages |
|---|---|---|---|
| Magnetic bead isolation | Not currently |
Highly pure end product Rapid |
Costly Low yield Depends on knowledge of specific surface markers Need to remove Evs from antibodies or other affinity agents, which may mask molecules required for target selection or effect. |
| Ultrafiltration | Yes | Concentrates large volumes | Potential losses under high pressure |
| Differential ultracentrifugation | No |
Commonly used method allowing comparison between studies Can be combined with concentration methods to produce large quantities |
Includes contaminants without additional isolation steps Evs may aggregate and lose functionality Pellet can be difficult to resuspend |
|
Density gradient ultracentrifugation |
No |
Commonly used method allowing comparison between studies Some gradient media, for example, iodixanol, impair EV function less than others Among the highest purity products |
Some media, for example, sucrose, may interfere with EV function Rotor size limits the total volume that can be processed. Lengthy: up to several days for this step alone (e.g., flotation gradients) |
| High performance liquid chromatography (size exclusion) | Yes | Ideal for large scale | Shown to preserve therapeutic activity |
| Size exclusion chromatography | Yes | Good separation, removing albumin, many lipoproteins | Post‐column concentration may be needed |
| Tangential flow filtration | Yes | Ideal for industrial manufacture | |
| Precipitation or “salting out” | Yes |
Does not require specialized equipment Rapid Polyethylene glycol (PEG) precipitation has been used to generate clinical grade Evs |
Relatively impure product PEG may interfere with some downstream assays and processes |
Abbreviation: EV, extracellular vesicles.
Harmonizing EV production methods across academic laboratories (while being careful not to stifle innovation) might have positive outcomes on its own by enhancing reproducibility. However, evolving academic production models must also consider the end goal of translation and production feasibility. As an example of larger‐scale production model, EVs from 25 to 50 liters of immortalized neural stem cell‐conditioned defined medium have been isolated using a hollow fiber‐based tangential flow filtration (TFF) system 40. EVs were first passed through hollow fibers with 100 nm pore size, then concentrated and separated from smaller aggregates using fibers with 300 kDa pore sizes, also allowing buffer exchange 40. Molecular fingerprinting (capillary gel electrophoresis, flow cytometry, and Western blotting) indicated protein composition differences of EVs recovered by standard stepped ultracentrifugation versus TFF, while TFF had relatively high batch‐to‐batch consistency and yield.
Another important issue is whether the method can be performed as a closed system. An alternative open system would have to be performed in class B or even class A rooms, adding substantially to cost. EVs, unlike cells, can be sterilized by filtration, although we recommend endotoxin testing of the end‐product to ensure that microbial contamination has had no influence. It remains prudent to select GMP‐compliant closed systems to protect from microbial contamination, avoid regulatory delays, and possibly circumvent expensive clean rooms.
In choosing an optimal EV isolation method, investigators should be aware that methods can be changed between Phase I and Phase II studies. Changes in methodology are even possible between Phase II and Phase III studies, although it could be a challenging task to explain such late changes to the satisfaction of the regulators. Investigators must justify the necessity for the change and provide adequate evidence of similarity of the therapeutically active substances used in the different phases. We recommend that clinical trials be conducted with medicinal products produced under the same conditions as those to be used for future manufacturing processes. For clinical studies, it is critical to have a reproducible and GMP compliant method with a reproducibility metric (e.g., molecular fingerprinting).
Identify optimal cell culture media components and cell culture conditions, for example, oxygen tension, for generating the EV type and potency most appropriate for the intended therapeutic use.
Standardize cell culture conditions for specific cell types. For example, in the MSC field, no standardized cell culture conditions have been defined, complicating comparison of results between different labs.
Optimize and use scalable (ideally closed) purification systems that preserve EV function.
Developing Tools for Preclinical and Clinical Studies
Rigorous in vitro and in vivo testing must precede approval of EV‐based therapeutics. There are currently shortcomings in the in vitro assays used to study EV‐based products, from quality control to MoA. We would therefore like to address opportunities to bridge the gap between preclinical and early clinical studies, as presented by three assay types (Table 2), safety testing, and comparison with current standards of care.
Table 2.
Definition of assay types
| Assay type | Description |
|---|---|
| Molecular fingerprinting | Identifies the components and composition of a potential drug substance (the molecule/s of interest) |
| Potency assays | Quantifies how well a potential drug substance elicits the desired biologic / therapeutic action or surrogate activity in vitro and in vivo |
| Mechanistic assays | Identifies how a potential drug substance interacts with the targets in the host organism to elicit the desired biologic/therapeutic action (mode or mechanism of Action, MoA) |
Fingerprinting Assays
Fingerprinting assays provide quality control and establish batch‐to‐batch consistency by examining a narrowly defined set of molecular surrogate markers of EV therapy that are expected to be present, absent, or to reach a specific threshold. Alone or with consideration of other characteristics such as size range, molecular markers should be measured at different stages of production and across batches. Although fingerprints have yet to be standardized and will likely be specific to each production process and clinical indication, their importance should not be underestimated as a key consideration in the regulatory process. Fingerprints alone may not prove functionality or identify MoA. Phase I and Phase II trials, although, will provide ample safety data and may allow correlation with certain effects of batch variation, helping to define a reliable molecular fingerprint of the final product.
Potency Assays
Like fingerprinting assays, potency assays are recommended to evaluate batch‐to‐batch variation and assess confidence in functionality. In vitro potency assays are desirable, as they can be performed more efficiently and cheaply than animal studies. Potency assays establish how well a substance elicits a desired action but do not necessarily reveal the underlying MoA. For example, T‐cells, NKT cells, and B‐cells contribute to antitumor properties of EVs 41, 42. A potency assay might determine which cells are activated by EVs and to what extent, but it may not provide mechanistic information about what signals are being provided to each cell type. However, the ideal potency assay, anchored in a proven MoA for a specific therapeutic use, will provide an accurate prediction of EV potency and will demonstrate whether or not the same cells and culture conditions consistently yield comparable EV products.
Potency assays may be required at different stages in the regulatory process, especially prior to clinical studies. The U.S. Code of Federal Regulations defines a potency assay as a quantitative test of a response to a product at a given dose; ideally, the assay should be based on the known or intended MoA of the product 43. In vitro potency assays do not necessarily predict a therapeutic effect and even less the patient outcome in clinical trials. Relevant national authorities may require potency assays specifically tailored to the intended clinical studies, and preclinical studies are expected to characterize therapeutic activity of an emerging novel product. Given that EVs are signal carriers, understanding the MoA and downstream signaling events will guide development of rigorous in vitro controls and assay readouts. Robust potency assays will contribute to assessing Proof of Function, or Proof of Concept, unlikely to come from Phase I trials with an emphasis on safety. Numerous existing potency assays for pharmaceutical compounds might be adapted to EV‐based products, but they are indication‐ and application‐specific. EV therapeutics may require tailored potency assays.
Mechanistic Assays
Mechanistic assays are important not only for future therapeutic indications, but also to establish positive and negative controls for potency and fingerprinting assays. While a proven MoA is not necessarily required prior to clinical testing, it is necessary to have a hypothesis related to the biology of treatment. Clinical studies enrolling large numbers of participants are a key tool for determining MoA, correlating EV variation with subject response. For later trials (Phase III and IV), a defined MoA or a reproducible clinical readout is critical to rigorous study design. Clinicians, who understand the comorbidities of the patient population, can aid in identifying the MoA and refining the selection of preclinical animal models.
Safety Testing
Unlike small molecule pharmaceutical compounds, there are no defined parameters or assays for safety testing of EV‐based therapeutics, which are defined as biological medicinal products 4. Understanding the biodistribution patterns of locally and systemically administered EVs is important to assessing safety. Novel labeling systems to track EVs in vivo using fluorescent dyes 44, 45 have been used to show biodistribution to the spleen, liver, lungs, and kidneys within 30 minutes of injection and to model half‐life and clearance via the hepatic and renal route in mice. An unanswered question, although, is what proportion of EVs, and what specific types of EVs within a preparation exert therapeutic functions. Possibly, EVs fulfilling a cellular “trash disposal” function have markers that provoke rapid clearance, while signaling EVs might pass these filters. Better single EV analysis, say, by advanced flow cytometry, is needed to answer this question. Closely related are the issues of dose and dose response, and more studies into the latter in particular are encouraged. The argument that exposure to high levels of EVs (e.g., in blood transfusions or other EV‐rich biologicals) demonstrates safety might be weakened if different effects are elicited by different subsets of EVs. Finally, EV source should also be considered in safety testing. While EVs may be derived from natural or even food substances (milk or plants), this does not equate with safety for injected use. Safety parameters for established drugs monitor a plethora of systems and organs and can be used as a model for EVs.
Comparison to Current Standard of Care
In our view, superiority and non‐inferiority determinations for EV‐based therapeutics are heavily dependent on dose and route of administration. Even so, the current standard of care must be considered when targeting indications for EV‐based therapeutics. For example, anti‐cancer therapeutics have a much lower current standard of care due to the urgent and unmet medical need, suggesting there is room for an effective EV‐based therapeutic to substantially improve patient care. In contrast, congested fields like cholesterol‐lowering drugs may present a higher burden of proof for EVs as an improvement over current products. Clinical readouts and head‐to‐head comparison will become necessary as EV‐based drugs move into clinical trials.
Action Items
Identify and describe molecular fingerprints of EVs for specific clinical indications.
Develop reliable, well characterized fingerprinting assays linked to manufacturing processes.
Identify and validate the parameters of potency assays for specific clinical indications.
Incorporate tailored potency assays to speed development of EV‐specific assays.
Identify MoA for EVs for specific clinical indications.
Use MoA data to inform and accelerate development of fingerprinting and potency assays.
Develop in vivo safety testing parameters for use in clinical trials.
Develop noninferiority and superiority testing parameters for use in clinical trials.
Role of Animal Models
Animal models of disease are important to EV therapeutics development, not least as we consider rigorous in vivo potency assays. Therefore, we would like to address some of the issues involved in selecting and utilizing animal models for EV studies.
Choosing an Animal Model
Appropriate selection of preclinical models will have implications for understanding how generalizable the effects of an EV‐based therapeutic are in later stage clinical trials. A non‐exhaustive list of frequently used animal models is provided in Table 3. An animal model must be specific to the intended use and clinical indication, some of which (e.g., wound healing and diabetes) have established models that might be adapted to EV studies. The model and species should provide all necessary aspects to mimic human disease and recovery, whether the disease state is natural or induced. For example, osteoarthritis requires both immunological and mechanical aspects that may not be replicated in animals. Genetic variability and co‐morbidities influence modeling of human clinical indications. For indications with lifestyle factors (e.g., weight gain), a heterogeneous animal population might be beneficial, mirroring comorbidities expected in a clinical study. However, a homogeneous animal population might mimic attempts in early‐stage clinical trials to limit intersubject variability. The economic sustainability of the model and cost per animal should be weighed against the scientific benefits. Finally, size matters: smaller models require fewer EVs.
Table 3.
Frequently used animal models
| Model system | Advantages | Disadvantages |
|---|---|---|
| Small animal models (rodents, rabbits) |
Relatively inexpensive compared with larger animals Shorter lifespan Tractable genetics (some) |
Immunologically distinct from humans, limits application (e.g., vaccination) Findings may require confirmation in other models |
| Companion animals (cats, dogs, horses) |
Mimic lifestyle diversity based on the owner's lifestyle Similar diseases and treatments (vs. human) Well established veterinary techniques |
Not permissible to euthanize the animals under most circumstances Costly |
| Laboratory cats or dogs |
Carnivores are phylogenomically closer to primates than to rodents 46
Controlled environment vs companion animals Relative genetic homogeny if desired (e.g., dog breeds) |
Can be euthanized if necessary Costly |
| Farm animals (sheep, goats, pigs, horses) |
Larger size = ease of product administration Strong models, for example, pig for cardiac and transplantation studies |
Costly Limited expertise For some, young animals preferable (otherwise too heavy) Imaging machines designed for humans needed because of size |
| Non‐human primates (Not required if sufficient evidence can be obtained from other models) | Most closely related to humans | Costly |
Use of Human EVs in Animal Models
An outstanding question is whether consensus had been reached regarding the use of human EVs in animal models. Indeed, this remains a point of discussion amongst the authors: some espouse the use of human EVs in animals, while others feel any such use is dependent on the indication and readouts from the model (especially immunological readouts). The argument could be made that, as human EVs are the eventual therapeutic goal, early preclinical development should focus on human EVs and attempt to establish low species specificity. Understanding the MoA in humans and the animal model(s) will allow an informed decision on whether human EVs are suitable for cross‐species applications.
Control Treatment
After selecting an appropriate model, what is the best control treatment for EV therapies? Proposals might include saline solution, sonicated EVs, cell lysate, or EVs from an alternative source (assumed to be inert). If possible, the control should be chosen based on the MoA. For example, if RNA cargo were thought to be the active therapeutic molecule, sonicated EVs might be an appropriate control (assuming RNA is exposed and degraded). Sonicated EVs might not be appropriate if the active therapeutic molecule were a surface protein, which might persist after sonication.
Limitations of Animal Models
We recognize that each animal model has limitations and that robust, rigorous in vitro potency assays using human samples might be sufficient or preferable in some cases. Notably, many animal models rely on inducing disease in otherwise healthy animals. Common comorbidity contexts of human patients may not be replicated; similarly, aged animals may need to be considered for some studies. Animal models are limited in their capacity for self‐reporting, and the clinical endpoints must therefore be carefully defined and reliable to distinguish treatment effects.
Animal Patients Rather Than Animal Models
Clinical work tends to be human‐centric, reflecting the priorities of many funding agencies. However, EVs may be tested in clinical applications in veterinary as well as in human medicine and provide robust and generalizable evidence of EV function and therapeutic efficacy. Companion dogs and cats may serve as good clinical models for evaluation of MSC‐derived EV therapies before advancing to human clinical trials, because companion animals are often exposed to similar lifestyle factors (e.g., active vs. sedentary) as their owners. Although data from animal patients might be produced under non‐GLP (good laboratory practice) conditions, unlike what would be expected for a preclinical animal study, such data could still be potentially useful.
Action Items
Identify robust animal models specific to EV‐related clinical indications under consideration
Define rigorous in vivo controls for use in animal models
Identify and characterize robust endpoints for animal studies that translate to human clinical trials
Consider the paradigm of testing EV therapeutics in animal patients, not models
Developments in Clinical Use of EV‐Based Therapeutics
General Overview
There is broad interest in utilizing EVs as therapeutics, whether as an active compound or delivery system. Searches of clinicaltrials.gov over time reveal an expanding number of trials associated with the search terms “extracellular vesicles,” exosomes,” and “microvesicles.” Trials utilizing the key words “extracellular vesicles and exosomes” are scarce by end of the year 2016, but it is expected that increasing numbers of studies testing EV‐based therapeutics will soon emerge. EVs are being developed primarily as tools for anti‐tumor therapies (with potentially reduced side‐effects compared with existing chemotherapies); vaccination (antigen presentation); regenerative therapies; and drug delivery. Exosome and exosome secretome studies (albeit for diagnostic purposes) are included as primary and secondary endpoints in numerous clinical trials for almost every indication, reflecting increasing recognition that EVs contribute to pathology in many different conditions. However, development of EV‐related diagnostic and prognostic markers has not been systematic to date. In many cases, a biological function for EVs in the conditions of interest has not been established. Given the high‐throughput mechanisms that are used to identify and characterize EVs and their protein components, this creates the risk of identifying correlations that do not establish causation. Therefore, cautious interpretation is needed, coupled with rigorous basic science studies to investigate role for EVs in the underlying disease mechanism early in the process of developing an EV diagnostic, prognostic, or therapeutic product.
Anti‐Tumor Applications
The application and role of EVs in tumor biology are well established 4. Therapeutic approaches can be broadly categorized as immunomodulatory or directly anti‐tumor. EV origin and composition can qualitatively change the immune response to EV‐expressed surface antigens. For example, Susanne Gabrielsson's group found that repeated injection of soluble antigens rendered responder cells anergic, whereas EVs loaded with immunostimulatory molecules evoked less immune exhaustion and robust natural killer and CD8+ T‐cell responses 47. EV subtypes such as microvesicles and exosomes can provoke different effects, possibly attributable to uptake or cargo.
Vaccination and Infectious Disease
EVs are released by antigen‐presenting cells to modulate immune responses 48, 49. They are also released by some microbes during infection, delivering virulence factors and eliciting immune responses 50, 51. Since EVs are not replication‐competent, they are attractive vaccine candidates. As discussed above, the composition and origin of EVs used in vaccine design are critical to eliciting a robust immune response. Participants of the ISEV‐SOCRATES Workshop discussed several new developments in this area, including anti‐Plasmodium vaccines 52.
Regenerative Therapies: Examples
Osteoarthritis
Cartilage injuries are major risk factors for osteoarthritis (OA). Human MSCs have demonstrated efficacy in cartilage repair, but the underlying MoA is not well understood 53. Wei Seong Toh's group reported that weekly injections of MSC‐EVs into immunocompetent rats enhanced neocartilage formation at 6 weeks, along with remodeling and hyaline cartilage and subchondral bone regeneration at 12 weeks 54. Cartilage regeneration was attributed to EV‐mediated enhanced cell survival and proliferation.
Myocardial Infarction
Ischemic heart disease is a leading cause of death worldwide. It was previously reported that large paracrine factors mediated protective effects of MSCs in myocardial infarction (MI) 55, 56. It is now apparent that EVs mediate these effects both in mouse and pig models. Furthermore, after cardiosphere‐derived cells (CDCs) were shown to produce therapeutic regeneration in the infarcted myocardium in rodent and porcine models and in clinical trials 57, the effects were found to be mediated largely by CDC‐derived EVs 58.
Action Items
Define and characterize the cargo required for maximal therapeutic efficacy, including assessment of proteins, lipids, metabolites, and others—not RNA alone—in mediating EV functions. (It is recognized that these analyses may not be straightforward or strictly necessary to advance clinical trials.)
Define the minimum required elements that can be packaged in an EV to direct the immune response to the desired outcome (e.g., immunostimulatory for vaccination or immunosuppressive for inflammation‐mediated applications).
Define the optimal antigen presentation package to elicit the desired immune effect for anti‐tumor and pathogen responses (whole protein, peptide loaded MHC, expressed nucleic acids, etc).
Challenges to Further Development of EV Therapies
In terms of the regulatory climate and challenges for developing EV‐based therapeutics, several points have become clear:
There are multiple components that may be unique to each preparation/isolation method.
The topology of EVs has become somewhat clearer in terms of what decorates the outside of the membrane versus what is inside (including nucleic acids that may in some circumstances associate with the outside of the EV).
Heterogeneity of EVs—or, more precisely, the contents of EV fractions—has become apparent, and methods are available and in development to analyze this.
What do these insights mean for developing EV‐based therapies? Clearly, the last few years have seen rapid advances in our knowledge that bring effective EV therapeutics closer to the clinic. Although questions remain, some of these are best classified as challenges for basic molecular science and not impediments to clinical development. EVs seem to exert comparable therapeutic effects to cell grafts; since they lack nuclei and can be sterilized by filtration, EVs have some advantages over cells as therapeutic agents and are likely to be managed differently in the regulatory environment. For now, it is not known how long EVs exerting the therapeutic effects remain in the circulation. It has to be considered that their half life time is much shorter than that of cells, resulting in the requirement of more repeated applications than for corresponding cellular therapeutics. The stages of clinical development are shown in Figure 1. Many previously approved products, including MSC products, blood products, antibodies, and insulin, may illustrate different paths through which EVs could be approved.
Figure 1.
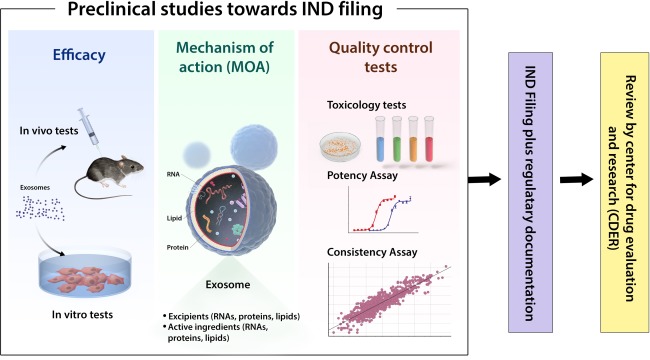
Preclinical studies toward Investigational New Drug filings.
Regulatory Climate
EVs are perhaps best categorized as “biological medicines.” In markets including Australia, the EU, and the U.S., biological medicines are regulated as part of the biologics class for pharmaceutical development. Within that class, multiple possible designations for EVs have different regulatory burdens (Table 4). EV‐based therapeutics have special challenges when it comes to defining the active ingredient or excipients compared with traditional therapeutics, as it is not clear in many cases whether the lipid membrane, the internal content, or a combination thereof is required for the therapeutic effect. Active research programs are required to physically characterize the active component of each EV‐based therapeutic (considering carefully that the EV itself might be the active component) and to describe the MoA. This ambiguity may require specific regulatory guidance for EV‐based therapeutics. However, as mentioned previously, MoA may be of secondary importance behind safety and efficacy, and translation may proceed while the optimal sources and manufacturing processes are developed. Independent of the MoA‐defining molecular component, categorization may depend on the genetic manipulation status of EV‐releasing cells. In principle, EVs could thus be grouped into three categories: (a) native EVs from nongenetically modified cells; (b) EVs from genetically modified cells, but where the EVs do not contain transgene products; and (c) EVs containing transgene products from modified cells (d). EVs of all three categories can be considered as biological medicine, however their subcategorization may be different. For example, EVs with biologically active transgene products should be considered advanced therapy medicinal products (ATMPs); EVs not containing these products would not fall within this subcategory.
Table 4.
Possible regulatory designations for EVs
| Possible designation for EV‐based therapeutics | Description (and see [4] for more information) |
|---|---|
| Biological medicinal products | A biological medicine is a medicine that contains one or more active substances made by or derived from a biological cell 59. This classification is appropriate for all EV‐based products but also for ATMPs (see below, which are either 1) cell therapy, 2) tissue engineered or 3) gene therapy products. |
| Advanced therapy medicinal products (ATMP) | Many EV‐based products are not categorized as ATMPs but as biological medicinal products, because EVs are not tissues or viable cells with a nucleus. The definitive designation of a particular EV‐product as an ATMP may depend on whether the Evs are produced by a genetically modified cell and whether the active substance exerts its therapeutic effects via the modified gene product (this makes them a gene therapy product, belonging to the pharmaceutical category of ATMPs). |
| Active compound | Suitable for situations in which the EV has membrane or surface components that are responsible for the therapeutic effect or is used for targeted delivery of an active compound to a specific tissue. Knowing the target of the supposed active molecular structure within the addressed tissue or organ substantiates argumentation of a proposed MoA. |
| Excipient | Suitable for when the EV is used as a necessary delivery vehicle for an active compound or molecule but is otherwise inert. |
| Tissues and cells | Regulations related to tissues and cells may serve as blueprints for EV‐based therapeutic products harvested from unmodified human cells (not passaged or manipulated in any way). |
| Vesicular paracrine factors (VPFs) | Secretome therapeutics likely contain EVs. |
Abbreviation: EV, extracellular vesicles.
Risk Assessment
Risk assessment is a crucial part of developing a novel therapeutic product. Novel pharmaceuticals are considered high‐risk if they meet any of three criteria (paraphrasing from 4): (a) particular knowledge of the MoA indicates high risk; (b) the nature of the target is unclear; or (c) the relevance of preclinical animal models is unclear. While most of these would seem to apply to EVs, there are equally compelling arguments that EVs are not high‐risk. As previously noted by ISEV 4: (a) autologous EVs occur naturally in the body; (b) EVs are passed through blood transfusions and have not caused major immunological problems; and (c) since there is substantial evidence that MSCs are safe for autologous or allogeneic therapy, it is likely that MSC‐derived EVs will also be safe. We strongly suggest that the body of evidence on safe use of EV‐replete tissues like blood and serum provides a robust basis for the general safety of EV treatments.
Both short‐ and long‐term risks must be addressed. Short‐term risks include immune responses to multiple injections of allogeneic EVs and altered glycosylation patterns on proteins from immortalized cells. Because some EVs may have potent clinical effects, overdose should also be considered. Long‐term risks include the potential for EVs from immortalized cells to carry oncogenic molecules that could epigenetically modify host cells. Some Workshop participants considered MSC‐derived EVs less risky for allogeneic use than, for example, DC‐derived EVs, as the former contain fewer antigen‐presenting molecules, and since MSCs themselves may inhibit pro‐inflammatory immune responses including maturation of B cells 60. However, based on current knowledge from animal models, the Workshop participants considered the risk of sensitization low.
Additional studies may be needed to address the following risk assessment concerns, which are likely to be raised by regulators:
Dose finding and quantification studies. How will the therapeutic unit be defined, for example, total RNA content, total protein content? And what is the relationship to MoA?
Safety studies for off‐target effects of EV‐based therapeutics.
Biodistribution and pharmacokinetics profiles.
Incidence and severity of allergic reactions.
Enrichment of harmful substances in EV‐based products purified from natural sources (e.g., prions from milk).
What standards should be implemented to ensure donor cells are safe?
What are the lead molecules in the product?
What are the quality assurance processes in place during manufacturing (including release criteria)?
Manufacturing Processes
An optimal manufacturing process would have the following attributes:
High capacity (albeit of less importance for highly potent EVs, as determined by dose‐response)
Closed system with defined, disposable components
High yield, reproducible purity
Serum‐free cell culture conditions
Good Manufacturing Practice (GMP) compliance
Regarding GMP compliance, it is important to note that GMP is meant to document each step in the production process and ensure consistency in the applied standard operating procedure. With that understanding in mind, it is up to scientists to develop the processes and establish the standards that will be deployed to ensure consistency. At the ISEV‐SOCRATES Workshop, Mario Gimona shared his experience with establishing an accredited GMP‐compliant manufacturing facility for MSC‐derived EVs at the Paracelsus Medical University of Salzburg, Austria. Cells used to produce EVs were grown in HPL‐containing media (with fibrinogen and platelet EVs removed) rather than serum. Platelet lysate supports cell expansion while eliminating the xenogeneic components of bovine serum. EVs are purified by ultracentrifugation combined with commercial size‐exclusion columns. Empirical optimization of protocols and rigorous potency testing are required as manufacturing processes are scaled, since it must be ensured that changes in the production process do not change product composition or efficacy.
Storage and transport conditions must also be optimized. For example, calcium phosphate crystals in resuspension buffers may confound particle counting, and EDTA can interact with surface molecules. Workshop participants noted that the most widely used buffers are not necessarily appropriate for all applications. Choice of buffer and other storage and transport parameters should be based on evidence and may require formal optimization studies.
Later steps in the manufacturing process are the final product release and stability testing over defined time periods. With no standardized release criteria to date that establish whether an EV preparation is ready for use and remains stable over time, release and stability testing will need to be developed for each application. Current EV counting methods do not necessarily distinguish EVs from other particles. However, we consider single EV‐based analysis by fluorescent labeling and advanced flow cytometry to be an up‐and‐coming technology to solve this problem. Other approaches, such as population‐based molecular fingerprinting or quantification of specific cargo (including but not limited to known active components) may also contribute to release standards.
Maximizing Research Participation
At this stage in the EV development cycle, many researchers perceive challenges to attracting large investments. Funding levels necessary to move from bench to bedside in Phase III trials are generally beyond the reach of academic laboratories, emphasizing the necessity of forming strong collaborative partnerships with industrial and clinical entities. This situation is expected to change positively in individual cases and with promising new results. Yet in the absence of supportive ties, some academic laboratories may not be well motivated to optimize products for clinical use: such research may have adverse effects on grant funding or even be antagonistic to traditionally defined progress, as solving production problems is unlikely to result in high‐impact publications. A combination of secured intellectual property and economic feasibility is often required to move from basic to translational science. One option to circumvent some of the perceived resource problems may be for academic laboratories to work toward applications in rare diseases, where lower regulatory hurdles could potentially allow more immediate testing. If successful, these studies could lead to more investment and broader applications. However, we would like to emphasize that funding on its own should not be the driving force behind scientific decisions; our suggestion is meant only to maximize research participation.
Action Items
Develop collaborations of academia, industry, and clinicians.
Provide additional evidence that EVs are safe and have reproducible effects.
Identify and define an EV production platform that can be used for many different applications.
Identify target applications (e.g., rare diseases or regenerative medicine) that may use native EVs or otherwise pose fewer hurdles to testing.
Author Contributions
A.T.R., K.W.W., B.G., E.R., and S.K.L.: wrote the manuscript, coordinating the input of all authors. M.G., D.dK., M.R., M.W., and V.K.Y contributed to writing, tables, and figures. All authors contributed to discussions surrounding the manuscript preparation and read and approved the final version.
Disclosure of Potential Conflicts of Interest
S.G., J.L., H.A.P., and S.K.L. are named inventors on one or more patents related to the therapeutic use of EVs. SG has research funding. J.dB. has an equity stake in Anjarium Biosciences. R.C is an employee of ReNeuron Group. A.G.I. is an employee of Capricor Therapeutics and is a patent holder. J.L. is an employee of, has research funding, and owns stock in Codiak BioSciences. H.T. is a founder and the board director of MiRTeL Co. LTD. and owns stock in MiRTeL Co. LTD. S.K.L. is a founder and board director of Paracrine Therapeutics Pte Ltd and owns stock in Paracrine Therapeutics Pte Ltd. H.A.P. is an employee of INNOVEX THERAPEUTICS S.L. and has research funding. The other authors indicated no potential conflicts of interest.
Acknowledgments
We acknowledge the contributions of all participants of the ISEV‐SOCRATES Therapeutics Workshop. We also thank Amanda Steele for assistance with manuscript preparation and editing.
Contributor Information
Eva Rohde, Email: e.rohde@salk.at.
Sai Kiang Lim, Email: saikiang.lim@imb.a-star.edu.sg.
References
- 1. Yáñez‐ Mó M, Siljander PR‐M, Andreu Z et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015;4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575–581. [DOI] [PubMed] [Google Scholar]
- 3. Tkach M, Théry C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016;164:1226–1232. [DOI] [PubMed] [Google Scholar]
- 4. Lener T, Gimona M, Aigner L et al. Applying extracellular vesicles based therapeutics in clinical trials ‐ an ISEV position paper. J Extracell Vesicles 2015;4:30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol 2013;200:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camussi G, Deregibus M‐C, Bruno S et al. Exosome/microvesicle‐mediated epigenetic reprogramming of cells. Am J Cancer Res 2011;1:98–110. [PMC free article] [PubMed] [Google Scholar]
- 7. Akers JC, Gonda D, Kim R et al. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus‐like vesicles, and apoptotic bodies. J Neurooncol 2013;113:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. György B, Hung ME, Breakefield XO et al. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu Rev Pharmacol Toxicol 2015. Jan;55:439–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kowal J, Arras G, Colombo M et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 2016;113:E968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoshino A, Costa‐Silva B, Shen T‐L et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fais S, O'Driscoll L, Borras FE et al. Evidence‐based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano 2016;10:3886–3899. [DOI] [PubMed] [Google Scholar]
- 12. Lai RC, Yeo RWY, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol 2015;40:82–88. [DOI] [PubMed] [Google Scholar]
- 13. Riazifar M, Pone EJ, Lötvall J et al. Stem cell extracellular vesicles: Extended messages of regeneration. Annu Rev Pharmacol Toxicol 2017;57:125–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang B, Yin Y, Lai RC et al. Immunotherapeutic potential of extracellular vesicles. Front Immunol 2014;5:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kordelas L, Rebmann V, Ludwig A‐K et al. MSC‐derived exosomes: A novel tool to treat therapy‐refractory graft‐versus‐host disease. Leukemia 2014;28:970–973. [DOI] [PubMed] [Google Scholar]
- 16. Hill AF, Pegtel DM, Lambertz U et al. ISEV position paper: Extracellular vesicle RNA analysis and bioinformatics. J Extracell Vesicles 2013;2. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24376909. Last accessed July 5, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Witwer KW, Buzas EI, Bemis LT et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles 2013;2. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24009894. Last accessed July 5, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lotvall J, Hill AF, Hochberg F et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gardiner C, Vizio D Di, Sahoo S et al. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J Extracell Vesicles 2016;5:32945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lacroix R, Judicone C, Mooberry M et al. Standardization of pre‐analytical variables in plasma microparticle determination: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J Thromb Haemost 2013; Available at: http://www.ncbi.nlm.nih.gov/pubmed/23551930. Last accessed July 5, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuana Y, Böing AN, Grootemaat AE et al. Handling and storage of human body fluids for analysis of extracellular vesicles. J Extracell Vesicles 2015;4:29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Meel R, Krawczyk‐Durka M, van Solinge WW et al. Toward routine detection of extracellular vesicles in clinical samples. Int J Lab Hematol 2014;36:244–253. [DOI] [PubMed] [Google Scholar]
- 23. Kim DJ, Linnstaedt S, Palma J et al. Plasma components affect accuracy of circulating cancer‐related microRNA quantitation. J Mol Diagn 2012;14:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuana Y, Bertina RM, Osanto S. Pre‐analytical and analytical issues in the analysis of blood microparticles. Thromb Haemost 2011;105:396–408. [DOI] [PubMed] [Google Scholar]
- 25. Atai NA, Balaj L, van Veen H et al. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J Neurooncol 2013;115:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Deun J, Mestdagh P, Agostinis P et al. EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 2017;14:228–232. [DOI] [PubMed] [Google Scholar]
- 27. Eitan E, Zhang S, Witwer KW et al. Extracellular vesicle–depleted fetal bovine and human sera have reduced capacity to support cell growth. J Extracell Vesicles 2015;4:26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shelke GV, Lässer C, Gho YS et al. Importance of exosome depletion protocols to eliminate functional and RNA‐containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles 2014;3 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25317276. Last accessed July 5, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schallmoser K, Bartmann C, Rohde E et al. Human platelet lysate can replace fetal bovine serum for clinical‐scale expansion of functional mesenchymal stromal cells. Transfusion 2007;47:1436–1446. [DOI] [PubMed] [Google Scholar]
- 30. Bartmann C, Rohde E, Schallmoser K et al. Two steps to functional mesenchymal stromal cells for clinical application. Transfusion 2007;47:1426–1435. [DOI] [PubMed] [Google Scholar]
- 31. Hemeda H, Giebel B, Wagner W. Evaluation of human platelet lysate versus fetal bovine serum for culture of mesenchymal stromal cells. Cytotherapy 2014;16:170–180. [DOI] [PubMed] [Google Scholar]
- 32. Burnouf T, Strunk D, Koh MBC et al. Human platelet lysate: Replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials 2016;76:371–387. [DOI] [PubMed] [Google Scholar]
- 33. Vader P, Mol EA, Pasterkamp G et al. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev 2016;106:148–156. [DOI] [PubMed] [Google Scholar]
- 34. Kooijmans SAA, Stremersch S, Braeckmans K et al. Electroporation‐induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release 2013;172:229–238. [DOI] [PubMed] [Google Scholar]
- 35. Heyes J, Palmer L, Bremner K et al. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release 2005;107:276–287. [DOI] [PubMed] [Google Scholar]
- 36. Webber J, Clayton A. How pure are your vesicles? J Extracell Vesicles 2013;2 Available at: http://www.ncbi.nlm.nih.gov/pubmed/24009896. Last accessed July 5, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schageman J, Zeringer E, Li M et al. The complete exosome workflow solution: From isolation to characterization of RNA cargo. Biomed Res Int 2013. ;2013:253957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brownlee Z, Lynn KD, Thorpe PE et al. A novel “salting‐out” procedure for the isolation of tumor‐derived exosomes. J Immunol Methods. 2014;407:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muller L, Hong C‐S, Stolz DB et al. Isolation of biologically‐active exosomes from human plasma. J Immunol Methods 2014;411:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vishnubhatla I, Corteling R, Stevanato L et al. The development of stem cell‐derived exosomes as a cell‐free regenerative medicine. J Circ Biomarkers 2014;3:2. [Google Scholar]
- 41. Zhang X, Pei Z, Chen J et al. Exosomes for immunoregulation and therapeutic intervention in cancer. J Cancer 2016;7:1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Naslund TI, Gehrmann U, Qazi KR et al. Dendritic cell‐derived exosomes need to activate both t and b cells to induce antitumor immunity. J Immunol 2013;190:2712–2719. [DOI] [PubMed] [Google Scholar]
- 43.CFR ‐ Code of Federal Regulations Title 21 ‐ Food and Drugs: Parts 1 to 1499. 2016. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm. Last accessed July 5,2017.
- 44. Lai CP, Mardini O, Ericsson M et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014;8:483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lai CP, Kim EY, Badr CE et al. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun 2015;6:7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cannarozzi G, Schneider A, Gonnet G. A phylogenomic study of human, dog, and mouse. PLoS Comput Biol 2007;3:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gabrielsson S, Gehrmann U, Hiltbrunner S et al. Exosomes loaded with α‐galactosylceramide amplify antitumor immunity via iNKT cells (75.5). J Immunol 2012;188(suppl 6):75.5. [Google Scholar]
- 48. Chaput N, Théry C. Exosomes: Immune properties and potential clinical implementations. Semin Immunopathol 2011;33:419–440. [DOI] [PubMed] [Google Scholar]
- 49. Greening DW, Gopal SK, Xu R et al. Exosomes and their roles in immune regulation and cancer. Semin Cell Dev Biol 2015;40:72–81. [DOI] [PubMed] [Google Scholar]
- 50. Brown L, Kessler A, Cabezas‐Sanchez P et al. Extracellular vesicles produced by the Gram‐positive bacterium Bacillus subtilis are disrupted by the lipopeptide surfactin. Mol Microbiol 2014;93:183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wolf JM, Espadas J, Luque‐Garcia J et al. Lipid biosynthetic genes affect Candida albicans extracellular vesicle morphology, cargo, and immunostimulatory properties. Eukaryot Cell 2015;14:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martin‐Jaular L, Nakayasu ES, Ferrer M et al. Exosomes from Plasmodium yoelii‐infected reticulocytes protect mice from lethal infections. PLoS 2011;6:e26588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Toh WS, Foldager CB, Pei M et al. Advances in mesenchymal stem cell‐based strategies for cartilage repair and regeneration. Stem Cell Rev 2014;10:686–696. [DOI] [PubMed] [Google Scholar]
- 54. Zhang S, Chu WC, Lai RC et al. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr Cartil 2016;24:2135–2140. [DOI] [PubMed] [Google Scholar]
- 55. Timmers L, Lim SK, Arslan F et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res 2008;1:129–137. [DOI] [PubMed] [Google Scholar]
- 56. Timmers L, Lim SK, Hoefer IE et al. Human mesenchymal stem cell‐conditioned medium improves cardiac function following myocardial infarction. Stem Cell Res 2011;6:206–214. [DOI] [PubMed] [Google Scholar]
- 57. Malliaras K, Makkar RR, Smith RR et al. Intracoronary cardiosphere‐derived cells after myocardial infarction: Evidence of therapeutic regeneration in the final 1‐year results of the CADUCEUS trial (CArdiosphere‐Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am Coll Cardiol 2014;63:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ibrahim AG‐E, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2014;2:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.European Medicine Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). 2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf. Last accessed July 5, 2017.
- 60. Asari S, Itakura S, Ferreri K et al. Mesenchymal stem cells suppress B‐cell terminal differentiation. Exp Hematol 2009;37:604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]