

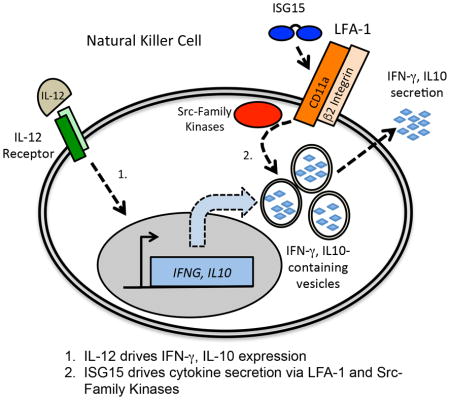
Summary
ISG15 is a ubiquitin-like protein that functions in innate immunity both as an intracellular protein modifier and as an extracellular signaling molecule that stimulates IFN-γ secretion. The extracellular function, important for resistance to mycobacterial disease, has remained biochemically uncharacterized. We have established an NK-92 cell-based assay for IFN-γ release, identified residues critical for ISG15 signaling, and identified the cell surface receptor as LFA-1 (CD11a/CD18; αLβ2 integrin). LFA-1 inhibition blocked IFN-γ secretion, splenocytes from CD11a−/− mice did not respond to ISG15, and ISG15 bound directly to the αI domain of CD11a in vitro. ISG15 also enhanced secretion of IL-10, indicating a broader role for ISG15 in cytokine signaling. ISG15 engagement of LFA-1 led to activation of SRC family kinases (SFKs) and SFK inhibition blocked cytokine secretion. These findings establish the molecular basis of the extracellular function of ISG15 and the initial “outside-in” signaling events that drive ISG15-dependent cytokine secretion.
Keywords: ISG15, IFN-γ, Mycobacteria, Natural Killer Cells, Integrin Receptor, LFA-1, CD11a/αL, SRC Family Kinases, UBAITs
eTOC Blurb
Swaim et al. have identified the receptor for extracellular ISG15 as the LFA-1 integrin. The direct binding of ISG15 to CD11a (αL) initiates “outside-in” signaling and activation of Src-Family Kinases, leading to secretion of IFN-γ and IL-10 from Natural Killer cells.
Introduction
ISG15 is a vertebrate ubiquitin-like protein (Ubl) and its expression is dependent on Type I Interferon (IFN-α/β) signaling (Farrell et al., 1979), indicative of a function in innate immune signaling (Hermann and Bogunovic, 2017; Morales and Lenschow, 2013a). It was the first Ubl to be described after ubiquitin (Haas et al., 1987; Loeb and Haas, 1992) and it is composed of two ubiquitin-like domains connected by a short linker (Narasimhan et al., 2005). The N- and C-terminal domains are 32% and 37% identical to ubiquitin, respectively, and its C-terminal LRLRGG sequence, critical for its conjugation function, is identical to that of ubiquitin. The primary sequence conservation of ISG15 across mammalian species is low compared to ubiquitin and most other Ubls, with human and mouse ISG15 being 66% identical.
The best-characterized function of ISG15 is as a post-translational modifier. It is conjugated to hundreds of proteins in IFN-α/β-stimulated cells, dependent on a single E1 activating enzyme (Ube1L/Uba7) (Yuan and Krug, 2001), a single E2 enzyme (UbcH8/Ube2L6) (Zhao et al., 2004), and a single major E3 enzyme (Herc5, a HECT domain ubiquitin ligase; Herc6 in mice) (Dastur et al., 2006; Ketscher et al., 2012; Oudshoorn et al., 2012). The expression of the conjugation enzymes is, like ISG15, dependent on IFN-α/β signaling, as is expression of Usp18 (mouse Ubp43), the ISG15-specific deconjugating enzyme (Malakhov et al., 2002). An interesting feature of the ISG15 conjugating system is that a single E3 enzyme (Herc5) mediates the ISGylation of hundreds of substrate proteins. A model to account for this is based on the finding that Herc5 cofractionates with polyribosomes and the 60S ribosomal subunit (Durfee et al., 2010). It was demonstrated that Herc5 modifies (ISGylates) proteins co-translationally in a seemingly stochastic manner. It was further proposed that, in the context of a Type I interferon response and viral infection, newly translated viral proteins, rather than cellular proteins, may be the biologically relevant targets of ISGylation. Poly-ISGylation (i.e., analogous to polyubiquitination) does not appear to occur to any appreciable extent and ISGylation is not a signal for proteasomal degradation (Liu et al., 2003). Based on studies with human papillomavirus pseudoviruses (Durfee et al., 2010) and modification of the influenza B nucleoprotein (NP) (Zhao et al., 2016), it has been proposed that ISGylation sterically interferes with the function of viral proteins, particularly those that form oligomers, where low-level (sub-stoichiometric) ISGylation can have a dominant-interfering effect on the entire pool of the oligomer.
Indications of a second non-canonical function of ISG15 came from early studies that reported that ISG15 could be found extracellularly, where it elicited IFN-γ secretion from lymphocytes (D’Cunha et al., 1996; Knight and Cordova, 1991). IFN-γ (Type II Interferon) is critical for activating the innate and adaptive immune responses to viral and intracellular bacterial infections. There was little follow-up to the early studies on extracellular ISG15 and a mechanism for signaling remained unidentified. This was revisited with the discovery of ISG15-deficient patients who acquired mycobacterial disease after vaccination with the live attenuated Bacillus-Calmette-Guérin (BCG) vaccine (Bogunovic et al., 2012). These patients were classified with other Mendelian Susceptibility to Mycobacterial Disease (MSMD) patients. Genetic deficiencies in eight different genes had been previously associated with MSMD: IL-12B (IL-12 subunit), IL-12Rβ1 (IL-12 receptor subunit), IFN-γR1 and IFN-γR2 (IFN-γ receptor subunits), STAT1, NEMO, CYBB, IRF8 (Bustamante et al., 2014). The products of these genes define the IL-12- and IFN-γ production and response axis, and therefore the identification of the ISG15-deficient MSMD patients led to a reexamination of its role in this pathway. It was confirmed that BCG induced IFN-γ secretion from normal control PBMCs (peripheral blood mononuclear cells), but not from PBMCs from the ISG15-deficient patients, and that extracellular IL-12 and ISG15 were synergistic in stimulating the release of IFN-γ from normal control PBMCs (Bogunovic et al., 2012). Within the PBMC population, Natural Killer (NK) cells and T lymphocytes secreted IFN-γ in response to IL-12 and ISG15, with NK cells responding most robustly, consistent with the role of NK cells as major producers of IFN-γ.
There are at least three central unresolved questions regarding the extracellular signaling function of ISG15: 1) the source and mechanism of delivery of ISG15 to the extracellular space, 2) the basis of ISG15 signaling and the identification of a putative cell surface receptor, and 3) the basis of synergism of ISG15 with IL-12. The first question was partially addressed by the finding that ISG15 is localized to gelatinase and secretory granules of granulocytes, suggesting that this may be a major source of extracellular ISG15 (Bogunovic et al., 2012). It has been reported that other cell types (epithelial-derived cell lines, fibroblasts, monocytes, lymphocytes, and neutrophils) are also capable of releasing ISG15 (Bogunovic et al., 2012; Knight and Cordova, 1991), although the mechanism of release in any of these cell types has not been characterized. Here, we focus on the second and third questions: the basis of ISG15 signaling and the identity of the ISG15 receptor, and the basis of synergy of ISG15 with IL-12. The results presented here reveal the molecular details of the signaling events initiated by extracellular ISG15, revealing a new function for integrin signaling in cytokine secretion and a role for ISG15 in cytokine secretion that includes but extends beyond IFN-γ.
Results
Surface residues of ISG15 necessary for signaling
It was previously shown that NK cells are the major cell type within PBMCs to produce IFN-γ in response to extracellular IL-12 and ISG15 (Bogunovic et al., 2012). To further characterize extracellular ISG15 signaling we sought a stable and homogenous source of NK-derived cells that would respond to IL-12 and ISG15. The NK-92 cell line, derived from a non-Hodgkin’s lymphoma, has an NK cell origin (Gong et al., 1994), and Figure 1A compares the response of PBMCs and NK-92 cells to purified IL-12 and ISG15 in an ELISA (enzyme-linked immunosorbent assay) for secreted IFN-γ. Normal PBMCs and NK-92 cells both produced a low level of IFN-γ secretion in the presence of IL-12 alone, whereas ISG15 by itself elicited no IFN-γ secretion. When added together with IL-12, ISG15 elicited a 5–10-fold increase in IFN-γ secretion over the level of IL-12 alone from both PBMCs and NK-92 cells. Ubiquitin elicited no increase in IFN-γ secretion above IL-12 alone. PBMCs reproducibly responded slightly more robustly than NK-92 cells, consistent with previous results that unsorted PBMCs responded better than fractionated primary NK or T cells (Bogunovic et al., 2012). Figure 1B shows that the response to ISG15 was dose-sensitive and that a non-conjugatable form of ISG15 (deleted of the terminal glycine residues; ΔGG) induced IFN-γ secretion from NK-92 cells similar to wild-type ISG15. While these results confirm that ISGylation is not required for the extracellular signaling function, a fusion protein consisting of ISG15 fused to the N-terminus of BirA (ISG15-BirA), which mimics an ISG15 conjugate, was synergistic with IL-12 in stimulating IFN-γ release. This result suggests that ISG15 conjugates produced intracellularly may be capable of stimulating IFN-γ secretion were they to be released into the extracellular space. ISG15 on a solid surface (GST-ISG15 on gluathione sepharose) also induced IFN-γ secretion similarly to soluble ISG15 (Supplemental Figure S2A), suggesting that cell surface-expressed ISG15 might also be capable of signaling.
Figure 1. NK-92 cells respond to IL-12 and ISG15 to secrete IFN-γ.

A. Normal human PBMCs and cultured NK-92 cells were treated with the indicated combinations of IL-12 and ISG15 or ubiquitin for 48 hours and cell culture supernatants were analyzed by ELISA for IFN-γ. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. B. Dose-response of NK-92 cells to wild type ISG15 (WT), ISG15-ΔGG, ubiquitin, or the ISG15-BirA fusion protein, all in the presence of IL-12. The ELISA for IFN-γ was performed as in (A); three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. See also Figure S1
We utilized NK-92 cells and a panel of ISG15 protein variants to identify the surface residues of ISG15 required for IFN-γ secretion. We initially altered 14 surface residues that are identical among mammalian ISG15 proteins but differ at the analogous position in ubiquitin; these residues were altered to that found in ubiquitin at that site. Based on results with these variants, additional ISG15 surface residues were altered to alanines. In total, 27 single-amino variants and two two-amino acid variants were scored for IFN-γ signaling (Figure 2A). All variants were built in the backbone of the C78S ISG15 variant, which alters a conserved cysteine residue in the linker. This mutation does not affect IFN-γ signaling or intracellular conjugation (Figure 2B), but the protein is expressed significantly better than wild-type ISG15 in bacteria; this variant was also used in determining the X-ray crystal structure of ISG15 (Narasimhan et al., 2005). As shown in Figure 2B, all alterations that resulted in a strong defect in signaling were located within the C-terminal domain of ISG15 (R99A, T101A, and T103A). Two other single alterations, Y96L and Q102D, resulted in weak stimulation of IFN-γ release (less than two-times more IFN-γ secretion than IL-12, alone), but the double variant, Y96L/Q102D, was completely defective. Consistent with these results, the isolated C-terminal domain of ISG15 retained signaling activity, and this was also abrogated by the Y96L/Q102D mutation. Figure 2C shows that the C-lobe residues identified in this analysis cluster on the surface of ISG15, comprising what we will refer to as the “IFN-γ patch” (see also Supplemental Figure S1A). The IFN-γ patch does not correspond to a region that mediates interaction with other known ISG15 binding proteins, including influenza virus NS1B (Guan et al., 2011) or USP18 (Basters et al., 2017), and does not correspond to the “hydrophobic patch” region of ubiquitin, which is the binding site for many ubiquitin binding proteins (Sloper-Mould et al., 2001) (Supplemental Figure S1B, C).
Figure 2. Surface residues of ISG15 required for IFN-γ signaling.

A. ISG15 sequences of human and three other mammalian species are shown, aligned with ubiquitin over both the N-and C-terminal ubiquitin-like domains. Residues in red are identical among all ISG15 sequences and residues in green are identical in two or three of the four ISG15 sequences. Red and green arrows indicate residues that, when altered, did or did not, respectively, result in a strong defect in IFN-γ secretion. B. ISG15 variant proteins were tested for their ability to stimulate IFN-γ release from NK-92 cells in the presence of IL-12. ISG15 mutations that led to secretion of less than two-times more the amount of IFN-γ than observed with IL-12 alone were considered to be signaling-defective. Signaling-competent variants are shown with green bars, and signaling-defective variants in red. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. C. The structure of ISG15 (PDB 1Z2M) is colored according to the results shown in (B); surface residues that, when altered, did or did not disrupt signaling are indicated in red and green, respectively. D. ISG15 was assayed for stable association with NK-92 cells. FLAG-ISG15 proteins, WT and the indicated variants, were incubated with intact NK-92 cells; cells were collected, washed, and total cell lysates were analyzed by SDS-PAGE and anti-FLAG immunoblotting. See also Figure S1 and S2.
To rule out the possibility that the IFN-γ patch mutations led to grossly misfolded proteins, the signaling-defective variants were tested for their ability to be conjugated intracellularly by co-transfection into HEK293T cells with the ISG15 E1, E2, and E3 enzymes (Uba7, Ube2L6, and Herc5). As shown in Supplemental Figure S2B, all of the signaling-defective mutants supported ISG15 conjugation. These variants were also subjected to a thermal denaturation analysis, which indicated that all of the signaling-defective mutants retained an unfolding temperature very similar to that of wild-type ISG15 (Supplemental Figure S2C).
Identification of the ISG15 receptor
To determine whether ISG15 might bind to a cell surface receptor, FLAG-tagged wild-type ISG15 and a subset of variants were assayed for their ability to stably associate with NK-92 cells. FLAG-ISG15 proteins were incubated with intact NK-92 cells for 1 hour and cells were collected by centrifugation and washed. The binding of ISG15 to cells was analyzed by preparing whole cell extracts and immunoblotting for FLAG-ISG15. As shown in Figure 2D, wild-type ISG15 and two signaling-competent mutants (R44A and S83A) stably associated with NK-92 cells, whereas four signaling-defective mutants did not (R99A, T101A, Y96L/Q102D, and T103A). The correlation between IFN-γ signaling and NK-92 cell association strongly suggested that ISG15 engages a cell surface receptor.
To identify the receptor we utilized an ISG15 UBAIT (Ubiquitin-Activated Interaction Trap) (O’Connor et al., 2015). UBAITs are fusions of a protein of interest (in this case ISG15) to a short flexible linker and ubiquitin (Figure 3A). Activation of the C-terminal ubiquitin moiety by E1 and E2 enzymes activates the UBAIT for a proximity ligation reaction, such that transiently interacting proteins can be trapped to the UBAIT via a stable amide bond; purification of the UBAIT via an N-terminal affinity tag and mass spectrometry protein identification then identifies the interacting proteins. We expressed and purified a FLAG-tagged ISG15 UBAIT, as well as a signaling-defective ISG15 UBAIT containing the Y96L/Q102D mutations, and a non-conjugatable control UBAIT (deleted of the terminal GG residues; ΔGG) (Figure 3B). UBAITs were charged in vitro by incubation with ATP, E1 ubiquitin activating enzyme (Uba1), and the yeast Ubc4 E2 enzyme, and added directly to intact NK-92 cells for 1 hour. Whole cell lysates were made under denaturing conditions and UBAITs and their conjugates were purified by anti-FLAG immunoprecipitation. The immunoprecipitates were size-fractionated by SDS-PAGE, excising the region of the gel that contained proteins that migrated with a greater apparent molecular weight than the UBAIT; this material was subject to in-gel trypsin digestion and LC/MS/MS to identify proteins conjugated to the UBAITs. Supplemental Table S1 shows the complete set of proteins identified in three biological repeats of experiment, and Figure 3C shows spectral counts for five proteins (CD11a, MYO1G, ESYT1, MCTP2, and OGFR) that met our initial criteria for being a putative ISG15 receptor: 1) they were isolated with the wild-type ISG15 UBAIT in all three repeats, with spectral counts that were at least 2-fold higher than observed with both the Y96L/Q102D UBAIT and the ΔGG UBAIT, and 2) they were transmembrane cell surface proteins. Of these five proteins, we were most intrigued by CD11a, as this was shown previously to be required for resistance to M. tuberculosis in a mouse model, and this was linked to an impaired IFN-γ response (Ghosh et al., 2006); none of the other four proteins had a known link to IFN-γ release or secretion.
Figure 3. The ISG15 UBAIT for receptor identification.

A. UBAITs are fusion proteins consisting of an affinity tag, a protein of interest, a short linker, and a C-terminal ubiquitin moiety (O’Connor et al., 2015). UBAITs can be activated by the E1 ubiquitin-activating enzyme and then transferred to an E2 protein. Proteins that interact transiently with the protein of interest can by covalently trapped to the UBAIT when an amine group of the interacting protein reacts with the E2-charged UBAIT. The covalent complexes are purified via the affinity tag of the UBAIT and the interacting protein(s) are identified by LC-MS/MS. B. Three UBAIT proteins were made: WT ISG15 with WT ubiquitin, the Y96L/Q102D ISG15 variant with WT ubiquitin, and WT ISG15 with a non-conjugatable form of ubiquitin (ΔGG). C. UBAITs were charged in vitro with purified E1 and E2 enzyme, added directly to intact NK-92 cells, and UBAIT conjugates were purified under denaturing conditions and analyzed by LC/MS/MS. Candidate ISG15 receptor proteins are shown, with total spectral counts from each of three biological replicates. Criteria for being a putative receptor were 1) spectral counts at least two-fold higher with the WT UBAIT than the Y96L/Q102D or ΔGG UBAITs, and 2) the proteins were membrane-associated surface proteins. See Supplementary Table for complete dataset. See also Figure S3.
CD11a (αL integrin) forms a heterodimeric complex with CD18 (β2 Integrin) to form the LFA-1 integrin receptor (Leukocyte Function-associated Antigen-1). LFA-1 is expressed on T cells, B cells, macrophages, neutrophils, and NK cells, and its best-characterized ligands are ICAMs (Intra-Cellular Adhesion Molecules) (Abram and Lowell, 2009). The interaction of LFA-1 with ICAMs is critical for adhesion and recruitment of immune cells to antigen-presenting cells and for cell killing by NK cells (Helander et al., 1996). ICAM1 is the major ligand for LFA-1 on T cells (Binnerts et al., 1994). To determine if CD11a and LFA-1 are involved in ISG15-dependent IFN-γ secretion, we first took advantage of antibodies (MHM24 and TS1/22) (Grönholm et al., 2016; Hildreth et al., 1983; Sanchez-Madrid et al., 1982) and a small molecule (A-286982) (Liu et al., 2000) that have been shown to block LFA-1 signaling. As shown in Figure 4A, increasing amounts of either of the CD11a-specific antibodies or A-286982 reduced the ability of ISG15 to stimulate IFN-γ secretion from NK-92 cells, while a control antibody against the transferrin receptor did not. We also determined whether binding of ISG15 could be conferred to HEK293T cells, an epithelial cell line that does not express LFA-1, by transfection of CD11a and/or CD18 expression plasmids (Kim et al., 2003). As shown in Figure 4B, 3xFLAG-ISG15 bound to intact HEK293T cells only when both CD11a and CD18 were expressed together; expression of the IL-12 receptor (IL-12R) did not confer ISG15 binding. CD18 can also heterodimerize with CD11b and CD11c, which are both approximately 35% identical to CD11a. Co-expression of either of these proteins with CD18 did not result in ISG15 binding to HEK293T cells (Figure 4B and Supplemental Figure S3A), suggesting that the ISG15 interaction is specific for CD11a/CD18.
Figure 4. Inhibitors of LFA-1 block ISG15 signaling.

A. MHM24 and TS1/22 antibodies, a small molecule inhibitor of LFA-1 (A-286982), or an antibody against the transferrin receptor (G1/221/12) were pre-incubated with NK-92 cells for one hour prior to addition of IL-12 and ISG15 for 48 hours, followed by an IFN-γ ELISA of the cell culture supernatants. The amount of added IgG antibody (ng) and the concentration of A-286982 (nM) are indicated. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. B. HEK293T cells, which do not express either CD11a or CD18, were transfected with plasmids expressing CD11a, CD11b, CD11c, with or without CD18, or the IL-12 receptor (IL-12R). FLAG-ISG15 was added to the culture media of the transfected cells and cells were assayed for their ability to stably associate with FLAG-ISG15, as in Figure 2D. See also Figure S3.
To confirm that LFA-1 mediates the extracellular signaling function of ISG15 we obtained splenocytes from CD11a−/− knockout mice (Ding et al., 1999). As with normal human PMBCs, mouse ISG15 and IL-12 were synergistic in stimulating IFN-γ secretion from the CD11a+/+ mouse splenocytes (Figure 5A). CD11a−/− splenocytes were completely defective in their response to ISG15. In addition to providing direct evidence for the role of CD11a in ISG15 signaling, this result further confirms that neither CD11b or CD11c can substitute for CD11a in ISG15-dependent IFN-γ secretion. ICAM1 interacts directly with the extracellular αI domain of CD11a (Shimaoka et al., 2003). To determine if ISG15 also interacts with this region of CD11a, purified GST-αI domain was assayed for binding to wild-type ISG15 as well as two signaling-competent variants (R44A and S84A) and four signaling-defective variants (R99A, T101A, Y96L/Q102D, T103A). As shown in Figure 5B, ISG15 and the signaling-competent variants all bound stably to GST-αI domain, while there was no detectable binding to any of the non-signaling variants. We therefore conclude that LFA-1 is the receptor for ISG15 and that the αI domain of CD11a contains the binding site for ISG15.
Figure 5. Splenocytes from CD11a −/− mice do not respond to ISG15.

A. Splenocytes from CD11a+/+ and CD11a −/− C57B6 mice were treated with mIL-12 and the indicated amounts of mISG15 for 48 hours (mISG15 alone was 117nM), and an IFN-γ ELISA of the cell culture supernatants was performed. IFN-γ release is shown as fold increase over that of untreated splenocytes. Three biological repeats of the experiment were conducted with spleens from three animals of each genotype, and the error bars represent the standard error of the mean. B. Purified GST-CD11a αI-domain (Qu and Leahy, 1995) on glutathione sepharose was incubated with FLAG-ISG15 or the indicated ISG15 variants. Beads were collected, washed, and analyzed for binding of FLAG-ISG15 by immunoblotting. See also Figure S4.
The equilibrium dissociation constant for the ISG15-αI interaction was determined with purified ISG15 and GST-αI domain proteins according to methods described by Pollard (Pollard, 2010). The Kd was determined to be 656 nM +/− 242 nM (Supplemental Figure S3B); this is similar to the Kd for the ICAM1-CD11a interaction (200–500 nM) (Shimaoka et al., 2001; Tominaga et al., 1998) and within the range for that reported for the ISG15-Influenza virus NS1B interaction (180 nM-1.9 μM) (Guan et al., 2011; Li et al., 2011). The binding of ICAM1 to the αI domain is dependent on the Metal Ion Dependent Adhesion Site (MIDAS), a group of residues that coordinate a magnesium ion at the ICAM1-αI interface (Shimaoka et al., 2003). MIDAS Mg2+ coordination residues (S139, S141, D239) and a residue that forms a salt bridge between ICAM1 and the αI domain (E241) were altered in various combinations to alanine residues, and none of these variants had a deleterious effect on ISG15 binding (Supplementary Figure S3C). This indicates that ICAM1 and ISG15 recognize distinct features of the αI domain. Consistent with this, purified ICAM1 did not compete for binding of ISG15 to the αI domain (Supplemental Figure S4A).
To determine whether ICAM1 might yet influence ISG15-dependent IFN-γ secretion splenocytes were obtained from ICAM1−/− knockout mice. As shown in Supplemental Figure S4B, ICAM1 deficiency had neither a negative or positive effect on ISG15-dependent IFN-γ secretion. In addition, a static cell adhesion assay was used determine whether ISG15 affected adhesion of Jurkat cells (a T cell line that expresses LFA-1) to ICAM1- or ICAM2-coated plates s(Strazza et al., 2014). As shown in Supplemental Figure S4C, increasing amounts of ISG15 (to a concentration 5-fold higher than the saturating amount of ISG15 in the IFN-γ secretion experiments) had no effect on cell adhesion. Together, these experiments indicate that the interaction of ISG15 with LFA-1 is biochemically and biologically separable from the interaction of ICAM proteins with LFA-1.
The basis of synergism between IL-12 and ISG15
IL-12 engages the IL-12 receptor to stimulate transcriptional activation of the IFN-γ gene (IFNG) (Chan et al., 1991). We therefore determined whether ISG15 influenced IFNG mRNA expression. Quantitative RT-PCR confirmed that IL-12 had a strong effect on expression of IFNG mRNA levels in NK-92 cells, while ISG15 did not significantly enhance IFNG mRNA either by itself or when combined with IL-12 (Supplemental Figure S5A). Consistent with this, IL-12 signaling resulted in STAT4 phosphorylation, which is critical for induction of IFNG expression, while ISG15 did not (Supplemental Figure S5B). To probe the relationship between IL-12 and ISG15, IFN-γ was expressed independently of IL-12 by transient transfection of an IFNG expression plasmid in Jurkat cells. As shown in Figure 6A, expression of IFNG, by itself, led to a low level of IFN-γ secretion relative to untransfected cells. The level of secreted IFN-γ was dramatically increased when the IFNG-transfected cells were treated with recombinant purified ISG15, but not when treated with the Y96L/Q102D signaling-defective variant. Together with the qRT-PCR results, these results indicate that IL-12 signaling and ISG15 signaling pathways are distinct and separable, with IL-12 driving expression of IFN-γ, and ISG15 driving enhanced IFN-γ secretion.
Figure 6. ISG15 signals independently of IL-12, and ISG15 induces secretion of IL-10.

A. Jurkat cells were transfected with an IFNG expression plasmid to induce IFN-γ expression independently of IL-12. Cells were then assayed for ISG15-dependent secretion of IFN-γ. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. B. NK-92 cells were treated with IL-12 and the indicated concentrations of ISG15 or the Y96L/Q102D variant, as indicated, and cell culture supernatants were assayed for secreted IL-10 by ELISA. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. See also Figure S5.
As IL-12 also induces the expression of the IL-10 cytokine (Morris et al., 1994), we determined whether extracellular ISG15 would also drive enhanced IL-10 secretion. As shown in Figure 6B, IL-12 and ISG15 were indeed synergistic in stimulating the secretion of IL-10. As with IFN-γ, the Y96L/Q1021D ISG15 variant did not enhance IL-10 secretion. This result indicates that ISG15 has a broader effect on cytokine secretion beyond its effects on IFN-γ.
Integrin receptors, including LFA-1, are subject to both “outside-in” and “inside-out” signaling (Abram and Lowell, 2009), however these signaling modes can be difficult to distinguish from each other because of the complex nature of leukocyte, cytokine, and cell-cell interactions. ISG15 engagement of LFA-1 appears to be a clear case of outside-in signaling, as ISG15-induced cytokine secretion occurred in the absence of other cell types or exogenous cytokines (Figure 6A). Activation of SRC family tyrosine kinases (SFKs) has been shown to be an early event associated with outside-in integrin signaling in leukocyte cell migration (Baruzzi et al., 2008), and we therefore examined whether ISG15 resulted in activation of SFKs. LYN, HCK, and FGR are three SFKs commonly expressed in myeloid cells, and all are expressed in NK-92 cells (Supplemental Figure S6A). Activation of SFK signaling was monitored with a phospho-specific antibody that detects the activated form of all three of these kinases (LYN, HCK, and FGR). As shown in Figure 7A, the addition of ISG15, alone, to NK-92 cells resulted in increased levels of SFK phosphorylation. IL-12 by itself did not result in SFK activation or enhance ISG15-dependent activation, and ISG15 did not activate LCK, an SFK associated with T cell receptor activation (Supplementary Figure S6B). As expected, the ability of ISG15 to activate SFKs was correlated with the ability of ISG15 to engage LFA-1: wild-type ISG15 and two signaling-competent ISG15 variants (R44A and S84A) induced SFK activation, while two signaling-defective variants (T101A and Y96A/Q102D) did not (Figure 7B). To determine if SFK activation was critical for ISG15-induced cytokine secretion we utilized PP2, a small molecule competitive inhibitor of ATP binding to LYN, HCK, and FGR (IC50 of 4–5 nM) (Bain et al., 2007). As shown in Figure 7C, PP2 effectively blocked the effect of ISG15 on secretion of both IFN-γ and IL-10. These results support a model in which ISG15 engagement of LFA-1 triggers an SFK-dependent mechanism of cytokine secretion.
Figure 7. Activation of SRC-family kinases (SFKs) mediates ISG15-dependent cytokine secretion.

A. NK-92 cells were treated for 10 minutes with IL-12 and/or ISG15 and total cell extracts were analyzed by immunoblotting with an antibody that recognizes the phosphorylated and activated form of three SRC family kinases expressed in NK-92 cells (LYN, HCK, FGR). B. As in (A.), phosphorylated and activated SFKs were analyzed by immunoblotting in response to the indicated ISG15 variants. C. IFN-γ ELISA of NK-92 cell culture supernatants in the presence of increasing amounts of ISG15, with and without addition of PP2 (10 μM), an inhibitor of LYN, HCK, and FGR. Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. D. IL-10 ELISA of NK-92 cell culture supernatants in the presence of increasing amounts of ISG15, with and without addition of PP2 (10 μM). Three biological repeats of the experiment were conducted and the error bars represent the standard error of the mean. See also Figure S6.
Discussion
ISG15 was presumed to be involved in innate immune responses since its earliest description as a Type I (IFN-α/β) interferon-induced protein (Farrell et al., 1979). The enzymatic machinery that mediates its canonical intracellular conjugation function has been elucidated and a model for how this is involved in antiviral responses has been proposed and tested (Durfee et al., 2010; Zhao et al., 2016). However, the most enigmatic aspect of the function of ISG15 centered on early reports that it had an extracellular signaling function that stimulated Type II (IFN-γ) release from lymphocytes (Knight and Cordova, 1991; Recht et al., 1991). Support for the biological significance of the extracellular function came with the discovery of ISG15-deficient patients susceptible to mycobacterial disease following exposure to the BCG vaccine strain (Bogunovic et al., 2012). As with other MSMD patients (Bustamante et al., 2014), this appeared to be a result of an inability to mount a robust IFN-γ response (Bogunovic et al., 2012). The results described here provide a biochemical basis for extracellular ISG15 signaling leading to IFN-γ secretion, identify the cell surface receptor for ISG15, and reveal the initial outside-in signaling events that stimulate LFA-1-dependent cytokine secretion.
All residues of ISG15 that were identified as being required for IFN-γ signaling were located on the C-terminal lobe of ISG15. Y96, T101, Q102, and T103 are very highly conserved among mammalian ISG15 sequences, while R99 is conserved among great ape ISG15s but is otherwise divergent. None of the IFN-γ signaling-defective variants negatively influenced intracellular conjugation, and these separation of function mutations should therefore prove useful in discerning which biochemical functions of ISG15 are critical in different biological settings (e.g., in response to specific viral or microbial infections).
The ISG15 UBAIT identified a CD11a as a candidate cell surface receptor, and this was validated with blocking antibodies to LFA-1, a small molecule inhibitor of LFA-1, mouse CD11a-deficient splenocytes, and direct in vitro binding of ISG15 to the αI domain of CD11a. While first described as reagents for identification of ubiquitin ligase substrates (O’Connor et al., 2015), UBAITs, as shown here, can be used for characterizing the interactome of virtually any protein of interest. UBAITs were previously used as intracellular reagents (Byun et al., 2017; O’Connor et al., 2015). In contrast, the ISG15-UBAIT, charged in vitro, was used as an extracellular reagent to identify cell surface interacting proteins. This approach may be generally useful for identification of receptors for other orphan ligands. It should be noted that because the ISG15-UBAIT is the fusion of one UBL to another (ubiquitin), an E2~charged ISG15 protein (without the ubiquitin moiety) could theoretically have been used directly as a proximity labeling reagent. However, this would have been unlikely to have been successful as ISG15 would have had to have been charged with its cognate E1 and E2 enzymes (UBE1L/UBA7 and UBCH8/UBE2L6, respectively), and UBCH8/UBE2L6, like its most closely related ubiquitin E2 enzyme, UBCH7/UBE2L3 (Wenzel et al., 2011), is expected to discharge very inefficiently to lysine resides.
The CD11a/αL subunit of LFA-1 has an extracellular region consisting of over 1000 amino acids, which includes the αI domain. The CD11a αI domain is the binding site for both ICAM1 (Shimaoka et al., 2003), and ISG15. Mutagenesis of the αI domain indicated that the determinants of the αI domain recognized by ISG15 and ICAM1 are distinct. While magnesium ion coordination residues are critical for ICAM1 binding (Shimaoka et al., 2003), alteration of these residues had no effect on ISG15 binding. Consistent with this, ICAM1 did not compete for in vitro binding of ISG15 to the αI domain. As ICAM1 is expressed at high levels on primary NK cells and NK-92 cells (Maki et al., 2001), we tested whether ICAM1 might play either a positive or negative role in ISG15-LFA-1 signaling using ICAM1−/− splenocytes. The ICAM−/− cells behaved identically to the ICAM+/+ cells, ruling out simple synergism or competition between ISG15 and ICAM1 for LFA-1. Similarly, we determined that ISG15 did not compete with or stimulate ICAM1- or ICAM2-mediated lymphocyte adhesion. Therefore, ISG15 and ICAM1 interactions with LFA-1 appear to be biochemically and biologically separable. At the same time, it is possible that there are biologic scenarios not represented by our cell-based assays where the two LFA-1 ligands might influence one another.
Integrin receptors can be activated by “inside-out” and “outside-in” signaling (Abram and Lowell, 2009). Inside-out signaling is dependent on upstream interactions with other receptors that lead to cytoplasmic signals that alter the conformation of the intracellular domains of the integrin receptor, triggering changes in the conformation of the extracellular domains (Luo et al., 2007). In the case of LFA-1, the extracellular conformational changes expose the CD11a αI domain, enabling high affinity binding to ICAM1. While outside-in is far less well understood, our results demonstrate that ISG15 can act directly on LFA-1 in the absence of any other prior activation, defining ISG15 as an outside-in signaling molecule. Consistent with an outside-in mechanism of action, the addition of ISG15, alone, resulted in activation of SRC family kinases (LYN, HCK, FGR), previously implicated in outside-in integrin signaling (Baruzzi et al., 2008; Giagulli et al., 2006).
The basis of synergism between IL-12 and ISG15 was not previously characterized. We ruled out the possibility that ISG15 enhanced the IL-12-dependent expression of the IFNG gene and demonstrated that IL-12 was unnecessary for ISG15-dependent secretion when IFNG expression was driven by plasmid expression in Jurkat cells. The fact that Jurkat cells express a defective IL-12 receptor (Ford et al., 2012) further cements the independence of ISG15 from effects on either extracellular or intracellular aspects of IL-12 signaling. These results lead to a model in which IL-12 and ISG15 signaling are distinct and independent, and that the basis of the apparent synergism is that IL-12 drives cytokine expression, while ISG15 drives enhanced cytokine secretion. It should be noted that it was previously reported that ISG15 induced a very low level of IFN-γ release from PMBCs in the absence of IL-12 (Bogunovic et al., 2012). Given the results presented here, the most likely explanation for this is that in the complex mixture of cell types that comprise PMBCs there was a source of IL-12 (or another cytokine) that supported a low level of IFNG mRNA expression.
We have identified the initial events in signaling by extracellular ISG15, however it remains unclear as to how ISG15 is released into the extracellular space or the cell source of extracellular ISG15. It was reported previously that ISG15 is localized in secretory granules of granulocytes, however a wide variety of other cell types, including fibroblasts and epithelial cells were also reported to release ISG15 (Bogunovic et al., 2012). We have shown that an artificial ISG15 conjugate (ISG15 in a translational fusion to BirA) ISG15 is capable of stimulating IFN-γ secretion with similar efficiency as free ISG15. This suggests that one source of extracellular ISG15 might be conjugates that are produced intracellularly as a result of Type I interferon signaling. ISGylated proteins might be trafficked to the cell surface, released due to cell death of pathogen-infected cells, or ISG15 might be incorporated onto surface proteins of released viruses or microbes. All of these sources of extracellular ISG15 conjugates could potentially engage LFA-1 on NK cells.
There was no prior indication that ISG15 might influence the secretion of additional cytokines, such as IL-10. However, in light of our model that IL-12 drives cytokine expression and ISG15 drives secretion, it is perhaps not surprising that ISG15 would enhance secretion of cytokines packaged in similar vesicles (Stow and Murray, 2013). The fact that ISG15 may impact a variety of cytokines suggests the possibility of using extracellular ISG15 in a therapeutic manner. With the advent of cell-based immunotherapies that rely on both genetic editing of host T cells (Lloyd et al., 2013) and the use of cytokines as immunotherapy agents (Lee and Margolin, 2011), ISG15 might be useful in augmenting cytokine secretion or as a trigger to induce cytokine secretion at specific times.
ISG15 is a complex multi-functional UBL involved in several aspects of innate immune responses to viral and microbial pathogens (Bogunovic et al., 2012; Morales and Lenschow, 2013b; Radoshevich et al., 2015; Speer et al., 2016; Zhang et al., 2015). In addition to its intracellular conjugation function and its extracellular signaling function, ISG15 has recently been shown to have an intracellular conjugation-independent function in modulating a non-catalytic function of USP18 (Zhang et al., 2015); this activity of USP18 directly regulates signaling through the Type I interferon receptor. Understanding the relationships between the extracellular function and the intracellular conjugation-dependent and -independent functions of ISG15 is likely to reveal critical connections among innate immune responses and provide opportunities for therapeutic modulation of responses to infectious diseases.
Star Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| M2 Anti-Flag | Sigma | CAT#F3165 |
| Anti-Phospho-Src family Y416 | Cell Signaling Technologies | CAT#2101 |
| MHM 24 | DSHB | CAT#MHM.24 |
| TS1/22 (TS1.22.1.1.13.3) | DSHB | CAT#TS1.22.1.1.13.3 |
| G1/221/12 | DSHB | CAT#G1/221/12 |
| Anti-Phospho-LCK Y394 | Thermo Scientific | CAT#pa5-37628 |
| Anti-LYN | Cell Signaling Technologies | CAT#2732 |
| Anti-HCK | Cell Signaling Technologies | CAT#14643 |
| Anti-IL12RB1 | Santa Cruz | CAT#Sc-658 |
| Anti-CD11a | Abcam | CAT#ab186873 |
| Anti-FGR | Cell Signaling Technologies | CAT#2755 |
| Bacterial and Virus Strains | ||
| BL21(DE3)pLysS E. coli were used for cloning and protein expression | Novagen | CAT#69388 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| A-286982 | Tocris | CAS#280749-17-9 |
| Mouse IL-12 | R&D Systems | CAT#419-ML |
| Human IL-12 | BD Pharmingen | CAT#554613 |
| PP2 | Adipogen | CAT# AG-CR1-35630-m001 |
| Recombinant ICAM1 | R&D Systems | CAT#ADP4-400 |
| Recombinate ICAM2 | Abcam | CAT#ab134869 |
| CFSE | Molecular Probes | CAT#C1157 |
| EASYTIDES ATP, g32P | PerkinElmer | CAT#BLU502A250UCSBG4 |
| cAMP-dependent protein kinase | Promega | CAT#V516A |
| IL-2 | Peprotech | CAT# 200-02 |
| Pierce™ Protease and Phosphatase inhibitor mini tablet EDTA-free | Thermo Scientific | CAT#88668 |
| PreScission Protease | GE Healthcare | CAT#27-0843-01 |
| Critical Commercial Assays | ||
| Human IFN-γ ELISA | Thermo Scientific | CAT#EHIFNG |
| Human IL-10 ELISA | Thermo Scientific | CAT#EHIL10 |
| Mouse IFN-γ ELISA | Thermo Scientific | CAT#EM1001 |
| Lonza SE nucleofection | Lonza | CAT#V4XC-2024 |
| Protein thermal shift kit | Thermo Scientific | CAT#4461146 |
| TaqMan® RNA-to-Ct™ | Thermo Scientific | CAT#4392656 |
| Bolt Bis-Tris gel 4–12% gradient | Thermo Scientific | CAT#NW04120BOX |
| Experimental Models: Cell Lines | ||
| NK-92 cells | Gong et al 1994 | M. Poenie Lab |
| Jurkat cells | Byun et al 2010C | J. Dudley Lab |
| HEK293T cells | ATCC | CAT#CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57B6 CD11a−/− mouse spleens | Jackson Laboratory | CAT#JR 5257 |
| C57B6 ICAM1−/− mouse spleens | Jackson Laboratory | CAT#JR2867 |
| C57B6 mouse spleens | Jackson Laboratory | CAT#JR 644 |
| Human PBMC | CTL Technologies | CAT# CTL-UP1 |
| Oligonucleotides | ||
| IFN-γ TaqMan® probe | Thermo Scientific | CAT#Hs00989291 |
| ActB TaqMan® probe | Thermo Scientific | CAT#Hs99999903 |
| Recombinant DNA | ||
| pGEM-T IFN-γ | Sino Biological | CAT#HG11725-G |
| pEYFP- N1 CD18 | Kim et al. 2003 | Addgene #8638 |
| pcDNA3- CD11a | This paper | |
| pcDNA3- CD11b FLAG | Sino Biologcial | CAT#HG12122-CF |
| pcDNA3- CD11c FLAG | Sino Biologcial | CAT#HG10494-CF |
| pGex-6pk- ISG15 WT | Durfee et al 2008 | |
| pGex-6pk- ISG15 Y96L Q102D | This paper | |
| pGEX-6p- ISG15 WT | Durfee et al 2008 | |
| pGEX-6p- ISG15 C78S | Durfee et al 2008 | |
| pGEX-6p- ISG15 I36A | This paper | |
| pGEX-6p- ISG15 K143A | This paper | |
| pGEX-6p- ISG15 K90A | This paper | |
| pGEX-6p- ISG15 L85F | This paper | |
| pGEX-6p- ISG15 N13A | This paper | |
| pGEX-6p- ISG15 Q102D | This paper | |
| pGEX-6p- ISG15 Q110A | This paper | |
| pGEX-6p- ISG15 Q34D | This paper | |
| pGEX-6p- ISG15 Q55E | This paper | |
| pGEX-6p- ISG15 R44A | This paper | |
| pGEX-6p- ISG15 R57A | This paper | |
| pGEX-6p- ISG15 R92A | This paper | |
| pGEX-6p- ISG15 R99A | This paper | |
| pGEX-6p- ISG15 S26E E27N | This paper | |
| pGEX-6p- ISG15 S62A | This paper | |
| pGEX-6p- ISG15 S83A | This paper | |
| pGEX-6p- ISG15 S94A | This paper | |
| pGEX-6p- ISG15 T101A | This paper | |
| pGEX-6p- ISG15 T103A | This paper | |
| pGEX-6p- ISG15 T95A | This paper | |
| pGEX-6p- ISG15 W123A | This paper | |
| pGEX-6p- ISG15 Y96L | This paper | |
| pGEX-6p- ISG15 Y96L Q102D | This paper | |
| pGEX-6p 3xFLAG- WT ISG15 | This paper | |
| pGEX-6p 3xFLAG- ISG15 R44A | This paper | |
| pGEX-6p 3xFLAG- ISG15 S83A | This paper | |
| pGEX-6p 3xFLAG- ISG15 T101A | This paper | |
| pGEX-6p 3xFLAG- ISG15 R99A | This paper | |
| pGEX-6p 3xFLAG- ISG15 T103A | This paper | |
| pGEX-6p 3xFLAG- ISG15 Y96L Q102D | This paper | |
| pGEX-6p CD11a αI domain | This paper | |
| pGEX-6p CD11a αI domain S139A S141A | This paper | |
| pGEX-6p CD11a αI domain S139A S141A D239A | This paper | |
| pGEX-6p CD11a αI domain D239A S241A | This Paper | |
| pcDNA3 3xFLAG- ISG15 WT | This paper | |
| pcDNA3 3xFLAG- ISG15 E97A | This paper | |
| pcDNA3 3xFLAG- ISG15 T101A | This paper | |
| pcDNA3 3xFLAG- ISG15 R99A | This paper | |
| pcDNA3 3xFLAG- ISG15 T103A | This paper | |
| pcDNA3 3xFLAG- ISG15 Y96L Q102D | This paper | |
| Software and Algorithms | ||
| Proteome Discoverer | Thermo Scientific | https://www.thermofisher.com/order/catalog/product/IQLAAEGABSFAKJMAUH |
| Protein Thermal Shift | Thermo Scientific | https://www.thermofisher.com/order/catalog/product/4466038 |
| Other | ||
| M2 FLAG affinity gel | Sigma | CAT#a2220 |
| Glutathione Sepharose | GE Healthcare | CAT#17075601 |
| Deposited Data (Mendeley) | doi:10.17632/djprp3p5ff.1 | |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jon Huibregtse (huibregtse@austin.utexas.edu).
Data and Software Availability
Dataset of raw images files for all figures is deposited at Mendeley.com (doi:10.17632/djprp3p5ff.1).
Experimental Model and Subject Details
Cell Culture
HEK-293T, Jurkat, and NK-92 cells were maintained at 37°C, 5% v/v CO2 in a humidified incubator. HEK-293T cells were maintained in DMEM (Corning) supplemented with 10% FBS (Sigma) and 1% Penicillin-Stretomycin (Corning). Jurkat cells were cultured in RPMI1640 (Lonza), 10% FBS, 1% Penicillin-Streptomycin. NK-92 cells were cultured in Alpha MEM without ribonucleosides and deoxyribonucleosides (Gibco) with 2 mM L-glutamine (Fisher), 1.5 g/l sodium bicarbonate (Sigma), 0.2 mM inositol (Sigma), 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid (Fisher), 100U/ml recombinant IL-2 (Peprotech), 12.5% horse serum (Gibco) and 12.5% FBS (Corning). Cryopreserved human PBMCs were obtained from CTL Technologies and were. Cells were thawed in CTL test media and plated in RPMI1640 at a cell density of 1×106 cells/mL at 37° C and 5% CO2.
Isolation of mouse splenocytes
Mouse spleens for C57B6 (CD11+/+, ICAM1+/+), CD11a−/− mice, and ICAM1−/− (JR664, JR5257, and JR2867, respectively) were purchased from the Jackson Laboratory and delivered in RPMI. Splenocytes were extracted and plated at a density of 1×106cells/ml and rested overnight in RPMI1640 at 37°C with 5% CO2.
Method Details
Protein purification
ISG15 proteins were purified as GST fusion proteins in BL21 E. coli. Overnight starter cultures were grown at 37° C for 16 hours. Cultures were diluted 1:10 and expanded for 2 hours at 37° C. Expression of proteins were induced with 100 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 hours at 30° C. Cells were collected by centrifugation, re-suspended in phosphate buffer saline (PBS) with 1% Triton, and sonicated for 30 seconds. Lysates were spun at 15000 × g for 10 minutes, and supernatants were incubated with Glutathione Sepharose (GE healthcare) for 2 hours with rotation at 4° C. Beads were collected and washed 3x with PBS 1% triton and 3x with 50mM Tris pH 8.0, 150mM NaCl, 0.01% triton, and 2.5mM EDTA. GST fusion proteins (on beads) were subjected to site-specific cleavage with PreScission Protease (GE healthcare) to remove the GST tag. Beads were removed and protein concentration in the supernatant was quantified by SDS-PAGE using a Licor Odyssey Imager. Input levels of proteins were normalized to the wild-type control sample for binding assays and Kd determinations.
pGex6pk-ISG15 (with a cAMP kinase acceptor site) was purified as above. Glutathione Sepharose-bound GST-pkISG15 was washed 3x with 20mM magnesium acetate, 40mM Tris pH 7.5. cAMP dependent protein kinase (Promega) was added to the GST-pkISG15 with 0.2mM [γ-32P]-ATP for 40 minutes at room temperature with rotation. Labeled GST-pkISG15 was collected and subjected to Prescission Protease cleavage. 32P-labeled pkISG15 concentration was determined by imaging of a coommassie blue stained gel and added to assays at the indicated concentrations.
GST-CD11a αI-domain was purified as described (Qu and Leahy, 1995) from BL21 E. coli. Overnight starter cultures were grown at 37°C for 16 hours. Cultures were diluted 1:20 and expanded for 2hrs at 37° C. Bacterial expression of GST fusion proteins were induced with 100 μM (IPTG) for 4 hours at 37° C. Cells were collected by centrifugation, resuspended in 50mM Tris pH 8.0, 200 mM NaCl, 1mM BME. Cells were lysed by sonication and insoluble material (containing GST-αI domain) was collected by centrifugation at 32,000 × g for 30 minutes. The pellet was re-suspended in 20mM Tris pH 8.0 and 7M Urea. Protein was refolded by dialysis in Buffer 1 (20mM glycine, pH 10.5, 15 mM DTT, 2M Urea, 5mM MgCl2) for 1 hour at 4° C, followed by dialysis in Buffer 2 (20mM glycine pH 10.5, 3mM DTT, 1M Urea, 5mM MgCl2, 1M NaCl) for 18 hours at 4°C, followed by dialysis in Buffer 3 (25mM Tris pH 8.0, 200mM NaCl, 5mM MgCl2, and 1mM DTT) for 1 hour at 4°C. Final dialysis was against PBS with 5mM MgCl2. Refolded GST-CD11a I-domain was bound to Glutathione beads (GE healthcare) overnight with rotation at 4°C. Beads were collected and washed 3x with PBS with 5mM MgCl2.
IFN-γ and IL-10 ELISAs
Prior to the start of the assays, NK-92 cells were cultured overnight in media without IL-2. Thawed human PBMCs and freshly isolated mouse splenocytes were rested overnight in media (described above) after plating at a density of 1×106 cells/ml. These cells were treated with 18 ng/mL IL-12 and/or ISG15 at the indicated concentrations for 48 hours. Antibodies were pre-incubated with cells for 1 hour before treating with IL12 and/or ISG15. PP2 (SRC family inhibitor) was added (10μM) to cells 15 minutes before treatment with ISG15 and IL-12. Cell culture supernatants were analyzed for human or mouse IFN-γor human IL-10 using Thermo Scientific ELISA kits (EHIFNG, EHIL-10, EM1001).
For Jurkat cell transfections, cells were seeded at 5×105 cells/mL two days before transfection. Cells were counted and resuspended at 1×106 cells/100μL of Lonza SE Nucleofection reagent with 2μg/sample of the indicated DNA. Cells were transfected with a Lonza Nucleofector 4d using program cl-120. Freshly nucleofected cells were rested for 3 hours in RMPI 1650 with serum, then plated at a density of 1×106cells/mL and treated with 59nM ISG15 for 48 hours. Cell culture supernatants were analyzed for human IFN-γby ELISA (EHIFNG Thermo Scientific).
Protein thermal shift assays
Protein thermal shift kit was obtained from Thermo Scientific. 1μg of ISG15 variants were analyzed as recommended by the manufacturer in a Viia7 thermocycler. Data was analyzed with protein thermal shift software for Tm (Thermo Scientific). Technical quadruplicates were performed on three biologic triplicates; error bars indicate standard error of the mean.
CD11a αI-Domain and MIDAS mutants Binding Assays
3xFLAG ISG15 was incubated with Glutathione-Sepharose beads with GST-CD11a αI-domain for 1 hour at 4°C in PBS with 5mM MgCl2. Beads were collected and washed 2x with the same buffer. Samples were run on a 13% gel and analyzed for the presence of 3xFLAG ISG15 by anti-FLAG immunoblot. Assays were performed in triplicate.
The Kd for the ISG15-CD11a αI domain interaction was determined according to methods described in Pollard (2010). A series of concentrations (250nM, 500nM, 667nM, 833nM, 1.16μM, 1.66μM, 3.33μM, and 5μM) of GST-CD11a αI-Domain on Glutathione Sepharose were incubated with 10nM 32P-labeled ISG15 for 1hr at 4°C. Beads were collected by low speed centrifugation and supernatants were directly removed to 1 ml scintillation vials and radioactivity was quantitated in a liquid scintillation counter (1 min). Kd was determined by calculating the depletion of input ISG15 in the supernatant, and analyzed using PRISM 6 one site total Nonlinear fit of Michaelis-Menten analysis. Three biological triplicates of the experiment were performed, each in technical triplicate.
ICAM1 competition binding assays were performed by incubating the GST-CD11a αI domain or the S139A S139A D239A MIDAS mutant (1.16μM) with 32P-labeled ISG15 (10nM) and increasing concentrations of ICAM1 (100nM, 190nM, 380nM, 760nM, and 1.9μM) for 1 hr at 4°C. Supernatants were collected and analyzed in a scintillation counter for depletion of ISG15 as described above. Three biological triplicates were performed, each in technical triplicate.
Static Cell Adhesion Assay
Cell adhesion assays were performed as described (Strazza et al.; 2010). Black clear bottom 96 well plates were coated with purified ICAM1 (R&D Systems) and ICAM2 (Abcam) proteins. Plates were washed with 50μL of PBS 2mM MgCl2 1mM CaCl2 wash solution and aspirated. 50μL of PBS 1mM CaCl2 2nM MgCl2, ICAM1 and ICAM2 10μg/mL coating solution was added to the plate for 1 hour at 37°C. Coating solution was aspirated and wells were washed one time with wash solution. 50μL of PBS 2mM MgCl2 1mM CaCl2 0.5% and BSA adhesion solution for 1 hour at 37°C. Plates were washed again one time with wash solution. Jurkat cells were serum starved for 2 hours and were labeled with Carboxyfluorescein Succinimidyl Ester (CFSE) and activated with PMA (10ng/ml) and treated with and without ISG15 at 21.3 μg/ml. Activated and treated cells were plated on ICAM1 and ICAM2 coated black clear bottom 96 well plates for 15 minutes at 37°C. Cells were washed three times with PBS 1mM CaCl2 2mM MgCl2 and 0.5% BSA. Total fluorescence was measured on a spectromax m3 plate reader with excitation 485nm emission 528nm. Total cell adhesion was calculated as the signal of washed and treated wells compared to signal from wells with no washes. Assay was performed in triplicate.
qRT-PCR
ISG15 and/or IL-12 treated NK-92 cells were collected for either two or six hours and RNA was extracted with Qiagen RNA extraction kit. A TaqMan® RNA-to-Ct™ 1-step kit was used with IFN-γTaqMan® probes Hs00989291. qPCR was run on a Viia7 qPCR machine. Assays were performed in triplicate.
ISG15 UBAITs
3xFLAG-ISG15-Ubiquitin was purified from BL21 bacteria activated with UBA1 (Boston Biochem) transferred to UBC4 (purified as a GST fusion protein as ISG15) in 5mM DTT, 5mM MgCl2, and 10mM ATP. This charged UBAIT was added to 24×106cells/mL NK-92 cells for 1hr at 37°c in serum free media. Cells were collected and lysed in 8M urea for 20 min. Urea was diluted to 1M with RIPA buffer and incubated with M2 FLAG beads (sigma) overnight at 4°C. Beads were collected and washed 3x in RIPA buffer. Samples were boiled in loading buffer containing 250mM Tris pH 6.8, 40%Glycerol, 8%, SDS, 100mM DTT and run on 10% SDS PAGE Gel. A gel slice was excised above the 25kDa size and sent for tryptic digestion and LC-MS/MS in the Thermo Orbitrap Fusion (UT Proteomics core). Proteins that had peptide spectral counts in three biological replicates were selected (supplemental table s1). Proteins that were at least two-fold higher in the WT ISG15 UBAIT than the ΔGG UBAIT or Y96L/Q102D UBAIT were considered possible ISG15 interactors colored gray. Further filtering identified 5 transmembrane proteins colored blue.
SRC family kinase analyses
NK-92 cells were treated with indicated protein for 10 min. Cells were collected and lysed in 100mM Tris pH 7.5, 100mM NaCl, 1% NP40 containing a Pierce™ Protease and Phosphatase inhibitor mini tablet EDTA-free (Thermo Scientific). Samples were normalized by Bradford and analyzed by western blot for the presence of phosphorylated Src family members. Assays were performed in triplicate.
ISG15 cell binding assays
1×106 transfected 293T cells or NK-92 cells were incubated with 1.2μg of the indicated FLAG tagged ISG15 mutant for 1 hour at 37°C with rotation. Cells were collected and washed 2 times with serum free media. A cell lysate was prepared and run on a 4–12% BOLT bis Tris gel (Thermo Scientific). ISG15 was detected by anti m2 FLAG western blot. Assay was performed in triplicate.
Cytokines and inhibitors
Cytokines and their sources were: mouse IL-12, and hICAM1 (R&D systems), human IL-12 (BD Pharmingen), human IL-2 (Peprotech), A-286982 (Tocris), PP2 src family inhibitor (Adipogen).
Antibodies and bead reagents
Anti Cd11a inhibitory antibodies MHM 24, TS1.22.1.1.13.3 and Tranferrin receptor antibodies G1/221/12 were obtained from the mouse hybridoma bank (DSHB) as cell culture supernatants. Other antibodies and their sources were: anti-M2 FLAG antibody (Sigma), anti-phospho LCK Y394 (Thermo scientific), anti-LYN (Cell Signaling Technologies), anti-HCK (Cell Signaling Technologies), anti-FGR (Cell Signaling Technologies), anti-phospho Src family Y416 (recognizing phosphor-LYN, HCK, and FGR; Cell Signaling Technologies), anti-M2 Flag Beads (Sigma), Glutathione Sepharose (GE healthcare).
Supplementary Material
Supplemental Figure S1. Structures and locations of protein interaction sites on ISG15; related to Figure 2.
Supplemental Figure S2. ISG15 on a surface retains signaling properties, and ISG15 variants retain the ability to be conjugated intracellularly and retain wild-type unfolding characteristics; related to Figure 2.
Supplemental Figure S3. Interaction of ISG15 with the αI domain of CD11a; related to Figures 3 and 4.
Supplemental Figure S4. The effect of ICAM1 on ISG15-LFA-1 binding, signaling, and cell adhesion; related to Figure 5.
Supplemental Figure S5. Quantitative RT-PCR of IFN-γ-encoding mRNA in NK-92; related to Figure 6.
Supplemental Figure S6. SRC family kinases in NK-92 cells; related to Figure 7.
Proteins purified in ISG15-UBAIT experiments; related to Figure 3.
Highlights.
The LFA-1 integrin is the receptor for extracellular ISG15
ISG15 triggers LFA-1 “outside-in” signaling
ISG15- and LFA-1-dependent IFN-γ and IL-10 secretion requires SRC family kinases
Ubiquitin-Activated Interaction Traps can be used to characterize ligand-receptor pairs
Acknowledgments
We thank Dan Leahy and Jaquelin Dudley for helpful discussions and reagents, Martin Poenie for reagents, and Hazel O’Connor and Sylvie Beaudenon-Huibregtse for critical reading of the manuscript. This work was supported by a grant to J. M. H. from the National Institutes of Health, National Institute for Allergy and Infectious Diseases (AI096090).
Footnotes
Author Contributions
C. D. S., L. A. C., and J. M. H. conceived and designed the research. C. D. S. and A. F. S. conducted all experiments and performed the analysis. C. D. S. and J. M. H. drafted the manuscript. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruzzi A, Caveggion E, Berton G. Regulation of phagocyte migration and recruitment by Src-family kinases. Cell Mol Life Sci CMLS. 2008;65:2175–2190. doi: 10.1007/s00018-008-8005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basters A, Geurink PP, Röcker A, Witting KF, Tadayon R, Hess S, Semrau MS, Storici P, Ovaa H, Knobeloch KP, et al. Structural basis of the specificity of USP18 toward ISG15. Nat Struct Mol Biol. 2017;24:270–278. doi: 10.1038/nsmb.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnerts ME, van Kooyk Y, Simmons DL, Figdor CG. Distinct binding of T lymphocytes to ICAM-1, -2 or -3 upon activation of LFA-1. Eur J Immunol. 1994;24:2155–2160. doi: 10.1002/eji.1830240933. [DOI] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. Mycobacterial disease and impaired IFN-γimmunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26:454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun H, Das P, Yu H, Aleman A, Lozano MM, Matouschek A, Dudley JP. Mouse Mammary Tumor Virus Signal Peptide Uses a Novel p97-Dependent and Derlin-Independent Retrotranslocation Mechanism To Escape Proteasomal Degradation. mBio. 2017;8 doi: 10.1128/mBio.00328-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Perussia B, Gupta JW, Kobayashi M, Pospísil M, Young HA, Wolf SF, Young D, Clark SC, Trinchieri G. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173:869–879. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem. 2006;281:4334–4338. doi: 10.1074/jbc.M512830200. [DOI] [PubMed] [Google Scholar]
- D’Cunha J, Knight EJ, Haas AL, Truitt RL, Borden EC. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc Natl Acad Sci U S A. 1996;93:211–215. doi: 10.1073/pnas.93.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol Baltim Md 1950. 1999;163:5029–5038. [PubMed] [Google Scholar]
- Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell PJ, Broeze RJ, Lengyel P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature. 1979;279:523–525. doi: 10.1038/279523a0. [DOI] [PubMed] [Google Scholar]
- Ford NR, Miller HE, Reeme AE, Waukau J, Bengtson C, Routes JM, Robinson RT. Inflammatory signals direct expression of human IL12RB1 into multiple distinct isoforms. J Immunol Baltim Md 1950. 2012;189:4684–4694. doi: 10.4049/jimmunol.1200606. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Chackerian AA, Parker CM, Ballantyne CM, Behar SM. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. J Immunol Baltim Md 1950. 2006;176:4914–4922. doi: 10.4049/jimmunol.176.8.4914. [DOI] [PubMed] [Google Scholar]
- Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, Laudanna C, Berton G. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol Baltim Md 1950. 2006;177:604–611. doi: 10.4049/jimmunol.177.1.604. [DOI] [PubMed] [Google Scholar]
- Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- Grönholm M, Jahan F, Bryushkova EA, Madhavan S, Aglialoro F, Soto Hinojosa L, Uotila LM, Gahmberg CG. LFA-1 integrin antibodies inhibit leukocyte α4β1-mediated adhesion by intracellular signaling. Blood. 2016;128:1270–1281. doi: 10.1182/blood-2016-03-705160. [DOI] [PubMed] [Google Scholar]
- Guan R, Ma LC, Leonard PG, Amer BR, Sridharan H, Zhao C, Krug RM, Montelione GT. Structural basis for the sequence-specific recognition of human ISG15 by the NS1 protein of influenza B virus. Proc Natl Acad Sci U S A. 2011;108:13468–13473. doi: 10.1073/pnas.1107032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AL, Ahrens P, Bright PM, Ankel H. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J Biol Chem. 1987;262:11315–11323. [PubMed] [Google Scholar]
- Helander TS, Carpén O, Turunen O, Kovanen PE, Vaheri A, Timonen T. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 1996;382:265–268. doi: 10.1038/382265a0. [DOI] [PubMed] [Google Scholar]
- Hermann M, Bogunovic D. ISG15: In Sickness and in Health. Trends Immunol. 2017;38:79–93. doi: 10.1016/j.it.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Hildreth JE, Gotch FM, Hildreth PD, McMichael AJ. A human lymphocyte-associated antigen involved in cell-mediated lympholysis. Eur J Immunol. 1983;13:202–208. doi: 10.1002/eji.1830130305. [DOI] [PubMed] [Google Scholar]
- Ketscher L, Basters A, Prinz M, Knobeloch KP. mHERC6 is the essential ISG15 E3 ligase in the murine system. Biochem Biophys Res Commun. 2012;417:135–140. doi: 10.1016/j.bbrc.2011.11.071. [DOI] [PubMed] [Google Scholar]
- Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- Knight E, Cordova B. IFN-induced 15-kDa protein is released from human lymphocytes and monocytes. J Immunol Baltim Md 1950. 1991;146:2280–2284. [PubMed] [Google Scholar]
- Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers. 2011;3:3856–3893. doi: 10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang D, Jiang Y, Sun J, Zhang S, Chen Y, Wang X. Crystal structure of human ISG15 protein in complex with influenza B virus NS1B. J Biol Chem. 2011;286:30258–30262. doi: 10.1074/jbc.C111.257899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Link JT, Pei Z, Reilly EB, Leitza S, Nguyen B, Marsh KC, Okasinski GF, von Geldern TW, Ormes M, et al. Discovery of novel p-arylthio cinnamides as antagonists of leukocyte function-associated antigen-1/intracellular adhesion molecule-1 interaction. 1 Identification of an additional binding pocket based on an anilino diaryl sulfide lead. J Med Chem. 2000;43:4025–4040. doi: 10.1021/jm0002782. [DOI] [PubMed] [Google Scholar]
- Liu M, Li XL, Hassel BA. Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J Biol Chem. 2003;278:1594–1602. doi: 10.1074/jbc.M208123200. [DOI] [PubMed] [Google Scholar]
- Lloyd A, Vickery ON, Laugel B. Beyond the antigen receptor: editing the genome of T-cells for cancer adoptive cellular therapies. Front Immunol. 2013;4:221. doi: 10.3389/fimmu.2013.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb KR, Haas AL. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem. 1992;267:7806–7813. [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10:369–383. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol. 2013a;425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol. 2013b;425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SC, Madden KB, Adamovicz JJ, Gause WC, Hubbard BR, Gately MK, Finkelman FD. Effects of IL-12 on in vivo cytokine gene expression and Ig isotype selection. J Immunol Baltim Md 1950. 1994;152:1047–1056. [PubMed] [Google Scholar]
- Narasimhan J, Wang M, Fu Z, Klein JM, Haas AL, Kim JJP. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J Biol Chem. 2005;280:27356–27365. doi: 10.1074/jbc.M502814200. [DOI] [PubMed] [Google Scholar]
- O’Connor HF, Lyon N, Leung JW, Agarwal P, Swaim CD, Miller KM, Huibregtse JM. Ubiquitin-Activated Interaction Traps (UBAITs) identify E3 ligase binding partners. EMBO Rep. 2015;16:1699–1712. doi: 10.15252/embr.201540620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudshoorn D, van Boheemen S, Sanchez-Aparicio MT, Rajsbaum R, Garcia-Sastre A, Versteeg GA. HERC6 is the main E3 ligase for global ISG15 conjugation in mouse cells. PloS One. 2012;7:e29870. doi: 10.1371/journal.pone.0029870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. A guide to simple and informative binding assays. Mol Biol Cell. 2010;21:4061–4067. doi: 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu A, Leahy DJ. Crystal structure of the I-domain from the CD11a/CD18 (LFA-1, alpha L beta 2) integrin. Proc Natl Acad Sci U S A. 1995;92:10277–10281. doi: 10.1073/pnas.92.22.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshevich L, Impens F, Ribet D, Quereda JJ, Nam Tham T, Nahori M-A, Bierne H, Dussurget O, Pizarro-Cerdá J, Knobeloch K-P, et al. ISG15 counteracts Listeria monocytogenes infection. eLife. 2015;4 doi: 10.7554/eLife.06848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht M, Borden EC, Knight E. A human 15-kDa IFN-induced protein induces the secretion of IFN-gamma. J Immunol Baltim Md 1950. 1991;147:2617–2623. [PubMed] [Google Scholar]
- Sanchez-Madrid F, Krensky AM, Ware CF, Robbins E, Strominger JL, Burakoff SJ, Springer TA. Three distinct antigens associated with human T-lymphocyte-mediated cytolysis: LFA-1, LFA-2, and LFA-3. Proc Natl Acad Sci U S A. 1982;79:7489–7493. doi: 10.1073/pnas.79.23.7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M, Lu C, Palframan RT, von Andrian UH, McCormack A, Takagi J, Springer TA. Reversibly locking a protein fold in an active conformation with a disulfide bond: integrin alphaL I domains with high affinity and antagonist activity in vivo. Proc Natl Acad Sci U S A. 2001;98:6009–6014. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, McCormack A, Zhang R, Joachimiak A, Takagi J, et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L. Distinct functional surface regions on ubiquitin. J Biol Chem. 2001;276:30483–30489. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7:11496. doi: 10.1038/ncomms11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow JL, Murray RZ. Intracellular trafficking and secretion of inflammatory cytokines. Cytokine Growth Factor Rev. 2013;24:227–239. doi: 10.1016/j.cytogfr.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Strazza M, Azoulay-Alfaguter I, Pedoeem A, Mor A. Static adhesion assay for the study of integrin activation in T lymphocytes. J Vis Exp JoVE. 2014 doi: 10.3791/51646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga Y, Kita Y, Satoh A, Asai S, Kato K, Ishikawa K, Horiuchi T, Takashi T. Affinity and kinetic analysis of the molecular interaction of ICAM-1 and leukocyte function-associated antigen-1. J Immunol Baltim Md 1950. 1998;161:4016–4022. [PubMed] [Google Scholar]
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, Huibregtse JM, Krug RM. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an. Proc Natl Acad Sci U S A. 2004;101:7578–7582. doi: 10.1073/pnas.0402528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Sridharan H, Chen R, Baker DP, Wang S, Krug RM. Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat Commun. 2016;7:12754. doi: 10.1038/ncomms12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Structures and locations of protein interaction sites on ISG15; related to Figure 2.
Supplemental Figure S2. ISG15 on a surface retains signaling properties, and ISG15 variants retain the ability to be conjugated intracellularly and retain wild-type unfolding characteristics; related to Figure 2.
Supplemental Figure S3. Interaction of ISG15 with the αI domain of CD11a; related to Figures 3 and 4.
Supplemental Figure S4. The effect of ICAM1 on ISG15-LFA-1 binding, signaling, and cell adhesion; related to Figure 5.
Supplemental Figure S5. Quantitative RT-PCR of IFN-γ-encoding mRNA in NK-92; related to Figure 6.
Supplemental Figure S6. SRC family kinases in NK-92 cells; related to Figure 7.
Proteins purified in ISG15-UBAIT experiments; related to Figure 3.