

Abstract
From the leaves of Nelumbo nucifera (an aquatic plant), one new compound, 24(R)-ethylcholest-6-ene-5α-ol-3-O-β-D-glucopyranoside (1), along with 11 known metabolites (2–12), were isolated and identified by spectroscopic methods including 1D- and 2D NMR. Antifungal activity for (R)-roemerine (3) (IC50/MIC = 4.5/10 μg/mL against Candida albicans) and antimalarial activity for (R)-roemerine (3) and N-methylasimilobine (5) (IC50 = 0.2 and 4.8 μg/mL for the D6 clone, respectively, and 0.4 and 4.8 μg/mL for the W2 clone, respectively) was observed. None of the compounds were cytotoxic to Vero cells up to a concentration of 23.8 μg/mL. NMR data for 10-eicosanol (7) and 7,11,15-trimethyl-2-hexadecanone (10) are presented for the first time. An analysis of the structure–activity relationship shows that the substituents in position C-1 and C-2 of aporphine alkaloids are crucial for the antimalarial activity.
Keywords: Nelumbo nucifera, Nelumbonaceae, 24(R)-Ethylcholest-6-ene-5α-ol-3-O-β-D-glucopyranoside, Roemerine, Antimalarial, SAR
1. Introduction
Nelumbo nucifera Gaertn. (Nelumbonaceae), commonly known as lotus, is a perennial aquatic plant grown and consumed throughout Asia. All parts of N. nucifera have been used for various medicinal purposes in oriental medicine. Lotus is reported to possess antidiarrheal, psychopharmacological, diuretic, antipyretic, antimicrobial and hypoglycemic activities (Rai, Wahile, Mukherjee, Saha, & Mukherjee, 2006). Previous work on the leaves of this plant resulted in the isolation of several alkaloids and other constituents (Kashiwada et al., 2005; Wassel, Saeed, Ibrahim, & El-Eraqy, 1996). As part of our on going search for antimicrobial and antimalarial compounds from higher plants, we have undertaken an investigation of the leaves of this plant. In this study, we describe the isolation, structure elucidation and biological activities of a new (1) and 11 (2–12) known compounds (Fig. 1) from the leaves of N. nucifera and some structure activity relationship (SAR) for the antimalarial activity of aporphine alkaloids.
Fig. 1.

Structures of some constituents of Nelumbo nucifera.
2. Results and discussion
Compound 1 was isolated as a white solid. Its molecular formula of C35H60O7, was determined by HRESIMS and indicated the presence of six degrees of unsaturation. 13C NMR and DEPT spectra showed 35 signals including 6 methyls, 11 methylenes, 15 methines and 3 quaternary carbons. Careful examination of the 1H-, 13C NMR and their 2D long-range correlations (Fig. 2) and comparison of aglycone values with literature indicated that compound 1 was a glycoside of the previously reported aglycone 24-ethyl-cholest-6-ene-3β,5α-diol (Greca, Fiorentino, Molinaro, Monaco, & Previtera, 1994). Cross-peak correlations for H-4 (δ 3.18) to C-2 (δ 30.3), C-3 (75.6) and with C-5 (δ 83.1) in the HMBC spectrum was used to place a further hydroxyl at the C-5 position and double bond protons present at δ 5.71 and 5.95 also showed HMBC correlations with C-5 (δ 83.1) indicating that there was a double bond between C-6 and C-7. The hydroxyl group at C-5 was determined to be α-oriented (Holland & Jahangir, 1983) from the signals observed in the 13C NMR for C-6 (δ = 133.9) and C-7 (δ = 132.2). The absolute configuration at C-24 was determined to be R (Wright et al., 1978) on the basis of the comparison of 13C NMR of 1 (δC = 46.4) and β-sitosterol-3-O-β-D-glucopyranoside (11) (δC = 46.4, having an R configuration at C-24) in pyridine-d5. The configuration of the anomeric carbon was defined as β from the coupling constant of 8.0 Hz. In situ acid hydrolysis of 1 afforded D-glucose. According to the molecular rotation formula (Klyne, 1950), the specific rotation of 1 ([α]D26: −14°) was multiplied by its molecular wt (m/z 592), the resulting value (−8288) was then divided by 100. The molecular rotation [M]Dα was found to be −82.9° and is comparable with levorotatory Me-β-D-glucopyranoside ([M]Dα = −66°) (Germonprez, Puyvelde, Maes, Tri, & Kimpe, 2004). According to the molecular rotation calculation, the glucose in 1 should possess the absolute configuration D-form, which is the common form for glucose existing in nature. The glycosidation position was unambiguously determined by a three-bond correlation between the glycosyl anomeric proton H-1′ (δH = 5.13) and C-3 (δC = 75.6) using HMBC. On the basis of the above evidence, the structure of 1 was established as 24(R)-ethyl-cholest-6-ene-5α-ol-3-O-β-D-glucopyranoside, a new steroid glucoside.
Fig. 2.
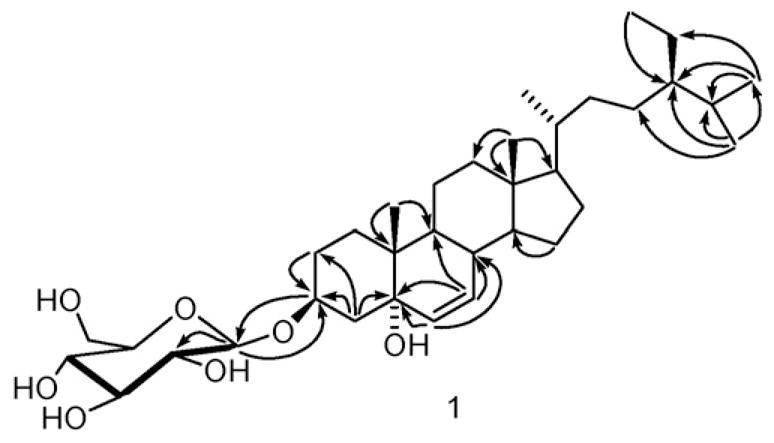
Key HMBC correlations for compound 1.
Eleven known compounds were identified as, dehydroroemerine (2), (R)-roemerine (3), nuciferine (4), N-methylasimilobine (5), and anonaine (6) (Guinaudeau, Leboeuf, & Cave, 1975, 1983), 10-eicosanol (7), 3,7,11,15-tetramethyl-1-hexadecen-3-ol (isophytol) (8) (Ahmad & Ali, 1991), 3,7,11,15-tetramethyl-2-hexadecen-1-ol (trans-phytol) (9) (Sims & Pettus, 1976), 7,11,15-trimethyl-2-hexadecanone (10) (Worner & Schreier, 1991), β-sitosterol-3-O-β-D-glucopyranoside (11) (Kojima, Sato, Hatano, & Ogura, 1990) and quercetin 3-O-β-D-glucopyranoside (12) (Markham, Ternai, Stanley, Geiger, & Mabry, 1978), by comparison of their spectral data with published values. This is the first report for the spectral data of 7, that was previously prepared synthetically (Churchward, Gibson, Meakins, & Mulley, 1950), and without any reference or spectral evidence isolated from Semiaquilegia adoxoides (Feng et al., 2006). The hydroxyl group of compound 7 was confirmed at position 10 by the GC–MS fragmentation pattern. Compound 10 was previously reported in the volatile fraction of Galium odoratum (Worner & Schreier, 1991). This is the first report of the isolation of 10 from N. nucifera and the first report of its 1H- and 13C NMR data.
The crude ethanolic extract along with fractions A–D (see Section 3) and all purified compounds except 2 and 6 were evaluated for in vitro antimalarial activity (against chloroquine sensitive (D6) and resistant (W2) clones of Plasmodium falciparum), cytotoxicity and for antifungal activity. Fractions A, C, D and compounds 3 and 5 exhibited activity against D6 (IC50 of 9.2, 3.6, 1.7, 0.2 and 4.8 μg/mL, respectively) and W2 clones (IC50 of 3.5, 3.2, 4.5, 0.4 and 4.8 μg/mL, respectively). The selectivity index of the antimalarial activity versus toxicity for compound 3 was 122 and 62 for D6 and W2 clones, respectively, as compared to a selectivity index of 5 for both clones for compound 5. Chloroquine and artemisinin were used as positive controls which showed IC50 values of 16.0 and 8.5 ng/mL (for D6) and IC50 of 150.0 and 9.0 ng/mL (for W2), respectively. None of the tested compounds or fractions had cytotoxic effects towards mammalian kidney fibroblasts (Vero cells) up to a concentration of 23.8 μg/mL. Only compound 3 had antifungal activity against Candida albicans with IC50/ MIC values of 4.5/10 μg/mL, respectively. The positive control amphotericin B gave IC50/MIC values of 0.2/0.6 μg/mL, respectively. This is the first report of the antimalarial activity of 3.
Compounds 3–5 have a similar aporphine alkaloid skeleton. The only difference is substitution at position C-1 and C-2. Compound 4 exhibited no activity and that the most potent metabolite (3) possessed a methylenedioxy moiety.
A literature survey for biological activity of 3 and 5 showed that 3 reverses a multidrug resistance phenotype and possessed weak cytotoxicity (You et al., 1995), mutagenicity (Nozaka et al., 1990), a relaxant effect (Chulia et al., 1995) and an inhibitory activity for CD45 protein tyrosine phosphatase (Miski et al., 1995), however 5 inhibited platelet aggregation (Chang, Wei, Teng, & Wu, 1998) and had a sedative effect (Han, Park, & Park, 1989).
3. Experimental
3.1. General experimental procedures
Optical rotations were determined with an AUTOPOL IV polarimeter. IR spectra were recorded on a JASCO 302-A spectrometer. The 1D- and 2D NMR spectra were run on a Varian 400 Mercury plus NMR spectrometer. Multiplicity determination (DEPT) and 2D NMR spectra (COSY, HMQC, and HMBC) were performed with standard pulse programs. GC–MS analysis was conducted using a Hewlett–Packard 5890 Series II plus/5972 MSD system, using a DB-5 capillary column. Column temperature, 150 °C (5 min) to 240 °C at 7 °C/min., injector temperature 240 °C, detector temperature 250 °C; helium was used as the carrier gas (39.9 cm/s). The mass spectra were obtained using an Agilent Series 1100 SL HPLC connected to a time of flight mass detector (model G1969A, Agilent Technologies) equipped with an ESI interface. Column chromatography was run using silica gel (40 μm, J.T. Baker), RP-18 (40 μm Bakerbond, J.T. Baker) and alumina (150 mesh, Sigma–Aldrich).
3.2. Plant material
The leaves of N. nucifera were purchased in 2004 from www.plumflowers.com, and authenticated in-house by Dr. Vaishali C. Joshi (taxonomist). A voucher specimen is deposited in the plant repository at The National Center for Natural Products Research, University of Mississippi (Voucher # NENUN 2449).
3.3. Extraction and isolation
The dried powdered material (700 g) was percolated with 95% EtOH (15 L). The ethanol extract was evaporated to dryness (75.44 g, 10.8%). Part of the extract (38.47 g) was fractionated using a published procedure (Kupchan, Dasgupta, Fujita, & King, 1963) to yield an alkaloidal fraction (0.54 g, fraction A) and a neutral/acidic fraction (20.0 g, fraction B). Part of fraction A (0.34 g) was separated into a phenolic alkaloidal fraction (0.14 g, fraction C) and a non-phenolic alkaloidal fraction (0.2 g, fraction D). Part of fraction D (0.14 g) was subjected to column chromatography using silica gel (13 g, 1 cm × 30 cm) eluted with a step gradient of petroleum ether/benzene and benzene/acetone to yield 14 fractions (D1–D14). Fraction D3 eluted with petroleum ether/ benzene (6:4, 65 mL) afforded 2 (1.3 mg). Fractions D10, D11, D12 and D14 eluted with benzene/acetone (96:4, 100 mL), (94:6, 150 mL), (93:7, 140 mL) and (82:18, 154 mL), respectively, afforded 3 (7 mg), 4 (16 mg), 5 (6 mg) and 6 (1.2 mg), respectively. Chromatography of fraction C (134 mg) over an alumina column (20 g, 1 cm × 28 cm) using a step gradient solvent system consisting of benzene/acetone, yielded an additional amount of 5 (29 mg).
Column chromatography of fraction B (20.0 g) on a silica gel column (350 g, 6.5 cm × 40 cm) eluting with petroleum ether (1.2 L), CHCl3 (2.0 L), CHCl3/EtOAc (1:1, 0.5 L), EtOAc (0.9 L), EtOAc/acetone (1:1, 0.6 L), and acetone (3.0 L) afforded four fractions: E–H (2.4, 11.2, 1.1, and 2.1 g, respectively). Column chromatography of fraction F (10.4 g) using silica gel (200 g, 3 cm × 60 cm) and eluting with a gradient solvent system consisting of hexane/CHCl3 with increasing polarity yielded seven fractions: F1–F7. Fraction F2 (1.5 g) was rechromatographed over a silica gel column (200 g, 3 cm × 60 cm) using hexane/EtOAc (98:2) to afford 10 (5 mg, 220 mL) and 7 (1 g, 1.4 L). Fractions F4 (29 mg) and F6 (15 mg) each containing one major spot, were purified on a silica gel column using hexane/EtOAc (99:1) to yield 8 (9 mg, 29 mL) and 9 (10 mg, 100 mL), respectively. Column chromatography of fraction G (1.1 g) using silica gel (80 g, 3 cm × 21 cm) and a step gradient solvent system consisting of CHCl3/EtOAc of increasing polarity, followed by EtOAc/MeOH mixtures, yielded 13 fractions (I1–I13). Fractions I10 and I11 were eluted with EtOAc/MeOH (99.5:0.5 and 99:1.0, 150 mL and 1 L, respectively) were pooled (80 mg). This residue was purified on a RP-18 column (18 g, 1 cm × 31 cm) eluting with MeOH to afford 1 (2.4 mg, 10 mL), and 11 (12 mg, 30 mL). Fractions I12 and I13, eluted with EtOAc/MeOH (98:2, 250 mL and 97:3, 200 mL) were pooled to yield 12 (12 mg).
3.4. 24(R)-Ethylcholest-6-en-5-α-ol-3-O-β-D-glucopyranoside (1)
White powder; [α] 14° (−)(c {0.1}, pyridine)D26; IR (cm−1): 3388 (–C–OH), 1591 (C C) neat max; 1H NMR (pyridine-d5, 400 MHz, δ): 5.95 (1H, dd, 2.4, 9.6, H-7), 5.71 (1H, d, 11.2, H-6), 5.13 (1H, d, 8.0, H-1′), 4.87 (1H, m, H-3), 4.48 (1H, dd, 2.4, 11.6, 1H-6′), 4.40 (1H, dd, 4.8, 11.6, 1H-6′), 4.27 (1H, m, H-3′), 4.30 (1H, m, H-4′), 4.09 (1H, t, 8.4, H-2′), 3.86 (1H, m, H-5′), 3.18 (1H, dd, 12.8, 4.4, 1H-4), 2.30 (1H, m, 1H-2), 1.95 (3H, m, 1H-4, 1H-8, 1H-12), 1.85 (2H, m, 1H-2, 1H-16), 1.72 (1H, m, 1H-1), 1.68 (1H, m, 1H-25), 1.52 (1H, m, 1H-15), 1.40 (6H, m, 1H-1, 2H-11, 1H-16, 1H-20, 1H-22), 1.30 (3H, m, 1H-15, 2H-28), 1.24 (2H, m, 2H-23), 1.20 (1H, m, 1H-12), 1.15 (1H, m, 1H-14), 1.10 (1H, m, 1H-22), 1.06 (1H, m, 1H-17), 1.00 (2H, m, 1H-9, 1H-24), 1.02 (3H, d, 6.4, 3H-27), 0.96 (3H, d, 5.6, 3H-21), 0.94 (3H, s, 3H-19), 0.85 (3H, d, 6.4, 3H-26), 0.89 (3H, t, 6.8, 3H-29), 0.67 (3H, s, 3H-18); 13C NMR (pyridine-d5, 100 MHz, δ): 29.0 (C-1), 30.3 (C-2), 75.6 (C-3), 34.0 (C-4), 83.1 (C-5), 133.9 (C-6), 132.2 (C-7), 39.4 (C-8), 44.6 (C-9), 39.0 (C-10), 21.5 (C-11), 40.6 (C-12), 44.0 (C-13), 54.1 (C-14), 24.4 (C-15), 29.5 (C-16), 56.5 (C-17), 12.5 (C-18), 15.7 (C-19), 36.8 (C-20), 19.2 (C-21), 34.5 (C-22), 26.8 (C-23), 46.4 (C-24), 29.8 (C-25), 20.3 (C-26), 19.6 (C-27), 23.7 (C-28), 12.5 (C-29), 104.0 (C-1′), 75.8(C-2′), 78.9 (C-3′), 72.0 (C-4′), 78.5 (C-5′), 63.1 (C-6′); HRESIMS (−), m/z 627.4040 [M+Cl]− (calcd. for C35H60O7Cl, 627.4027).
3.5. Acid hydrolysis of 1
Compound 1 and an authentic sugar sample (D-glucose) were spotted on a silica gel TLC plate and hydrolyzed in situ by exposure to HCl vapor at 70 °C for 25 min. The TLC plate was then developed with CHCl3/MeOH/AcOH/H2O (14:6:2:1) and sprayed with 0.5% vanillin in 5% ethanolic sulfuric acid for visualization. The hydrolyzed glucose moiety matched the Rf (0.28) of the standard D-glucose.
3.6. 10-Eicosanol (7)
White solid, optically inactive; IR (cm−1): 2849, 2916 (C–H), 3332 (–C–OH) neat max; 1H NMR (CDCl3, 400 MHz, δ): 0.85 (3H, d, J = 5.2 Hz, CH3-1 or CH3-20), 0.86 (3H, d, J = 5.2 Hz, CH3-20 or CH3-1), 1.23 (26H, brs, H-2-7 and 13–19), 1.34 (8H, brs, H-8, 9, 11, 12), 3.55 (1H, s, H-10); 13C NMR (CDCl3, 100 MHz, δ): 72.3 (C-10), 37.7 (C-9, 11), 32.1 (C-3, 18, ω-2), 29.9 (C-5, 6, 7, 13, 14, 15, 16), 29.5 (C-4, 17, ω-3), 25.9 (C-8, 12), 22.9 (C-2, 19, ω-1), 14.4 (C-1, 20); GC, RT 56.0 min; GC/MS m/z 297 [M−H]+, 157, 139, 125, 111, 97, 83, 69 and 55; m/z 297 [M−H]+ (calcd. for C20H41O, 297).
3.7. 7,11,15-Trimethyl-2-hexadecanone (10)
Colorless liquid, optically inactive; IR (cm−1) 2953, 2923, 2850 (C–H), 1719 (〉C=O) neat max; 1H NMR (CDCl3, 400 MHz, δ) 2.37 (2H, t, 7.2, 2H-3), 2.10 (3H, s, 3H-1), 1.54 (2H, m, 2H-4), 1.48 (1H, m, 1H-15), 1.34 (2H, m, 1H-7, 1H-11), 1.22 (10H, m, 2H-5, 2H-8, 2H-9, 2H-10 and 2H-12), 1.01–1.14 (6H, m, 2H-6, 2H-13, 2H-14), 0.84 (6H, d, 6.4, 3H-15a, 3H-16), 0.82 (6H, d, 3H-7a, 3H-11a); 13C NMR (pyridine-d5, 100 MHz, δ): 30.1 (C-1), 209.6 (C-2), 44.4 (C-3), 21.6 (C-4), 29.9 (C-5), 36.7 (C-6), 32.9 and 33.0 (C-7 or C-11, values may interchanged), 19.8 (C-7a), 37.4, 37.5 and 37.6 (C-8, C-10, C-12, values may interchanged), 24.6 (C-9), 20.0 (C-11a), 25.0 (C-13), 39.6 (C-14), 28.2 (C-15), and 22.9 (C-15a, C-16); ESI-MS m/z 305 [M+Na]+ (calcd. for C19H38ONa, 305).
3.8. Determination of in vitro antimalarial and cytotoxic activities
The in vitro antimalarial activity of test samples was determined against two strains of P. falciparum (D6: chloroquine-sensitive; W2: chloroquine-resistant). The assay was based on the determination of plasmodial LDH activity using Malstat™ reagent and was performed in 96-well plates as described previously (Jain et al., 2005). The level of in vitro cytotoxicity of each sample was also determined towards mammalian kidney fibroblasts (VERO cells) as described earlier (Jain et al., 2005) and the selectivity index (SI) was calculated as the ratio of IC50 in Vero cells and IC50 in P. falciparum. Two standard antimalarial agents chloroquine and artemisinin were used as positive controls and DMSO was used as a vehicle control.
3.9. Determination of antifungal activity
Susceptibility testing was performed using a modified version of the CLSI (formerly NCCLS) methods (NCCLS, 2002; Jain et al., 2005). C. albicans ATCC 90028 was obtained from the American Type Culture Collection (Manassas, VA). Amphotericin B (ICN Biomedicals, Ohio) was included as a positive control. The MIC was defined as the lowest test concentration that allows no detectable growth.
Acknowledgments
The authors thank Dr. Bharathi Avula for providing mass spectroscopic data, Frank T. Wiggers, Dr. Zulfiqar Ali and Dr. Rahul S. Pawar for NMR data and Ms Marsha Wright for conducting the antimicrobial testing, which was supported by NIH, NIAID Division of AIDS, Grant no. AI 27094. This work was supported in part by the United States Department of Agriculture, Agricultural Research Service, Specific Cooperative Agreement no. 58-6408-2-0009 and Grant number P20RR021929 from the National Center for Research Resources.
References
- Ahmad VU, Ali MS. Terpenoids from marine red alga Laurencia pinnatifida. Phytochemistry. 1991;30:4172–4174. [Google Scholar]
- Chang F, Wei J, Teng C, Wu Y. Two new 7-dehydroaporphine alkaloids and antiplatelet action aporphines from the leaves of Annona purpurea. Phytochemistry. 1998;49:2015–2018. doi: 10.1016/s0031-9422(98)00376-8. [DOI] [PubMed] [Google Scholar]
- Chulia S, Ivorra MD, Cave A, Cortes D, Noguera MA, D’Ocon MP. Relaxant activity of three aporphine alkaloids from Annona cherimolia on isolated aorta of rat. Journal of Pharmacy and Pharmacology. 1995;47:647–650. doi: 10.1111/j.2042-7158.1995.tb05852.x. [DOI] [PubMed] [Google Scholar]
- Churchward VR, Gibson NA, Meakins RJ, Mulley JW. The preparation of a series of (±)-n-eicosanols and n-eicosanones. Journal of Chemical Society. 1950:959–960. [Google Scholar]
- Feng N, Zheng C, Hai-Tao C, Yong J, Fa-Kui C, Peng-Fei T. Constituents from the roots of Semiaquilegia adoxoides. Chinese Journal of Chemistry. 2006;24:1788–1791. [Google Scholar]
- Germonprez N, Puyvelde LV, Maes L, Tri MV, Kimpe ND. New pentacyclic triterpene saponins with strong anti-leishmanial activity from the leaves of Maesa balasae. Tetrahedron. 2004;60:219–228. [Google Scholar]
- Greca MD, Fiorentino A, Molinaro A, Monaco P, Previtera L. Hydroperoxysterols in Arum italicum. Natural Product Letters. 1994;5:7–14. [Google Scholar]
- Guinaudeau H, Leboeuf M, Cave A. Aporphine alkaloids. Lloydia. 1975;38:275–338. [PubMed] [Google Scholar]
- Guinaudeau H, Leboeuf M, Cave A. Aporphinoid alkaloids III. Journal of Natural Products. 1983;46:761–835. doi: 10.1021/np50002a001. [DOI] [PubMed] [Google Scholar]
- Han BH, Park MH, Park JH. Chemical and pharmacological studies on sedative cyclopeptide alkaloids in some Rhamnaceae plants. Pure and Applied Chemistry. 1989;61:443–448. [Google Scholar]
- Holland HL, Jahangir Reactions of steroidal 4,5- and 5,6-epoxides with strong bases. Canadian Journal of Chemistry. 1983;61:2165–2170. [Google Scholar]
- Jain M, Khan SI, Tekwani BL, Jacob MR, Singh S, Singh PP, et al. Synthesis, antimalarial, antileishmanial, and antimicrobial activities of some 8-quinolinamine analogues. Bioorganic & Medical Chemistry. 2005;13:4458–4466. doi: 10.1016/j.bmc.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Kashiwada Y, Aoshima A, Ikeshiro Y, Chen Y, Furukawa H, Itoigawa M, et al. Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure–activity correlations with related alkaloids. Bioorganic & Medical Chemistry. 2005;13:443–448. doi: 10.1016/j.bmc.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Klyne W. The configuration of the anomeric carbon atoms in some cardiac glycosides. Journal of Biochemistry. 1950;47:xli–xlii. [PubMed] [Google Scholar]
- Kojima H, Sato N, Hatano A, Ogura H. Sterol glucosides from Prunella vulgaris. Phytochemistry. 1990;29:2351–2355. [Google Scholar]
- Kupchan SM, Dasgupta B, Fujita E, King ML. The alkaloids of American lotus, Nelumbo lutea. Tetrahedron. 1963;19:227–232. [Google Scholar]
- Markham KR, Ternai B, Stanley R, Geiger H, Mabry TJ. Carbon-13 NMR studies of flavonoids-III. Tetrahedron. 1978;34:1389–1397. [Google Scholar]
- Miski M, Shen X, Cooper R, Gillum AM, Fisher DK, Miller RW, et al. Aporphine alkaloids, CD45 protein tyrosine phosphatase inhibitors, from Rollinia ulei. Bioorganic & Medicinal Chemistry Letters. 1995;5:1519–1522. [Google Scholar]
- NCCLS. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard M27-A2. 15. Vol. 22. Villanova, PA: National Committee on Clinical Laboratory Standards; 2002. [Google Scholar]
- Nozaka T, Watanabe F, Tadaki S, Ishino M, Morimoto I, Kunitomo J, et al. Mutagenicity of isoquinoline alkaloids, especially of the aporphine type. Mutation Research, Genetic Toxicology Testing. 1990;240:267–279. doi: 10.1016/0165-1218(90)90077-f. [DOI] [PubMed] [Google Scholar]
- Rai S, Wahile A, Mukherjee K, Saha BP, Mukherjee PK. Anti-oxidant activity of Nelumbo nucifera (sacred lotus) seeds. Journal of Ethnopharmacology. 2006;104:322–327. doi: 10.1016/j.jep.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Sims JJ, Pettus JA., Jr Isolation of free cis- and trans-phytol from the red alga Gracilaria andersoniana. Phytochemistry. 1976;15:1076–1077. [Google Scholar]
- Wassel G, Saeed A, Ibrahim N, El-Eraqy W. Flavonoids of Nelumbo nucifera Gaertn and biological evaluation. Egyptian Journal of Pharmaceutical Science. 1996;37:585–596. [Google Scholar]
- Worner M, Schreier P. Uber die Aromastoff-Zusammensetzung von Waldmeister (Galium odoratum L. Scop) Zeitschrift fur Lebensmittel-Untersuchung und-Forschung. 1991;193:317–320. [Google Scholar]
- Wright JLC, McInnes AG, Shimizu S, Smith DG, Walter JA, Idler D, et al. Identification of C-24 alkyl epimers of marine sterols by 13C nuclear magnetic resonance spectroscopy. Canadian Journal of Chemistry. 1978;56:1898–1903. [Google Scholar]
- You M, Wickramaratne DBM, Silva GL, Chai H, Chagwedera TE, Farnsworth NR, et al. (−)-Roemerine, an aporphine alkaloid from Annona senegalensis that reverses the multidrug-resistance phenotype with cultured cells. Journal of Natural Products. 1995;58:598–604. doi: 10.1021/np50118a021. [DOI] [PubMed] [Google Scholar]