

Abstract
Pain is both a major clinical and economic problem, affecting more people than diabetes, heart disease, and cancer combined. While a variety of prescribed or over-the-counter (OTC) medications are available for pain management, opioid medications, especially those acting on the μ-opioid receptor (μOR) and related pathways, have proven to be the most effective, despite some serious side effects including respiration depression, pruritus, dependence, and constipation. It is therefore imperative that both academia and industry develop novel μOR analgesics which retain their opioid analgesic properties but with fewer or no adverse effects. In this review we outline recent progress towards the discovery of safer opioid analgesics.
Signaling Pathways of the μOR
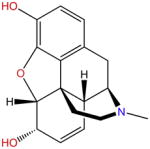
The opium poppy was known to possess powerful analgesic (see Glossary) properties even in ancient times [1]. It was not until the 19th century that one of its potent analgesic ingredients, morphine, was successfully isolated (Box 1). However, morphine was also shown to have adverse effects on both the respiratory and gastrointestinal (GI) systems. Addiction and tolerance caused by this substance led to strict government regulations for its production, use, and distribution [2]. Pharmacological studies later revealed that opioid receptors trigger a series of intracellular responses which are responsible for their pharmacological outcomes [3]. The μ opioid receptor (μOR) is a well-known member of this receptor family (Box 2). Many morphine analogs are believed to target μORs via two distinct downstream signaling pathways that are simultaneously stimulated. These two pathways are independently associated with the analgesic properties and undesired side effects of opioids [4].
Box 1. The History of Painkiller Development.
Opioids extracted from opium poppies have been used to treat pain for thousands of years. In the early 19th century morphine was first extracted in a pure form and applied widely as a painkiller during wartime. In 1830 the naturally occurring methylated morphine, codeine, was first isolated by Jean-Pierre Robiquet to replace raw opium for medical applications [47]. In 1843 Dr Alexander Wood administered morphine by injection for the first time [48]. Charles Romley Wright, an English scientist, synthesized heroin in 1874 and sold it to the Bayer Company in 1898 [49]. Salicylic acid was first isolated in 1828 by Johann Andreas Buchner, and was formulated by Frederick Bayer and Felix Hoffman in 1895 [50]. In an effort to develop less-addictive painkillers, chemists synthesized compounds such as codeine and methadone in the mid-20th century. By the late 20th century a new generation of painkillers was introduced: synthetic opiates which mimicked the above natural painkillers. These included Vicodin, OxyContin, and Percocet (1999) [51].
Box 2. The Family of Opioid Receptors.
ORs are the primary targets of opioid painkillers. ORs are distributed widely in the brain, and are also found in the spinal cord and digestive tract [52]. There are five different types of OR: δOR, κOR, μOR, the nociceptin receptor (ORL1), and ζOR. δORs are mainly distributed in the brain and peripheral sensory neurons. They mediate analgesic, antidepressant, and convulsant effects [53–55]. κORs are located in both peripheral sensory neurons and the spinal cord. These are involved in analgesia, anticonvulsant effects, depression, diuresis, dysphoria, and stress [56]. μORs are found in the brain, spinal cord, peripheral sensory neurons, and intestinal tract. They are responsible for analgesia, physical dependence, miosis, euphoria and GI tract motility [53]. Nociceptin ORL1 receptors in the brain and spinal cord are associated with anxiety and depression. ζORs distributed in the brain, heart, liver, and kidney are involved in tissue growth [57]. Currently, μORs are the most attractive target for painkiller drug discovery within the OR family owing to their special pharmacological properties [58].
Decades of research have gradually uncovered the downstream signaling pathways associated with the analgesic and adverse effects of opioids (Figure 1 and Box 3) [5]. Analgesia is achieved via a classical G-protein pathway which suppresses neuronal excitability and promotes the hyperpolarization of neurons [6]. An agonist-induced conformational change in the μOR instigates the binding of the Gi protein, and results in the dissociation of its α subunit from the β and γ subunit complex [7]. The α subunit inhibits the activity of adenylyl cyclase, reducing the production of intracellular cAMP [8] (Figure 1). The cyclic nucleotide-gated ion channels then remain closed, hampering the influx of Na+ and thereby suppressing the excitability of neurons. Meanwhile, the βγ subunits not only inhibit T-type calcium channels, preventing Ca2+ influx and neuronal depolarization, but also activate the G-protein inwardly rectifying potassium (GIRK) channels, promoting K+ efflux and hyperpolarization [8,9] (Figure 1).
Figure 1.

Signaling Pathways of the μ-Opioid Receptor (μOR). μOR can activate the heterotrimeric G protein, Gi. G protein-coupled receptor kinases (GRKs) together with protein kinases C (PKCs) catalyze the phosphorylation of agonist-bound receptors, which can subsequently either bind to arrestin, undergo internalization, or signal through MAP kinase and other pathways. μORs exhibit basal agonist-independent activation of Gi. Molecules that can suppress basal activity are called inverse agonists [9,46]. Neutral antagonists block the binding of other ligands without imposing a biological response. There are two categories of agonists: full agonists and partial agonists [9,46]. Full agonists produce a full biological response whereas partial agonists only produce a partial biological response even at saturating concentrations [9,46]. These properties are independent of ligand affinities.
Box 3. Mechanisms of Nociception and Analgesia.
There are two different target areas for painkiller development: the dorsal horn and periphery (Figure I). CNS neurons located at the dorsal horn are targets for analgesic development. In this area, several GPCRs (such as opioid receptors, serotonin receptors, and cannabinoid receptors) and ion channels (such as GABA and NMDA receptors) are responsible for nerve signaling. In peripheral areas, GPCRs work together with ion channels and other receptors, such as the potassium channel (Kv), sodium channel (Nav), calcium channel (Cav), transient receptor (TRP), and purinoceptor (P2X), to execute neuronal sensing. Numerous analgesics with increased selectivity for receptors/ion channels, or with biased agonism for a downstream pathway, have been designed to reduce side effects.
Figure I.

Targets Involved in Modern Nociception and Analgesia Drug Design. (A) Peripheral targets including Kv, Nav, Cav, TRP, P2X, and GPCRs. (B) Their locations in the dorsal horn and periphery. (C) Dorsal horn targets including opioid, serotonin, cannabinoid, GABA, and NMDA receptors.
By contrast, most undesirable opioid-mediated effects are related to the β-arrestin pathway, which regulates the desensitization and internalization of the opioid receptor [6,10]. Of the four arrestin subtypes, arrestin-1 and arrestin-4 are visual arrestins that bind to activated and phosphorylated rhodopsin and cone opsin, and terminate phototransduction [11]. The other two, arrestin-2 and arrestin-3 (also known as β-arrestin 1 and β-arrestin 2, respectively), are responsible for regulating the activities of many non-visual G protein-coupled receptors (GPCRs) (Box 4) [12]. In the classical view of this pathway, an activated receptor exposes its domains for phosphorylation, a process mediated by G-protein receptor kinases (GRKs) and protein kinase C (PKC) [11,13]. Specific domains of the arrestins recognize this phosphorylated and activated state of the receptor, resulting in receptor–arrestin binding [12]. Bound arrestin sterically precludes G-protein coupling and attenuates G protein-dependent signaling [12]. Arrestin also acts as a scaffolding protein that promotes internalization of the receptor. During internalization the receptor is transported into the cytoplasm as an endosome. Subsequently, the receptor will either be degraded by lysosomes or recycled to the cell membrane [14].
Box 4. Brief Introduction to GPCRs.
GPCRs are seven transmembrane proteins that represent a primary class of drug targets. GPCRs can detect molecules outside the cell that activate internal cellular responses. When an agonist binds to a GPCR it causes a series of conformational changes [59,60]. Following that, the G protein α subunit dissociates from the β and γ subunits to further affect intracellular signaling proteins [61]. According to their unique structures and functions, GPCRs comprise five different classes (Figure I). More than 800 GPCRs are expressed in the human body [62] and are responsible for cellular signaling. By contrast, there are only five different types of G proteins [26], including Gs, Gi, Go, Gq/11, and Gβ, and these bind to activating GPCRs [63]. GPCRs are involved diverse physiologically significant processes and constitute the most popular targets for drug discovery. More than 35% of marketed drugs are estimated to target GPCRs [64].
Figure I.

The GPCR Family. (A) The phenotypic tree of GPCRs composed of five different families. White circles, GPCRs without crystal structures. Red circles, GPCRs with crystal structures. (B) Structures of different GPCR classes.
Activation Mechanism of μOR
Both antagonist-bound and agonist-bound μOR crystal structures are now available. In the inactive complex (PDB: 4DKL) [15], an irreversible antagonist, β-funaltrexamine (β-FNA), locates at the orthosteric site of the receptor (Figure 2). In the agonist-bound structure, BU72 binds to μOR in a similar way (PDB: 5C1M) [4]. The amino acid conformations in the ligand-binding regions differ subtly. However, the side chain of a highly conserved residue W2936.48 [16], identified as a switch for forming a continuous water channel (Figure 2B,C) [17–19], ‘flips’ upon agonist binding. Specifically, when antagonist β-FNA binds to μOR, the cyclopropylmethyl group of β-FNA forms a tight edge-to-face σ–π stacking interaction with W2936.48 (Figure 2C), stabilizing the conformation of W2936.48 which then blocks the formation of a continuous water channel [20,21]. By contrast, the agonist BU72 forms an edge-to-edge hydrophobic contact with the W2936.48 side chain, leaving an empty space in the binding pocket which facilitates the formation of a continuous water channel (Figure 2C). In addition to the molecular switching of W2936.48, structural rearrangements (Figure 2E) occur in the extracellular core triad which consists of I1553.40, P2445.50, and F2896.44 [4]. Such rearrangements have also been observed in two additional crystal structures of activated GPCRs: the β2AR adrenergic receptor (PDB: 3SN6) [22] and the M2 muscarinic acetylcholine receptor (PDB: 4MQS) [23]. The extracellular switch rearrangements result in intracellular molecular switches in both Y2525.58 and Y3367.53 (Figure 2D). In addition, the highly conserved ionic lock [17,24–26] between D1643.49 and R1653.50 is disrupted following agonist binding (Figure 2D). With the molecular switches in both extracellular and intracellular regions, the receptor attains its characteristic activated state in which the transmembrane helices (TMs) undergo unique movements (Figure 2A), with TM5 moving inward by ~3 Å, TM6 outward by ~10 Å, and TM7 inward by ~2 Å [4]. This leads to a large void in the cytoplasmic zone, and this allows the binding of G protein.
Figure 2.

Activation Mechanism of the μ-Opioid Receptor (μOR). (A) Superimposed structures of inactive μOR (grey cartoon) and activated μOR (green cartoon). Transmembrane helices (TM) V, VI, and VII undergo unique movements upon agonist binding. (B) Molecular switches in the orthosteric site at the extracellular region. (C) Binding modes of the antagonist β-FNA (grey ball-and-stick) and agonist BU72 (green ball-and-stick). β-FNA forms a tight stacking interaction with highly conserved W2936.48, whereas BU72 leaves a large void in the orthosteric site (yellow circle). (D) Molecular switches in the intracellular region. (E) Rearrangements of the PIF core. Left, antagonist-bound μOR; right, agonist-bound μOR.
Different Strategies in Designing Safer μOR Analgesics
PZM21: A G-Protein Biased Agonist
While a G-protein biased agonist is bound to μOR, it can induce G protein-mediated analgesia and alleviate undesirable effects caused by the arrest in pathway[27]. The structure-based drug design strategyofPZM21 revealed new binding modes that are worthy of attention. Despite the comment from Manglik et al. that ‘some of the properties of PZM21 (Box 5) were likely to be fortuitous’ [28], PZM21 with its in vivo activities apparently exemplifies a success in rational drug design.
Box 5. Summary of Different Painkillers.
Most opioids/opiates (Table I) produce their anti-nociceptive effects via activation of μORs in the CNS. μORs activate Gi/o proteins that inhibit the activity of adenylyl cyclase. Consequently, reduction of intracellular cAMP levels hampers the opening of cAMP-sensitive sodium channels, leading to reduced excitabilities of neurons. The G-protein pathway also inhibits calcium channels and promotes potassium influx. Apart from the G-protein pathway, activation of μORs also affects the arrestin pathway that accounts for most of the adverse effects of opioids/opiates. Unlike morphine and codeine, novel μOR agonists, such as PZM21 [28] and TRV130 [65], retain most the opioid analgesic effects but with reduced toxic side effects, in part because of their biased agonism for the G-protein pathway.
Oxycodone is a semi-synthetic opioid that is used to treat pain from various causes. It appears to have a higher affinity for κOR than for μOR [66]. Methadone is an agonist of μOR. It also blocks the NMDA receptor and inhibits the reuptake of serotonin and noradrenaline. It has been used as maintenance treatment for opioid dependence and chronic pain. Buprenorphine is believed to be a partial agonist for μOR. It exhibits a U-shaped dose–response curve such that the anti-nociceptive effect increases at low dose, but diminishes at higher doses. One explanation for this phenomenon is that high-dose buprenorphine also activates N/OP receptors which counteract the μ-opioid-mediated effects. BU08028 [32] is a structural analog of buprenorphine that targets both μOR and N/OP receptors, and this improves its safety profile.
Synthetic opioids, including fentanyl, remifentanil, and pethidine, offer fast onset and recovery and are often used as surgical analgesics. Remifentanil may also activate the NMDA receptor, and paradoxically induces hyperalgesia. Pethidine has a pronounced anticholinergic effect. NFEPP is an analog of fentanyl and is active at a lower pH than the physiological environment. Because the pH is reduced in injured tissues, NFEPP may have a more site-specific action there [40].
Tramadol is a synthetic opioid for treating mild to severe acute and chronic pain. It also inhibits the reuptake of serotonin and noradrenaline.
Despite its long history of clinical application, the precise mechanism of paracetamol action is not fully understood (Table II). It is thought to inhibit the peroxidase activity of cyclooxygenases 1 and 2, and reduce the level of tyrosine radicals required for prostaglandin biosynthesis [67]. Its anti-nociceptive effects may be attributed to its metabolite, AM404, which inhibits the anandamide membrane transporter and increases the levels of endogenous cannabinoids in the CNS [67]. Paracetamol evidently is also involved in the central serotonergic pathway, where its analgesic effect is abolished when coadministered with 5HT3 selective antagonists [68].
NSAIDs (Table III) block the hydrophobic channel of cyclooxygenase, preventing arachidonic acid from reaching the catalytic site of this enzyme, and thereby inhibiting the formation of prostaglandins [69]. According to their selectivity towards cyclooxygenase isoforms, NSAIDs can be divided into non-selective and COX-2 selective. Non-selective NSAIDs can be further subdivided into smaller groups based on their chemistry: in other words salicylates (aspirin, salicylic acid, diflunisal, etc.), propionic acids (naproxen, ketoprofen, ibuprofen, etc.), acetic acids (sulindac, indo-methacin, diclofenac, ketorolac, etc.), fenamic acids (mefenamic acid, tolfenamic acid, flufenamic acid, etc.), oxicams (piroxicam, meloxicam, etc.), and non-acidic groups (naphthylbutanone).
COX-1 is responsible for cytoprotective activity, whereas COX-2 is inducible in case of inflammation [69]. As a result, non-selective NSAIDs often demonstrate more pronounced GI and renal side effects. Designing COX-2 inhibitors was intended to avert these GI complications. However, there was concern about the cardiovascular risks associated with COX-2 inhibitors [70], leading to the withdrawal of rofecoxib in 2004 [71]
NSAIDs were also reported to affect COX-independent pathways. For example, aspirin (high-dose), ibuprofen, indomethacin, flurbiprofen, and sulindac inhibit the activation of nuclear factor-κB [69], an important transcription factor that induces gene expression of various proinflammatory cytokines. NSAIDs also can inhibit the adhesion of leukocytes to endothelial cells and suppress extravasation events [69].
Glucocorticoids (Table IV; hydrocortisone, prednisolone, methylprednisolone, dexamethasone, betamethasone, beclomethasone, fludrocortisone) exert analgesic and anti-inflammatory effects via both non-genomic and genomic pathways. Their non-genomic effect rapidly reduces glutamate release but increases the release of endocannabinoids and γ-aminobutyric acid, resulting in significant reduction in neuronal excitability and anti-hyperalgesia [72]. A genomic pathway is related to their persistent effect on chronic pain. Glucocorticoids first bind to glucocorticoid receptors in the cytoplasm. These complexes then translocate to the nucleus and dimerize. The dimerized complexes bind to the glucocorticoid-responsive elements of DNA that express anti-inflammatory cytokines (transactivation). Meanwhile, both the complex monomers and dimers bind to nuclear factor-κB elements on DNA, inhibiting the expression of proinflammatory cytokines (transrepression) [73].
Noting that dimer-dependent transactivation can lead to other metabolic side effects, whereas transrepression can be achieved by glucocorticoid receptor monomers, researchers focused on designing dissociative ligands that favor monomer activity (e.g., RU-24858 and RU-24782). Molecules without a steroidal scaffold such as (+)-ZK216348 were developed as selective agonists of glucocorticoid receptors to avoid binding to mineralocorticoid and hormonal receptors. Selective glucocorticoid receptor agonists, such as mapracorat and fosdagrocorat, have entered Phase II clinical trials for treating ocular inflammation [74] and rheumatoid arthritis [75].
Lidocaine, prilocaine, bupivacaine, and mepivacaine (Table V) are used as local anesthetics. These molecules inhibit sodium channels in the periphery and prevent neuron firing. However, systemic administration of these molecules may employ sodium channel-independent pathways for anti-nociceptive effects [76].
Anti-epileptic drugs that inhibit sodium channels (e.g., carbamazepine and lacosamide) also possess analgesic properties, notably in the treatment of trigeminal neuralgia or other neuropathic pain [77].
Despite structural their similarity to γ-aminobutyric acid, gabapentin and pregabalin do not seem to bind to the GABAB receptor [78]. Instead, they modulate glutaminergic levels and bind to the α2δ subunit of voltage-sensitive calcium channels [78]. Ziconotide is an antagonist of calcium channel Cav2.2 [78].
Tricyclic antidepressants (Table VI) such as amitriptyline have a long history in the treatment of neuropathic pain. Their mechanism of action likely involves the inhibition of serotonin and noradrenaline reuptake [79].
Starting with over 3 million commercially available lead-like compounds, putative docking configurations against the inactive μOR were computationally generated and later prioritized using a physics-based energy function. Docking poses with strained ligand conformations were manually removed. Interestingly, many newly sampled ligands not only used their cationic amines to ion-pair with D1473.32 at the orthosteric site, a canonical interaction between most opioid ligands and opioid receptors, but also employed their urea amide groups to hydrogen-bond with the same aspartate anchor. This dual hydrogen bond interaction is relatively novel for opioid ligands [28]. Preliminary data at this stage showed that the μOR binding affinities of high-scoring ligands had already reached the μM range. After a few steps of lead optimization by docking, a potent lead compound was found to activate Gi/o with low levels of arrestin-3 recruitment [28]. Structure-guided optimization of the lead was further performed. The introduction of a phenol hydroxyl group into the lead compound then yielded PZM21, which was designed to utilize a water-meditated hydrogen bond with H2976.52 [28]. This interaction was also observed in other protein–ligand complexes of μOR, δOR [29], and κOR [30]. PZM21 showed a strong binding affinity to μOR in radioligand binding assays and promising efficacy in a Gi/o activation assay [28]. Notably, substituting the thiophene moiety of PZM21 with a larger benzothiophene did not impair its activity. This moiety was modeled to occupy the more open and specific region of μOR, and might contribute to the specificity of PZM21 over other opioid receptor subtypes: PZM21 is a κOR antagonist, and also a very weak δOR agonist. Signaling studies showed that this molecule is highly Gi/o biased [28]. At its maximal concentration, arrestin-3 recruitment was undetectable and μOR internalization was minimal compared to DAMGO or morphine. Despite the dependence of arrestin recruitment on the expression level of G protein-coupled receptor kinase 2 (GRK2) [31], arrestin recruitment for PZM21 remained weak even in the presence of overexpressed GRK2 [28].
Regarding analgesia, PZM21 displayed a few unique properties among known opioid analgesics. For example, it demonstrated dose-dependent analgesia in a mouse hotplate assay, but not in the tail-flick assay [28]. Behavioral responses in the hotplate experiment can further be subdivided into either afferent or reflexive, and PZM21 exerts analgesia solely towards the affective component of pain [28]. Nevertheless, the analgesic response of μOR knockout mice was completely abolished in the hotplate assay, suggesting that PZM21 analgesia is rooted in μOR activation [28].
PZM21 also displays fewer side effects than morphine, with a substantially weaker constipating effect and minimal respiratory depression. In the case of morphine, respiratory depression persists even after the resolution of its analgesic effect, reflecting a differential recruitment of arrestin-3 at later timepoints following drug administration. Conversely, respiratory depression induced by PZM21 remains minimal at later timepoints, again suggesting that it activates a small amount of arrestin-3 signaling in vivo [28]. Reinforcement and addiction are the major drawbacks of many opioids, which may also activate the dopaminergic reward circuits. In in vivo experiments, hyperlocomotion is taken as an endpoint of mesolimbic dopamine activation. Interestingly, PZM21 had no apparent hyperlocomotive effect versus vehicle at a nearly equi-analgesic dose. It also did not induce a conditioned place preference response [28].
BU08028: A Dual-Function Molecule
Another strategy to reduce the side effects of analgesics is to design ligands that act on multiple opioid or opioid-like receptors [32]. The nociceptin/orphanin FQ peptide (NOP) receptor, that is activated by the endogenous ligand nociception/orphanin FQ (N/OFQ), is known to counteract some undesirable opiate properties [33,34] and to block the morphine-induced reward pathway [35]. A mixed μOR/NOP receptor agonist could be an ideal candidate to leverage the undesirable effects of opioid-mediated analgesia. Buprenorphine, a successful analgesic with a reduced risk of addiction, is often used as an alternative to methadone to minimize the withdrawal symptoms of heroin. Despite uncertainty as to whether it is a rewarding compound, most of its pharmacological profile is attributed to its partial agonism of μOR [32]. Interestingly, buprenorphine shows dose-dependent anti-nociception at lower doses that is reversed at higher doses [32]. An explanation for this phenomenon is that buprenorphine is also a NOP receptor agonist with lower binding affinity and efficacy, and therefore its μOR-mediated effect diminishes only at higher doses [32]. Despite its complex properties, buprenorphine serves as a good starting point for medicinal chemists in search of a bivalent analog that demonstrates higher efficacy towards NOP than μOR.
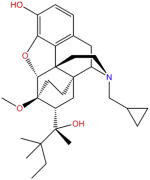
BU08028 (Box 5), an analog of buprenorphine, is the first universal opioid ligand that shows high binding affinities to all three opioid receptors (μ, δ, and κ) [32]. It shares the most common chemical scaffold of the morphine class of compounds, and is believed to interact with opioid receptors similarly to the ligand observed in the crystal structure [36]. Because BU08028 competes for μOR and NOP with radiolabeled endogenous ligands in binding assays, it is fair to postulate that BU08028 also binds to the orthosteric sites of these two receptors. Moreover, BU08028 and other morphine-like molecules have a basic piperidine ring scaffold that forms an ionic lock with μOR. The structural similarity of these molecules suggests that they might bind to μOR at the same site in an identical manner. Regarding the activity of BU08028, [35S]GTPγS binding assays demonstrated that BU08028 is comparable to buprenorphine in activating μOR, but shows no effect on δ- and κ-receptors. Regarding the NOP receptor, the activity of BU08028 is approximately sixfold higher than that of buprenorphine [32]. NOP activation also triggers the Gi/o pathway and produces peripheral anti-nociception. However, at the supra-spinal level, it counteracts opioid-mediated effects by suppressing the descending inhibitory control circuitry [37].
BU08028 also displays interesting differences between mice and primate models. BU08028 produces long-lasting anti-nociceptive effects in both models [36]. However, contributions of μOR and NOP receptors to the anti-nociceptive effects may differ. In mice, the μOR antagonist SB612111 produced a statistically significant potentiation of BU08028-induced anti-nociceptive effects, but only when a high dose of BU08028 was used, or when BU08028 was introduced at much later timepoints after SB612111 injection. This result suggests that low doses of BU08028 do not induce sufficient anti-nociception as a result of its NOP receptor agonist activity [32]. Conversely, in the primate model, dose–response curves for BU08028-induced anti-nociception shift to the right to a similar extent in the presence of the μOR antagonist naltrexone or the NOP antagonist J-113397. These findings indicate that both receptors contribute to the effects of BU08028 [36]. Moreover, BU08028 produced a conditioned place preference (CPP) in mice and failed to attenuate the CPP induced by cocaine [32].
Meanwhile, BU08028 did not cause a statistically significant increase in the number of self-administered drug injections in monkeys, suggesting that BU08028 does not have reinforcing effects [36]. In addition, the monkey study offers more clinically relevant data on the effects of BU08028. Thus, systemic BU08028 caused a dose- and time-dependent alleviation of capsaicin-induced thermal allodynia [36]; higher doses of BU08028 did not significantly affect respiratory and cardiovascular functions [36], and repeated administration of BU08028, followed by a combination of naltrexone and J-113397, did not precipitate withdrawal signs and therefore does not produce acute physical dependence [36]. Taken together, these results suggest that the development of BU08028 represents an important step in the design of an effective opioid drug with significantly reduced side effects [38].
NFEPP: A pH-Sensitive Molecule
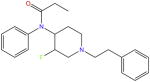
As an analog of the known μOR agonist, fentanyl [39,40], the compound (±)-N-(3-fluoro-1-phenethylpiperidin-4-yl)-N-phenyl propionamide (NFEPP, Box 5), was designed to selectively activate peripheral μORs at the source of pain generation [40]. Similarly to other morphine-like compounds, NFEPP contains a basic piperidine ring [4,36]. Because the Y326 mutation in rat μOR significantly reduces the binding affinities of morphine, fentanyl, and other antagonists, NFEPP might bind to the same region of μOR in a similar fashion [41]. Given the different pH conditions in normal (pH = 7.4) and inflamed (pH = 5–7) tissues, a safer agonist would activate μOR exclusively at low pH in inflamed sites.
With the help of computational methods, development of NFEPP started from estimations of the pKa and binding Gibbs free energy (ΔG) of fluorinated fentanyl derivatives in protonated/deprotonated forms under acidic/neutral conditions. NFEPP was selected for further study because it has a calculated pKa of 6.7, with potentially favorable binding to the receptor under acidic conditions. Furthermore, both in vitro and in vivo experiments were carried out to test the efficacy of NFEPP. In the binding experiments, NFEPP was found to compete with the radiolabeled endogenous ligand [3H] DAMGO in the binding site, suggesting that it is likely to target the orthosteric site of μOR. Further, NFEPP was believed to display a lower affinity at physiological pH than under acidic conditions [40]. In G-protein activation tests, NFEPP was found to activate Gi protein, and was anti-nociceptive, similarly to fentanyl. Furthermore, it caused a significant decrease in FRET (related to G protein dissociation upon μOR stimulation) only at low pH, 6.5 [40]. This clearly contrasts with fentanyl which caused a FRET decrease under both pH conditions.
To examine analgesic efficacy in vivo, fentanyl and NFEPP were tested in clinically relevant rat models of persistent or acute inflammatory pain. At low doses, NFEPP produced dose-dependent analgesia only in inflamed (‘injured’) paws, whereas fentanyl produced analgesia in inflamed and contralateral (‘non-injured’) paws. At a higher dose, fentanyl induced respiratory depression, while NFEPP did not cause obvious respiratory depression or sedation. Naloxone-methiodide (NLXM), a μOR antagonist that does not cross the blood–brain barrier (BBB), reversed the analgesic effects of NFEPP and partially those of fentanyl, suggesting that NFEPP might act exclusively on peripheral receptors. In addition, typical side effects mediated by central opioid receptors were not induced by NFEPP.
In general, NFEPP is likely to act solely on peripheral μOR in injured tissue to produce analgesia via selective activation. Studies of NFEPP therefore provide a general strategy to target ‘disease-specific’ conditions and/or conformations of opioid receptors for the discovery of new and safer painkillers.
ST034307: An AC1-Selective Inhibitor
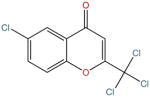
Obtained from the screening of a chemical library that contains natural compounds and derivatives, ST034307 (Box 5) illustrates a different strategy to achieve the selective treatment of pain. The therapeutic target AC1 is an isoform of the adenylyl cyclase (AC) family of enzymes which catalyze the production of cAMP from ATP. It was suggested by Brust et al. [42] that, given the distinct distributions and functions of AC isoforms, selective inhibition of AC1 might reduce adverse effects [42].
Inspired by forskolin, a natural nonselective AC activator commonly used to increase cAMP, Brust et al. [42] employed a screening platform to test over 3000 natural compounds derived from the NDL-3000 library. ST034307 was one of the potential AC1 inhibitors, identified from a search for compounds that inhibit A23187 + forskolin-stimulated cAMP accumulation in cells. The selectivity of ST034307 was first verified in tests of AC1 and the closely related isoform AC8, and further confirmed by evidence of low-level inhibition of cAMP accumulation in HEK cells expressing all other AC isoforms [42]. Notably, the exact binding mode of ST034307 to AC1 is still undetermined. Unlike other non-competitive and uncompetitive inhibitors (e.g., NKY80) which demonstrate more robust inhibitory effects when AC is more active, ST034307 showed no change or a decrease of inhibitory efficacy in the presence of the AC activators A23187 or forskolin. This result hints that ST034307 may have a different binding mode than other AC inhibitors [42].
The mechanism of ST034307 action on signaling pathways was investigated in various cellular and in vitro contexts. This demonstrated a direct inhibition of AC1 by ST034307, rather than an upstream or downstream process or a noncompetitive reaction [42]. Furthermore, ST034307 enhanced μOR-mediated inhibition of AC1 in short-term inhibition cellular assays, and also blocked heterologous sensitization of AC1 caused by chronic μOR activation. Thus, selective AC1 inhibition likely can prevent and suppress opioid dependence. Analgesic properties of ST034307 were further tested in a mouse model. ST034307 caused significant relief of inflammatory pain, with an effect comparable to that of the μOR agonist DAMGO [42].
Based on various tests and assays indicating selective inhibition of AC1 while producing analgesia, ST034307 holds promise for treating inflammatory and/or chronic pain, either as a standalone drug or in complementary use with μOR agonists. Indeed, selective inhibition of AC1 provides an additional strategy for finding safer painkillers.
Concluding Remarks
Around the globe, pain remains a clinical and economic problem, such that designing safer analgesics has become a vital challenge to both academia and industry. Recent advances in structural and computational biology have allowed the discovery of potentially safer drug candidates which target μOR signaling pathways by different means. Such strategies include the use of G-protein biased molecules such as PZM21, dual functional modulators such as BU08028, pH-sensitive molecules such as NFEPP, and adenylyl cyclase AC1 modulators such as ST034307. Additional strategies can reduce the toxicity of opioid drugs. In particular, the development of functionally selective κOR agonists targeting peripheral sensory neurons can significantly reduce adverse effects normally mediated through the central nervous system (CNS) [43]. Alternatively, polyethylene glycol (PEG)-conjugated μOR antagonists with increased half-lives [44] cannot penetrate the BBB and alleviate constipation during opioid pain management [44,45]. Although the long-term safety of these new candidates requires further evaluation and clinical studies, these exciting breakthroughs are indeed encouraging in the pursuit of next-generation safer painkillers (see Outstanding Questions).
Outstanding Questions.
Recently designed painkillers are ‘non-toxic’ in terms of respiratory depression, pruritus, dependence, and constipation. However, are they also ‘non-toxic’ in other aspects such as affecting hERGs, cytochromes, and so on?
Can we combine the four mentioned strategies together to design a more effective painkiller with much less toxicity?
Can we design a painkiller that activates receptors and ion channels in both the dorsal horn and the periphery?
What are the key structural elements responsible for the G protein biased signaling pathway of μORs?
When will the first safer μOR-mediated painkiller appear on the market?
Table I.
Opioids
| Name | Molecular structure |
|---|---|
| PZM21 |
|
| BU08028 |
|
| NFEPP |
|
| ST034307 |
|
| Morphine |
|
| Codeine |
|
| Oxycodone |
|
| Methadone |
|
| TRV130 |
|
| Buprenorphine |
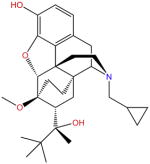
|
| Fentanyl |
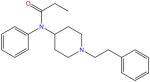
|
| Pethidine |
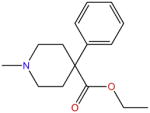
|
| Remifentanil |
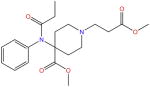
|
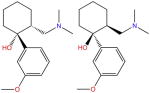
| Tramadol |
|
Table II.
Paracetamol-type
| Name | Molecular structure |
|---|---|
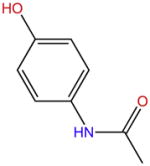
| Paracetamol |
|
Table III.
NSAIDs
| Name | Molecular structure |
|---|---|
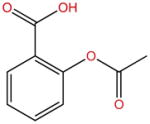
| Aspirin |
|
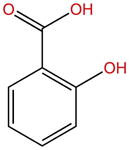
| Salicylic acid |
|
| Di3unisal |
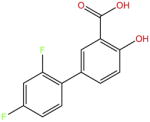
|
| Naproxen |
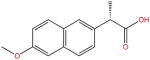
|
| Ketoprofen |
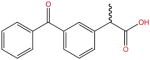
|
| Ibuprofen |
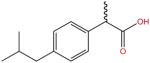
|
| Sulindac |
|
| Indomethacin |
|
| Diclofenac |
|
| Ketorolac |
|
| Mefenamic acid |
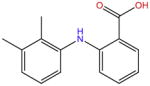
|
| Tolfenamic acid |
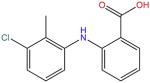
|
| Flufenamic acid |
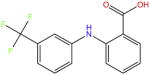
|
| Piroxicam |
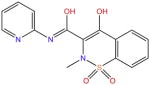
|
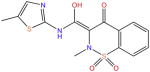
| Meloxicam |
|
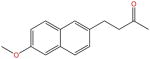
| Nabumetone |
|
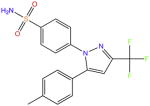
| Celecoxib |
|
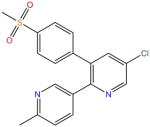
| Etoricoxib |
|
| Parecoxib |
|
Table IV.
Glucocorticoids
| Name | Molecular structure |
|---|---|
| Hydrocortisone |
|
| Prednisolone |
|
| Methylprednisolone |
|
| Dexamethasone |
|
| Betamethasone |
|
| Beclomethasone |
|
| Fludrocortisone |
|
| RU-24858 |
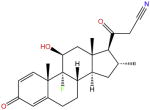
|
| RU-24782 |
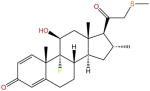
|
| (+)-ZK216348 |
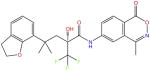
|
| Mapracorat |
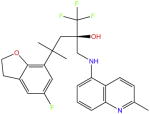
|
| Fosdagrocorat |
|
Table V.
Ion Channel Blockers
| Name | Molecular structure |
|---|---|
| Lidocaine |
|
| Prilocaine |
|
| Bupivacaine |
|
| Mepivacaine |
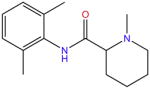
|
| Carbamazepine |
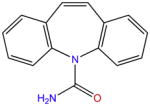
|
| Lacosamide |
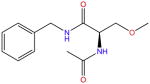
|
| Gabapentin |
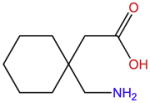
|
| Pregabalin |
|
| Ziconotide |
|
Table VI.
Tricyclic Antidepressants
| Name | Molecular structure |
|---|---|
| Amitriptyline |
|
Trends.
Pain is a major clinical and economic problem. Owing to the serious side effects associated with current painkil-lers, the discovery of less toxic medications is an imperative in both academia and industry.
Owing to recent advances in computational and structural biology, several μOR-mediated painkillers with fewer side effects have been successfully designed.
Glossary
- Agonist
a molecule that binds to a receptor which subsequently produces a biological response.
- Analgesics
drugs used clinically for pain control. Depending on their mechanism of action and molecular structure, analgesics can be categorized into different classes. Some prototypical examples are provided here. Paracetamol and its structurally related analogs form a commonly used analgesic class. Non-steroidal anti-inflammatory drugs (NSAIDS) and glucocorticoids inhibit the syntheses of proinflammatory substances which sensitize nociceptive nerve endings. Opioids, opiates, and local anesthetics suppress the excitability of sensory neurons in different parts of the body. Antidepressants, especially the tricyclic group, are used for treatment of neuropathic pain.
- Antagonist
a molecule that binds to a receptor, blocking or mitigating agonist-evoked responses.
- G protein-coupled receptor (GPCR)
GPCRs are proteins that share a seven-transmembrane domain (TM) and can couple to heterotrimeric G proteins. They play a crucial role in cellular signal transduction and represent a primary class of drug targets. Acting by direct binding, drugs can modulate GPCR activity and influence the signaling pathways associated with numerous diseases. GPCRs are grouped into five different classes according to their structures and functions.
- Inverse agonist
a molecule that binds to a receptor, blocking or mitigating agonist-evoked responses, and further depressing the basal response of the receptor.
- Opiates
natural compounds with pharmacological activities found in opium poppies.
- Opioids
natural or synthetic compounds which bind to ORs to exert their pharmacological actions.
- Opioid receptors (ORs)
these include several subtypes which couple with Gi/o proteins and exert their actions when opioids or opiates (e.g., codeine is transformed into its active metabolite, morphine) bind. ORs are expressed widely in the brain as well as in other parts of the central nervous system (CNS).
References
- 1.Kosten TR, George TP. The neurobiology of opioid dependence: implications for treatment. Sci Pract Perspect. 2002;1:13–20. doi: 10.1151/spp021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Contet C, et al. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Stein C. Opioid receptors. Annu Rev Med. 2016;67:433–451. doi: 10.1146/annurev-med-062613-093100. [DOI] [PubMed] [Google Scholar]
- 4.Huang W, et al. Structural insights into mu-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siuda ER, et al. Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr Opin Pharmacol. 2016;32:77–84. doi: 10.1016/j.coph.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson H. Biological membranes. Essays Biochem. 2015;59:43–69. doi: 10.1042/bse0590043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manglik A, et al. Structural insights into the dynamic process of beta2-adrenergic receptor signaling. Cell. 2015;161:1101–1111. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 12.Tobin AB. G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol. 2008;153(Suppl 1):S167–S176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukla AK, et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cahill CM, et al. Allostatic mechanisms of opioid tolerance beyond desensitization and downregulation. Trends Pharmacol Sci. 2016;37:963–976. doi: 10.1016/j.tips.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manglik A, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trzaskowski B, et al. Action of molecular switches in GPCRs – theoretical and experimental studies. Curr Med Chem. 2012;19:1090–1109. doi: 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan S, et al. The molecular mechanism of P2Y1 receptor activation. Angew Chem Int Ed Engl. 2016;55:10331–10335. doi: 10.1002/anie.201605147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan S, et al. Mechanistic studies on the stereoselectivity of the serotonin 5-HT1A receptor. Angew Chem Int Ed Engl. 2016;55:8661–8665. doi: 10.1002/anie.201603766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoddart LA, et al. Effect of a toggle switch mutation in TM6 of the human adenosine A(3) receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br J Pharmacol. 2014;171:3827–3844. doi: 10.1111/bph.12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAllister SD, et al. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 21.Yuan S, et al. W246 opens a gate for a continuous intrinsic water pathway during activation of the adenosine A receptor. Angew Chem Int Ed Engl. 2014;54:556–559. doi: 10.1002/anie.201409679. [DOI] [PubMed] [Google Scholar]
- 22.Rasmussen SGF, et al. Crystal structure of the beta2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruse AC, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanni S, et al. Observation of ‘ionic lock’ formation in molecular dynamics simulations of wild-type beta 1 and beta 2 adrenergic receptors. Biochemistry. 2009;48:4789–4797. doi: 10.1021/bi900299f. [DOI] [PubMed] [Google Scholar]
- 25.Isberg V, et al. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol Sci. 2015;36:22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katritch V, et al. Structure–function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Link A, Muller CE. G-protein-coupled receptors: sustained signaling via intracellular megaplexes and pathway-specific drugs. Angew Chem Int Ed Engl. 2016;55:15962–15964. doi: 10.1002/anie.201609015. [DOI] [PubMed] [Google Scholar]
- 28.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenalti G, et al. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat Struct Mol Biol. 2015;22:265–268. doi: 10.1038/nsmb.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nickolls SA, et al. Co-expression of GRK2 reveals a novel conformational state of the micro-opioid receptor. PLoS One. 2013;8:e83691. doi: 10.1371/journal.pone.0083691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khroyan TV, et al. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J Pharmacol Exp Ther. 2011;336:952–961. doi: 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reinscheid RK, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 34.Toll L, et al. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol Rev. 2016;68:419–457. doi: 10.1124/pr.114.009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy NP, et al. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999;832:168–170. doi: 10.1016/s0006-8993(99)01425-0. [DOI] [PubMed] [Google Scholar]
- 36.Ding H, et al. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci U S A. 2016;113:E5511–E5518. doi: 10.1073/pnas.1605295113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert DG. The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov. 2008;7:694–710. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- 38.Li JX. Buprenorphine analogue BU08028 is one step closer to the Holy Grail of opioid research. Proc Natl Acad Sci U S A. 2016;113:10225–10227. doi: 10.1073/pnas.1612752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filizola M, et al. Differentiation of delta, mu, and kappa opioid receptor agonists based on pharmacophore development and computed physicochemical properties. J Comput Aided Mol Des. 2001;15:297–307. doi: 10.1023/a:1011187320095. [DOI] [PubMed] [Google Scholar]
- 40.Spahn V, et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science. 2017;355:966–969. doi: 10.1126/science.aai8636. [DOI] [PubMed] [Google Scholar]
- 41.Mansour A, et al. Key residues defining the mu-opioid receptor binding pocket: a site-directed mutagenesis study. J Neurochem. 1997;68:344–353. doi: 10.1046/j.1471-4159.1997.68010344.x. [DOI] [PubMed] [Google Scholar]
- 42.Brust TF, et al. Identification of a selective small-molecule inhibitor of type 1 adenylyl cyclase activity with analgesic properties. Sci Signal. 2017;10:aah5381. doi: 10.1126/scisignal.aah5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jamshidi RJ, et al. Functional selectivity of kappa opioid receptor agonists in peripheral sensory neurons. J Pharmacol Exp Ther. 2015;355:174–182. doi: 10.1124/jpet.115.225896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang JS, et al. Emerging PEGylated drugs. Expert Opin Emerg Drugs. 2009;14:363–380. doi: 10.1517/14728210902907847. [DOI] [PubMed] [Google Scholar]
- 45.Weber HC. Opioid-induced constipation in chronic non-cancer pain. Curr Opin Endocrinol Diabetes Obes. 2016;23:11–17. doi: 10.1097/MED.0000000000000220. [DOI] [PubMed] [Google Scholar]
- 46.Nygaard R, et al. The dynamic process of beta2-adren-ergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warolin C. Pierre-Jean Robiquet. Rev Hist Pharm. 1999;47:97–110. (in French) [PubMed] [Google Scholar]
- 48.Howard-Jones N. A critical study of the origins and early development of hypodermic medication. J Hist Med Allied Sci. 1947;2:201–249. doi: 10.1093/jhmas/ii.2.201. [DOI] [PubMed] [Google Scholar]
- 49.Erickson D. Painkiller. There’s still room for luck in industrial chemistry. Sci Am. 1991;265:101. [PubMed] [Google Scholar]
- 50.De La Cruz JP, et al. Differences in the effects of extended-release aspirin and plain-formulated aspirin on prostanoids and nitric oxide in healthy volunteers. Fundam Clin Pharmacol. 2003;17:363–372. doi: 10.1046/j.1472-8206.2003.00137.x. [DOI] [PubMed] [Google Scholar]
- 51.Van Zee A. The promotion and marketing of oxycontin: commercial triumph, public health tragedy. Am J Public Health. 2009;99:221–227. doi: 10.2105/AJPH.2007.131714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waldhoer M, et al. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 53.Castro JD, et al. Regional Opioid Analgesia: Physiophar-macological Basis, Drugs, Equipment, and Clinical Application. Kluwer Academic Publishers; 1991. [Google Scholar]
- 54.Benedetti C, et al. Opioid Analgesia: Recent Advances in Systemic Administration. Raven Press; 1990. [Google Scholar]
- 55.Oldendorf WH, et al. Blood–brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science. 1972;178:984–986. doi: 10.1126/science.178.4064.984. [DOI] [PubMed] [Google Scholar]
- 56.De Leo JA, et al. Immune and Glial Regulation of Pain. IASP Press; 2007. [Google Scholar]
- 57.Zagon IS, et al. The biology of the opioid growth factor receptor (OGFr) Brain Res Brain Res Rev. 2002;38:351–376. doi: 10.1016/s0165-0173(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 58.Pasternak G, Pan YX. Mu opioid receptors in pain management. Acta Anaesthesiol Taiwan. 2011;49:21–25. doi: 10.1016/j.aat.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rose AS, et al. Role of structural synamics at the receptor G protein interface for signal transduction. PLoS One. 2015;10:e0143399. doi: 10.1371/journal.pone.0143399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geppetti P, et al. G protein-coupled receptors: dynamic machines for signaling pain and itch. Neuron. 2015;88:635–649. doi: 10.1016/j.neuron.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 61.Heng BC, et al. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol Adv. 2013;31:1676–1694. doi: 10.1016/j.biotechadv.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 62.Katritch V, et al. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rees S, et al. GPCR drug discovery through the exploitation of allosteric drug binding sites. Receptors Channels. 2002;8:261–268. [PubMed] [Google Scholar]
- 65.Soergel DG, et al. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 66.Ordóñez Gallego A, et al. Oxycodone: a pharmacological and clinical review. Clin Transl Oncol. 2007;9:298–307. doi: 10.1007/s12094-007-0057-9. [DOI] [PubMed] [Google Scholar]
- 67.Ghanem CI, et al. Acetaminophen from liver to brain: new insights into drug pharmacological action and toxicity. Pharmacol Res. 2016;109:119–131. doi: 10.1016/j.phrs.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klinger RY, Habib AS. Acetaminophen and ondansetron: the central serotonergic connection. J Clin Anesth. 2017;40:101–102. doi: 10.1016/j.jclinane.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 69.Diaz-Gonzalez F, Sanchez-Madrid F. NSAIDs: learning new tricks from old drugs. Eur J Immunol. 2015;45:679–686. doi: 10.1002/eji.201445222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hampton T. Cox-2 inhibitors and heart risks. JAMA. 2012;307:2247–2247. [Google Scholar]
- 71.Krumholz HM, et al. What have we learnt from Vioxx? BMJ. 2007;334:120–123. doi: 10.1136/bmj.39024.487720.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romundstad MDL, Stubhaug MDPDA. Glucocorticoids for acute and persistent postoperative neuropathic pain. What is the evidence? Anesthesiology. 2007;107:371–373. doi: 10.1097/01.anes.0000279487.27940.5c. [DOI] [PubMed] [Google Scholar]
- 73.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 74.Monica B, Santi S. Mapracorat, a novel non-ateroidal selective glucocorticoid receptor agonist for the treatment of allergic conjunctivitis. Inflamm Allergy – Drug Targets. 2014;13:289–298. doi: 10.2174/1871528113666141106101356. [DOI] [PubMed] [Google Scholar]
- 75.Stock T, et al. Improved disease activity with fosdagrocorat (PF-04271327), a partial agonist of the glucocorticoid receptor, in patients with rheumatoid arthritis: a Phase 2 randomized study. Int J Rheum Dis. 2017;20:960–970. doi: 10.1111/1756-185X.13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeilhofer HU, Schmelz M. Local anesthetics take a central action in analgesia. Pain. 2015;156:1579–1580. doi: 10.1097/j.pain.0000000000000233. [DOI] [PubMed] [Google Scholar]
- 77.Zakrzewska JM, Linskey ME. Trigeminal neuralgia. BMJ. 2014;348:g474. doi: 10.1136/bmj.g474. [DOI] [PubMed] [Google Scholar]
- 78.Offord J, Isom LL. Drugging the undruggable: gabapentin, pregabalin and the calcium channel alpha2delta subunit. Crit Rev Biochem Mol Biol. 2015;51:246–256. doi: 10.3109/10409238.2016.1173010. [DOI] [PubMed] [Google Scholar]
- 79.Gilron I, et al. Neuropathic pain: principles of diagnosis and treatment. Mayo Clin Proc. 2015;90:532–545. doi: 10.1016/j.mayocp.2015.01.018. [DOI] [PubMed] [Google Scholar]