

Abstract
Computational models of enhancer function generally assume that transcription factors (TFs) exert their regulatory effects independently, modeling an enhancer as a “bag of sites”. These models fail on endogenous loci that harbor multiple enhancers, and a “two-tier” model appears better suited: in each enhancer TFs work independently, and the total expression is a weighted sum of their expression readouts. Here we test these two opposing views on how cis-regulatory information is integrated. We fused two Drosophila blastoderm enhancers, measured their readouts, and applied the above two models to this data. The two-tier mechanism better fits these readouts, suggesting these fused enhancers comprise multiple independent modules, despite having sequence characteristics typical of single enhancers. We show that short-range TF-TF interactions are not sufficient to designate such modules, suggesting unknown underlying mechanisms. Our results underscore that mechanisms of how modules are defined and how their output is combined remain to be elucidated.
Graphical abstract
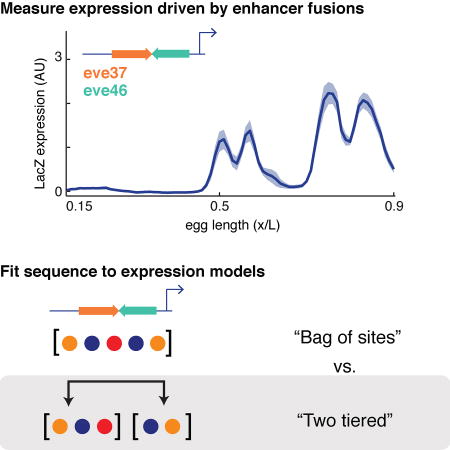
Introduction
An important goal in regulatory genomics is to understand how gene transcription is regulated by enhancers – classically defined as “discrete DNA elements that contain specific sequence motifs with which DNA-binding proteins interact and transmit molecular signals to genes” (Blackwood and Kadonaga, 1998). Experiments can identify the transcription factors (TFs) that bind to an enhancer and provide insight on how TFs influence the transcriptional readout of an enhancer. However, we need computational models to unify this experimental knowledge into a mechanistic framework to predict the transcriptional readouts of arbitrary sequences. Such models have been under development in multiple prokaryotic and eukaryotic systems for decades (Ay and Arnosti, 2011). The majority of models have focused on readouts of single enhancers, although the field has recently begun to address how multiple enhancers control the expression of a single gene (Bothma et al., 2015; Perry et al., 2010; Staller et al., 2015). Below, we describe the critical distinctions between modeling sequence readouts at the level of a single enhancer, or at the level of a locus that contains multiple enhancers.
Enhancer-level models assume that enhancers function independent of their genomic context (Banerji et al., 1981; Bulger and Groudine, 2011; Khoury and Gruss, 1983), giving rise to their common designation as cis-regulatory “modules”. This assumption greatly simplifies our task in building a model, since we can focus on TF binding to the enhancer while ignoring TF binding to the rest of the genome. But even after this simplification, we must decide how TFs bound to sites within an enhancer exert their regulatory effects together. This too is a question of modularity (Dynan, 1989). The simplest assumption is that each bound TF acts independently and the enhancer behaves as a ‘bag of sites’. This ‘bag of sites’ assumption and its extension to incorporate local TF-TF interactions underlie a range of enhancer-level models, e.g., log-linear models (Bussemaker et al., 2001; Wunderlich et al., 2012), logistic regression models (Ilsley et al., 2013; Kazemian et al., 2010), activator site occupancy models (Fakhouri et al., 2010; Zinzen et al., 2006), and more complex thermodynamics-based models (He et al., 2010; Segal et al., 2008).
To build locus-level models that predict the readout of an arbitrary locus, which can harbor multiple enhancers, we must also define modularity over longer DNA length scales. In the simplest case, we could extend the ‘bag of sites’ assumption to the entire locus, treating the entire locus as one long enhancer, but we have shown that this is not effective (Samee and Sinha, 2014). In (Kim et al., 2013), local interactions between TFs were sufficient to model even-skipped (eve) expression in Drosophila from known regulatory sequences in the eve locus. In (Samee and Sinha, 2014), we found it necessary to delineate the binding sites in a locus as being partitioned into separate enhancers and assume that each enhancer acts as a module, independently from the rest of the locus. We modeled the readouts of ~30 Drosophila developmental genes from their loci utilizing this model. We address the differences in these modeling approaches in the Discussion.
How do we decide when to apply these various assumptions about how TFs control gene expression? The ‘bag of sites’ model works well on individual enhancers, but not when modeling an entire locus. Assuming that TFs interact locally may improve models, but it is not clear whether it can predict locus level regulation in all cases. Resolving this issue is critical not only for modeling entire loci, but also for modeling enhancers themselves. Enhancer lengths vary between ~100 and ~1000 bps, making it formally possible that a long enhancer sequence contains multiple independent sub-modules. If these sub-modules exist within a long enhancer, does it require that the enhancer be modeled using the same principles found to be effective to model an entire locus?
Here we challenged thermodynamic models that operate at the enhancer level and the locus level by constructing a series of reporter constructs driven by enhancer fusions, and fitting these models to the resulting quantitative data. We concatenated two enhancers, resulting in a contiguous piece of regulatory DNA that resembles a single enhancer in length (~1 Kbp long) and binding site distribution; we then asked if their readout follows the enhancer-level model, validating the bag of sites assumption, or the two-tier model, supporting the assumption that an enhancer might function as a collection of sub-modules.
We chose to work with eve37 and eve46, two enhancers of the developmental gene even-skipped (eve), a textbook example of enhancer modularity (Clyde et al., 2003; Fujioka et al., 1999; Struffi et al., 2011) (Figure 1A). These two enhancers contain binding sites for the same nine TFs (Figure S1), but differ primarily in the affinity of the binding sites for two repressor TFs, Hunchback (Hb) and Knirps (Kni) (Figure 1B). Due to this special property, which distinguishes our study from a previous study of fused enhancers (Small et al., 1993), it is not clear what expression pattern an eve37/eve46 fusion should direct. If an enhancer-level model is operational, one would expect two broad stripes as the readout. This is because the enhancer-level model interprets the collection of Hb sites in the fusion construct as a single repressive influence (of strength dependent on the number and affinities of those sites) on gene expression, and likewise for the Kni sites present in the construct. If a two-tier model is operational in the fusion constructs, with the two constituent enhancers as the two independent sub-modules, then one would expect four stripes (Figure 1C).
Figure 1. eve37 and eve46 respond differently to the same repressors.

(A) Seven-striped expression of the even-skipped (eve) gene (top) and the genomic region containing the enhancers known to drive this pattern (bottom). Each enhancer is annotated with the stripe where it drives eve expression. (B) eve is regulated by the repressors Hb (green) and Kni (blue); these TFs have different spatial concentrations in the blastoderm embryo. The boundaries of eve37 and eve46 expression (black peaks) are set by differential sensitivities to Hb and Kni. (C) Information integration at multiple length scales predicts different outputs of enhancer fusions. If the enhancer-level model is operational and the fused enhancers are read as a “bag of sites”, we expect two broad stripes (top). If a two-tier model is operational and the component enhancers remain autonomous, we expect four stripes (bottom). The mechanism under a two-tier model for maintaining inter-stripe gaps between stripes 3 and 4 and stripes 6 and 7 is unknown.
We contextualize the output of our reporter constructs with three different thermodynamics-based models, each of which makes a different assumption about the existence of sub-modules within an enhancer and about the mechanisms to delineate those sub-modules. We use the GEMSTAT model (He et al., 2010) which operates under the ‘bag of sites’ assumption, the GEMSTAT-SRR model which allows for the delineation of sub-modules based on distance-dependent interactions between TFs, and the two-tier model GEMSTAT-GL (GEMSTAT for Gene Locus) (Samee and Sinha, 2014) which delineates transcriptionally active segments in a gene locus and linearly combines the readouts of the active segments to model gene expression.
We found that GEMSTAT fits the readouts of the individual enhancers of eve with high accuracy, but fails completely to model the fusion constructs even though they are similar in length to a typical enhancer. Given the expressiveness of GEMSTAT and the flexibility allowed in estimating the model parameters (Methods), we interpreted this as implying that current enhancer-level models are insufficient to explain the readouts of these enhancer-length sequences. We attempted next to fit the data using GEMSTAT-GL. GEMSTAT-GL uses all the thermodynamic parameters from GEMSTAT and uses additional parameters to model the weights of the independent regulatory segments mentioned above. To reduce the possibility that GEMSTAT-GL overfits data by leveraging its additional parameters, we adopted a constrained strategy for parameter estimation (Methods). Despite such constraints, for each of the fusion constructs GEMSTAT-GL selected independent regulatory segments whose readouts when linearly aggregated fit the construct’s readout with much higher accuracy, thus resolving the motivating question of this study. An enhancer-length regulatory sequence may indeed have within it sub-modules that are best thought of as functioning independently and ought to be modeled as such.
Our observation of multiple independent sub-modules within an enhancer-length sequence suggests regulatory effects of DNA-bound TFs are somehow localized. One well-studied mechanism that results in such localization is that of ‘quenching’ or ‘short-range repression’ (SRR) (Kulkarni and Arnosti, 2005), whereby a repressor TF inhibits activator binding within a short range (~100–150 bps). We investigated this mechanism within our modeling framework and noted that it improves fits compared to the enhancer-level GEMSTAT model but fails to capture several salient features of data that GEMSTAT-GL was able to model. In summary, our results of modeling a unique dataset strongly argue that even enhancer-length sequences might work through independently functioning smaller segments present within the sequence.
Results
An enhancer-level model explains the eve enhancer readouts but fails to explain readouts of fusion constructs
We fused eve37 and eve46 together in multiple orientations driving expression of a LacZ reporter gene (Figure 2, see Experimental Methods). All four possible junctions between the two enhancers are represented. The constructs drive patterned expression when placed next to a reporter gene (Figure 2A). Each enhancer fusion drove an expression pattern with more than two stripes; fusion C drove a pattern with four distinct stripes. Because our goal was to test the limits of short-range interactions to define which transcription factor binding sites are included in a module, we also constructed two additional versions of fusion C; one with a 200bp spacer (‘Fusion C200’) and one with a 1000bp spacer (‘Fusion C1000’), each of which should place the enhancers beyond the range of short-range repressors (typically 150bp). These fusion constructs with spacers drive highly similar patterns to Fusion C (Figure 2B). All of these sequences are ~1.5–2.5 Kbp in length, comparable to the average length of other developmental enhancers in Drosophila (Figure 2C).
Figure 2. Expression profiles from fused enhancers.

(A) We measured LacZ expression normalized to a hkb co-stain driven by eve37/eve46 enhancer fusions in multiple configurations. Average expression for each transgenic line is displayed as a function of AP position with the shadow representing standard error of the mean (SEM). (B) For one fusion configuration, ‘FusionC,’ we placed 200bp and 1000bp spacers between the enhancers and measured LacZ expression as above. (C) The length and binding site content of these synthetic constructs (orange) are comparable to other Drosophila developmental enhancers (blue).
We then tested whether the ‘bag of sites’ assumption underlying enhancer-level models holds for six fusion constructs, by applying GEMSTAT to these data (Figure 3). The enhancer-level GEMSTAT model can accurately model the readouts of ~40 A/P enhancers of the Drosophila embryo, using a common parameter setting (He et al., 2010); it can also accurately model all five A/P enhancers of eve (He et al., 2010; Samee and Sinha, 2014) (Figure 3A).
Figure 3. The enhancer-level GEMSTAT fails to explain readouts of fused enhancers.

(A) We applied GEMSTAT to three single eve enhancers. In all panels, experimentally measured expression profiles are shown in blue and GEMSTAT output in red. For this approach, we first fit the model simultaneously on ~30 developmental enhancers, excluding all enhancers of eve. We then fit the model on three enhancers of eve, starting from P initial parameters and letting Q denote the optimized parameters following a constrained strategy (see Data and Methods). (B) We used GEMSTAT to fit a separate model for each of the fused enhancers, starting from Q as the initial estimate and using a constrained fitting strategy. (C) We also used GEMSTAT to fit a separate model for six fused enhancers using an unconstrained fitting strategy.
In our first strategy, we fit GEMSTAT on the eve3/7, eve5, and eve4/6 enhancers and utilized the trained parameters as the initial parameter estimates to optimize GEMSTAT individually for each of the four fusion constructs. Since all sequences being modeled here were eve enhancers and derivatives thereof, we followed a constrained optimization strategy in both steps of this exercise (Methods). Although the endogenous enhancer readouts were fit reasonably well, for all fusion constructs GEMSTAT predicted only two broad domains of expression spanning eve stripes 3–4 and 6–7, thus failing to capture any salient feature of those readouts (Figure 3B, Table S2).
To confirm that this failure of enhancer-level assumptions was not an artifact of the parameter optimization process, we next fit GEMSTAT individually for each fusion construct following an unconstrained strategy (Methods). Although this setup provided considerable flexibility in parameter fitting (in comparison to a typical enhancer modeling setup it is flexible to an extent that might lead to data over-fitting), GEMSTAT failed again to model any construct’s readout with satisfactory accuracy (Figure 3C, Table S2). This failure to model the readout of any fusion construct, despite an unconstrained fitting procedure and rigorous search over the parameter space, strongly suggests that assumptions of an enhancer-level model do not hold for these constructs.
A two-tier model can explain readouts of the fused enhancers by identifying independent regulatory segments within each construct
The failure of the enhancer-level GEMSTAT model, as noted above, is similar to our previous failure to model the expression pattern of a gene from its intergenic locus using GEMSTAT (Samee and Sinha, 2014). For genes expressed in multiple stripes (e.g., eve in Figure 1A), the GEMSTAT model in that study predicted a broad expression pattern spanning all stripes. This led us to develop the GEMSTAT-GL model, which assumes that a gene locus comprises several independent segments (i.e., putative enhancers) and models the readout of the locus as a weighted sum of the readouts of the segments. Readouts of individual segments are modeled using GEMSTAT, and GEMSTAT-GL has the ability identify the segments in course of modeling the data.
Noting the similar nature of failures, we asked if GEMSTAT-GL could model the six fusion constructs. Note that applying the GEMSTAT-GL model to these data amounts to assuming that a fusion construct may function more like a gene locus, with independently functioning sub-modules within, even though the construct’s length is typical of a single enhancer. By also testing constructs with spacers, we can address the length scale beyond which a sequence starts functioning as a collection of sub-modules. We optimized the GEMSTAT-GL parameters under a constrained setting to avoid over-fitting (Methods), and obtained improved fits to the readouts of all constructs (Figure 4).
Figure 4. The two-tier GEMSTAT-GL model captures the readouts of enhancer fusions.

We applied GEMSTAT-GL (for gene locus) to the six fusion constructs. Experimentally determined expression profiles (blue) and model predictions (red) are shown as a function of egg length along the AP axis.
GEMSTAT-GL was successful in capturing both inter-stripe gaps observed in constructs ‘Fusion C’, FusionC 200’, and FusionC 1000’ and the broad domains of overlapping stripe patterns observed in constructs ‘Fusion A’, ‘Fusion B’ and ‘Fusion D’. Each fusion construct thus appears to comprise two independent sub-modules: Hb and Kni binding sites mediate repression only within the corresponding sub-module. The efficacy of this model supports the idea that there are two levels of information integration in regulatory DNA—one where modules are defined and another where the information from modules is integrated—even when the regulatory DNA in question resembles a single enhancer in its length and binding site content.
Interestingly, GEMSTAT-GL always selected one contributing regulatory segment from each of the two constituent enhancers in a fusion construct (Table S3). That it did not select multiple segments from any of the two constituent enhancers is consistent with the literature: prior experimental attempts to identify smaller functional segments within these constituent enhancers showed loss of function (Fujioka et al., 1999). The GEMSTAT-GL model never selected a segment that straddles across the boundary of the constituent enhancers, even though its optimization procedure had that flexibility. This raises the intriguing possibility of yet unidentified mechanisms that confine the sub-modules within the individual enhancers although they were fused without any spacer.
A model where repressors act only over short ranges can explain readouts of fusion constructs with spacer sequences but fails on other constructs
The above exercise utilizing GEMSTAT-GL points to the existence of at least two independent regulatory segments within each construct. However, it is not clear how these segments are determined. Previous studies have suggested that some repressor TFs, by inhibiting activator binding within short ranges around their binding sites (typically 100–150 bps), may give rise to sub-modules within enhancer-length sequences (Gray and Levine, 1996; Gray et al., 1994; Kim et al., 2013; Small et al., 1993). We therefore considered this short-range repression (SRR) mechanism as a potential explanation for the existence of independent segments within our fusion constructs. In a synthetic construct, two enhancers will function independently under the SRR mechanism when they are sufficiently far apart so that repressors bound to one enhancer do not interact with activators in the other enhancer. In the fusions, however, the constituent enhancers may interfere with each other’s regulatory effect, as has been shown in (Kim et al., 2013; Small et al., 1993). In our previous work with the two-tier model we found the SRR mechanism to be insufficient to model a gene’s expression pattern as the readout of its locus. A different implementation of the SRR model was used in (Kim et al., 2013) to explain the seven-striped pattern of eve from its locus, but the authors had chosen to model only those pieces of the eve locus that have known enhancer activity for eve. Hence, it was not clear to us a priori if the SRR mechanism would prove to be a satisfactory explanation for the independent action of the two enhancers in our fusions.
We therefore asked whether GEMSTAT implemented with SRR could explain the following two features of our dataset. First, the four fusion constructs without spacers produce more than two peaks, indicative of underlying sub-modules, but the stripes are not always entirely distinct. This may be due to local interference between TFs bound at the junction of the enhancers. Second, fusion construct C does produce four stripes, and this is maintained with spacers, i.e., Fusion C200 and Fusion C1000. To fit these data, we allowed GEMSTAT-SRR the same flexibilities in unconstrained training as were allowed to the baseline GEMSTAT model (Methods). We found that for the constructs with spacers, especially the construct with 1000bp spacer sequence, the model’s accuracy was comparable to that of the two-tier GEMSTAT-GL model (Figure 5). However, in cases where the two constituent enhancers were fused adjacent to each other, GEMSTAT-SRR’s fits were not as accurate as GEMSTAT-GL. ‘FusionC’ is a particular example where GEMSTAT-SRR significantly fell short of modeling the data. For this construct, GEMSTAT-SRR failed to model both the expression at the domain of stripe 3 and the gap between stripes 3 and 4. Overall, GEMSTAT-SRR improved over the baseline GEMSTAT model for these constructs, yet the fits were not so accurate that we could attribute the disrupted stripe formation to SRR-driven interference of TF activities at the junctions.
Figure 5. GEMSTAT with short-range repression (GEMSTAT-SRR) less accurately predicts salient features of fused enhancer expression.

We applied GEMSTAT-SRR to the six fusion constructs and compared the predictions to those made by GEMSTAT-GL. Experimentally determined expression profiles in blue, GEMSTAT-SRR predictions in red and GEMSTAT-GL predictions in gray dashes.
These observations imply that the SRR model, at least as implemented in GEMSTAT, is not mechanistically rich enough to explain the expression patterns driven by these fusion constructs. We therefore favor the GEMSTAT-GL model’s findings as the explanation to the readouts of our fusion constructs: linear aggregation of individual enhancer output gives rise to the observed expression patterns.
Discussion
We have identified a gap in our current assumptions about enhancer readout by applying computational analysis to quantitative measurements of output from a set of fused enhancers. In current models of enhancer readout (enhancer-level model), TFs bound to an enhancer are assumed to act as a ‘bag of sites’, exerting their effects independently. The effects of spacing between sites and their relative arrangement are also sometimes included, such that the TFs act somewhat coordinately within an enhancer. Although two decades of quantitative studies have championed this model, we found it failed for a set of fused enhancers. To explain the readouts of these fusions, we instead needed to assume that an enhancer might comprise multiple segments where information from bound TFs is interpreted independently within each segment. This is analogous to the two-tier model we proposed in previous work (Samee and Sinha, 2014), where the output of entire loci is comprised of a weighted sum of output from its component enhancers. Here we show that this same two-tier mechanism captures the output of enhancer-length sequences as well (Figure 6).
Figure 6. A “two-tiered” mechanism may define and integrate sub-modules of regulatory sequence at the level of single enhancers and entire loci.

(Top) Enhancer sequences contain binding sites for different TFs that function by activating or repressing their target gene. Enhancer-level models capture each TF input independently, representing the enhancer as one “bag of sites”. Two-tier models, such as GEMSTAT-GL, can also be applied to enhancer-length sequences by first separating TF inputs into multiple regulatory segments and then integrating their weighted output to predict expression. (Bottom) Two-tier and enhancer-level models can be applied to an entire locus. Enhancer-level models consider TF binding across the locus as a large “bag of sites”, without considering individual enhancers as separate regulatory entities. We can also apply the two-tiered model to a gene locus. This approach first subdivides the regulatory sequence around a gene into smaller modules and then integrates the regulatory information from each module to predict expression.
It is important to consolidate this idea with previous reports on computational modeling of enhancer sequences where short-range effects were assumed for repressors (Kim et al., 2013; Small et al., 1993). We must first clarify where the short-range repression model stands in relation to the enhancer-level and the two-tier models we use here. It has been proposed that short-range repression could effectively partition bound TFs into multiple enhancers (Gray and Levine, 1996; Gray et al., 1994; Kim et al., 2013; Small et al., 1993); thus it could be considered in the middle of a hierarchy of models between our enhancer-level and two-tier models. We showed that our enhancer-level model fails to explain the fusions, but that our two-tier model is successful. Therefore it is a relevant question whether short-range repression can account for the effective partitioning of the fusions into multiple segments, each of which adheres to enhancer-level rules. A related study found that short-range repression was sufficient to explain the output of part of a locus, implying that it may serve as a mechanism to partition enhancers from longer sequences (Kim et al., 2013). However, we found here that short-range repression was inadequate to explain the partitioning of some of the fusion constructs in our dataset.
For practical considerations, it is also important to discuss when one class of modeling assumptions, e.g., “bag of sites” in GEMSTAT, the “two-tier” approach of GEMSTAT-GL, or the short-range repression assumption of GEMSTAT-SRR, should be considered more suitable than the other two classes. As with any other type of statistical modeling, the choice depends on the type of questions being pursued; and the answers should be analyzed with a clear understanding of the limitations of the chosen model. We note that, GEMSTAT-GL is more flexible than GEMSTAT and GEMSTAT-SRR in terms of selecting/redefining active modules, and was successful in modeling locus-level gene expression when both GEMSTAT and SRR had failed. However, GEMSTAT-GL cannot identify any mechanistic event that could have demarcated the modules it selected. The SRR model is more mechanistically grounded than both GEMSTAT and GEMSTAT-GL, but implements only one possible mechanism of distance-dependent repressor function. We speculate that it may be due to this limited view of repressor function that SRR underperformed compared GEMSTAT in our previous study on ~40 developmental enhancers (He et al., 2010). In our experience, GEMSTAT is often good as a first approximation. The same has been shown in many publications using different realizations of the bag of sites model (Bussemaker et al., 2001; Ilsley et al., 2013; Kazemian et al., 2010; Segal et al., 2008; Wunderlich et al., 2012). However, as we obtain more data on enhancer activity and improve our understanding of TF-TF and TF-DNA interactions, we believe mechanistically grounded models like SRR will eventually improve and may become more appropriate than the two other models for most data sets.
Our study suggests mechanisms other than short-range repression delineate active modules within enhancer-length sequences. What mechanisms might delineate such segments? We do not know of any direct interaction between the relevant TFs that can explain such delineations, despite nearly two decades of study of these proteins. DNA shape itself may impose some modularity on enhancer sequences at short length scales (Peng and Sinha, 2016). Another possibility is that insulator and architectural proteins play a role. Weak binding sites for such proteins are prevalent throughout the eve enhancers; our fusion constructs do not create any new sites for Trl/GAF, su(Hw), CTCF, Cp190, or BEAF-32 at the junctions (Figure S2). Finally, indirect interaction between TFs could occur through binding to large co-factor complexes, such as mediator or CBP (Merika et al., 1998) or through chromatin state (Voss et al., 2011).
Our results are relevant for interpreting the increasing volume of high-throughput functional genomic data annotating active regulatory DNA in the genome; in particular it will be important to assess whether contiguous sequences act as a single enhancer, or as multiple functional sub-modules, like our fusions. In general, we emphasize that future computational modeling of enhancer readouts should not overlook the examples where the models perform poorly, and rather should systematically consider the existence of unknown mechanisms of enhancer function, such as the two-tiered effects we reveal here.
Experimental methods
Transgenic fly lines
We used RedFly to identify coordinates of the D. mel even-skipped stripe 3/7 and stripe 4/6 enhancers (Gallo et al., 2011). The eve_stripe_3+7 element is 510bp (Release 5 coordinates 2R: 5863006–5863516) (Small et al., 1996), while the eve_stripe4_6 element is 800bp (Release 5 coordinates 2R:5871404–5872203) (Fujioka et al., 1999). Note that the stripe 4/6 enhancer coordinates from REDfly contain an extra 208bp on the 3’ end compared to the construct tested in Fujioka et al., 1999 (Fujioka et al., 1999). Enhancers were PCR amplified from genomic DNA from w118 Drosophila melanogaster flies, sequence verified and inserted into the multiple cloning site of the pBOY vector (Hare et al., 2008) using isothermal assembly (Gibson et al., 2009), which leaves scar-less junctions. A list of all enhancers and spacer sequences are given in Text S1. pBOY contains an eve core promoter 20bp downstream of the multiple cloning site that drives an eve/ lacZ fusion transcript. The vector also contains an attB site for phiC31 site specific integration (Fish et al., 2007) and the mini-white gene for selection of transformants. Each plasmid was injected into attP2 flies (Markstein et al., 2008) by BestGene and homozygosed using the mini-white eye color marker.
Embryo collection and in situ hybridization
Embryo collection and whole mount in situ hybridization was performed as previously described (Luengo Hendriks et al., 2006). Briefly, 0–4hr embryos (25C) were collected, dechorionated in 50% bleach, fixed in a 1:4 mixture of 10% formaldehyde to heptane, and devitellinized in heptane and methanol by shaking. Embryos were post-fixed in formaldehyde and a formaldehyde based hybridization buffer. Hybridizations were performed at 56°C with three full length cDNA probe s: a DIG-labeled probe for fushi tarazu (ftz), a DNP-labeled lacZ or eve probe and a DNP-labeled probe against huckebein (hkb). The probes were detected by successive antibody staining using anti-DIG-HRP (anti-DIG-POD; Roche, Basel, Switzerland) and anti-DNP-HRP (Perkin-Elmer TSA-kit, Waltham, MA, USA), and labeled by reactions with coumarin- and Cy3-tyramide (Perkin-Elmer). Embryos were treated with RNase and incubated with Sytox Green (Invitrogen, Carlsbad, CA, USA) to stain nuclei. Finally, embryos were dehydrated in ethanol and mounted in DePex (Electron Microscopy Sciences, Hatfield, PA, USA), using #1 coverslips to form a bridge to preserve 3D embryo morphology.
Imaging and image processing
Embryos were imaged and computationally segmented for further analysis (Fowlkes et al., 2008). A three-dimensional image stack of each embryo was acquired on a Zeiss LSM Z10 with a plan-apochromat 20×0.8 NA objective using 2-photon microscopy. Embryos were binned into six time points of approximately 10 minute windows using the extent of membrane invagination under phase-microscopy as a morphological marker. Time points correspond to 0–3%, 4–8%, 9–25%, 26–50%, 51–75% and 76–100% membrane invagination along the side of the embryo that has progressed most. We imaged embryos from all age ranges and, except where noted, display data from the early blastoderm (4% – 31% membrane invagination) when hkb normalization is used. Image files were processed into PointCloud representations containing the coordinates and fluorescence levels for each nucleus.
hkb normalization
Normalization to a hkb co-stain was performed to test the variation in absolute levels of expression across reporter lines (Wunderlich et al., 2014). Embryos were stained with a mixture of lacZ- or eve-DNP and hkb-DNP probe. For each embryo, background was calculated as the mode of the fluorescence distribution. After subtracting background, mean hkb fluorescence was calculated as the geometric mean of the anterior and posterior expression domains. We noted that eve stripe 7 overlaps slightly with the posterior expression domain of hkb, and so chose to use the geometric mean of anterior and posterior rather than solely the posterior domain as in (Wunderlich et al., 2014) to limit the impact of overlapping expression. The fluorescence in each nucleus was then divided by the mean hkb fluorescence to yield a normalized expression level.
Input data for models
Our sequence dataset, expression readouts, TF motifs, and TF concentrations are described in Text S1. Of note, since eve stripes 3 to 7 span 40–95% of the anterior-posterior (A/P) axis of the Drosophila embryo, here we have considered sequence readouts and TF concentrations only within this spatial range. Below we outline the previously published GEMSTAT and GEMSTAT-GL models (He et al., 2010; Samee and Sinha, 2014), and detail our parameter estimation strategy. We optimized all models utilizing weighted pattern generating potential (w-PGP) as the objective function. w-PGP was introduced in (Samee and Sinha, 2014), we give an overview of w-PGP in Text S1.
GEMSTAT and GEMSTAT-GL
In GEMSTAT, three major components interact to regulate gene expression during transcription: (a) DNA sequence, (b) TF molecules, and (c) the basal transcriptional machinery (BTM). A TF molecule may bind the sequence at any binding site, with site-specific affinity computed from the TF’s PWM (Text S1). The BTM may initiate transcription when bound at the core promoter of the gene. Interactions between bound TF molecules and the BTM determine the occupancy, i.e., probability of binding, of the BTM at the promoter. The level of gene expression is assumed proportional to BTM occupancy. The model fits two free parameters for each TF. The first parameter is a product of two unknowns: the TF’s strength of DNA binding and a proportionality constant that scales the relative concentration of the TF to an absolute value. Note that our TF concentration data is derived from fluorescence intensity and thus is a relative measurement. The second parameter represents a bound TF’s potency for activating or repressing BTM binding. The model can also include cooperative TF binding which requires one free parameter for each pair of cooperatively binding TFs. In the default configuration of GEMSTAT, repressors exert their effects by acting over long-range to directly reduce BTM occupancy at the promoter. In an alternate configuration, repressors act only over short ranges by interfering with activator occupancy at neighboring sites. We call the latter model GEMSTAT-SRR (short-range repression).
GEMSTAT-GL (GEMSTAT for Gene Locus) models a gene’s expression as a weighted summation of expression driven by several enhancers within its locus, where each enhancer’s output is predicted by GEMSTAT. From the intergenic locus of a gene, GEMSTAT-GL automatically selects a handful of segments that together generate the gene’s expression. The number, lengths, and locations of contributing segments, as well as the weight of each segment’s contribution are free parameters in the model. A constrained parameter estimation strategy, as described below, is adopted to avoid over-fitting.
Parameter estimation
In estimating our model parameters, we followed either a “constrained” or an “unconstrained” strategy. In the constrained strategy (adopted while modeling the eve enhancers and the fusion constructs collectively, and for fitting the GEMSTAT-GL model; see below) we first construct 1000 models for a dataset comprising ~30 A/P enhancers whose readouts fall within 40–95% of the A/P axis. Of note, we had excluded all eve enhancers from this dataset. For this dataset, we first randomly sampled ~1 million points (each denoting a different parameterization of the model) from the parameter space (Samee et al., 2015). Then we considered each of the top 1000 models from the sampled collection, one at a time, as the initial parameterization of GEMSTAT and re-estimated parameters to optimize the model for the ~30 enhancers. The optimized parameters of these models then become the initial parameters for optimizing GEMSTAT or GEMSTAT-GL on eve enhancers and/or the fusion constructs. For GEMSTAT we then construct 1000 models starting from each of the 1000 models optimized on the ~30 enhancers; since optimization of GEMSTAT-GL is time-consuming, we optimize 10 GEMSTAT-GL models starting from the top 10 among the 1000 models. In course of optimization, we constrain the value of each parameter to vary by at most two-fold from its initial value. In GEMSTAT-GL we additionally constrain each window’s weight to vary by at most two-fold from the weights of the other windows. As we had discussed in (Samee and Sinha, 2014) this strategy can ensure that the final model trained on the enhancers of a single gene is largely consistent with a model that reflects other regulatory parts of the genome.
In the unconstrained strategy, we do not use the ~30 enhancers mentioned above: we randomly sample ~1 million points directly for the enhancer being modeled. Then, starting from the top 1000 models from the sampled collection, one at a time, we optimize 1000 models for the enhancer. We do not impose any constraints during this optimization.
Supplementary Material
Acknowledgments
NIH 1 R01 GM114341 A (SS). The DePace lab acknowledges generous funding from the NIH (U01GM103804, K99HD73191, R21HD072481, R01GM122928), the NSF (CAREER-iOS 1452557), the Giovanni Armenise Foundation, the Jane Coffin Childs Foundation, the Novartis Fellows Fund and the McKenzie Family Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, TLM and AHD; Methodology, MAHS, TLM, KMB, AHD, and SS; Software, MAHS; Validation, KMB; Formal Analysis, MAHS, TLM, KMB, AHD, SS, BJV; Investigation, MAHS, TLM, KMB, MDB, KBE, ZW, JE, AHD, and SS; Writing - Original Draft, MAHS, AHD, and SS; Writing - Review & Editing, MAHS, KMB, AHD, SS; Visualization, MAHS, KMB, AHD, and SS; Supervision, AHD and SS; Funding Acquisition, AHD, ZW, and SS.
References
- Ay A, Arnosti DN. Mathematical modeling of gene expression: a guide for the perplexed biologist. Crit. Rev. Biochem. Mol. Biol. 2011;46:137–151. doi: 10.3109/10409238.2011.556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- Bothma JP, Garcia HG, Ng S, Perry MW, Gregor T, Levine M. Enhancer additivity and non-additivity are determined by enhancer strength in the Drosophila embryo. Elife. 2015;4 doi: 10.7554/eLife.07956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- Clyde DE, Corado MSG, Wu X, Paré A, Papatsenko D, Small S. A self-organizing system of repressor gradients establishes segmental complexity in Drosophila. Nature. 2003;426:849–853. doi: 10.1038/nature02189. [DOI] [PubMed] [Google Scholar]
- Dynan WS. Modularity in promoters and enhancers. Cell. 1989;58:1–4. doi: 10.1016/0092-8674(89)90393-0. [DOI] [PubMed] [Google Scholar]
- Fakhouri WD, Ay A, Sayal R, Dresch J, Dayringer E, Arnosti DN. Deciphering a transcriptional regulatory code: modeling short-range repression in the Drosophila embryo. Mol. Syst. Biol. 2010;6:341. doi: 10.1038/msb.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish MP, Groth AC, Calos MP, Nusse R. Creating transgenic Drosophila by microinjecting the site-specific phiC31 integrase mRNA and a transgene-containing donor plasmid. Nat. Protoc. 2007;2:2325–2331. doi: 10.1038/nprot.2007.328. [DOI] [PubMed] [Google Scholar]
- Fowlkes CC, Hendriks CLL, Keränen SVE, Weber GH, Rübel O, Huang M-Y, Chatoor S, DePace AH, Simirenko L, Henriquez C, et al. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm. Cell. 2008;133:364–374. doi: 10.1016/j.cell.2008.01.053. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Emi-Sarker Y, Yusibova GL, Goto T, Jaynes JB. Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multistripe positioning by gap gene repressor gradients. Development. 1999;126:2527–2538. doi: 10.1242/dev.126.11.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo SM, Gerrard DT, Miner D, Simich M, Des Soye B, Bergman CM, Halfon MS. REDfly v3.0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 2011;39:D118–D123. doi: 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gray S, Levine M. Short-range transcriptional repressors mediate both quenching and direct repression within complex loci in Drosophila. Genes Dev. 1996;10:700–710. doi: 10.1101/gad.10.6.700. [DOI] [PubMed] [Google Scholar]
- Gray S, Szymanski P, Levine M. Short-range repression permits multiple enhancers to function autonomously within a complex promoter. Genes Dev. 1994;8:1829–1838. doi: 10.1101/gad.8.15.1829. [DOI] [PubMed] [Google Scholar]
- Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008;4:e1000106. doi: 10.1371/journal.pgen.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Samee MAH, Blatti C, Sinha S. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLoS Comput. Biol. 2010;6 doi: 10.1371/journal.pcbi.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilsley GR, Fisher J, Apweiler R, De Pace AH, Luscombe NM. Cellular resolution models for even skipped regulation in the entire Drosophila embryo. Elife. 2013;2:e00522. doi: 10.7554/eLife.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Blatti C, Richards A, McCutchan M, Wakabayashi-Ito N, Hammonds AS, Celniker SE, Kumar S, Wolfe SA, Brodsky MH, et al. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G, Gruss P. Enhancer elements. Cell. 1983;33:313–314. doi: 10.1016/0092-8674(83)90410-5. [DOI] [PubMed] [Google Scholar]
- Kim A-R, Martinez C, Ionides J, Ramos AF, Ludwig MZ, Ogawa N, Sharp DH, Reinitz J. Rearrangements of 2.5 Kilobases of Noncoding DNA from the Drosophila even-skipped Locus Define Predictive Rules of Genomic cis-Regulatory Logic. PLoS Genet. 2013;9:e1003243. doi: 10.1371/journal.pgen.1003243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni MM, Arnosti DN. cis-regulatory logic of short-range transcriptional repression in Drosophila melanogaster. Mol. Cell. Biol. 2005;25:3411–3420. doi: 10.1128/MCB.25.9.3411-3420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo Hendriks CL, Keränen SVE, Fowlkes CC, Simirenko L, Weber GH, DePace AH, Henriquez C, Kaszuba DW, Hamann B, Eisen MB, et al. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution I: data acquisition pipeline. Genome Biol. 2006;7:R123. doi: 10.1186/gb-2006-7-12-r123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 2008;40:476–483. doi: 10.1038/ng.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFNβ Enhanceosome Is Required for Synergistic Activation of Transcription. Mol. Cell. 1998;1:277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- Peng P-C, Sinha S. Quantitative modeling of gene expression using DNA shape features of binding sites. Nucleic Acids Res. 2016;44:e120. doi: 10.1093/nar/gkw446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MW, Boettiger AN, Bothma JP, Levine M. Shadow enhancers foster robustness of Drosophila gastrulation. Curr. Biol. 2010;20:1562–1567. doi: 10.1016/j.cub.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samee MAH, Sinha S. Quantitative modeling of a gene’s expression from its intergenic sequence. PLoS Comput. Biol. 2014;10:e1003467. doi: 10.1371/journal.pcbi.1003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samee MAH, Lim B, Samper N, Lu H, Rushlow CA, Jiménez G, Shvartsman SY, Sinha S. A Systematic Ensemble Approach to Thermodynamic Modeling of Gene Expression from Sequence Data. Cell Syst. 2015;1:396–407. doi: 10.1016/j.cels.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Small S, Arnosti DN, Levine M. Spacing ensures autonomous expression of different stripe enhancers in the even-skipped promoter. Development. 1993;119:762–772. [PubMed] [Google Scholar]
- Small S, Blair A, Levine M. Regulation of Two Pair-Rule Stripes by a Single Enhancer in the Drosophila Embryo. Dev. Biol. 1996;175:314–324. doi: 10.1006/dbio.1996.0117. [DOI] [PubMed] [Google Scholar]
- Staller MV, Vincent BJ, Bragdon MDJ, Lydiard-Martin T, Wunderlich Z, Estrada J, DePace AH. Shadow enhancers enable Hunchback bifunctionality in the Drosophila embryo. Proc. Natl. Acad. Sci. U. S. A. 2015;112:785–790. doi: 10.1073/pnas.1413877112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struffi P, Corado M, Kaplan L, Yu D, Rushlow C, Small S. Combinatorial activation and concentration-dependent repression of the Drosophila even skipped stripe 3+7 enhancer. Development. 2011;138:4291–4299. doi: 10.1242/dev.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung M-H, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell. 2011;146:544–554. doi: 10.1016/j.cell.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich Z, Bragdon MD, Eckenrode KB, Lydiard-Martin T, Pearl-Waserman S, DePace AH. Dissecting sources of quantitative gene expression pattern divergence between Drosophila species. Mol. Syst. Biol. 2012;8:604. doi: 10.1038/msb.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich Z, Bragdon MD, DePace AH. Comparing mRNA levels using in situ hybridization of a target gene and co-stain. Methods. 2014;68:233–241. doi: 10.1016/j.ymeth.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen RP, Senger K, Levine M, Papatsenko D. Computational models for neurogenic gene expression in the Drosophila embryo. Curr. Biol. 2006;16:1358–1365. doi: 10.1016/j.cub.2006.05.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.