

Abstract
The interplay between platelets and tumor cells is known to play important roles in metastasis by enhancing tumor cell survival, tumor-vascular interactions, and escape from immune surveillance. However, platelet-covered circulating tumor cells (CTC) are extremely difficult to isolate due to masking or downregulation of surface epitopes. Here we describe a microfluidic platform that takes advantage of the satellite platelets on the surface of these “stealth” CTCs as a ubiquitous surface marker for isolation. Compared to conventional CTC enrichment techniques which rely on known surface markers expressed by tumor cells, platelet-targeted isolation is generally applicable to CTCs of both epithelial and mesenchymal phenotypes. Our approach first depletes unbound, free platelets by means of hydrodynamic size-based sorting, followed by immunoaffinity-based capture of platelet-covered CTCs using a herringbone micromixing device. This method enabled the reliable isolation of CTCs from 66% of lung and 60% of breast cancer (both epithelial) patient samples, as well as in 83% of melanoma (mesenchymal) samples. Interestingly, we observed special populations of CTCs that were extensively covered by platelets, as well as CTC-leukocyte clusters. Because these cloaked CTCs often escape conventional positive and negative isolation mechanisms, further characterization of these cells may uncover important yet overlooked biological information in blood-borne metastasis and cancer immunology.
Introduction
A large body of evidence has accumulated suggesting the involvement of the coagulation system in the hematogenous dissemination of cancer. As early as 1865, the association between venous thromboembolism and cancer has been reported which is now referred to as the Trousseau’s Syndrome1. In 1878, Billroth found histological evidence of tumor cells within a thrombus, suggesting that thrombosis may play a mechanistic role in the spread of distant metastases2. It is now recognized that both high platelet count and expression of coagulation factors by cancer cells are negative prognostic markers in many different types of cancers3–5. In vitro and animal experiments have shed light on how platelets are involved in various steps of the metastatic cascade. Platelets contain a variety of angiogenic and vascular remodeling factors in granules that are released upon activation, regulating the tumor vasculature through which circulating tumor cells (CTCs) disseminate5,6. The physical interaction between platelets and tumor cells, frequently enhanced by tumor-derived tissue factor and thrombin7,8, confers a number of survival advantages in the circulation. For instance, platelets (together with fibrin) can protect tumor cells from natural killer cell-mediated lysis by physical shielding9,10, or by conferring a “pseudonormal” phenotype via the transfer of platelet-derived MHC class I to the tumor cell surface11. Platelets may also aid in the establishment of metastases by promoting tumor cell adhesion under shear flow12 and guide the formation of early metastatic niches13. The central role that platelets play in these processes is evident in several independent animal studies that demonstrated inhibition of metastasis by platelet depletion and the restoration of metastatic potential by platelet reconstitution14–16.
Over the past two decades, numerous technologies have emerged to isolate rare CTCs from blood of cancer patients for a range of scientific and clinical applications (for a comprehensive review, refer to reference 17). To this end, our group pioneered the development of microfluidic technologies that isolate CTCs by immunoaffinity-based positive enrichment18,19 or “negative” selection (i.e., by depleting hematologic cells)20, or by taking advantage of the size and asymmetry of CTC clusters21,22. However, the isolation of platelet-covered CTCs is extremely difficult and current technologies have not been designed to detect this particular CTC population. This difficulty derives from the inherent biological and physical interactions between platelets and CTCs. For instance, platelet-derived growth factors and the direct contact between platelets and tumor cells may induce epithelial-mesenchymal transition (EMT)23, which downregulates the surface expression of epithelial cell adhesion molecule (EpCAM)—an extensively used marker for the isolation and identification of CTCs of epithelial origin17. In addition, the platelet cloak on the CTCs may shield them from being captured by antibodies. Further, because CTCs are often clustered with leukocytes19,21 and this interaction may be enhanced by activated platelets and clotting proteins24–26, “negative” enrichment by leukocyte depletion20 likely removes a significant fraction of CTCs.
Here we address this gap by treating platelets as a ubiquitous cell-surface marker for the isolation of platelet-cloaked CTCs from patient blood. We target this CTC population using a positive enrichment approach that captures platelet-covered cells. Due to the massive number of free, unbound platelets that are present in whole blood on the scale of 108 per mL, their efficient depletion prior to cell isolation is critical to prevent the saturation of capture antibodies. We implement a two-step microfluidic strategy that first depletes free platelets by size20 using principles of deterministic lateral displacement (DLD)27, followed by the isolation of platelet-covered CTCs and clusters using the herringbone CTC chip (HB-Chip)19 which induces microvortices28 to enhance cell-capture surface interactions. Using blood from patients with lung cancer, we compare this approach to a conventional one that uses EpCAM-based capture, and formally show the presence of a CTC population that is coated with platelets in clinical samples. Further, we demonstrate a critical advantage of this approach in isolating CTCs from both epithelial (lung & breast) and mesenchymal (melanoma) cancer types without the a priori knowledge about their surface tumor marker expression. This work enables the isolation of a CTC subpopulation that is correlated with high metastatic potential but yet overlooked by existing CTC isolation technologies.
Results and Discussion
Microfluidic CTC isolation from patients with metastatic lung cancer
We employed a two-stage microfluidic approach to isolate platelet-covered CTCs based on immunoaffinity to platelets (Fig. 1). Because platelets are abundant in whole blood (~108/mL), we first processed whole blood through a microfluidic debulking device which consists of arrays of microposts to remove free platelets—together with red blood cells (RBCs)—by hydrodynamic size-based sorting (DLD)20. This process achieves a >2-log depletion of free platelets while retaining nucleated cells. The product containing CTCs and leukocytes was then processed through a microfluidic herringbone mixing device functionalized with antibodies to human platelets (CD41 HB-Chip) or EpCAM (EpCAM HB-Chip) for the isolation of CTCs.
Figure 1.

Schematic of microfluidic blood processing. Whole blood is first enriched for nucleated cells based on size using a DLD micropost array. The product is then processed using the herringbone micromixing chip (HB-Chip) functionalized with CD41 or EpCAM antibodies to achieve affinity-based CTC capture.
We applied this platform to process 32 blood specimens from 24 independent metastatic (stage IV) lung cancer patients. All captured cells on the HB-Chips were immunostained to score CTCs, which are identified by the positive staining of tumor markers EpCAM and cadherin-11 while negative for the leukocyte marker CD45. A platelet marker (CD61) was also included. Using the CD41 HB-Chip, CTCs were found in 21 out of 32 cases (66%) with counts ranging from 0.4 to 8.5 CTCs/mL of blood (mean 1.68 ± 2.04 CTCs/mL; Fig. 2A). In comparison, the EpCAM HB-Chip resulted in lower capture rates in terms of both positivity of CTCs (29% of patients) as well as CTC count (range 0.4–1.2 CTCs/mL; mean 0.17 ± 0.31 CTCs/mL; p = 0.0013; Fig. 2A). In control blood specimens from healthy individuals, we detected only one false-positive event in our 5 control samples using the CD41 HB-Chip (Fig. 2A), demonstrating the specificity of this approach. A larger cohort of healthy donor control samples will be required to characterize the rate of false-positive detection for clinical diagnostic applications.
Figure 2.

Lung CTC isolation using platelet- and EpCAM-targeted capture. (A) CTC counts in blood samples obtained from healthy donor (HD) and metastatic lung cancer patients (Lung pt) that were captured using CD41 or EpCAM antibodies. (B) Fluorescent images of CTCs captured using CD41 (top) and EpCAM antibodies (bottom). The cells were immunostained for EpCAM/cadherin-11 (CTC marker; Alexa Fluor 488; green), CD45 (leukocyte marker; Alexa Fluor 647; red) and CD61 (platelet marker; Alexa Fluor 568; gold) respectively. Scale bar, 10 μm.
CD61 staining on captured CTCs confirmed the presence of platelets specific to the CD41 HB-Chip, whereas the traditional EpCAM-targeted approach reveals little to no platelet coverage (Fig. 2B). The degree of platelet coverage around CTCs exhibited a wide spectrum including cells that are completely soaked with platelets (Fig. 2B & Fig. 3B). This finding may partially explain the lower capture rates of CTCs using the EpCAM HB-Chip, as the platelet cloak may have masked the EpCAM on cell surface and thereby reducing capture efficiency. We also note that there is a population of platelet-cloaked cells on the CD41 HB-Chip that remain unidentified with the current staining protocol. We attempted to include cytokeratin staining as an additional tumor marker; however, the required fixation and permeabilization procedures were incompatible with our live-cell membrane staining protocol. Further optimization of immunostaining and spectral separation of epithelial and mesenchymal markers will be critical in characterizing the heterogeneous CTCs along the EMT spectrum.
Figure 3.

(A) CTC count from breast cancer and melanoma patient blood using platelet-targeted capture. (B) Representative fluorescent images of CTCs from lung and breast cancer and melanoma patient samples. Lung and breast CTCs were immunostained for EpCAM/Cadherin-11 (Alexa Fluor 488; green). Melanoma CTCs were immunostained for CSPG4, MCAM, TYRP1, and α-SMA (Alexa Fluor 488; green). Leukocytes and platelets were stained with antibodies to CD45 (Alexa Fluor 647; red) and CD61 (Alexa Fluor 568; gold) respectively. Note the extensive platelet coating on both CTCs and leukocytes. Scale bar, 10 μm.
CTC isolation from patients with breast cancer and melanoma
We extended this microfluidic platform to isolate CTCs from other cancer types that are of epithelial (breast, n = 5) and non-epithelial (melanoma, n = 6) origins. Breast CTCs were identified using the same protocol as lung patient samples, while melanoma CTCs were stained with an antibody cocktail that targets melanoma specific antigens (CSPG4, MCAM, TYRP1, and α-SMA) as previously described29. In a small cohort of patients, we have demonstrated reliable CTC capture (Fig. 3A) in breast cancer (3 of 5 patients; range 1.0–2.7 CTCs/mL; mean 1.1 ± 1.2 CTCs/mL) and melanoma (5 of 6 patients; range 1.4–17.3 CTCs/mL; mean 4.9 ± 6.5 CTCs/mL). Similar to the CTCs from lung cancer patients, all CTCs captured with this approach are associated with different degrees of platelet coverage (Fig. 3B). Furthermore, across all three cancer types analyzed in this study, we did not observe any association between the type of cancer treatment (chemotherapy, targeted therapy, or immunotherapy) and the number of platelet-coated CTCs. Together, these results suggest that platelet-CTC interaction is a general phenomenon, and demonstrate the applicability of platelet-targeted approach in isolating CTCs from both epithelial and mesenchymal origins without requiring knowledge of their surface epitopes.
CTC-leukocyte clusters
In addition to single CTCs, we have also identified cell clusters comprising CTCs and leukocytes using platelet-targeted isolation. Indeed, over 40% of captured CTCs were in the form of CTC-leukocyte clusters (Fig. 4A), and the number of associated leukocytes appear to correlate with the extent of platelet coverage around the CTCs (Fig. 4B–D). These findings provide support that platelets mediate CTC-leukocyte interactions, and implicate their potential roles in establishing early metastatic niches13, promoting tumor-vascular interactions12,30,31, and facilitating metastatic growth32,33. Whereas these pro-tumorigenic effects are typically conferred by granulocytic and monocytic cells, the interaction of T cells and natural killer cells with CTCs may also originate from anti-tumor immune responses. The capability of our approach to isolate platelet-cloaked CTCs and CTC-leukocyte clusters—both of which are difficult to detect using conventional affinity-based selection methods—enables the comprehensive characterization of CTCs that are highly relevant in blood-borne metastasis and tumor immunology. Further, given the pro-survival signals conferred by platelets and certain types of leukocytes, some of these CTCs may possess a much higher chance of ex vivo expansion, and thereby overcome a critical bottleneck in establishing patient-derived CTC lines for personalized drug screening without the need of invasive biopsies34.
Figure 4.

CTC-leukocyte clusters. (A) CTC counts in the form of single cells and clusters that contain 1–2 or >2 leukocytes (WBCs). (B–D) Representative fluorescent images of CTC-leukocyte clusters from lung cancer patient samples. Cells were stained with antibodies to EpCAM/cadherin-11 (Alexa Fluor 488; green), CD45 (Alexa Fluor 647; red), and CD61 (gold). Scale bar, 10 μm.
CTC purity with platelet-targeted isolation
A key issue that potentially limits the performance of our approach is the relatively low purity of CTCs compared with other technologies. Because size-based enrichment of nucleated cells followed by platelet-targeted capture does not preferentially isolate CTCs over leukocytes, we observed a large number of contaminating leukocytes on the CD41 HB-Chip (~105/mL), compared to the conventional EpCAM HB-Chip (~104/mL; p = 0.0046; Fig. 5). As leukocytes also interact with platelets in the circulation35, such undesired leukocyte capture is different from non-specific binding events as observed in other isolation strategies and presents significant engineering challenges. To minimize the impact of leukocyte contamination in downstream molecular analyses, selective release of captured CTCs can be achieved by localized degradation of polymeric nanocoating in the microfluidic device36.
Figure 5.
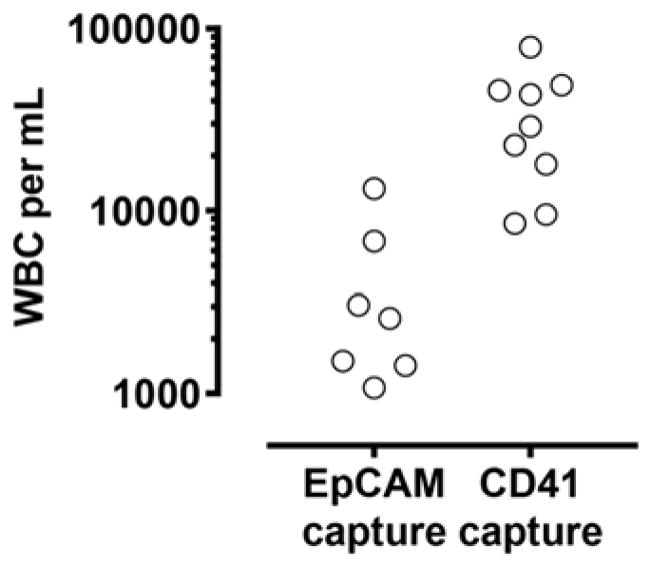
Leukocyte (WBC) contamination in EpCAM- and platelet-targeted CTC capture.
Conclusions
In this work, we present a two-stage microfluidic approach that isolates CTCs by targeting platelets that satellite on the surface of the tumor cells without requiring the a priori knowledge of CTC surface marker expression. We show proof-of-principle that platelet-coated CTCs indeed exist in significant numbers in metastatic cancer patients of both epithelial (lung and breast) and non-epithelial (melanoma) tumor origins. We report robust capture of these cells in the form of both single cells and clusters, including an interesting population that associates extensively with leukocytes. The ability to isolate these “stealth” CTCs opens up new avenues for non-invasive cancer diagnostics as well as investigations in blood-borne metastasis and tumor immunology. More work will be needed to molecularly characterize this special CTC population to understand the interplay between EMT and interactions with platelets and leukocytes, and to determine their clinical significance.
Methods
Patient samples and blood processing
Patients with advanced lung, breast and melanoma cancer (all stage IV) were recruited according to protocol DF/HCC 05-300 approved by the institutional review board of Massachusetts General Hospital. Informed consent was obtained for all donors and all experiments were performed in compliance with the relevant laws and institutional guidelines. All specimens were collected into vacutainer (Becton-Dickinson) tubes containing the anticoagulant EDTA and were processed using the microfluidic chips within 4 hours of blood draw.
Microfluidic device fabrication and operation
The first-stage hydrodynamic sorting (DLD) devices were designed at Massachusetts General Hospital and fabricated by Silex with deep reactive ion etching on silicon wafers as described previously20,37. The DLD array consists of 24 parallel channels that are 150 μm in depth. The gap size between posts and the vertical pitch are both 32 μm, the horizontal pitch is 56 μm, and the row shift fraction is 1/60. The device was sealed with anodically bonded glass cover to form the microfluidic chamber. A custom polycarbonate manifold was used to form the fluidic connections to the microchip. The second-stage HB-Chips were made of PDMS bonded to glass substrates using soft lithography techniques, and functionalized with anti-EpCAM (20 μg/mL, R&D Systems) or anti-CD41 (20 μg/mL, Abcam) using avidin-biotin chemistry as described19.
Whole blood was first processed through the DLD chip with co-flow of buffer (1% w/v Pluronic F-68, Sigma; in PBS). The product contains highly enriched nucleated cells in buffer with complete blood counts (Sysmex KX-21N) that indicate >2 log-depletion of platelets. This product was then processed using the CD41 or EpCAM HB-Chip at 1.14 ± 0.24 mL/hr for CTC isolation.
Immunostaining of captured cells
For lung and breast patient samples, captured cells on the HB-Chips were stained with fluorochrome-conjugated antibodies as follows: anti-EpCAM (3:100, Alexa Fluor 488 conjugate, Cell Signaling Technologies), anti-cadherin-11 (1:10, Alexa Fluor 488 conjugate, R&D Systems), anti-CD45 (1:20, Alexa Fluor 647 conjugate, BD Biosciences), and anti-CD61 (1:100, Abcam) that was conjugated to Alexa Fluor 568 via amine groups (Alexa Fluor 568 Antibody Labeling Kit; Life Technologies). We chose to group together epithelial and mesenchymal markers into the same color in order to maximize the sensitivity of CTC detection while simultaneously including markers for platelets (CD61) and leukocytes (CD45). The cells were then fixed with 2% paraformaldehyde for 1 hour after which nuclei were stained with DAPI. For melanoma patient samples, cells were fixed with 4% paraformaldehyde, permeabilized with 1% NP-40 (Sigma-Aldrich), and immunostained as previously described29. The primary antibodies used were: anti-CSPG4 (1:100, mouse IgG2a, Abcam), anti-MCAM (1:200, mouse IgG2a, BioLegend), anti-TYRP1 (1:200, mouse IgG2a, Abcam), anti-SMA (1:400, mouse IgG2a, Sigma), anti-CD45 (1:400, rat IgG, Santa Cruz Biotechnology), and anti-CD61 (1:100, mouse IgG1, Abcam). Secondary antibodies were Alexa Fluor 488-conjugated goat anti-mouse IgG2a, Alexa Fluor 568-conjugated goat anti-mouse IgG1, and goat anti-Rat Alexa Fluor 647 (all from Invitrogen).
Statistical analyses
Numerical data are expressed as mean ± standard deviation. Pair-wise comparisons used two-tailed t test. Comparisons of grouped data used one-way ANOVA followed by Tukey’s posttest for multiple comparisons. All tests were performed with Prism 7 (GraphPad).
Acknowledgments
We thank Octavio Hurtado for training and assistance in microfabrication, and Elaina Chan and Bailey Grinnell for help with clinical studies. We also thank healthy volunteers and patients who donated blood specimens. This work was supported by funding from the US National Institutes of Health (NIH) P41 BioMEMS Resource Center (EB002503; M.T.), NIH National Institute of Biomedical Imaging and Bioengineering (EB012493; M.T.), and the Howard Hughes Medical Institute (D.A.H.). X.J. was supported by a postdoctoral fellowship from American Cancer Society.
References
- 1.Varki A. Blood. 2007;110:1723–9. doi: 10.1182/blood-2006-10-053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Billroth T. Lectures on surgical pathology and therapeutics: a handbook for students and practitioners. The New Syndenham Society; London: 1878. [Google Scholar]
- 3.Nash GF, Turner LF, Scully MF, Kakkar AK. Lancet Oncol. 2002;3:425–30. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 4.Erpenbeck L, Schön MP. Blood. 2010;115:3427–3436. doi: 10.1182/blood-2009-10-247296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gay LJ, Felding-Habermann B. Nat Rev Cancer. 2011;11:123–34. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabrkhany S, Griffioen AW, Oude Egbrink MGA. Biochim Biophys Acta. 2011;1815:189–96. doi: 10.1016/j.bbcan.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Nierodzik ML, Karpatkin S. Cancer Cell. 2006;10:355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Ruf W, Yokota N, Schaffner F. Thromb Res. 2010;125(Suppl):S36–S38. doi: 10.1016/S0049-3848(10)70010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieswandt B, Hafner M, Echtenacher B, Männel DN. Cancer Res. 1999;59:1295–300. [PubMed] [Google Scholar]
- 10.Palumbo JS, Talmage KE, Massari JV, LaJeunesse CM, Flick MJ, Kombrinck KW, Jirousková M, Degen JL. Blood. 2005;105:178–85. doi: 10.1182/blood-2004-06-2272. [DOI] [PubMed] [Google Scholar]
- 11.Placke T, Örgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, Salih HR. Cancer Res. 2012;72:440–448. doi: 10.1158/0008-5472.CAN-11-1872. [DOI] [PubMed] [Google Scholar]
- 12.Konstantopoulos K, Thomas SN. Annu Rev Biomed Eng. 2009;11:177–202. doi: 10.1146/annurev-bioeng-061008-124949. [DOI] [PubMed] [Google Scholar]
- 13.Labelle M, Begum S, Hynes RO. Proc Natl Acad Sci U S A. 2014;111:E3053–61. doi: 10.1073/pnas.1411082111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasic GJ, Gasic TB, Stewart CC. Proc Natl Acad Sci U S A. 1968;61:46–52. doi: 10.1073/pnas.61.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karpatkin S, Pearlstein E, Ambrogio C, Coller BS. J Clin Invest. 1988;81:1012–9. doi: 10.1172/JCI113411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Blood. 2004;104:397–401. doi: 10.1182/blood-2004-02-0434. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira MM, Ramani VC, Jeffrey SS. Mol Oncol. 2016;10:374–394. doi: 10.1016/j.molonc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–9. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stott SL, Hsu C, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Proc Natl Acad Sci U S A. 2010;107:18392–7. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen P, Morgan B, Trautwein J, Kimura A, Sengupta S, Stott SL, Karabacak NM, Barber TA, Walsh JR, Smith K, Spuhler PS, Sullivan JP, Lee RJ, Ting DT, Luo X, Shaw AT, Bardia A, Sequist LV, Louis DN, Maheswaran S, Kapur R, Haber DA, Toner M. Sci Transl Med. 2013;5:179ra47. doi: 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, Luo X, Bardia A, Wittner BS, Ramaswamy S, Shioda T, Ting DT, Stott SL, Kapur R, Maheswaran S, Haber DA, Toner M. Nat Methods. 2015;12:685–91. doi: 10.1038/nmeth.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Au SH, Edd J, Stoddard AE, Wong KHK, Fachin F, Maheswaran S, Haber DA, Stott SL, Kapur R, Toner M. Sci Rep. 2017;7:2433. doi: 10.1038/s41598-017-01150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labelle M, Begum S, Hynes RO. Cancer Cell. 2011;20:576–90. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber C, Springer TA. J Clin Invest. 1997;100:2085–93. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL, Bugge TH. Blood. 2000;96:3302–3309. [PubMed] [Google Scholar]
- 26.Kitamura T, Qian BZ, Pollard JW. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang LR, Cox EC, Austin RH, Sturm JC. Science. 2004;304:987–90. doi: 10.1126/science.1094567. [DOI] [PubMed] [Google Scholar]
- 28.Stroock AD, Dertinger SKW, Ajdari A, Mezic I, Stone HA, Whitesides GM. Science. 2002;295:647–51. doi: 10.1126/science.1066238. [DOI] [PubMed] [Google Scholar]
- 29.Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, Hoang MP, Brannigan BW, Lawrence DP, Flaherty KT, Sequist LV, McMahon M, Bosenberg MW, Stott SL, Ting DT, Ramaswamy S, Toner M, Fisher DE, Maheswaran S, Haber DA. Cell Rep. 2014;7:645–653. doi: 10.1016/j.celrep.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Int J Cancer. 2009;125:1298–1305. doi: 10.1002/ijc.24409. [DOI] [PubMed] [Google Scholar]
- 31.Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, Ferri LE. Cancer Res. 2012;72:3919–27. doi: 10.1158/0008-5472.CAN-11-2393. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, Yan J, Hua Y, Tiede BJ, Lu X, Haffty BG, Pantel K, Massagué J, Kang Y. Cancer Cell. 2011;20:701–14. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Q, Zhang XHF, Massagué J. Cancer Cell. 2011;20:538–49. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maheswaran S, Haber DA. Cancer Res. 2015;75:2411–5. doi: 10.1158/0008-5472.CAN-15-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Totani L, Evangelista V. Arterioscler Thromb Vasc Biol. 2010;30:2357–2361. doi: 10.1161/ATVBAHA.110.207480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reátegui E, Aceto N, Lim EJ, Sullivan JP, Jensen AE, Zeinali M, Martel JM, Aranyosi AJ, Li W, Castleberry S, Bardia A, Sequist LV, Haber DA, Maheswaran S, Hammond PT, Toner M, Stott SL. Adv Mater. 2015;27:1593–9. doi: 10.1002/adma.201404677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karabacak NM, Spuhler PS, Fachin F, Lim EJ, Pai V, Ozkumur E, Martel JM, Kojic N, Smith K, Chen P, Yang J, Hwang H, Morgan B, Trautwein J, Barber TA, Stott SL, Maheswaran S, Kapur R, Haber DA, Toner M. Nat Protoc. 2014;9:694–710. doi: 10.1038/nprot.2014.044. [DOI] [PMC free article] [PubMed] [Google Scholar]